

Abstract
The efficacy and pharmacokinetics of a new parenteral formulation of halofantrine were studied in mice infected with Plasmodium berghei. The formulation consisted of nanocapsules with an oily core, prepared from either poly(d,l-lactide) (PLA) homopolymer or PLA that was surface modified with grafted polyethylene glycol chains. They were compared with a previously described intravenous halofantrine preparation. No toxic effects were observed with halofantrine in form of nanocapsules after intravenous administration for doses of up to 100 mg/kg, whereas the solubilized form in polyethylene glycol-dimethylacetamide was toxic at this dose. The halofantrine-loaded nanocapsules showed activity that was similar to or better than that of the solution in the 4-day test and as a single dose in severely infected mice, with only minimal differences between the two nanocapsule formulations. Halofantrine pharmacokinetics were determined in parallel with parasite development in severely infected mice. Nanocapsules increased the area under the curve for halofantrine in plasma more than sixfold compared with the solution throughout the experimental period of 70 h. Furthermore, nanocapsules induced a significantly faster control of parasite development than the solution in the first 48 h posttreatment. While the parasitemia fell more rapidly with PLA nanocapsules, the effect was more sustained with the surface-modified ones. This is consistent with surface-modified nanocapsules remaining longer in the circulation. These results suggest that nanocapsule formulations could provide a more favorable halofantrine profile in the plasma and reduce the intravenous dose necessary and therefore the toxicity, thus suggesting the use of halofantrine by a parenteral route in severe malaria.
The emergence of chloroquine resistance in Plasmodium falciparum has spurred the search for new antimalarial drugs. One of these, halofantrine hydrochloride, is a 9-phenanthrenemethanol, one of over 300,000 potential antimalarial compounds tested by the Walter Reed Institute and further developed by SmithKline Beecham as Halfan. It is one of the most active antimalarial drugs against P. falciparum in vitro, with a long half-life which lessens the need for repeated administration. Its antimalarial activity is mediated partly by the parent drug and partly by its N-desbutyl metabolite (2, 7). It has a place in treatment of multiresistant and chloroquine-resistant strains of P. falciparum when administered orally. However, halofantrine has serious cardiotoxicity, prolonging the QT interval, which also causes the cardiac side effects of quinine and mefloquine. Furthermore, cross-resistance between quinoline antimalarial drugs may occur.
Severe malaria is a medical emergency, during which the rapid achievement of therapeutic drug concentrations in the blood is a priority and parenteral administration is the rule (21). In order to provide a fast-acting treatment for severe malaria cases, an intravenous (i.v.) formulation of halofantrine has been tested clinically in humans (8). However, it was not without adverse effects, including severe local irritation with strong erythema (probably related to the toxicity of the solvents used in preparation) as well as serious cardiac side effects (QT interval prolongation), which bring into question its routine use. Furthermore, this formulation has to be given as a perfusion, whereas direct bolus injection would be more practical.
We have developed a colloidal drug delivery system, a long-circulating formulation of nanocapsules (NC) with an oily core in which halofantrine free base was entrapped (13). This type of formulation shows reduced uptake by the mononuclear phagocyte system and therefore is able to circulate longer after i.v. administration because of steric stabilization afforded by polyethylene glycol (PEG) chains linked to the NC surface (14, 15). These surface-modified NC would be expected to act as circulating reservoirs of antimalarial drug, releasing the drug progressively and therefore increasing its availability to parasitized red blood cells (RBC).
The components used to produce NC, i.e., biodegradable and well defined polymers such as poly(d,l-lactic acid) (PLA), poloxamer 188, and soy lecithin as surfactants and Miglyol 810 as the central oily core, are all generally considered safe for use in parenteral formulations. The PLA-PEG copolymer (20) has not yet undergone extensive toxicity testing but would be expected to be cleaved by esterases to release PEG, which can be eliminated by renal filtration (23). Halofantrine can be entrapped within the oily core at high concentrations, leading to a readily dispersible formulation of the drug. The easy preparation of NC by inexpensive and rapid methods is another advantage (4, 9). Furthermore, the encapsulation of halofantrine within NC without surface modification by PEG was able to greatly reduce the prolongation of the QT interval in anesthetized rats compared with free halofantrine (10). This can be explained by the retention of the drug within the core of the NC, reducing its contact with tissues.
In preliminary studies, NC formulations containing halofantrine were evaluated in vitro against P. falciparum and showed the same activity as free drug; that is, in a standard 48-h test the 50% inhibitory concentrations for the free and encapsulated forms were the same (unpublished results). This indicates that intact halofantrine could be released from NC in a medium containing serum proteins.
The aim of the present work was to assess the activity of halofantrine-loaded NC in vivo, in Plasmodium berghei-infected mice. Several different experimental protocols were chosen. Two different NC formulations were tested: one bearing PEG chains at the surface and therefore expected to be long-circulating and another without PEG, which would be expected to be captured rapidly by the liver and spleen. These were compared with the i.v. preparation of the drug described by Krishna et al. (8). In an attempt to relate circulating drug concentrations to efficacy, the pharmacokinetic profile of halofantrine was measured at the same time as disease progression in severely infected mice.
MATERIALS AND METHODS
Chemicals.
Soy lecithin (soy phosphatidylcholine [∼70% phosphatidylcholine]) Epikuron 170 was purchased from Lucas Meyer (Le Blanc Mesnil, France), and poloxamer 188 (Synperonic F68) was purchased from ICI (Cleveland, United Kingdom). PLA (average molecular weight, 42,000) was supplied by Phusis (Saint Ismier, France), and PLA-PEG 45-20 copolymer (PLA molecular weight, 45,000; PEG molecular weight, 20,000), synthesized and characterized as described previously (20), was provided by R. Gref (UMR 8612, Chatenay-Malabry, France). Miglyol 810 was a gift from Hulls (Marl, Germany). Halofantrine hydrochloride (racemic mixture), desbutylhalofantrine hydrochloride, and the internal standard 2,4-dichloro-9-(2-dibutylamino-1-hydroxy)ethyl-6-trifluoromethylphenanthrene hydrochloride were kindly supplied by the manufacturer (SmithKline Beecham, Welwyn, United Kingdom). Acetonitrile and methanol (Prolabo, Paris, France) high-performance liquid chromatography (HPLC) grade. Triethylamine and tert-butyl methyl ether (HPLC grade) were purchased from Sigma (Saint Quentin Fallavier, France). All other chemicals and solvents were analytical reagent grade. Water was purified by reverse osmosis (Milli-Q; Millipore).
Parasite.
P. berghei NK-173, a strain free of contamination with Eperythrozoon coccoides and sensitive to chloroquine, was used for antimalarial evaluation in vivo. This strain is known to induce high mortality in mice, providing a good model to estimate survival and antimalarial efficacy in reducing parasitemia, and is sensitive to all currently used antimalarial drugs. It was kindly supplied, frozen in nitrogen, by I. Landau, Muséum National d'Histoire Naturelle, Paris, France.
Animals.
Animal experiments were carried out according to the Principles of Laboratory Animal Care and legislation in force in France. Female outbred eperythrozoon-free Swiss albino mice (CD1) weighing 20 to 24 g were obtained from Charles River, L’Arbresle, France. They were kept in a normal diurnal cycle and had free access to food and water throughout the experiments.
NC preparation.
Halofantrine free base was prepared by treating a solution of the salt as previously described (19). NC were obtained by interfacial polymer deposition following solvent displacement as previously described (4, 15). Soy lecithin (0.75%, wt/vol) and Miglyol 810 (2.5%, wt/vol) as the hydrophobic surfactant and oil phase, respectively, were used with PLA and poloxamer 188 as a hydrophilic surfactant to prepare unmodified NC (PLA-POLOX NC). PLA-PEG 45-20 diblock copolymer was used to obtain surface-modified NC (PLA-PEG NC) at 30% (wt/wt) PEG of total polymer (5.1% of NC total weight). Briefly, 60 mg of total polymer, 75 mg of lecithin, 250 μl of Mygliol 810, and 1 mg of halofantrine base were dissolved in 10 ml of acetone. This organic solution was poured into 20 ml of external aqueous phase (containing 0.375% [wt/vol] poloxamer 188 in the case of PLA NC only) under agitation. The solvents was evaporated to 10 ml under reduced pressure. For the dose-response study, the amount of halofantrine in the acetone phase was increased so as to give different doses of the drug with the same polymer concentration. The mean size and polydispersity index of the NC were determined as previously described (16). For comparison, a parenteral solution of halofantrine was prepared by dissolving the hydrochloride in dimethylacetamide-PEG 400 at 40:60 (vol/vol) as previously described (8) and further diluting in 5% (wt/vol) glucose. The NC were also diluted in 5% (wt/vol) glucose before i.v. injection into mice (1 mg/kg of body weight in 200 μl for NC or 100 μl for the solution).
Antimalarial activity in P. berghei-infected mice. (i) Four-day screening test.
The 4-day test was performed as described by Peters et al. (18). An infective inoculum was prepared from a previously infected donor mouse with rising parasitemia (20%). On day 0 the mice were infected i.v. with 106 P. berghei-parasitized RBC in 0.2 ml of phosphate-buffered saline. They were randomly divided into seven groups of 10 and treated once daily, by the i.v. route or orally by gavage, with the different formulations of halofantrine for four consecutive days (days 0 to 3). For oral administration, 0.2 ml of micronized halofantrine suspension (0.2% [wt/vol] carboxymethyl cellulose and 0.04% [wt/vol] Tween 80) was given by gavage to each mouse at a dose of 4 mg of halofantrine/kg/day. By the i.v. route, halofantrine-treated groups received a dose of 1 mg/kg/day. Control groups received 0.2 ml of unloaded NC per day at the same concentration as the halofantrine-loaded NC preparations (6 mg of polymer/kg/day).
Thin blood smears were made from tail blood from untreated controls and from treated animals on days 4, 7, and 16 after infection. Levels of parasitemia were measured in Giemsa-stained smears, and RBC numbers were determined on the same days.
(ii) Dose-response study.
For the dose-response study, severely infected mice were used in a protocol adapted from that described by Osdene et al. (17). One million P. berghei-parasitized RBC (prepared as described above) were injected intraperitoneally. The mice were randomly divided into 15 treatment groups and 1 control group of 10 mice each on day 0. Groups were treated 3 days after infection by the i.v. route with a single dose of halofantrine (1 to 100 mg/kg) in different formulations.
Thin blood smears were made from tail blood on days 3 (before treatment), 5, 6, 10, 14, 32, and 60 after infection. Levels of parasitemia were determined on Giemsa-stained smears (both ring stages and schizonts were counted). If mice were free from Plasmodium after at least 200 fields (magnification, ×1,000) were checked, they were considered cured but were kept under observation for 60 days in case of relapse.
(iii) Correlation between efficacy and halofantrine pharmacokinetics.
The study of the correlation between efficacy and halofantrine pharmacokinetics also used the protocol of Osdene et al. (17). One million P. berghei-parasitized RBC from a previously infected donor mouse with rising parasitemia (20%) were diluted in 0.2 ml of phosphate-buffered saline and injected intraperitoneally. The mice were immediately randomly divided into eight groups of 6 to 10 mice for each formulation and a separate control group of 8 mice (no treatment). The groups were treated 5 days after infection with a single dose of halofantrine in different formulations (1 mg/kg) by the i.v. route. Thin blood smears were made from tail blood on day 5 after infection (before treatment) and at various times after treatment for each mouse. The mice were then anesthetized by intraperitoneal injection of 100 μl of sodium pentobarbital (35 mg/kg) and bled by cardiac puncture. