

Abstract
G-protein-coupled receptors (GPCRs) are cell membrane protein receptors that transduce signals across the cell membrane and are important targets for therapeutic interventions. As members of the GPCR superfamily, chemokine receptors such as CXCR4 play critical roles in normal physiology as well as the pathology of many human diseases including cancer, inflammation, autoimmune diseases, and human immunodeficiency virus (HIV) infection. Here we report the discovery and study of a novel peptide ligand of CXCR4 using D-amino acids and bivalent ligand approach. This peptide, DV1-K-(DV3), shows very high affinity for CXCR4 with an IC50 of 4 nM in anti-CXCR4 monoclonal antibody (mAb) 12G5 competitive assay, which is more potent than full length natural ligand SDF-1α, even though the peptide is less than half of the number of residues of SDF-1α. This peptide can block the calcium influx stimulated by SDF-1α and inhibit cancer cell migration in vitro via CXCR4, thus functioning as a CXCR4 antagonist. Furthermore, DV1-K-(DV3) peptide displayed anti-HIV activity by inhibiting HIV-1 infection mediated by CXCR4. With its high receptor affinity and stability from D-amino acids, this peptide may be a new probe of CXCR4 functions in physiology and pathology and promising lead for therapeutic development.
Keywords: CXCR4, Chemokine receptors, SDF-1α, Peptide antagonist, Bivalent ligand, Protein-protein interaction, Cell migration, HIV infection
Introduction
Chemokine receptors are seven transmembrane (7TM) helical proteins which belong to the superfamily of G-protein-coupled receptors (GPCRs). Chemokine receptor CXCR4 is expressed on many types of cells, such as leukocytes, hematopoietic stem cells (HSCs), endothelial cells, and tumor cells. In recent years, there has been intensive biological and chemical research of CXCR4, highlighted and exemplified by the recent determination of the three-dimensional crystal structures of CXCR4 in complex with small molecule and peptide ligands [1], to study the mechanisms of chemokine receptor-ligand interactions and functions [2,3,4]. CXCR4 and its natural ligand stromal cell-derived-factor-1 (SDF-1α) are involved in number of human diseases, such as cancer, inflammation, auto-immune diseases, and acquired immunodeficiency syndrome (AIDS) [5,6]. The development of new ligands targeted to CXCR4 has become a promising therapeutic approach for cancer, AIDS, and other diseases [7,8].
Like many GPCRs, CXCR4 can form dimers or even oligomers in the active state [4,9]. However, the role of CXCR4 dimerization in normal physiology and disease still awaits further investigation. The use of a linker to conjugate synthetically two identical or different ligand molecules to form a bivalent, dimeric ligand molecule as a probe of the function of the receptor dimer is becoming an important chemical biology strategy [10,11]. Previously, we reported the CXCR4 antagonist peptides DV1 and DV3 [12], both of which are D-amino acid peptides derived from the N-terminal sequences of viral macrophage protein-II (vMIP-II) and display moderate affinity to CXCR4 with IC50 of 236 nM and 440 nM, respectively, in an anti-CXCR4 mAb 12G5 competitive assay. We have applied the bivalent ligand approach to synthesize a high affinity peptide DV1-K-(DV3) by chemically linking DV1 and DV3 through the side chain of an added Lys residue. DV1-K-(DV3) competes with the CXCR4 binding of mAb 12G5 with an IC50 value of ~ 4 nM, which is 59 and 110 fold more potent than monomeric DV1 and DV3, respectively. It is noteworthy that the CXCR4 receptor affinity of DV1-K-(DV3) as measured by mAb 12G5 competitive binding assay is even higher than the natural ligand of CXCR4, SDF-1α, whereas DV1-K-(DV3) is much smaller in size (less than half of the residues in SDF-1α) and more amenable for synthesis and further chemical modifications.
Material and Methods
Peptide Synthesis
All the peptides were prepared by manual solid-phase synthesis using a TentaGel S RAM (0.24 mmol/g) resin as the solid support and 9-Fluorenylmethoxy carbonyl (Fmoc) chemistry. A 5-fold excess of Nα-Fmoc-amino acid, diisopropyl- carbodiimide (DIC), hydroxybenzotriazole (HOBt) was used in every coupling reaction step. Removal of the N-terminal Fmoc group was accomplished by 20% piperidine in dimethylformamide (DMF) for two cycles (5min. and 15min.). Dde (N-[1-(4, 4-dimethyl-2, 6-dioxocyclohex-1-ylidene) ethyl]) group was deprotected with 2% hydrazine in DMF for 2 min, and the step was repeated three times. The bivalent peptide was synthesized starting from a DV3/DV1 peptide with a C-terminal Lys. The second DV3 moiety was synthesized from C-terminal to N-terminal by coupling it through the ε-amine group of the Lys side chain. The cleavage of a peptide from the resin was carried out with cleavage cocktail comprised of water (5%, vol/vol), thiophenol (5%, vol/vol), and triuoroacetic acid (TFA) (90%, vol/vol) for 2 hrs at room temperature with gentle stirring. The peptides were precipitated by adding ice-cold diethyl ether and washed repeatedly in cold diethyl ether. The crude peptides were dissolved in 20% acetonitrile in deionized water before being lyophilized. Then the crude peptides were dissolved in water/acetonitrile and purified using semi-preparative reverse-phase high-performance liquid chromatography (RP-HPLC) [13]. The fractions containing the peptides were pooled together and lyophilized. The purity of the final products was assessed by analytical reverse-phase HPLC [13], and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS). All peptides were at least 95% pure.
CXCR4 Competitive Binding Assay
HEK293 cells were grown in RPMI1640 medium with 10% (v/v) FBS, 100 IU penicillin, 0.1 mg/mL streptomycin and 2 mM L-glutamine. The presence of 400 μg/mL geneticin was used as a selection agent to make the HEK293 cells stably express CXCR4. Before performing the assay, HEK293 cells were collected and washed twice with FACS buffer (0.5% BSA, 0.05% sodium azide in PBS) by centrifugation. The cells were then seeded in v-shaped 96-well plates at 5×105 cells/well, and co-incubated with various concentrated peptides and primary antibody (1:3000, mouse anti-human CD184 antibody, BD Biosciences, USA) for 40 min on ice. After incubation, cells were washed twice with assay buffer by centrifugation, and then co-incubated with secondary antibody (1:250, anti-mouse IgG-FITC antibody, Sigma, USA) for 30 min on ice. After incubation, the cells were washed twice with assay buffer by centrifugation and 50 μL FACS buffer was added per well finally. The fluorescence (485EX/528EM) was recorded using a Synergy 2 microplate reader (BioTek, USA). The mean values of fluorescence were expressed as a percentage of the control group values. CXCR4 competitive binding assays were performed in duplicate and the results were presented as means ± SEMs. Experimental data were generated from at least three independent experiments. Binding curves were fitted using a sigmoidal dose-response model, and the IC50 values were calculated using GraphPad Prism 4.
CXCR4 Chemotaxis Assay
SUP-T1 cells were grown in RPMI1640 medium with 10% (v/v) FBS, 100 IU penicillin, 0.1 mg/mL streptomycin and 2 mM L-glutamine. Before performing the chemotaxis assay, SUP-T1 cells were collected and washed twice with assay buffer (RPMI1640 medium with 0.5% BSA) by centrifugation. A sample containing 1×106 cells were first co-incubated with variant concentrated of peptides for 2 hrs., and then were seeded in the upper chambers of HTS transwell 96-well plates with 5 μm pore size (Corning, USA). The upper chambers were placed into the lower chambers, which contained 200 μL assay buffer and 1 nM SDF-1α as chemoattractant. Background groups were run by adding only assay buffer in the lower chambers. The transwell plate was placed in a 37°C cell culture incubator in a 5% CO2 for 3 hrs while the cells were allowed to migrate. After incubation, the upper chambers were removed and SUP-T1 cells that had migrated to the lower chambers were quantified by Cell Titer-Blue reagent (Promega, USA).
Calcium Mobilization Assay
SUP-T1 cells were collected by centrifugation and washed twice with assay buffer (HBSS buffer with 20 mM HEPES) by centrifugation. Probenecid sodium (5 μM, Sigma, USA) and Fura-2 AM dye (2 μM, Molecular Probes, USA) were added to the cells and incubated in 37°C cell culture incubator in a 5% CO2 for 30 minutes with gently vortex every 5 min. After incubation, the cells were washed twice with assay buffer and resuspended at cell density of 1×106/mL. The assays were performed by pre-incubating the cells with CXCR4 antagonist peptides at variant concentrations for 2~3 min, followed by stimulation with 50 nM SDF-1α. The fluorescence (340EX/510EM) was measured by a fluorescence spectrometer measurement program.
HIV Drug Screening Assay
Antiviral activity was evaluated by inhibition of the replication of HIV as measured by the production of p24 antigen in culture supernatants (Perkin Elmer’s Alliance HIV-1 P24 ELISA kit, catalog #NEK050001KT). Non-adherent CEM cells were infected in suspension with the NL4-3 strain of HIV-1 at an MOI of 0.001 for 2 hours at 37°C. The cells were washed and centrifuged three times (300g, 20°C, 6 minutes) with Dulbecco’s PBS and then diluted with plus 2 μg/ml of Polybrene (Sigma, catalog N028) to a cell density of 500,000 cells/mL, and dispensed into 96 well plates (Falcon round bottom clear plate, catalog 353077) at 100 μL/well. Each drug was diluted to a 2× concentration and 100 μL dispensed into each respective well. The infected cells were incubated at 37°C for 7 days. 50 μL of supernatant were collected and stored at -20°C. The supernatants were assayed for p24 at a 1/100 dilution with PBS. The percent compound inhibition represented the percent p24 value relative to infected cells not exposed to drug.
Drug Cytotoxicity Assay
The Live Dead assay (Invitrogen/Molecular Devices, Catalog L3224) was used to assess cytotoxicity from drug exposure through perturbation of fluorescent dyes. The conditions mimicked the HIV drug screen (minus the virus) and the cell viability was determined after 7 days of exposure to the two highest concentrations of drug tested in the drug inhibition assay, 10 μM and 100 μM. The protocol followed manufacturers’ specifications and the dyes Calcein (ex/em - 485/530nm) and Ethidum homodimer-1 (ex/em – 530/645nm) were applied to the test cells for 1.5 hrs then the values read on a Molecular Devices Spectra Max 96 well reader. Subsequently, the values were normalized relative to internal controls cells without drug exposure as a percent of the control.
Results and Discussion
Dramatic Enhancement of CXCR4 Binding Affinity of DV1-K-(DV3)
Bivalent ligand DV1-K-(DV3) contains two CXCR4 monomer ligands, DV1 and DV3 linked chemically via the side chain of a Lys residue. DV1 is an all D-amino acid peptide derived from the N-terminus (residues 1-21) of vMIP-II and displays moderate binding affinity with CXCR4 and anti-HIV entry activity [12]. The mutation of DV1 peptide residues revealed that the N-terminal 1-10 amino acids were critical for CXCR4 binding, while the C-terminal 11-21 amino acids provided additional binding to the receptor[12]. A peptide containing the first 10 residues of DV1 peptide was synthesized and named as DV3, which showed an IC50 value of 440 nM in 12G5 CXCR4 competitive binding assay and was about two-fold less potent than DV1 (236nM). We synthetically conjugated the DV1 sequence with the DV3 sequence through the additional C-terminal D-Lys ε-amine group with this Lys side chain’s four-carbon linker. This generated a new heterodimeric ligand called DV1-K-(DV3). As a comparison, we also synthesized a DV3 dimer peptide, differing from DV1-K-(DV3) only in that the DV3 dimer did not have the C-terminal 11-21 residues of the DV1 peptide (Table 1). We hypothesized that this design of DV1-K-(DV3) would be able to create a “branch-like” peptide, which could present DV1 and DV3 to reach two CXCR4 receptors simultaneously.
Table 1.
Sequences or chemical structure and IC50 values of CXCR4 ligands. The IC50 value of DV3 was from the previously published results [12]
| Name | Peptide Sequenceα | IC50 (nM) |
|---|---|---|
|
| ||
| SDF-1α | KPVSLSYRCPCRFFESHVARANVKHLKILNTPNCALQI | 14 |
| VARLKNNNRQVCIDPKLKWIQEYLEKALNK | ||
|
| ||
| RCP168 | LGASWHRPDKCCLGYQKRPLPQVLLSSWYPTSQLCSK | 40 |
| PGVIFLTKRGRQVCADKSKDWVKKLMQQLPVTAR | ||
|
| ||
| Plerixafor |
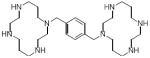
|
180 |
|
| ||
| DV1 | LGASWHRPDKCCLGYQKRPLP | 236 |
|
| ||
| DV3 | LGASWHRPDK | 440b |
|
| ||
| DV3 Dimer |
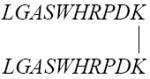
|
133 |
|
| ||
| DV1-K-(DV3) |
|
4 |
D-Amino acids are shown in italics.
The IC50 value of DV3 was from the previously published results [12]
To determine the binding affinity of these two new bivalent peptides, DV1-K-(DV3) and DV3 dimer, we performed an antibody competitive binding assay using HEK293 cell that stable expressed CXCR4. The natural ligand SDF-1α, the synthetic chemokine RCP168, the small molecule Plerixafor reported by others, and DV1 were used for comparison. We found a dramatic increase in the CXCR4 binding affinity of DV1-K-(DV3) bivalent peptide in these assays. The IC50 value of DV1-K-(DV3) was 4 nM, which was 59 fold more potent than DV1 monomeric peptide. DV3 dimer peptide also showed some increase in CXCR4 binding and was about 3 fold more potent than monomeric DV3 (Table 1, Figure 1). The degree of potency enhancement of DV3 dimer is much lower than that of DV1-K-(DV3). These results suggested that DV1-K-(DV3) has more optimal interaction with CXCR4 than monomeric DV1, DV3 or the dimeric DV3 peptide.
Figure 1.
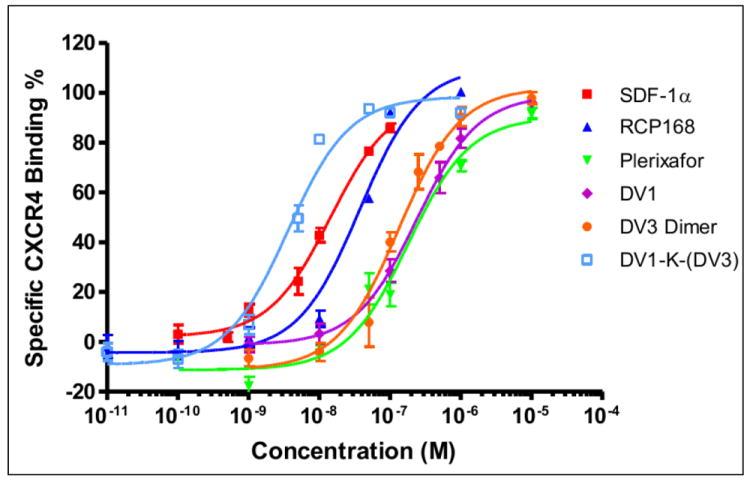
Binding activities of CXCR4 ligands. The IC50 values of these ligands were determined by 12G5 antibody competition binding assays. SDF-1α (■), RCP168 (▲), DV1 (◆) and Plerixafor (▼) were used as controls. DV3 dimer was shown in solid dot (●) and DV1-K-(DV3) was shown as hollow square (□). The results are the mean values of at least three independent experiments.
Functional Characterization of DV1-K-(DV3) Peptide
We determined the functional properties of DV1-K-(DV3) by conducting CXCR4 chemotaxis assays using SUP-T1 cells. The DV1-K-(DV3) peptide could block the cell migration induced by SDF-1α (Figure 2A) and the IC50 value for the chemotaxis assay was 500 nM (Figure 2B). This result indicated that DV1-K-(DV3) acts as a CXCR4 antagonist. We also performed a calcium mobilization assay for DV1-K-(DV3). As we expected, this peptide, at concentration of 1 μM, almost completely inhibited the calcium influx induced by SDF-1α (Figure 3).
Figure 2.
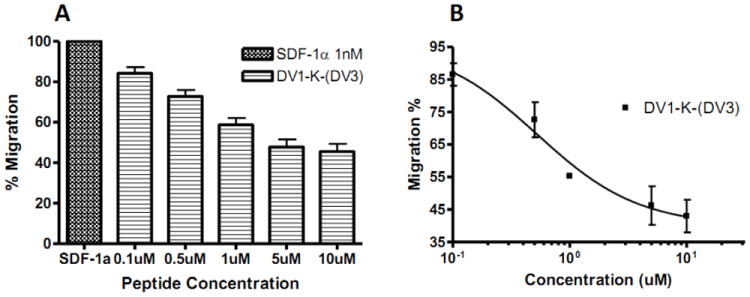
Inhibitory activity of bivalent peptide DV1-K-(DV3) in blocking chemotaxis activated by SDF-1α. Chemotaxis was expressed as the percentage of SUP-T1 cells initially seeded in the transwells that migrated into the bottom chambers following stimulation by 1 nM SDF-1α. A. Histogram for experiment data, B. The inhibition curve was fitted by sigmoidal dose-response model. The results are mean values of at least three independent experiments.
Figure 3.
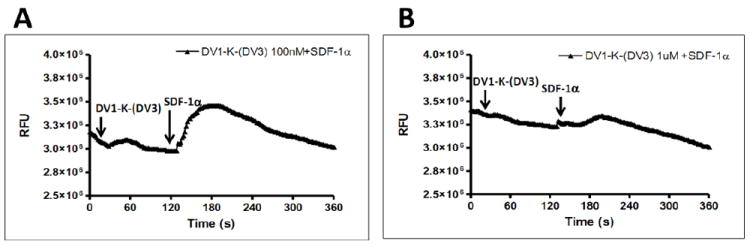
Inhibitory activity of bivalent peptide DV1-K-(DV3) in blocking calcium influx triggered by SDF-1α. The influx of Ca2+ from endoplasmic reticulum into cytoplasm was measured in response to treatment with SDF-1α and the peptide. The SUP-T1 cells were pre-incubated with various concentrations of the peptide for 2~3 min, followed by addition of 50 nM SDF-1α to stimulate the Ca2+ influx.
Potent Anti-HIV Activity of DV1-K-(DV3) Peptide
DV1-K-(DV3) was a potent CXCR4 antagonist in functional assays. We then tested whether this peptide could block HIV-1 infection via its co-receptor CXCR4. By performing the HIV drug screen assay using the NL4-3 virus strain, we found that DV1-K-(DV3) peptide inhibited of HIV-1 infection with an IC50 value of around 1 μM (Figure 4A). DV1-K-(DV3) peptide showed no cytotoxicity in the LIVE DEAD assay even at the highest concentrations tested (Figure 4B). These results demonstrate that DV1-K-(DV3) peptide is a potent HIV-1 entry inhibitor.
Figure 4.
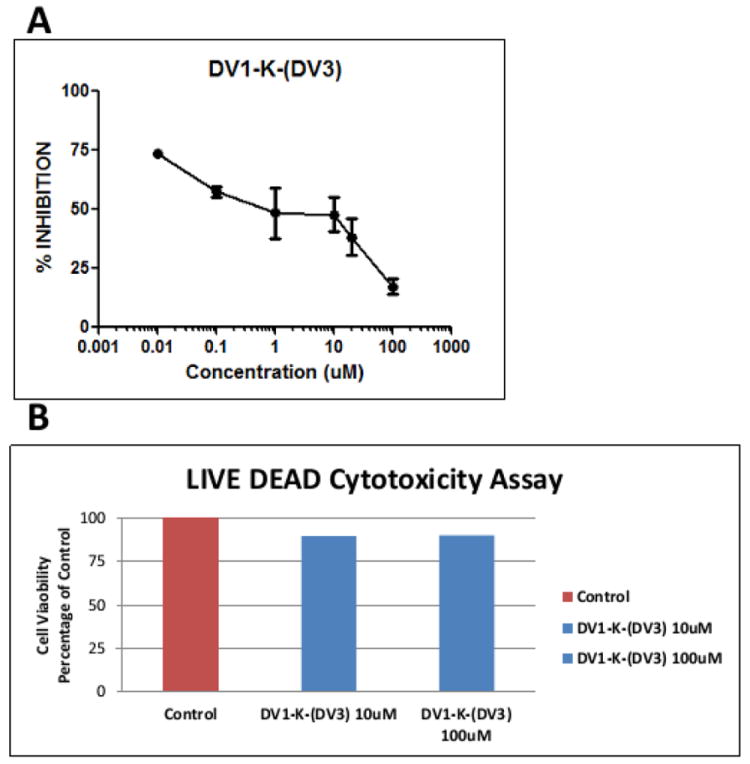
Bivalent peptide DV1-K-(DV3) showed significant anti-HIV activity and no cytotoxicity. A. HIV-1 inhibition in the drug screening assay. B. No cellular toxicity in the LIVE DEAD cytotoxicity assay.
In summary, we designed and synthesized a new bivalent, dimeric peptide antagonist DV1-K-(DV3) of CXCR4. DV1-K-(DV3) showed very high binding affinity to CXCR4 and could block the cell migration and receptor signaling as an antagonist. More importantly, DV1-K-(DV3) showed inhibition of the infection of CXCR4 co-receptor dependent HIV-1 virus. With its high receptor affinity and composition of D-amino acids which are known to be enzymatically stable, DV1-K-(DV3) may be a useful probe to study the functional role of CXCR4 dimerization and a new lead to develop therapeutics for diseases in which CXCR4 plays a role.
Highlights.
We designed, synthesized and studied a new bivalent D-peptide antagonist of CXCR4.
This peptide shows very high binding affinity for CXCR4.
This peptide inhibits calcium influx and cancer cell migration triggered by SDF-1α
This peptide inhibits HIV-1 infection and is not cytotoxic.
This peptide is a new probe of CXCR4 function and lead for therapeutic development.
Acknowledgments
This work was supported by grants from the National Institutes of Health. Y. X. was supported in part by the Chinese Scholarship Council. This work was supported by the Department of Veterans Affairs, the Translational Virology Core of the UCSD Center for AIDS Research AI36214, and the James B. Pendleton Charitable Trust.
Footnotes
Author Contributions
YX performed biological experiments and computer modeling studies. SD designed and synthesized the peptides. SE performed the antiviral and cytotoxicity assays. DDR designed and analyzed the antiviral and cytotoxicity work. JA and ZH oversaw the design, synthesis, biological and computer modeling studies.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, Hamel DJ, Kuhn P, Handel TM, Cherezov V, Stevens RC. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. 2010;330:1066–1071. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Choi WT, Kumar S, Madani N, Han X, Tian S, Dong CZ, Liu D, Duggineni S, Yuan J, Sodroski JG, Huang Z, An J. A novel synthetic bivalent ligand to probe chemokine receptor CXCR4 dimerization and inhibit HIV-1 entry. Biochemistry. 2012;51:7078–7086. doi: 10.1021/bi2016712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lane JR, Sexton PM, Christopoulos A. Bridging the gap: bitopic ligands of G-protein-coupled receptors. Trends Pharmacol Sci. 2013;34:59–66. doi: 10.1016/j.tips.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 4.Harrison C. G protein-coupled receptors: Insights into chemokine receptors. Nat Rev Drug Discov. 2010;9:920. doi: 10.1038/nrd3331. [DOI] [PubMed] [Google Scholar]
- 5.Choi WT, Duggineni S, Xu Y, Huang Z, An J. Drug discovery research targeting the CXC chemokine receptor 4 (CXCR4) J Med Chem. 2012;55:977–994. doi: 10.1021/jm200568c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pease J, Horuk R. Chemokine receptor antagonists. J Med Chem. 2012;55:9363–9392. doi: 10.1021/jm300682j. [DOI] [PubMed] [Google Scholar]
- 7.Zhou N, Luo Z, Luo J, Hall JW, Huang Z. A novel peptide antagonist of CXCR4 derived from the N-terminus of viral chemokine vMIP-II. Biochemistry. 2000;39:3782–3787. doi: 10.1021/bi992750v. [DOI] [PubMed] [Google Scholar]
- 8.Mason JS, Bortolato A, Congreve M, Marshall FH. New insights from structural biology into the druggability of G protein-coupled receptors. Trends Pharmacol Sci. 2012;33:249–260. doi: 10.1016/j.tips.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 9.Vila-Coro AJ, Rodriguez-Frade JM, Martin De Ana A, Moreno-Ortiz MC, Martinez AC, Mellado M. The chemokine SDF-1alpha triggers CXCR4 receptor dimerization and activates the JAK/STAT pathway. FASEB J. 1999;13:1699–1710. [PubMed] [Google Scholar]
- 10.Tanaka T, Nomura W, Narumi T, Masuda A, Tamamura H. Bivalent ligands of CXCR4 with rigid linkers for elucidation of the dimerization state in cells. J Am Chem Soc. 2010;132:15899–15901. doi: 10.1021/ja107447w. [DOI] [PubMed] [Google Scholar]
- 11.Birnkammer T, Spickenreither A, Brunskole I, Lopuch M, Kagermeier N, Bernhardt G, Dove S, Seifert R, Elz S, Buschauer A. The bivalent ligand approach leads to highly potent and selective acylguanidine-type histamine H(2) receptor agonists. J Med Chem. 2012;55:1147–1160. doi: 10.1021/jm201128q. [DOI] [PubMed] [Google Scholar]
- 12.Zhou N, Luo Z, Luo J, Fan X, Cayabyab M, Hiraoka M, Liu D, Han X, Pesavento J, Dong CZ, Wang Y, An J, Kaji H, Sodroski JG, Huang Z. Exploring the stereochemistry of CXCR4-peptide recognition and inhibiting HIV-1 entry with D-peptides derived from chemokines. J Biol Chem. 2002;277:17476–17485. doi: 10.1074/jbc.M202063200. [DOI] [PubMed] [Google Scholar]
- 13.Duggineni S, Mitra S, Lamberto I, Han X, Xu Y, An J, Pasquale EB, Huang Z. Design and synthesis of potent bivalent peptide agonists targeting the EphA2 receptor. ACS Med Chem Lett. 2013;4:344–348. doi: 10.1021/ml3004523. [DOI] [PMC free article] [PubMed] [Google Scholar]