

Abstract
Osteoprotegerin (OPG) is a key regulator of bone remodeling. Mutations and variations in the OPG gene cause many human diseases that are characterized by not only skeletal abnormalities but also poorly understood hearing loss: Paget’s disease, osteoporosis, and celiac disease. To gain insight into mechanisms of hearing loss in OPG deficiency, we studied OPG knockout (Opg −/−) mice. We show that they develop sensorineural hearing loss, in addition to conductive hearing loss due to abnormal middle-ear bones. OPG deficiency caused demyelination and degeneration of the cochlear nerve in vivo. It also activated ERK, sensitized spiral ganglion cells (SGC) to apoptosis, and inhibited proliferation and survival of cochlear stem cells in vitro, which could be rescued by treatment with exogenous OPG, an ERK inhibitor, or bisphosphonate. Our results demonstrate a novel role for OPG in the regulation of SGC survival, and suggest a mechanism for sensorineural hearing loss in OPG deficiency.
Keywords: osteoprotegerin, spiral ganglion cells, cochlear neurons, sensorineural hearing loss, auditory stem cell
Introduction
Osteoprotegerin (OPG), also known as tumor necrosis factor receptor superfamily member 11b (TNFRSF11B), is a key regulator of bone remodeling. It functions as a soluble, neutralizing antagonist that competes with receptor activator of NF-κB (RANK) on preosteoclasts and osteoclasts for RANK ligand (RANKL) produced by osteoblasts to inhibit osteoclast formation and function (Simonet et al., 1997; Khosla, 2001). Altered expression of OPG has been described in a variety of human diseases that are associated not only with skeletal abnormalities, but also with hearing loss of poorly understood etiology. Loss of function mutations in the OPG gene account for the majority of cases of Juvenile Paget’s disease (Whyte et al, 2003; Daroszewska and Ralston, 2006), an autosomal recessive osteopathy characterized by a generalized increase in bone turnover leading to widespread skeletal deformities in childhood, bone pain and deafness. Genetic variation at the OPG locus is a risk factor for adult-onset Paget’s disease (Daraszewska et al., 2004) and osteoporosis (Richards et al, 2008). Osteoporosis is associated with otosclerosis (Clayton et al. 2004) – a common hearing disorder. Neutralizing autoantibodies against OPG cause the development of high-turnover osteoporosis in celiac disease (Riches et al., 2009), an autoimmune malabsorptive disorder of the small intestine associated with hearing loss (Hizli et al., 2011).
In general, hearing loss can be categorized as conductive, due to impaired conduction of sound to the inner ear, or sensorineural, due to damage of delicate mechanosensory structures in the inner ear, cochlear nerve, or higher order auditory centers. While conductive hearing loss in some OPG-related disorders can be attributed, at least in part, to abnormal function of middle-ear bones that transmit acoustic vibrations to the inner ear (Whyte et al., 2002; Daroszewska and Ralston., 2006), mechanisms of sensorineural hearing loss in these disorders are poorly understood (Bahmad and Merchant, 2007; Karosi et al., 2011).
The OPG null mouse (Opg −/−) provides a unique opportunity to decipher mechanisms of hearing loss due to a variety of OPG-related human diseases (Bucay et al., 1998; Mizuno et al., 1998). Opg −/− mice are known to have progressive hearing loss due to resorption of ossicles in the middle ear (Kanzaki et al., 2009, Zehnder et al., 2005; Zehnder et al., 2006); a recent study suggested an additional sensorineural component (Qin et al., 2010). Here we elucidate mechanisms of the sensorineural hearing loss due to OPG deficiency by complementing functional tests of hearing with detailed histopathologic analyses, and pharmacologic studies in cultured spiral ganglion cells (SGC) and stem cells. We show that OPG plays a role in function and maintenance of the auditory nerve.
Materials and Methods
Animals
Homozygous Opg −/− mice in the C57BL/6 background were generated by targeted gene disruption of exon 2 in the Opg locus (Mizuno et al., 1998), and were obtained from CLEA-Japan, Inc. Since the littermates of Opg −/− mice were not available, age-matched wild type (WT) C57BL/6 mice were obtained from Jackson laboratory (Bar Harbor, ME). The C57BL/6 mice are of the same genetic background, the colonies were maintained by non-sibling mating, and our re-examination of the inner ear tissue generated 8 years ago from the same colonies and by different investigators (Zehnder et al., 2005, 2006) revealed the same features that we have discovered during the course of the current study. All animal procedures were approved by the Animal Care and Use Committee of the Massachusetts Eye and Ear Infirmary.
Reagents and antibodies
Antibodies (anti-ERK, anti-p-ERK, anti-JNK, anti-p-JNK, anti-p38, anti-p-p38, anti-β actin and anti-cleaved caspase 3) were obtained from Cell Signaling. Anti-BrdU antibody was obtained from Sigma, anti-TuJ antibody was from Covance, anti-S100 antibody was from Dako, and anti- MBP was from Novus Biologicals. OPG and RANKL were from R & D Systems. PD 98059 was from Sigma-Aldrich. Zoledronate was from Novartis.
Plastic embedding for histopathological examination
Animals were intracardially perfused with 2.5 % glutaraldehyde and 1.5% paraformaldehyde in 0.1M phosphate buffer (PB). Animals of the following ages were studied: 3 weeks (wk), 6 wk, 8 wk and 10 wk. A total of 4–13 ears from 4–10 animals per age were examined. Cochleae were extracted, the round and oval window membranes were pierced and flushed with fixative to ensure perfusion of the entire cochlea, and the cochleae were post-fixed overnight. Cochleae were incubated in 1% osmium tetroxide for 60 min, rinsed with ddH2O before decalcification in 0.12M EDTA in 0.1M PB with 1% glutaraldehyde (pH 7) for 3–4 d on a shaker at room temperature. The samples were dehydrated with 70, 95, and 100% ethanol and incubated with propylene oxide (PO) for 30 min. Cochleae were embedded in araldite-PO (1:1) for 1 h followed by araldite-PO (2:1) overnight, degassed in vacuum for 2 h, and placed in a 60°C oven for at least 2 d. Cochleae were cut into 20 μm midmodiolar sections and examined under a light microscope using Normarski DIC optics.
