

Abstract
In kidneys, each tubular epithelial cell contains a primary cilium that protrudes from the apical surface. Ciliary dysfunction was recently linked to acute kidney injury (AKI) following renal ischemia-reperfusion. Whether ciliary regulation is a general pathogenic mechanism in AKI remains unclear. Moreover, the ciliary change during AKI and its underlying mechanism are largely unknown. Here we examined the change of primary cilium and its role in tubular cell apoptosis and AKI induced by cisplatin, a chemotherapy agent with notable nephrotoxicity. In cultured human proximal tubular HK-2 epithelial cells, cilia became shorter during cisplatin treatment, followed by apoptosis. Knockdown of Kif3a or Polaris (cilia maintenance proteins) reduced cilia and increased apoptosis during cisplatin treatment. We further subcloned HK-2 cells and found that the clones with shorter cilia were more sensitive to cisplatin-induced apoptosis. Mechanistically, cilia-suppressed cells showed hyperphosphorylation or activation of ERK. Inhibition of ERK by U0126 preserved cilia during cisplatin treatment and protected against apoptosis in HK-2 cells. In C57BL/6 mice, U0126 prevented the loss of cilia from proximal tubules during cisplatin treatment and protected against AKI. U0126 up-regulated Polaris, but not Kif3a, in kidney tissues. It is suggested that ciliary regulation by ERK plays a role in cisplatin-induced tubular apoptosis and AKI.
Keywords: Cisplatin, Cilia, ERK, Apoptosis, Acute Kidney Injury
1. Introduction
Cilia are tiny, membrane-enclosed organelles that protrude from the apical side of the cell. Structurally, cilia contain nine sets of microtubule doublets arranged in a circular pattern with (9+2) or without (9+0) a central pair of microtubule singlets. In human body including kidneys, most cells have a single, non-motile (9+0) cilium called primary cilium. Ciliogenesis is a complex process involving microtubule polymerization and intraflagellar transport (IFT). IFT is a two-parallel process of anterograde transport towards the ciliary tip of the axoneme by the heterotrimeric kinesin-2 motor protein complex (Kif3a, Kif3b, Kap) and retrograde transport facilitated by the motor protein dynein [1]. Dozens of factors have been reported to regulate the length of cilia [2]. The functions of primary cilia have been obscure for decades, however recently cilia have been recognized to play pivotal roles in embryo development, and cell and tissue homeostasis. Primary cilia may be the signaling hub housing the receptors of multiple signaling molecules such as Wnt, Hedgehog, and Notch [2, 3]. At the cellular level, primary cilia may participate in the regulation of cell differentiation, proliferation, and apoptosis [4, 5]. Notably, ciliary dysfunction contributes critically to the pathogenesis of a large spectrum of human genetic diseases, named ciliopathies [6]. In mice, deletion of KIF3a leads to stunted primary cilia resulting in polycystic kidney disease (PKD) [7].
Interestingly, several recent studies have suggested a connection between ciliary dysfunction and acute kidney injury (AKI). On one hand, the loss of cilia seems to sensitize renal ischemia-reperfusion-induced AKI and on the other, ischemic AKI can promote or accelerate PKD in mice with ciliary defects [8-12]. Despite these notable observations, it is unclear whether ciliary regulation is involved specifically in ischemia-reperfusion injury or in other types of AKI as well. Moreover, the ciliary change during AKI and its underlying mechanism are largely unknown.
Cisplatin is a widely used chemotherapy drug for cancer treatment [13]. However, the anti-cancer efficacy of cisplatin is limited by its side effects in normal tissues and organs, especially the kidneys [14-19]. Nephrotoxicity or AKI occurs in about one-third of patients undergoing cisplatin treatment [15]. The pathogenesis of cisplatin AKI involves the activation of multiple signaling pathways that cause tubular cell injury and death, accompanied by inflammation and vascular damage in kidney tissues [14-19]. Mitogen-activated protein kinases (MAPK), DNA damage response, and oxidative stress are among the important signaling pathways that contribute to kidney tubular injury during cisplatin nephrotoxicity[14-19]. As such, a variety of approaches targeting these pathways have been taken for renoprotection [14, 15]. For example, U0126, the MEK inhibitor that blocks ERK activation, has been reported to attenuate cisplatin-induced kidney injury in mice and in cultured tubular epithelial cells [10, 20-23].
In the present study, we demonstrate that cilia are suppressed during cisplatin treatment of HK-2 cells in vitro and mice in vivo. One of the ciliary proteins that is reduced in expression is Polaris. The cells with ciliary defects are sensitized to cisplatin-induced apoptosis. U0126 can up-regulate Polaris and preserve cilia during cisplatin treatment, accompanied by the prevention of tubular apoptosis and AKI. Together, the results suggest a pathogenic role of ERK-mediated ciliary dysfunction in AKI.
2. Materials and methods
2.1. Reagents and antibodies
Acetyl tubulin (#T7451) and β-actin (#A2228) antibodies were purchased from Sigma (St. Louis, MO). Kif3a (#611508) and Polaris (#13967-1-AP) antibodies were acquired from BD Transduction Laboratory (Franklin Lakes, NJ) and Proteintech Group (Chicago, IL). Antibodies to MAPK (P-ERK at Thr202/Tyr204, #9101) and p53 (P-p53 at Ser15, #9284; p53, #9282) pathways were obtained from Cell Signaling Technologies (Cambridge, MA). Carbobenzoxy-Asp-Glu-Val-Asp-7-amino-4-trifluoromethyl coumarin (DEVD-AFC) and 7-amino-4-trifluoromethyl coumarin (AFC) were bought from Enzyme Systems Products (Livermore, CA). Cyclophilin B (CypB) antibody (#ab16045) was purchased from Abcam (Cambridge, MA). Secondary antibodies for Western blot were from Jackson ImmunoResearch (West Grove, PA) and for immunofluorescence staining from Chemicon (Temecula, CA) respectively. For apoptosis assay by flow cytometry, a kit (#556420) containing annexin V-FITC and propidium iodide (PI) was obtained from BD Pharmingen (San Jose, CA). Other reagents were from Sigma unless specifically indicated.
2.2. Cisplatin-induced AKI in mice
Eight-week old C57BL/6 male mice, purchased from the Jackson Laboratories, were housed for one week before experiments in the animal facility of the Charlie Norwood VA Medical Center at Augusta. These mice had free access to water and regular chow. Animal experiments were conducted according to the guidelines approved by the Institutional Animal Care and Use Committee of the Charlie Norwood VA Medical Center and Medical College of Georgia. Three groups of mice (4 mice/group) were set up for experiments: Group I was injected with saline as control; Group II was injected with 30 mg/kg cisplatin intraperitoneally; Group III was injected with 10 mg/kg U0126 via tail vein and one hour later was injected with 30 mg/kg cisplatin intraperitoneally. Mice were sacrificed at 72 hrs after cisplatin administration to collect blood for measurement of the blood urea nitrogen (BUN) and serum creatinine (SCr) respectively using a commercial kit (Stanbio Laboratories, Boerne, TX) and the picric acid method established in this lab [24]. Kidneys were also harvested for histological analysis and immunoblotting [25].
2.3. Subcloning of HK-2 cells
HK-2 cells from the ATCC (CRL-2190) were cultured in the DMEM/F12 media and passaged at a dilution concentration of 500 cells in 1 ml. After 5 weeks of growth, a total of 40 single colonies was picked up and amplified. After 3-day postconfluence, cells were stained with anti-acetyl tubulin antibody. Cilia frequency (cells with cilia/total cells) was calculated and compared among all the clones. Four clones of cells (C4, 28, 32, 39) demonstrated >75% cilia frequency while in five clones (C3, 8, 10, 13, 14) cilia frequency was <25%. Cilia frequency ranged from 25% to 75% in the rest of clones.
2.4. Establishment of Kif3a and Polaris knockdown cells
The HK-2 C4 clone with good cilia was used for establishment of Kif3a and Polaris knockdown cells. Briefly, four Kif3a shRNA constructs and five Polaris shRNA constructs were individually mixed well with the MISSION Lentiviral Packaging Mix (SHP001, Sigma), and transfected into HEK293FT cells by Lipofectamine 2000 (Invitrogen, Grand Island, NY). Culture media were replaced with fresh media 24 hrs after transfection, and collected at 36 hrs. 100 μl of culture media containing virion particles were added to HK-2 cells for selection by puromycin (1 μg/ml). Meanwhile, cells transduced with the MISSION Non-Target shRNA Control Transduction Particles (SHC002V, Sigma) were used as the control. Knockdown clones were confirmed by immunoblotting with respective antibodies.
2.5. Immunostaining and quantification of the cilia length and frequency in kidney tissues and cultured cells
Tissue processing
After 24 hrs of fixation in 4% paraformaldehyde, mouse kidneys were transferred to 70% ethanol, processed with Shandon Citadel 100 (Thermal Fisher Scientific, Waltham, MA) overnight, and then embedded in paraffin. Kidneys were sectioned at 7 μm (Leica RM2025, Germany) and dried overnight before deparaffinization and rehydration through graded concentrations of ethanol. Antigen retrieval was done by putting slides in the boiling retrieval buffer (10 mM sodium citrate pH 6.0, 0.05% tween-20).
Cell fixation
Cells were fixed in 4% paraformaldehyde/2% sucrose solution and permeabilized with 0.1% Triton X-100. Immunostaining was described in detail previously [26]. Briefly, cells and tissue sections were blocked by bovine serum albumin (BSA) for 1 hour. After incubation with primary antibodies for 1 hour, cells and tissue sections were washed with phosphate buffered saline (PBS), and then incubated with secondary antibodies for another hour. Slides were mounted by the Prolong Gold antifade reagent with DAPI (4′-6-diamidino-2-phenylindole) (P36931, Sigma). Alternatively, nuclei were stained with Hoechst 33342 (H-1399, Invitrogen). All the procedures were performed at room temperature. For the image analysis, Zeiss Axio fluorescence and confocal microscopes (Carl Zeiss Inc., Thornwood, NJ) were used. The confocal microscope was equipped with the LSM Image analysis system and used for the measurement of the cilia length and quantification of cilia frequency.
2.6. Protein extraction from tissue and cell samples and immunoblotting
Proteins from kidney tissues and cells were extracted with the SDS lysis buffer as previously shown [25]. Frozen kidney tissues were homogenized in SDS buffer on ice. Similar to protein extraction from cells, samples were centrifuged at 15,000 rpm for 10 min. Supernatants were used for protein analysis. Protein samples were electrophoresed in a 10% Bis-Tris gel and transferred to the PVDF nylon membranes (Bio-Rad, Hercules, CA). After blocking with 5% non-fat dry milk (Lab Scientific, Livingston, NJ) in PBS for 1 hour, the filter was incubated with the primary antibody and washed with PBS/0.1% Tween 20 (MP Biomedicals, Santa Ana, CA). The filter was finally incubated with the secondary immunoglobulins conjugated with the horseradish peroxidase. The bound antibodies were detected with the Detection Reagent (Pierce, Rockford, IL). Quantification of the band intensity was accomplished with the NIH Image J software.
2.7. Cisplatin treatment, morphologic analysis, and caspase activity assay in HK-2 cells
Morphologic analysis of apoptotic cells and caspase activity assay were done as described previously [27]. For morphologic analysis of the apoptotic cells, cells treated with cisplatin were stained with Hoechst 33342 for 5 min before microscope examination. Cellular and nuclear morphology was evaluated with phase-contrast and fluorescence microscopy. The observable features of apoptotic cells included the cellular shrinkage, nuclear condensation and fragmentation, as well as the formation of apoptotic bodies. Five fields with approximately 200-300 cells per field were examined to estimate the percentage of apoptotic cells. For the caspase activity assay, 50 to 100 μg of cell lysates was added to an enzymatic reaction containing a fluorogenic substrate for caspases. Emitted fluorescence during reaction was quantified at excitation 360 nm/emission 530 nm. Caspase activity was calculated based upon a standard curve and expressed as the nanomolar amount of the liberated AFC by 1mg protein of cell lysates.
2.8. Apoptosis analysis with annexin-V-FITC and propidium iodide
Apoptosis was further quantified by fluorescence-activated cell sorting (FACS) after annexin V-FITC and propidium iodide (PI) staining as described before [27]. Cisplatin-treated cells were trypsinized and centrifuged together with supernatants harvested. Pelleted cells were washed with cold PBS, resuspended in 100 μl Binding Buffer at a concentration of 1×106/ml, and transferred into a 1.5 ml Eppendorf tube. Annexin V-FITC and PI were added for 15-minute incubation in the dark at room temperature. Finally, 400 μl Binding Buffer was added to each tube for analysis by a BD FACSCalibur flow cytometer (BD Biosciences, San Jose, CA).
2.9. Statistics
Values were expressed as mean ± standard deviation (SD). Student’s T-Test and Chi-Square Test were used for comparison. Spearman correlation was used to calculate the correlation coefficient of two factors. p < 0.05 was considered statistically significant.
3. Results
3.1. Suppression of cilia during cisplatin treatment of HK-2 cells
To study the effect of cisplatin on cilia length and frequency, we applied cisplatin into HK-2 cells in a time course manner. After 2- and 4 hours of cisplatin treatment, cilia length and frequency remained unchanged. However, cisplatin treatment for 8-16 hours led to significant decreases in cilia length and frequency as compared with the control group (Figure 1A). Cilia length was shortened to about half of control at 8-hour treatment and was close to zero at 16-hours of treatment. Likewise, cilia frequency was reduced from ~70% in control to ~5% after 16-hour treatment. Consequently, cilia were not barely detected.
Figure 1. Decreases of cilia length and frequency during cisplatin treatment.

HK-2 cells were cultured with or without 100 μM cisplatin for 2, 4, 8, and 16 hrs for immunostaining with anti-acetyl tubulin antibody. Nuclei were stained with DAPI. (A) Representative images. Scale bar, 10 μm. (B) Cilia length. Cilia length was measured in n>30 cells/group to calculate the average values. After measurement, cilia were divided into different length ranges to calculate their length distribution (n>30 cells/group). (C) Cilia frequency. Cilia frequency was indicated by the percentage of cells with detectable cilia. For each group, the cells in 5 different view fields of microscope were evaluated. Open bar, DMSO-treated control; filled bar, cisplatin-treated group. * p <0.001 vs. DMSO-treated control.
3.2. Establishment of cilia-suppressed cell clones
To study the relationship between cilia and apoptosis, we first sub-cloned HK-2 cells and examined cilia by immunostaining to divide the cell clones into three groups, i.e., the cilium-long (Cilium-L), cilium-short (Cilium-S), and cilium-middle. A big difference in cilia length and frequency was observed between the Cilium-L (C4, C28) and Cilium-S (C13, C14) cells (Figure 2A). Additionally, we knocked down Kif3a and Polaris with shRNAs (Figure 2B). By immunoblotting, it was shown that high efficiency of knockdown was achieved in Kif3a-2 and Polaris-2 and -3 clones. Compared with the non-target shRNA transfected control, the knockdown cells exhibited significantly shorter cilia, although their cilia frequency was only marginally reduced as revealed by immunostaining (Figure 2B).
Figure 2. Establishment of cilia-suppressed cell clones.

(A) Subcloning of cells with long and short cilia. HK-2 cells were subcloned to select cell clones with long cilia (eg. C4 and C28) and short cilia (eg. C13 and C14) revealed by immunofluorescence of acetyl tubulin. Cilia were measured in different cell clones (n>20 cells/group) and cilia frequency determined from >4 different view-fields of microscopy. (B) Establishment of stable Kif3a or Polaris knockdown clones. Constructs of four Kif3a and five Polaris shRNAs as well as one non-target shRNA sequence were separately transduced into HK-2 cells using a retroviral system. Cilia length (n>20 cells/group) and frequency (5 different view-fields) in knockdown cells were reduced as compared with the non-target sequence transfected control cells. Knockdown efficiency was confirmed by immunoblotting with respective antibodies. * p <0.001 Cilia-S (C13, C14) vs. Cilia-L (C4, C28), and knockdown (Kif3a-2, Polaris-2, Polaris-3) vs control. Nuclei were stained with DAPI. Scale bar, 10 μm.
3.3. Cilia-suppressed cells are more sensitive to cisplatin treatment
We tested the sensitivity difference of Cilium-S and Cilium-L cells to cisplatin treatment. Compared with Cilium-L cells, more Cilia-S cells underwent apoptosis after 16 hours of cisplatin treatment at a concentration of 100 μM (Figure 3A). In order to confirm this observation, we used three doses of cisplatin (50, 100, 200 μM) to treat Kif3a and Polaris knockdown cells. As shown in Figure 3B, in each of the cisplatin concentrations a higher percentage of apoptotic cells were observed in Kif3a or Polaris knockdown cells than the non-target shRNA transfected cells. By FACS analysis of annexin V-FITC/PI staining, we further confirmed that more apoptosis was induced by cisplatin in Polaris knockdown cells than the non-target shRNA transfected cells (not shown). Together, the results indicate that ciliary suppression sensitizes HK-cells to cisplatin-induced apoptosis.
Figure 3. Cilia-suppressed cells are more sensitive to cisplatin treatment.

(A) Cisplatin-induced apoptosis in long cilia C4 and short cilia C13 cells. The cells were treated with cisplatin at 100 μM for 16 hrs and then stained by Hoechst 33342. (B) Cisplatin-induced apoptosis in Kif3a and Polaris knockdown cells and control shRNA transfected cells. The cells were treated with 50, 100 or 200 μM cisplatin for 16 hrs and then stained by Hoechst 33342. Cisplatin-representative cell and nuclear images were collected by phase (×20) and fluorescence microscopy (top × 20; bottom ×40). For quantification, apoptotic cells were counted from >4 different view fields of microscopy. p * <0.05, ** <0.01, Cilia-S vs Cilia-L, or knockdown vs control.
3.4. Heightened ERK1/2 activation in cilia-suppressed cells during cisplatin treatment
MAPKs, especially ERK1/2, play important roles in tubular cell apoptosis and kidney injury during cisplatin treatment [23, 28]. Thus, to understand the mechanism of cisplatin sensitivity of cilia-suppressed HK2 cells, we analyzed ERK1/2 phosphorylation in Cilium-L and –S cells. As shown in Figure 4A, cisplatin-induced ERK1/2 phosphorylation or activation in Cilia-S cells was markedly higher than that in Cilia-L clones. In line with this observation, Kif3a and Polaris knockdown cells showed significantly higher ERK1/2 phosphorylation than control shRNA-transfected cells following cisplatin treatment (Figure 4B).
Figure 4. Heightened ERK activation by cisplatin in cilia-suppressed cells.

Cells were treated with cisplatin (100 μM) for 16 hrs to collect whole cell lysate for immunoblotting of P-ERK and cyclophilin B (CypB: loading control). Densitometry was conducted for semi-quantification. (A) P-ERK in long cilia (C4, 28) cells and short cilia (C13, 14) clones. The experiments were repeated twice. For densitometry, C4 and C28 values were combined as Cilia-L, while C13 and C14 as Cilia-S. (B) P-ERK in Kif3a and Polaris knockdown clones (n=3/each clone). p # <0.01 Cilia-S vs Cilia-L; * <0.05 Kif3a or Polaris knockdown vs control.
3.5. Inhibitory effect of U0126 on cisplatin-induced caspase activation in cilia-suppressed cells
Based on the above results, we hypothesized that the heightened ERK activation in cilia-suppressed cells may underlie their sensitivity to cisplatin-induced apoptosis. To pursue this possibility, we examined the effect of U0126, a specific pharmacological inhibitor of MEK that blocks ERK1/2 phosphorylation and activation. We initially tested five different doses of U0126 in HK-2 cells and found that 5μM was able to block ERK phosphorylation during cisplatin treatment (Figure 5A). To quantify apoptosis, we measured caspase activity. As shown in Figure 5B, the cells with short cilia (Cilia-S) were more sensitive to cisplatin-induced caspase activation or apoptosis and importantly, U0126 largely abrogated the injury sensitivity of Cilium-S cells. Similarly, U0126 blocked cisplatin-induced caspase activation in Kif3a and Polaris knockdown cells, while only marginal effects were shown in non-targetshRNA transfected cells (Figure 5C). These observations support a role of heightened ERK activation in the sensitivity of ciliary defective cells.
Figure 5. Inhibitory effect of U0126 on cisplatin-induced caspase activation in cilia-suppressed cells.
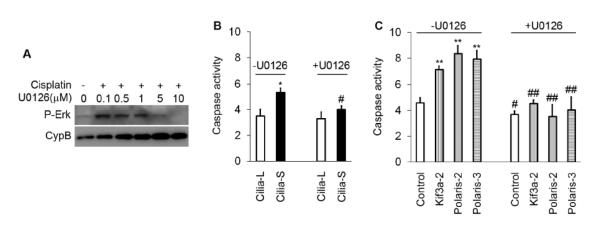
(A) Effects of various doses of U0126 on ERK phosphorylation during cisplatin treatment of HK-2 cells. U0126 of 0.1-10 μM were added during 100 μM cisplatin treatment to collect lysate at 16 hrs to analyze P-ERK and cyclophilin B. (B) (C) HK-2 cell clones were treated with 100 μM cisplatin treatment for16 hrs in the absence or presence of 5 μM U0126. Cell lysate was collected to measure caspase activity. Caspase activity was markedly reduced by U0126, especially in cilia-suppressed cells including both Cilia-S and Kif3a/Polaris knockdown cells. p * <0.05, ** <0.01 cilia-S vs cilia-L, or knockdown (Kif3a or Polaris) vs control; # <0.05, ## <0.01 U0126 vs non-U0126.
3.6. U0126 increases cilia length and frequency in HK-2 cells
To study whether U0126 protects HK-2 cells by promoting ciliogenesis, we quantified the length and frequency of cilia. In untreated HK-2 cells, U0126 significantly increased the cilia length and frequency (Figure 6A). In Cilia-L (C28) and Cilia-S (C13) cells treated with cisplatin, U0126 increased cilia length from almost none to 0.5-1.5 μm, and frequency to 10-20% (Figure 6B). Likewise, cilia length and frequency was increased by U0126 to some extent in Polaris knockdown cells (Figure 6C).
Figure 6. Inhibition of ERK by U0126 increases cilia length and frequency in HK-2 cells.

(A) Short cilia HK-2 C28 cells were incubated with or without 5 μM U0126 for 16 hrs. Immunofluorescence of acetyl tubulin was analyzed to reveal cilia. Cilia were measured to determine their length and the % cells with cilia was calculated to indicate the frequency of cilia. (B) Short cilia C28 and long cilia C13 cells were treated with 100 μM cisplatin for 16 hrs in the absence or presence of 5 μM U0126. Cilia were revealed by immunofluorescence of acetyl tubulin. After cisplatin treatment, the cells had no or very short cilia. U0126 increased cilia length and frequency in both C28 and C13 cells during cisplatin treatment. (C) Polaris knockdown and non-target shRNA transfected control cells were treated with 100 μM cisplatin in the absence or presence of 5 μM U0126. Cilia were revealed by immunofluorescence of acetyl tubulin. U0126 preserved cilia during cisplatin treatment as indicated by cilia length and frequency. For cilia length measurement, 20 to 60 cells were counted. Cells from >4 different view fields of microscopy were calculated to determine the cilia frequency. p * <0.01, ** 0.001 cisplatin+U0126 vs cisplatin. Scale bar, 10μm.
3.7. Inverse correlation between cilia length and caspase activity
To study the correlation of the cilia length and caspase activity, we measured the changes of cilia length and caspase activity during cisplatin treatment and calculated the correlation coefficient. As shown in Figure 7A, cilia length change appeared to precede the increase of caspase activity. Upon U0126 treatment, Cilia length inversely correlated with caspase activity (correlation coefficient r=-0.6) (Figure 7B). The most effective dose of U0126 in lengthening cilia was achieved at 1 μM (Figure 7C).
Figure 7. Inverse correlation of cilia length and caspase activation during cisplatin treatment of HK-2 cells.
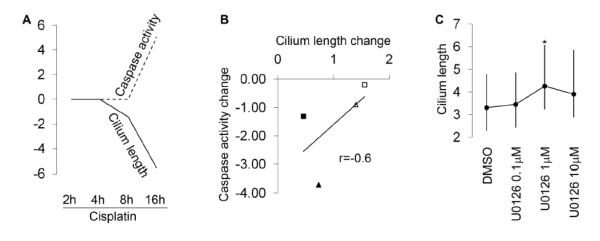
(A) Time-dependent reduction of cilia length and caspase activation during cisplatin treatment. Dashed line-Increase in caspase activity; solid line-decrease in cilia length. (B) Inverse correlation of cilia length and caspase activity changes upon U0126 treatment. Empty triangle, Cilia-L; filled triangle, Cilia-S; empty square, non-target control; filled square, knockdown clones. Each point represented the average of respective values. (C) Increase of cilia length after 0.1-10 μM U0126 treatment in HK-2 cells (>25 cells/group). p * <0.05 U0126 vs control.
3.8. U0126 inhibits ERK activation, preserves cilia, and protects against cisplatin-induced kidney injury in mice
As reported previously [20], cisplatin induced ERK phosphorylation or activation in kidney tissues in mice, which was suppressed markedly by U0126 (Figure 8A). As expected, cisplatin induced AKI as indicated by increases in BUN and serum creatinine, which were also partially suppressed by U0126 (Figure 8B). We further analyzed the changes in cilia in proximal tubules. As shown in Figure 9A, primary cilia were rarely observed in the injured dilated proximal tubules after cisplatin treatment, although the cilia in normal-looking proximal tubules did not show significant changes (Figure 9B). Interestingly, in mice pre-treated with U0126, a significantly higher frequency of cilia was detected in injured tubules of kidneys than those in the only cisplatin-treated mice (Figure 9C). These results suggest that by inhibiting ERK, U0126 may preserve cilia and protect kidney tubules and tissues during cisplatin treatment.
Figure 8. Inhibition of ERK activation by U0126 protects against cisplatin-induced kidney injury.

(A) ERK activation in kidney tissues during cisplatin treatment and the inhibitory effect of U0126. Total 11 C57/Bl6 mice (male, 8 weeks) were divided into 3 groups: Group I had 4 mice injected with saline as control; Group II had 3 mice injected with 30mg/kg cisplatin; and Group III had 4 mice injected with 30mg/kg cispaltin and 10 mg/kg U0126. After 3 days of treatment, kidney tissues were collected for immunoblotting of phospho-ERK and cyclophilin B (CypB). The blots were also subjected to densitometry to semiquantify P-ERK/CypB to indicate ERK activation. p * <0.001 cisplatin vs control; # <0.01 cisplatin+U0126 vs cisplatin. (B) Effect of U0126 on cisplatin-induced decrease of renal function. Blood samples were collected at the end of 3 days of treatment to measure BUN and serum creatinine (SCr). p * <0.01 ** <0.001 cisplatin vs control; # <0.05 ## <0.01 cisplatin+U0126 vs cisplatin.
Figure 9. Preservation of cilia by U0126 in dilated kidney tubules during cisplatin treatment of mice.

Experimental conditions were as described in Figure 8. Kidney tissues were co-stained for cilia (acetyl tubulin immunofluorescence-red), proximal tubules (LTA-green), and cell nuclei (DAPI-blue). (A) Representative image of kidney tissues. In normal mice (control), the fine structure of primary cilia in proximal tubules was observed (white arrow). However, cilia were rarely found in the injured/dilated tubules of cisplatin-treated mice (n>20) (A-right side images), whereas primary cilia appeared normal in non-dilated proximal tubules (A-left side images). With U0126 treatment, an appreciable proportion of primary cilia survived the cisplatin treatment in the dilated tubules (white arrow). Please note, LTA failed to label the dilated proximal tubules due to the loss of brush borders. One representative kidney section from each group was presented. G, glomerulus. Scale bar, 30 μm. (B) Cilia length. (C) Cilia frequency in dilated tubules. p * <0.01 cisplatin+U0126 vs cisplatin.
3.9. Cisplatin suppresses the expression of Polaris in mouse kidneys and HK-2 cells: effect of U0126
To explore the mechanisms of ciliary suppression upon cisplatin treatment, we examined the expression of Polaris and Kif3a, key proteins in the maintenance of ciliary homeostasis. Following cisplatin treatment, Kif3a was up-regulated in kidney tissues, whereas Polaris was mildly suppressed (Figure 10A). Consistently, in HK-2 cells cisplatin significantly reduced Polaris expression after 8 to 16 hours of treatment (Figure 10B). Notably, U0126 increased the expression of Polaris in kidney tissues during cisplatin-treatment in mice (Figure 10A), which was accompanied by the preservation of cilia and renoprotection (Figures 8, 9).
Figure 10. Cisplatin suppresses the expression of Polaris in mouse kidneys and HK-2 cells: effect of U0126.

(A) Effect of cisplatin and U0126 on Kif3a and Polaris expression in mouse kidneys. Kidney tissues were collected after 3 days of saline control, cisplatin, or cisplatin+U0126 treatment for immunoblot analysis and subsequent densitometry. Following cisplatin treatment, Kif3a was markedly induced, while Polaris expression was marginally suppressed. U0126 reversed the suppression of cisplatin on Polaris. p * <0.01 cisplatin vs control; # <0.05 cisplatin+U0126 vs cisplatin. (B) Time-dependent decreases in Polaris during cisplatin treatment of HK-2 cells. Upon 8- and 16-hour treatment of cisplatin, the expression of Polaris was reduced significantly. p *<0.05 cisplatin vs control.
4. Discussion
Ciliary dysfunction has been linked to AKI following renal ischemia-reperfusion [8-12]. However, it is unclear whether this is specific for ischemic AKI or a general pathogenic process in kidney injury. Moreover, the ciliary change during AKI and its underlying mechanism are largely unknown. In this study, we demonstrated ciliary disruption using cisplatin treatment of HK-2 cells in vitro and mice in vivo. Using subcloned as well as Kif3a and Polaris knockdown HK-2 cells, we further showed that ciliary defective cells are sensitized to cisplatin-induced apoptosis. Mechanistically, our results indicate that ERK1/2 may be responsible for ciliary disruption during cisplatin treatment by suppressing specific ciliary maintenance proteins such as Polaris.
Ciliogenesis mainly depends on microtubule polymerization, which is orchestrated by cargo transport facilitated by kinesin-2 complex towards the tip and cytoplasmic dynein towards the base of cilia. In this process, IFTs play essential roles. Indeed, loss or mutations of various IFT proteins lead to shortened cilia in mice [2, 7, 29]. Our study demonstrates for the first time that cisplatin reduces the cilia length and frequency not only in cultured human HK-2 cells but also in proximal tubular cells in mouse kidneys. Interestingly, while Kif3a was shown to be markedly up-regulated during cisplatin treatment, Polaris was decreased. Although these results do not exclude a regulation of other IFT or ciliary proteins, it is suggested that ciliary disruption during cisplatin treatment may result from the in-balance between these proteins. We propose that upon cell stress or injury, key ciliary proteins are destabilized or become non-functional, resulting in the resorption or disassembly of cilia. In support of this possibility, U0126 increased Polaris and preserved cilia in kidney tissues during cisplatin treatment in the present study.
At the cellular level, cilia are known to play important roles in the regulation of cell cycle and cell proliferation [4, 5]. However, relatively less is known about the involvement of cilia in the regulation of cell death or survival. Our data indicate that the decrease of cilia length and frequency preceded caspase activation during cisplatin treatment of HK-2 cells, suggesting that ciliary reabsorption may be an early event in apoptosis. Notably, the cells with ciliary suppression were shown to be more sensitive to cisplatin-induced apoptosis, supporting a role of cilia and related signaling in cell survival. Consistently, in vivo in mouse kidneys cilia became shortened following cisplatin treatment and U0126 preserved cilia, which was accompanied by the protection of kidney tissues and renal function. It remains unclear how cilia promote cell survival and maintain tissue integrity. Nevertheless, compared to the cells with normal or long cilia, during cisplatin treatment cilia-suppressed cells showed higher phosphorylation or activation of ERK, an injurious signaling protein kinase in cisplatin nephrotoxicity [23, 28]. It is proposed that by suppressing injury signaling and enhancing survival pathways, cilia may help cells cope with stress and pathological conditions. As tiny organelles protruding from cell surface, cilia have been proposed to be the sensors to physical, chemical, and biological signals. It is therefore not far-fetched to speculate that in response to cell stress, cilia may function to sense the signals from the environment, including those of growth factors, for cell survival. As a result, the loss or dysfunction of cilia may sensitize cells to apoptosis.
Our study reveals an interesting relationship between ERK and cilia. On one hand, suppression of cilia resulted in hyper-activation of ERK in HK-2 cells, suggesting that cilia play a role in quenching ERK. On the other hand, inhibition of ERK by U0126 resulted in the preservation of cilia during cisplatin treatment, suggesting that ERK contributes to ciliary disruption in this pathological condition. It is unclear how cilia modulate ERK, but as suggested above, one possibility is that cilia may sequester ERK and/or key proteins responsible for ERK activation. For the role of ERK in ciliary disruption, we speculate that ERK may destabilize some of the key IFT proteins to disrupt ciliary homeostasis. In support of this, cisplatin induced ERK activation and concomitant decreases in Polaris in kidney tissues and HK-2 cells. Moreover, inhibition of ERK by U0126 enhanced the expression of Polaris and preserved cilia (Figures 8-10). Obviously, further investigations are needed to analyze other ciliary or IFT proteins that may be subjected to ERK regulation. In addition, it is important to delineate the molecular mechanism whereby ERK regulates these proteins. Indeed, similar role of ERK in regulation of cilia length has been shown in endothelial cells [30, 31].
The protective effect of U0126 in cisplatin-induced tubular cell apoptosis and AKI in animals has been demonstrated by several previous studies. Mechanistically, U0126 may modulate mitochondrial function, sodium transport as well as decreasing inflammation and apoptosis [15, 20, 21]. Although detailed analysis of apoptosis in vivo by methods such as measuring caspase activity was not conducted in the current study, the pathogenic role of apoptosis in cisplatin-induced AKI and the effect of U0126 on caspase activation in HK2 cells (Figure 5) suggest that a major effect of U0126 on cisplatin-induced AKI is through suppressing apoptosis. The results suggest a new cellular mechanism of the cytoprotective effects: by attenuating ERK, U0126 may protect cells and tissues against injury and cell death by maintaining the primary cilia in proximal tubular cells. Certainly, these mechanisms are not mutually exclusive, suggesting the ERK may target multiple pathways or cellular processes to induce cell death and tissue damage during AKI. Nonetheless, a protective role of ERK has also been demonstrated in renal systems under certain circumstances, such as ischemic preconditioning and adenosine –mediated protection [28, 32, 33]. Although the exact underlying mechanism remains unclear, the dichotomous role of ERK may be related to the differences of the injury/stress and the cell/tissue types examined.
In conclusion, our results indicate that ciliary disruption may be a general pathogenic process in AKI. During cisplatin treatment, ciliary disruption may result from ERK-mediated suppression of ciliary proteins such as Polaris. Ciliary disruption sensitizes cells to injurious signaling and apoptosis in renal tubular cells, contributing to kidney tissue damage. Preserving cilia may offer a new therapeutic strategy for AKI.
Highlights.
1. Cilia-suppressed cells are sensitized to cisplatin-induced apoptosis.
2. Cilia are suppressed during cisplatin treatment of kidney cells and tissues.
3. Cilia suppression during cisplatin treatment is mediated by ERK.
4. ERK inhibition up-regulates Polaris, preserves cilia, and rescues renal function during cisplatin treatment.
Acknowledgements
This work was supported in part by the grants from the National Institutes of Health and Veterans’ Administration (VA) of USA and The Second Xiangya Hospital at Central South University of China.
Footnotes
Conflict of interest The authors do not have any conflicts of interest to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Kozminski KG, Beech PL, Rosenbaum JL. The Chlamydomonas kinesin-like protein FLA10 is involved in motility associated with the flagellar membrane. J Cell Biol. 1995;131:1517–1527. doi: 10.1083/jcb.131.6.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Avasthi P, Marshall WF. Stages of ciliogenesis and regulation of ciliary length. Differentiation. 2012;83:S30–42. doi: 10.1016/j.diff.2011.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Nachury MV, Seeley ES, Jin H. Trafficking to the ciliary membrane: how to get across the periciliary diffusion barrier? Annu Rev Cell Dev Biol. 2010;26:59–87. doi: 10.1146/annurev.cellbio.042308.113337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Nauli SM, Zhou J. Polycystins and mechanosensation in renal and nodal cilia. Bioessays. 2004;26:844–856. doi: 10.1002/bies.20069. [DOI] [PubMed] [Google Scholar]
- [5].Irigoin F, Badano JL. Keeping the balance between proliferation and differentiation: the primary cilium. Curr Genomics. 2011;12:285–297. doi: 10.2174/138920211795860134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med. 2011;364:1533–1543. doi: 10.1056/NEJMra1010172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lin F, Hiesberger T, Cordes K, Sinclair AM, Goldstein LS, Somlo S, Igarashi P. Kidney-specific inactivation of the KIF3A subunit of kinesin-II inhibits renal ciliogenesis and produces polycystic kidney disease. Proc Natl Acad Sci U S A. 2003;100:5286–5291. doi: 10.1073/pnas.0836980100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Patel V, Li L, Cobo-Stark P, Shao X, Somlo S, Lin F, Igarashi P. Acute kidney injury and aberrant planar cell polarity induce cyst formation in mice lacking renal cilia. Hum Mol Genet. 2008;17:1578–1590. doi: 10.1093/hmg/ddn045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zhou J, Ouyang X, Schoeb TR, Bolisetty S, Cui X, Mrug S, Yoder BK, Johnson MR, Szalai AJ, Mrug M. Kidney Injury Accelerates Cystogenesis via Pathways Modulated by Heme Oxygenase and Complement. J Am Soc Nephrol. 2012 doi: 10.1681/ASN.2011050442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Bastos AP, Piontek K, Silva AM, Martini D, Menezes LF, Fonseca JM, Fonseca, Germino GG, Onuchic LF. Pkd1 haploinsufficiency increases renal damage and induces microcyst formation following ischemia/reperfusion. J Am Soc Nephrol. 2009;20:2389–2402. doi: 10.1681/ASN.2008040435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Prasad S, McDaid JP, Tam FW, Haylor JL, Ong AC. Pkd2 dosage influences cellular repair responses following ischemia-reperfusion injury. Am J Pathol. 2009;175:1493–1503. doi: 10.2353/ajpath.2009.090227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Takakura A, Contrino L, Zhou X, Bonventre JV, Sun Y, Humphreys BD, Zhou J. Renal injury is a third hit promoting rapid development of adult polycystic kidney disease. Hum Mol Genet. 2009;18:2523–2531. doi: 10.1093/hmg/ddp147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22:7265–7279. doi: 10.1038/sj.onc.1206933. [DOI] [PubMed] [Google Scholar]
- [14].Pabla N, Dong Z. Cisplatin nephrotoxicity: Mechanisms and renoprotective strategies. Kidney Int. 2008;73:994–1007. doi: 10.1038/sj.ki.5002786. [DOI] [PubMed] [Google Scholar]
- [15].Arany I, Safirstein RL. Cisplatin nephrotoxicity. Semin Nephrol. 2003;23:460–464. doi: 10.1016/s0270-9295(03)00089-5. [DOI] [PubMed] [Google Scholar]
- [16].Price PM, Safirstein RL, Megyesi J. The cell cycle and acute kidney injury. Kidney Int. 2009;76:604–613. doi: 10.1038/ki.2009.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Miller RP, Tadagavadi RK, Ramesh G, Reeves WB. Mechanisms of Cisplatin nephrotoxicity. Toxins (Basel) 2010;2:2490–2518. doi: 10.3390/toxins2112490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Sanchez-Gonzalez PD, Lopez-Hernandez FJ, Lopez-Novoa JM, Morales AI. An integrative view of the pathophysiological events leading to cisplatin nephrotoxicity. Crit Rev Toxicol. 2011;41:803–821. doi: 10.3109/10408444.2011.602662. [DOI] [PubMed] [Google Scholar]
- [19].dos Santos NA, Rodrigues M.A. Carvalho, Martins NM, dos Santos AC. Cisplatin-induced nephrotoxicity and targets of nephroprotection: an update. Arch Toxicol. 2012;86:1233–1250. doi: 10.1007/s00204-012-0821-7. [DOI] [PubMed] [Google Scholar]
- [20].Jo SK, Cho WY, Sung SA, Kim HK, Won NH. MEK inhibitor, U0126, attenuates cisplatin-induced renal injury by decreasing inflammation and apoptosis. Kidney Int. 2005;67:458–466. doi: 10.1111/j.1523-1755.2005.67102.x. [DOI] [PubMed] [Google Scholar]
- [21].Nowak G. Protein kinase C-alpha and ERK1/2 mediate mitochondrial dysfunction, decreases in active Na+ transport, and cisplatin-induced apoptosis in renal cells. J Biol Chem. 2002;277:43377–43388. doi: 10.1074/jbc.M206373200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Bae IH, Kang SW, Yoon SH, Um HD. Cellular components involved in the cell death induced by cisplatin in the absence of p53 activation. Oncol Rep. 2006;15:1175–1180. [PubMed] [Google Scholar]
- [23].Arany I, Megyesi JK, Kaneto H, Price PM, Safirstein RL. Cisplatin-induced cell death is EGFR/src/ERK signaling dependent in mouse proximal tubule cells. Am J Physiol Renal Physiol. 2004;287:F543–549. doi: 10.1152/ajprenal.00112.2004. [DOI] [PubMed] [Google Scholar]
- [24].Jiang M, Wei Q, Dong G, Komatsu M, Su Y, Dong Z. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int. 2012 doi: 10.1038/ki.2012.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Pabla N, Dong G, Jiang M, Huang S, Kumar MV, Messing RO, Dong Z. Inhibition of PKCdelta reduces cisplatin-induced nephrotoxicity without blocking chemotherapeutic efficacy in mouse models of cancer. J Clin Invest. 2011;121:2709–2722. doi: 10.1172/JCI45586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wang S, Zhang J, Nauli SM, Li X, Starremans PG, Luo Y, Roberts KA, Zhou J. Fibrocystin/polyductin, found in the same protein complex with polycystin-2, regulates calcium responses in kidney epithelia. Mol Cell Biol. 2007;27:3241–3252. doi: 10.1128/MCB.00072-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Jiang M, Wang CY, Huang S, Yang T, Dong Z. Cisplatin-induced apoptosis in p53-deficient renal cells via the intrinsic mitochondrial pathway. Am J Physiol Renal Physiol. 2009;296:F983–993. doi: 10.1152/ajprenal.90579.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zhuang S, Schnellmann RG. A death-promoting role for extracellular signal-regulated kinase. J Pharmacol Exp Ther. 2006;319:991–997. doi: 10.1124/jpet.106.107367. [DOI] [PubMed] [Google Scholar]
- [29].Pazour GJ, Baker SA, Deane JA, Cole DG, Dickert BL, Rosenbaum JL, Witman GB, Besharse JC. The intraflagellar transport protein, IFT88, is essential for vertebrate photoreceptor assembly and maintenance. J Cell Biol. 2002;157:103–113. doi: 10.1083/jcb.200107108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Nauli SM, Jin X, AbouAlaiwi WA, El-Jouni W, Su X, Zhou J. Non-motile primary cilia as fluid shear stress mechanosensors. Methods Enzymol. 2013;525:1–20. doi: 10.1016/B978-0-12-397944-5.00001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Abdul-Majeed S, Moloney BC, Nauli SM. Mechanisms regulating cilia growth and cilia function in endothelial cells. Cell Mol Life Sci. 2012;69:165–173. doi: 10.1007/s00018-011-0744-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].di Mari JF, Davis R, Safirstein RL. MAPK activation determines renal epithelial cell survival during oxidative injury. Am J Physiol. 1999;277:F195–203. doi: 10.1152/ajprenal.1999.277.2.F195. [DOI] [PubMed] [Google Scholar]
- [33].Joo JD, Kim M, Horst P, Kim J, D’Agati VD, Emala CW, Sr., Lee HT. Acute and delayed renal protection against renal ischemia and reperfusion injury with A1 adenosine receptors. Am J Physiol Renal Physiol. 2007;293:F1847–1857. doi: 10.1152/ajprenal.00336.2007. [DOI] [PubMed] [Google Scholar]