Paraffin embedding for in situ hybridization and immunohistochemistry
Animals were intracardially perfused with 4% paraformaldehyde in 0.1M PB. Animals of the following ages were studied: 6, 10, and 16 wk. A total of 6 ears from 3 animals per age were examined. In situ hybridization for Opg was performed on 10 μm thick paraffin-embedded cochlear sections as previously described using the anti-sense probe from nucleotide 133 to 668 of Opg cDNA (Brookler et al., 1997, Stankovic et al., 2010). Briefly, the digoxigenin (DIG)-labeled single stranded antisense RNA probes were prepared using T7 RNA polymerase with the presence of DIG-dUTP (digoxigenin DNA labeling mixture; Roche, Basel, Switzerland) according to the manufacturer’s protocol. The DIG-labeled single stranded sense RNA probes were prepared using T3 RNA polymerase and digoxigenin DNA labeling mixture. Sections were deparaffinized with xylene, washed in 100% ethanol, rehydrated through a series of graded ethanol, and treated with 3% H2O2 for 20 min to reduce endogenous peroxidase activity. Sections were placed in 4% formaldehyde for 20 min, washed with PBS, digested with proteinase K (10μg/ml) in PBS for 7 min, placed again in 4% formaldehyde for 20 min, immersed in triethanolamine and acetic anhydride solution for 10 min, and washed in PBS. The hybridization mixture containing DIG labeled probe was applied to each section for 16 h at 50°C. Sections were washed sequentially with 5X SSC, 2x SSC and 0.2X SSC, washed with maleic acid buffer (Roche), blocked in blocking solution (Roche) for 1 h, incubated with anti-DIG-AP antibodies (Roche) for 1 h, washed with DIG wash buffer (Roche) and incubated with detection buffer (Roche). RNA signals were developed using NBT/BCIP solutions (Thermo Scientific). Slides were mounted with Vectorshield mounting medium (Vector Laboratories), and analyzed with bright field microscopy. Serial sections of tissues hybridized with sense probes served as negative controls.
Immunohistochemistry was performed using anti-TuJ or anti-MBP primary antibodies, and 568 Alexa Fluor anti-rat or anti-mouse secondary antibodies, similar to a previously described protocol (Stankovic et al., 2004).
OPG ELISA
SGC were grown in culture plates. After 24h, the culture medium was collected and the cell debris was cleared by spinning at 14,000 g for 10 min at 4°C. Quantification of the OPG levels in the culture media was performed according to the manufacturer’s manual (Mouse Quantikine OPG/TNFSRSF11b Immunoassay, R&D Systems).
Cellular viability assay
For nuclear condensation assays, cells were cultured on glass cover slips in 24-well plates. After overnight treatment with 500 μM H2O2, cells were washed three times with PBS and fixed with 4% paraformaldehyde. Apoptotic cells were detected by staining with 1 μg/ml Hoechst 33342 (Sigma) for 5 min and observed by fluorescence microscopy (Zeiss).
Spiral ganglion cell culture
Cochleae were retrieved from postnatal day 3–5 mice and placed in ice-cold Hank’s balanced salt solution (HBSS, Invitrogen). The cartilaginous otic capsule was peeled away and the modiolus was isolated from the surrounding tissue, cut into pieces, and transferred to an enzymatic solution containing trypsin (2.5mg/ml) at 37°C for 20 min. The enzymatic digestion was terminated by adding culture medium. The tissue was dissociated by gentle mechanical trituration with a pipette. The cell suspension was sequentially plated onto dry non-coated 35 mm dishes. The final plating was transferred into poly-L-ornithine coated cell culture plates. The culture medium contained Dulbecco’s modified eagle’s medium (DMEM) and F-12 (1:1 v:v), 10% FBS, 5% horse serum, NT-3 (20ng/ml), BDNF (5ng/ml), and 2% B-27 supplement. Some cultures were pre-treated for 3h and then co-treated with OPG (100ng/l), PD98059 (20nM) or zoledronate (10μM) 24h prior to treatment with 500 μM H2O2. To eliminate neurons, cultures were treated with 1 μM β-bungarotoxin for 3d then changed to 0.5 μM for 3 more days. To selectively culture fibroblasts as a control, cells that attached to the culture dish after the first plating were cultured in DMEM supplemented with 10 % FBS.
Functional assessment of auditory phenotype
The auditory brainstem response (ABR) was measured in response to tones presented into the ear canal at half octaves, at the following frequencies 5.66, 8, 11.32, 16, 22.64, 32, and 45.25 kHz, and 5 dB steps from 15 to 80 dB SPL. Responses were measured using subdermal electrodes: positive behind the ipsilateral pinna, negative at the vertex, and ground at the tail. For each frequency and sound level, 512 responses were recorded and averaged using custom LabVIEW data acquisition software run on a PXI chassis (National Instruments Corp., Austin, Texas). Auditory threshold was defined as the first level at which a repeatable wave was detected.
Distortion product otoacoustic emissions (DPOAE) were measured by introducing two tones, of frequencies f1and f2, into the external auditory canal and simultaneously recording the acoustic signal emitted from the canal. Tone 2 was presented at half octave intervals (i.e. at 5.66, 8, 11.32, 16, 22.64, 32, and 45.25 kHz) and intensities of 10–80 dB SPL scaled in 5 dB steps. Tone 1 was controlled such that f1 = f2/1.2 and presented at a level 10 dB greater than f2. Auditory threshold was defined to occur at the level of tone 2 where the magnitude of the 2f1-f2 distortion product tone exceeded 0 dB SPL (the system noise floor was ~−10 dB SPL across the frequencies tested).
Culture and analysis of neurospheres
For each experiment, spiral ganglia of four to six, 1–3d old, WT or Opg−/− mice were dissected in HBSS. The spiral ganglion cells (neurons and glia) were dissociated using trypsin (0.25%) for 13 min in PBS at 37°C. The enzymatic digestion was stopped by adding 10% FBS in DMEM-high glucose medium. The tissue was washed twice and gently triturated. The cell suspension was passed through a 70μm cell strainer (BD Labware). Single cells were cultured in DMEM-high glucose and F12 (mixed 1:1) supplemented with N2 and B27 (Invitrogen), EGF (20ng/mL; Chemicon), bFGF (10ng/mL; Chemicon), IGF-1 (50ng/mL; Chemicon), and heparan sulfate (50ng/mL; Sigma). Newly formed spheres were cultured in ultra-low-cluster plates (Costar) for 4 days, and were termed first generation spheres. Spheres were subsequently passaged every 4 d until the third generation, when 5–10 spheres were collected and dissociated (Oshima et al. 2007). Spheres were passaged at a clonal level for 3 more times. Before each passage, the sphere morphology was assessed and the spheres were counted using the Metamorph counting software. At the third generation, spiral ganglion spheres from WT and Opg−/− mice were separated into 4 groups and treated with RANKL (100 ng/ml), OPG (100 ng/ml) or zoledronate (1 μM). The control group was untreated. After 3 d of treatment, the spheres were passaged and treated again for 3 d. Twelve hours before plating, proliferating cells in the spheres were labeled with BrdU (3μg/ml). After 12 h, the spheres were plated for 1 h on poly-L-lysine (0.01%, Cultrex)-coated glass coverslips (Marienfeld GmbH, Germany), fixed in 4% paraformaldehyde for 10 min, treated with 1 N HCl for antigen retrieval for BrdU staining, incubated for 1 h in blocking solution (0.3% Triton, 15% goat serum in 1x PBS), and incubated overnight with either anti-cleaved caspase 3 antibody to assess cell death, or anti-BrdU antibody to assess cell proliferation. After 3 washing steps with 1x PBS, the spheres were incubated in secondary antibody for 2 h (568 Alexa fluor anti rat or anti mouse). Nuclei were stained with DAPI. Staining was visualized by epifluorescence microscopy (Axioskop 2 Mot Axiocam, Zeiss). Counting was done with the Metamorph software.
Statistical analysis
Data within a group were compared using the paired t test and data between groups were compared using the analysis of variance. Differences were considered significant if P<0.05.
Results
Degeneration of the cochlear nerve in OPG-deficient mice
Examination of osmium-stained cochlear sections of 3 to10 wk old Opg −/− and WT mice revealed that the early pathologic signs in Opg −/− mice were demyelination of typically myelinated cell bodies of spiral ganglion neurons, and clustering of neuronal cell bodies into aggregates with poorly defined cellular boundaries (Fig. 1B, C). Similar pathology has been described in aging mice (Hequembourg and Liberman, 2001) and in Ly5.1 mice, which carry a differential Ly5 allelic form and show a high degree of the “human-like” feature of unmyelinated spiral ganglion neurons that aggregate into neural clusters (Jyothi et al., 2010). The region primarily occupied by cell bodies of spiral ganglion neurons (Figure 1B) was fitted with three non-overlapping square frames whose sides measured 136 μm. Within each frame, we traced the outlines of neuronal aggregates (marked with asterisks in Fig. 1B), added their areas across all three frames, and expressed the cumulative area as a fraction of the total area covered by the frames. This quantification of the “degenerating area” was focused on the apical turn of the cochlea (Fig. 1D) because the pathologic changes within the spiral ganglion showed an earlier and more severe onset in the apical than in the basal turn of the cochlea. The degenerating area of spiral ganglion neurons increased in size from 3 to 10 wk (Fig. 1D) in Opg −/− re age-matched WT mice. At these time points, we did not observe any obvious abnormality in the organ of Corti (Fig. 1E), stria vascularis or other intracochlear structures, consistent with a prior report in older mice (Zehnder et al.; 2006).
Figure 1. Degenerative changes in the cochlear nerve in OPG deficient mice.

(A) A schematic of a cochlear cross section depicting 3 regions that were studied, and sound frequencies that these regions are tuned to. The boxed region in the apex indicates the spiral ganglion that contains somata of cochlear neurons shown in (B, C) and Fig. 2A. (B, C) Osmicated, plastic-embedded sections of 10 wk old cochlear neurons showed that cell bodies of most WT neurons were individually surrounded by osmiophilic Schwann cells (left) whereas cell bodies of Opg−/− neurons formed degenerating aggregates with poorly defined cellular boundaries (marked with white asterisks in B and black arrow in C). Scale bar: 50 μm (B), 10 μm (C). The white arrows point to normal neurons (C, right). (D) The area of aggregates of degenerating neuronal cell bodies, when expressed as a fraction of the total area occupied by neuronal cell bodies of a cochlear apical half turn was larger and increased faster with age in Opg−/− than in WT mice. The error bars indicate the standard errors of the results from 4–13 ears from 4–10 animals for each age. (E) Osmicated, plastic-embedded sections of 10 wk old WT (left) and Opg−/− (right) mice showed that there was no obvious abnormality in the organ of Corti, which includes sensory inner hair cells (IHC) and outer hair cells (OHC), as well as supporting cells comprising Deiters cell (DC), pillar cell (PC) and Hensen’s cell (HC). Scale bar: 25 μm.
To investigate if there is a correlation between demyelination and neuronal degeneration, we performed immunohistochemistry with antibodies against β-III tubulin (TuJ), or Schwann cell specific myelin basic protein (MBP) on cochlear sections from WT and Opg −/− mice at 6, 10 (Fig. 2A) and 16 wk. Because red was used to label TuJ+ neurons, and green to label MBP+ Schwann cells, yellow staining identified myelinated neurons. Whereas cell bodies of most WT spiral ganglion neurons were myelinated (i.e. yellow in the merged image in Fig. 2A), those of Opg−/− mice within the pathologic aggregates were unmyelinated (i.e. red in the merged image in Fig. 2A). Stained cells were counted in 3 cochlear regions schematized in Figure 1A. The number of demyelinated neurons in Opg −/− cochleae significantly increased by 10 wk in the cochlear apex (Fig. 2B), and progressed to involve the mid base and base by 16 wk (Fig. 2C). These results suggest that OPG deficiency leads to acceleration of age-related changes and demyelination in SGC.
Figure 2. Loss of myelination in OPG deficient cochlea.

(A) Immunohistochemistry for TuJ and MBP at 10 wk of age confirmed demyelination of pathologic neural aggregates. DAPI is a nuclear stain. Scale bar: 50 μm. (B) The fraction of demyelinated neurons compared to the total number of neurons significantly increased with age in Opg−/− compared to the age-matched WT mice in the cochlear apex. This fraction refers to the Tuj positive neurons that are not surrounded by the MBP signal compared to TuJ positive neurons that are surrounded by the MBP signal. (C) The fraction of demyelinated neurons compared to the total number of neurons significantly increased with age in Opg−/− compared to age-matched WT mice in the cochlear base.*P<0.05. For bar graphs, gray indicates WT and black Opg−/− in this and other figures. Data expressed as mean +/− standard error of the mean in all figures. The error bars in (B) and (C) are based on measurements from 3 different animals per group.
OPG deficiency leads to adolescent onset hearing loss
To determine whether the observed histological changes in Opg −/− mice correlated with progressive sensorineural hearing loss, auditory brainstem response (ABR) and distortion product otoacoustic emission (DPOAE) tests were performed on Opg −/− and age-matched WT mice at 6, 8, 10, and 16 wk of age. ABR is a neural response, which is routinely used to assess function of the auditory pathway, whereas DPOAEs are generated by hair cells, and assess function of the middle ear and cochlear amplifier. By comparing shifts in ABR and DPOAE thresholds, it is possible to infer relative contributions of conductive vs. sensorineural hearing loss (Qin et al., 2010). This is because sound energy passes through the middle ear once to generate ABR and twice to generate DPOAE. Consequently, conductive pathology tends to produce changes in DPOAE thresholds that are about double the size of changes in ABR thresholds, whereas DPOAE and ABR threshold shifts are similar in sensorineural pathology originating in the inner ear, and ABR threshold shifts exceed DPOAE changes if the pathology is localized in the neurons (Qin et al., 2010).
Due to system limitations at low frequencies and age-related hearing loss at high frequencies, we focused on three middle frequency points (11.32, 16, and 22.64 kHz) and plotted the changes in threshold shift, averaged over the 3 frequencies, in ABR and DPOAE measurements for Opg −/− vs. WT mice as a function of age (Fig. 3A). We found that threshold shifts in ABR grew consistently as a function of age whereas DPOAE threshold shifts slowed down after 8 wk. These observations suggest a growing sensorineural hearing loss in aging Opg −/− mice (beginning after ~ 8 wk) superimposed upon a conductive hearing loss, which presents earlier (before 8 wk).
Figure 3. Sensorineural hearing loss in OPG deficient mice.
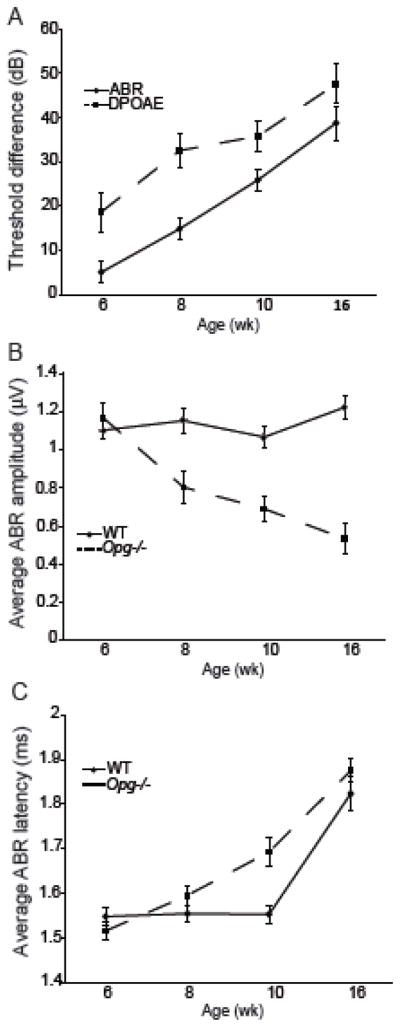
Measurements of ABR and DPOAE determined threshold shifts (A) in Opg−/− compared to WT mice as a function of age. Data are the average thresholds from 11.32, 16, and 22.64 kHz. Error bars present the standard error of the difference between sample means. N=7, 4, 8, and 5 (Opg−/− mice) or 7, 8, 7, and 5 (WT mice) for 6, 8, 10, and 16 wk, respectively. ABR wave 1 amplitude (B) data are the average responses to 11.32, 16, and 22.64 kHz tones presented at 60, 70, and 80 dB SPL. ABR wave 1 latency values (C) are the averaged peak latencies values in response to 11.32, 16, and 22.64 kHz tones presented at 5, 10, 15 and 20 dB above threshold.
Another way to assess neuronal degeneration that precedes neuronal loss is to quantify changes in ABR wave 1 amplitude and latency. Previous work has shown that fractional decrements in wave 1 amplitude match eventual fractional loss of cochlear neurons (Kujawa and Liberman, 2009). Here we presented the ABR wave 1 amplitude as the average response to tones presented at 60, 70 and 80 dB SPL at each of the 3 frequencies. ABR wave 1 amplitude in Opg −/− mice substantially decreased with age (Fig. 3B), while responses in WT mice remained stable. This observation is consistent with a growing sensorineural hearing loss in Opg −/− mice, which preceded, and roughly matched in magnitude, the increasing size of the degenerating areas in the cochlear neurons (Fig. 1C). Threshold-adjusted ABR wave 1 peak latency measurements, averaged across 11.32, 16, and 22.64 kHz tones presented at 5, 10–15 and 20 dB above threshold, demonstrate that latency increased as a function of age in Opg −/− mice, but remained stable until 10 weeks in WT mice. By 16 weeks of age, the WT latency nearly matched the Opg −/− latency. Importantly, the age range where Opg −/− latency is substantially larger than WT latency (Figure 3C) coincides with the period when DPOAE threshold shift increases at its slowest rate (Figure 3A), implying that the latency shift cannot be explained simply by an increasing conductive hearing loss. Instead, growing auditory nerve dysfunction is contributing to the latency shift, consistent with our histological observations (Fig. 1D).
Cochlear neurons and Schwann cells express and secrete OPG
To identify cochlear cells that express Opg mRNA, we performed in situ hybridization and found that the strongest Opg signal was in cochlear neurons and the associated Schwann cells (collectively termed spiral ganglion cells, SGC) (Fig. 4A). To test whether OPG was secreted by SGC, the OPG level in the medium of cultured SGC was measured with ELISA (Fig. 4B). Cultured SGC consisted of ~90% Schwann cells (S100 positive) and ~10% neurons (TuJ positive, Supplemental Fig. 1). OPG was abundantly secreted by WT SGC but was not detected in the culture medium from Opg −/− cells. Although fibroblasts secreted some OPG, the level was substantially lower than that of SGC (p=0.009). We next tested whether OPG suppressed apoptosis in cochlear SGC by treating the cells with H2O2 to produce oxidative stress. Oxidative stress is known to cause degeneration of the cochlear nerve following acoustic trauma (van Campen et al. 2002; Wang et al 2002). Nuclear condensation, a late-stage marker of apoptosis, significantly increased (p = 0.003) in Opg −/− compared to that in WT SGC after H2O2 treatment (Fig. 4C).
Figure 4. Expression and secretion of OPG by cochlear neurons and Schwann cells.

(A) In situ hybridization for Opg showed strong signal in cochlear neurons (white arrow) and Schwann cells (black arrow) in WT cochlea (left), and no signal with the control (sense) probe (right). Scale bar: 100 μm. (B) Measurements of secreted OPG in culture medium of WT SGC and fibroblasts showed that OPG was abundantly secreted by WT SGC but not secreted by Opg−/− cells. The error bars indicate the standard errors of three replicates. ND: not detectable. (C) Treatment of cultured SGC with H2O2 resulted in more nuclear condensations in Opg−/− cells than in WT cells, as shown in the representative images (left, white arrows), and bar graphs (right); cells in 10 random fields in each independent experiment were counted. The error bars indicate the standard errors of 4 replicates. The nuclei were stained with Hoechst 33342.
Loss of OPG expression sensitizes cultured SGC to apoptosis induced by oxidative stress
To investigate the mechanisms that regulate apoptosis caused by the loss of OPG in SGC, we studied ERK 1/2 (hereafter referred to as ERK), p38, and JNK signaling pathways. These pathways are known to be regulated by OPG in bone (Khosla, 2001; Lahne and Gale, 2008), and have been implicated in hearing loss due to acoustic trauma or ototoxic drugs (Zine and van der Water, 2004). After H2O2 treatment, ERK was substantially activated in Opg −/− SGC, as evidenced by the presence of phospho-ERK (p-ERK), while p38 and JNK were only slightly affected (Fig. 5A). The magnitude of p-ERK activation was substantially larger in Opg −/− than WT SGC (Fig. 5A), demonstrating enhanced sensitivity of Opg −/− SGC to oxidative stress. The activation of ERK signaling in Opg −/− SGC was suppressed by either exogenous OPG, an ERK inhibitor (PD 98059), or zoledronate, a bisphosphonate used to treat osteoporosis and lytic bone lesions due to metastases (Lipton et al., 2002), and known to target the ERK pathway in endothelial cells (Hasmim et al., 2007) (Fig. 5B). Exogenous OPG, PD 98059, and zoledronate not only suppressed ERK activation but also rescued Opg −/− SGC from death caused by H2O2 induced oxidative stress (Fig. 5C).
Figure 5. Sensitization of OPG deficient SGC to oxidative stress and apoptosis.

(A) After overnight treatment with 500 μM H2O2, Opg−/− SGC showed increased ERK activation, reflected in increased expression of phosphorylated ERK, which was substantially larger than ERK activation in the WT SGC. (B) Treatment of cultured Opg−/− SGC with H2O2 induced substantial ERK activation, while p38 and JNK activation was only slightly affected. ERK activation was suppressed by pre-treatment for 3 hours and co-treatment with either exogenous OPG (100 ng/L), the ERK inhibitor PD 98059 (PD, 20nM), or zoledronate (Zole, 10μM). This is a representative result from three replicates. (C) Exogenous OPG, the ERK inhibitor, and zoledronate rescued Opg−/− SGC from death. Pretreatment followed by co-treatment of SGC with OPG, PD 98059, or zoledronate rescued H2O2 induced oxidative cell death. NT: non-treated. The error bars indicate the standard errors of three replicates.
OPG deficient auditory stem cells have decreased proliferative capacity and altered morphology
Since degenerative changes in cochlear SGC were first detected at 3 wk (Fig. 1C) and the ERK pathway is known to influence progenitor cell expansion in the nervous system (Burdon et al., 2002), we tested the possibility that OPG deficiency affects progenitor cells in the spiral ganglion. Progenitor cells isolated from the ganglion by sphere formation have the capacity for self-renewal and for differentiation into neurons and glia (Martinez-Monedero et al., 2008). Neurospheres from WT and Opg −/− spiral ganglion were propagated as described in the Materials and Methods and the number of spheres was counted after each passage. Neurospheres from Opg −/− did not proliferate well (Fig. 6A), and were much smaller (Fig. 6B) than WT spheres. OPG deficiency also resulted in increased death of neurospheres as shown by increased cleaved caspase 3 staining compared to WT spheres (Fig. 6C).
Figure 6. Reduced proliferation and survival of floating spheres from Opg−/− spiral ganglion.

(A) Ten spheres were picked (hence there were no error bars for the first propagation) at the third passage and propagated twice. Opg−/− spheres proliferated slower than WT spheres. The error bars indicate the standard errors of three replicates. (B) Opg−/− spheres were smaller than WT spheres. Scale bar: 50 μm. (C) Floating spheres from Opg −/− mice were significantly more apoptotic than WT spheres, as reflected in expression of cleaved caspase 3 detected by immunocytochemistry per DAPI positive nuclei in spheres. The error bars indicate the standard errors of 4 replicates. For each replicate, cells were counted in 4 randomly selected fields of each plate. (D) Counting of floating Opg −/− spheres treated with BrdU revealed that OPG and zoledronate treatment increased, whereas RANKL treatment had a tendency to reduce, the number of BrdU positive cells per DAPI positive nuclei in spheres. The error bars indicate the standard errors of three replicates. The fraction of BrdU+ cells was counted similar to the counting in (C). Representative images of the cells were shown in (F) – top panel. (E) Cleaved caspase 3 immunostaining showed that Rankl treatment increased whereas OPG and zoledronate treatment decreased the number of cleaved caspase 3 positive apoptotic cells per DAPI positive nuclei in spheres. The error bars indicate the standard errors of 4 replicates. The fraction of caspase 3 + cells was counted as in (C). Representative images of the cells were shown in (F) – bottom row. (F) Representative images of Opg−/− spheres stained with DAPI to label nuclei and anti-BrdU antibody to label proliferating cells (top panel) or anti-cleaved caspase 3 antibody to label apoptotic cells (bottom panel) in response to no treatment (control), or treatment with RANKL, OPG, or zoledronate. Scale bar applies to all figures and represents 50 μm.
OPG promotes proliferation and suppresses apoptosis in OPG deficient auditory stem cells
Treatment with exogenous OPG or zoledronate significantly enhanced proliferation of Opg −/− neurospheres while the addition of RANKL had no significant effect, as shown by the BrdU staining (Fig. 6D, F – top panel). Exogenous OPG and zoledronate also suppressed apoptosis in Opg −/− spheres, as analyzed by staining for cleaved caspase 3 (Fig. 6E, F – bottom panel). Although treatment of Opg −/− spheres with RANKL did not significantly affect proliferation, it increased apoptosis (Fig. 6E, F). The addition of RANKL, OPG, or zoledronate did not affect organ of Corti spheres isolated from Opg −/− mice (data not shown). These results suggest that OPG and zoledronate have an anti-apoptotic role in neural progenitor cells, in addition to the anti-apoptotic role in adult SGC.
Discussion
We have shown that OPG, a bone related cytokine, promotes survival of neurons and neural stem cells in the cochlea. Our findings provide a mechanism for sensorineural hearing loss due to OPG deficiency as observed in juvenile Paget’s disease and related common disorders of adult onset Paget’s disease, otosclerosis, osteoporosis, and celiac disease. Although others have suggested that sensorineural hearing loss observed in these osteopathies is secondary to bone remodelling, due to diffusion of inflammatory cytokines from the remodelling cochlear bone into the cochlear soft tissues (Lindsay and Suga, 1976; Adams, 2002; Zehnder et al., 2006; Bahmad and Merchant, 2007), our data suggest an additional, direct mechanism for primary neuronal degeneration due to OPG deficiency. We show that OPG protects SGC from apoptosis by inhibiting ERK, and that OPG deficiency leads to degeneration of SGC. ERK is known to be an important regulator of myelination during development (Newbern et al., 2011) and after nerve injury (Tsuda et al., 2011). Our finding of ERK activation by oxidative stress in cochlear SGC is consistent with the reported activation of ERK by reactive oxygen species in a variety of cultured glia and neurons (de Bernardo et al., 2004; Chen et al., 2009; Chen et al., 2009), and with the demyelination associated with ERK activation in leprosy (Tapinos et al., 2006).
Our findings, combined with the published data on the critical role that OPG plays in bone remodeling (Simonet et al., 1997; Zehnder et al., 2005), suggest that the interplay between bone and neurons, which is known to be important during development (Garcia-Castellano et al., 2000; Jones et al., 2004), continues in adulthood mediated, at least in part, by OPG. The ability of nerves to secrete OPG, which promotes their survival and inhibits bone remodeling, may be advantageous because nerves within the central nervous system are surrounded by bone and pass through osseous foramina to reach their distal targets. Anti-apoptotic properties of OPG are also thought to limit immune-mediated damage in the nervous system.
Although separation of the conductive and sensorineural components of a mixed hearing loss is straightforward in human adults and adolescents, it is challenging in animals because behavioral audiometry is not possible for measurement of bone-conduction (Qin et al., 2010). Nonetheless, a relative comparison of ABR and DPOAE threshold shifts, as well as ABR amplitudes and latencies can provide insight in animals (Qin et al., 2010), especially when combined with detailed histologic analyses. While our results are in general agreement with the reported hearing tests in Opg −/− mice of 7 and 16 wk (Kanzaki et al., 2009), or 8 and 21 wk (Zehnder et al., 2006), we present a more thorough characterization of the auditory phenotype as it develops. Kanazaki et al. (2009) only measured ABR, not DPOAE, and concluded that ABR shifts in Opg −/− mice were purely conductive due to age-related resorption of middle-ear ossicles. However, our hearing tests combined with the other results of this study suggest that degeneration of SGC plays a previously unrecognized role in the auditory phenotypes due to OPG deficiency. Future screening for mutations in OPG among people with seemingly unrelated types of sensorineural hearing loss would help decipher the role OPG plays in human hearing.
Opg −/− mice used in our experiments are known to develop skeletal abnormalities very similar to human osteoporosis (Bucay et al., 1998; Mizuno et al., 1998). A prior detailed examination of Opg −/− temporal bone (Zehnder et al., 2006), which houses the inner ear, found many similarities with human otosclerotic temporal bone, including abnormal bony remodelling, cavitation within some remodelling foci, and thickening of middle ear mucosa. There were also significant differences between Opg −/− mice and clinical otosclerosis (Zehnder et al., 2006), as Opg −/− mice do not develop stapes fixation, which is a hallmark of clinical otosclerosis, and Opg −/− mice demonstrate active remodelling throughout the entire skeleton, including the otic capsule, malleus and incus, whereas clinical otosclerosis is typically limited to the otic capsule and rarely involves the malleus and incus. Based on these findings, Zehnder et al. (2006) concluded that human otosclerosis is a different process than that observed in Opg −/− mice. Our findings in Opg −/− mice are more consistent with histopathologic findings in human Paget’s disease, which includes multifocal bony involvement, remodelling of the otic capsule, and loss of cochlear neurons (Bahmad and Merchant, 2007; Lindsay and Suga, 1976). A significant difference is that clinical Paget’s disease is typically associated with thickening of the cochlear bone, whereas Opg −/− mice develop thinning of the cochlear bone due to unopposed activity of osteoclasts. It may be that Juvenile Paget’s disease is best modelled by Opg −/− mice, because it is known to result from loss of function mutations in the OPG gene (Whyte et al, 2003; Daroszewska and Ralston, 2006), and it is characterized by a generalized increase in bone turnover leading to widespread skeletal deformities in childhood, bone pain and deafness. Histopathology of the human temporal bone in juvenile Paget’s disease has not been described.
Our result that OPG deficiency inhibits proliferation and promotes apoptosis of auditory stem cells is consistent with the role that has been observed for OPG in other signaling pathways. Although OPG per se has never before been studied in the context of neural stem cells, the proliferative and pro-survival effects of OPG parallel the effects of Notch, which increases proliferation of inner ear stem cells (Jeon et al., 2011) and inhibits osteoclastogenesis (Yamada et al., 2003). Wnt/β-catenin signaling also leads to neural stem cell proliferation (Shi et al., 2010) and increases expression of OPG (Glass et al., 2005).
Our study suggests that therapeutic interventions to increase OPG levels or to inhibit ERK activation, such as with bisphosphonates, may prevent degeneration of the cochlear nerve and subsequent sensorineural hearing loss in OPG-related osteopathies, as well as in demyelinating diseases. Consistent with our results, a recent study has shown that inhibition of ERK activation in vivo attenuates cisplatin-induced ototoxicity and the associated hearing loss (Lee et al., 2010). Our experiments suggest a possible novel indication for bisphosphonates, i.e. the prevention of SGC degeneration and the resulting sensorineural hearing loss, in addition to their effect on conductive hearing loss due to the resorption of the ossicles in the middle ear. Consistent with this prediction, a recent study has reported that zoledronate, the bisphosphonate we studied in vitro, ameliorates sensorineural hearing loss in humans in vivo (Quesnel et al, 2012). The reported improvement in word recognition after zoledronate treatment (Quesnel et al, 2012) is a hallmark of neural restoration. Our results motivate future exploration of possible neuroprotective effects of drugs designed to target bone remodeling, and possible skeletal side effects of neuroprotective drugs.
Supplementary Material
Highlights.
Osteoprotegerin (OPG) deficiency causes sensorineural and conductive hearing loss.
OPG deficiency sensitizes spiral ganglion cells (SGC) to apoptosis.
OPG, an ERK inhibitor and zoledronate promote survival of SGC.
OPG promotes survival and proliferation of inner ear stem cells.
Acknowledgments
This work was supported by grants from the National Institute on Deafness and Other Communication Disorders (NIH – NIDCD K08 DC010419), the Massachusetts Life Sciences Center, the Boston Foundation and the Bertarelli Foundation to K.M.S. We thank Drs. Charles Liberman and Gabriel Corfas for helpful comments.
Abbreviations
- OPG
osteoprotegerin
- SGC
spiral ganglion cells
- RANK
receptor activator of NF-κB
- RANKL
RANK ligand
- WT
wild type
- ERK
extracellular signal-regulated kinase
- JNK
c-Jun N-terminal kinase
- MBP
myelin basic protein
- ABR
auditory brain response
- DPOAE
distortion product otoacoustic emission
- Zole
zoledronate
Footnotes
Author contributions: S-Y. K., A.S.E., and K.M.S. designed research; S.-Y. K., J.S.K., J.B.J., D. P.-F., and A.C.L performed research; S.-Y. K., J.S.K., J.B.J., A.C.L.; A.S.E., and K.M.S analyzed data; and S.-Y.K., J.S.K., J.B.J., A.L., and K.M.S. wrote the paper.
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams JC. Clinical implications of inflammatory cytokines in the cochlea: a technical note. Otol Neurotol. 2002;23(3):316–22. doi: 10.1097/00129492-200205000-00015. [DOI] [PubMed] [Google Scholar]
- Bahmad F, Jr, Merchant SN. Paget disease of the temporal bone. Otol Neurotol. 2007;28:1157–1158. doi: 10.1097/MAO.0b013e31802ce49e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookler KH, Tanyeri H. Etidronate for the the neurotologic symptoms of otosclerosis: preliminary study. Ear Nose Throat J. 1997;76:371–6. 9–81. [PubMed] [Google Scholar]
- Bucay N, et al. Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998;12:1260–1268. doi: 10.1101/gad.12.9.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdon T, et al. Signaling, cell cycle and pluripotency in embryonic stem cells. Trends Cell Biol. 2002;12:432–438. doi: 10.1016/s0962-8924(02)02352-8. [DOI] [PubMed] [Google Scholar]
- Chen J, et al. Dopamine promotes striatal neuronal apoptotic death via ERK signaling cascades. Eur J Neurosci. 2009;29:287–306. doi: 10.1111/j.1460-9568.2008.06590.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, et al. p38 and ERK, but not JNK, are involved in copper-induced apoptosis in cultured cerebellar granule neurons. Biochem Biophys Res Commun. 2009;379:944–948. doi: 10.1016/j.bbrc.2008.12.177. [DOI] [PubMed] [Google Scholar]
- Clayton AE, et al. Association between osteoporosis and otosclerosis in women. J, Laryngol, Otol. 2004;118:617–621. doi: 10.1258/0022215041917790. [DOI] [PubMed] [Google Scholar]
- Daroszewska A, et al. Susceptibility to Paget’s disease of bone is influenced by a common polymorphic variant of osteoprotegerin. J Bone Miner Res. 2004;19:1506–1511. doi: 10.1359/JBMR.040602. [DOI] [PubMed] [Google Scholar]
- Daroszewska A, Ralston SH. Mechanisms of disease: genetics of Paget’s disease of bone and related disorders. Nat Clin Pract Rheumatol. 2006;2:270–277. doi: 10.1038/ncprheum0172. [DOI] [PubMed] [Google Scholar]
- de Bernardo S, et al. Role of extracellular signal-regulated protein kinase in neuronal cell death induced by glutathione depletion in neuron/glia mesencephalic cultures. J Neurochem. 2004;91:667–682. doi: 10.1111/j.1471-4159.2004.02744.x. [DOI] [PubMed] [Google Scholar]
- García-Castellano JM, et al. Is Bone a target tissue for the nervous system? new advances on the understanding of their interactions. Iowa Orthop J. 2000;20:49–58. [PMC free article] [PubMed] [Google Scholar]
- Glass DA, 2nd, et al. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev Cell. 2005;28:751–764. doi: 10.1016/j.devcel.2005.02.017. [DOI] [PubMed] [Google Scholar]
- Hasmim M, et al. Zoledronate inhibits endothelial cell adhesion, migration and survival through the suppression of multiple, prenylation-dependent signaling pathways. J Thromb Haemost. 2007;5:166–173. doi: 10.1111/j.1538-7836.2006.02259.x. [DOI] [PubMed] [Google Scholar]
- Hequembourg S, Liberman MC. Spiral ligament pathology: a major aspect of age-related cochlear degeneration in C57BL/6 mice. J Assoc Res Otolaryngol. 2001;2:118–129. doi: 10.1007/s101620010075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hizl S, et al. Sensorineural hearing loss in pediatric celiac patients. Int J Pediatr Otorhinolaryngol. 2011;75:65–68. doi: 10.1016/j.ijporl.2010.10.009. [DOI] [PubMed] [Google Scholar]
- Jeon SJ, et al. Notch signaling alters sensory or neuronal cell fate specification of inner ear stem cells. J Neurosci. 2011;31:8351–8358. doi: 10.1523/JNEUROSCI.6366-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KB, et al. Bone and Brain: A review of neural, hormonal, and musculoskeletal connections. Iowa Orthop J. 2004;24:123–132. [PMC free article] [PubMed] [Google Scholar]
- Jyothi V, et al. Unmyelinated auditory type I spiral ganglion neurons in congenic Ly5.1 mice. J Comp Neurol. 2010;518:3254–3271. doi: 10.1002/cne.22398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanzaki S, et al. Bisphosphonate therapy ameliorates hearing loss in mice lacking osteoprotegerin. J Bone Miner Res. 2009;24:43–49. doi: 10.1359/jbmr.080812. [DOI] [PubMed] [Google Scholar]
- Karosi T, et al. Osteoprotegerin expression and sensitivity in otosclerosis with different histological activity. Eur Arch Otorhinolaryngol. 2011;268:357–365. doi: 10.1007/s00405-010-1404-y. [DOI] [PubMed] [Google Scholar]
- Khosla S. Minireview: the OPG/RANKL/RANK system. Endocrinol. 2001;142:5050–5055. doi: 10.1210/endo.142.12.8536. [DOI] [PubMed] [Google Scholar]
- Kujawa SG, Liberman MC. Adding insult to injury: cochlear nerve degeneration after “temporary” noise-induced hearing loss. J Neurosci. 2009;29:14077–14085. doi: 10.1523/JNEUROSCI.2845-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahne M, Gale JE. Damage-induced activation of ERK1/2 in cochlear supporting cells is a hair cell death-promoting signal that depends on extracellular ATP and calcium. J Neurosci. 2008;28:4918–4928. doi: 10.1523/JNEUROSCI.4914-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, et al. Epicatechin protects the auditory organ by attenuating cisplatin-induced ototoxicity through inhibition of ERK. Toxicol Lett. 2010;199:308–316. doi: 10.1016/j.toxlet.2010.09.013. [DOI] [PubMed] [Google Scholar]
- Lindsay JR, Suga F. Paget’s disease and sensorineural deafness: temporal bone histopathology of Paget’s disease. Laryngoscope. 1976;86(7):1029–42. doi: 10.1288/00005537-197607000-00019. [DOI] [PubMed] [Google Scholar]
- Lipton A, et al. The new bisphosphonate, Zometa (zoledronic acid), decreases skeletal complications in both osteolytic and osteoblastic lesions: a comparison to pamidronate. Cancer Invest. 2002;20:s45–s54. doi: 10.1081/cnv-120014886. [DOI] [PubMed] [Google Scholar]
- Martinez-Monedero R, et al. Differentiation of inner ear stem cells to functional sensory neurons. Dev Neurobiol. 2008;68:669–684. doi: 10.1002/dneu.20616. [DOI] [PubMed] [Google Scholar]
- Mizuno A, et al. Severe osteoporosis in mice lacking osteoclastogenesis inhibitory factor/osteoprotegerin. Biochem Biophys Res Commun. 1998;247:610–615. doi: 10.1006/bbrc.1998.8697. [DOI] [PubMed] [Google Scholar]
- Newbern JM, et al. Specific functions for ERK/MAPK signaling during PNS development. Neuron. 2011;69:91–105. doi: 10.1016/j.neuron.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Z, et al. Measurement of conductive hearing loss in mice. Hear Res. 2010;263:93–103. doi: 10.1016/j.heares.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quesnel AM, et al. Third-generation bisphosphonates for treatment of sensorineural hearing loss in otosclerosis. Otol Neurotol. 2012;33:1308–1314. doi: 10.1097/MAO.0b013e318268d1b3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards JB, et al. Bone mineral density, osteoporosis, and osteoporotic fractures: a genome-wide association study. Lancet. 2008;371:1505–1512. doi: 10.1016/S0140-6736(08)60599-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riches PL, et al. Osteoporosis associated with neutralizing autoantibodies against osteoprotegerin. N Engl J Med. 2009;361:1459–1465. doi: 10.1056/NEJMoa0810925. [DOI] [PubMed] [Google Scholar]
- Shi F, et al. Beta-catenin up-regulates Atoh1 expression in neural progenitor cells by interaction with an Atoh1 3′ enhancer. J Biol Chem. 2010;285:392–400. doi: 10.1074/jbc.M109.059055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonet WS, et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997;89:309–319. doi: 10.1016/s0092-8674(00)80209-3. [DOI] [PubMed] [Google Scholar]
- Stankovic K, et al. Survival of adult spiral ganglion neurons requires erbB receptor signaling in the inner ear. J Neurosci. 2004;24:8651–8661. doi: 10.1523/JNEUROSCI.0733-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankovic KM, et al. Differences in gene expression between the otic capsule and other bones. Hear Res. 2010;265(1–2):83–9. doi: 10.1016/j.heares.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapinos N, et al. ErbB2 receptor tyrosine kinase signaling mediates early demyelination induced by leprosy bacilli. Nat Med. 2006;12:961–966. doi: 10.1038/nm1433. [DOI] [PubMed] [Google Scholar]
- Tsuda Y, et al. Axonal outgrowth is associated with increased ERK 1/2 activation but decreased caspase 3 linked cell death in Schwann cells after immediate nerve repair in rats. BMC Neurosci. 2011;12:12. doi: 10.1186/1471-2202-12-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Campen LE, et al. Oxidative DNA damage is associated with intense noise exposure in the rat. Hear Res. 2002;64:29–38. doi: 10.1016/s0378-5955(01)00391-4. [DOI] [PubMed] [Google Scholar]
- Wang Y, et al. Dynamics of noise-induced cellular injury and repair in the mouse cochlea. J Assoc Res Otolaryngol. 2002;3:248–268. doi: 10.1007/s101620020028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte MP, et al. Osteoprotegerin deficiency and juvenile Paget’s disease. New Eng J Med. 2002;347:175–184. doi: 10.1056/NEJMoa013096. [DOI] [PubMed] [Google Scholar]
- Yamada T, et al. Regulation of osteoclast development by Notch signaling directed to osteoclast precursors and through stromal cells. Blood. 2003;101:2227–2234. doi: 10.1182/blood-2002-06-1740. [DOI] [PubMed] [Google Scholar]
- Zehnder AF, et al. Osteoprotegerin in the inner ear may inhibit bone remodeling in the otic capsule. Laryngoscope. 2005;115:172–177. doi: 10.1097/01.mlg.0000150702.28451.35. [DOI] [PubMed] [Google Scholar]
- Zehnder AF, et al. Osteoprotegrin knockout mice demonstrate abnormal remodeling of the otic capsule and progressive hearing loss. Laryngoscope. 2006;116:201–206. doi: 10.1097/01.mlg.0000191466.09210.9a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zine A, van de Water TR. The MAPK/JNK signaling pathway offers potential therapeutic targets for the prevention of acquired deafness. Curr Drug Targets CNS Neurol Disord. 2004;3:325–332. doi: 10.2174/1568007043337166. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.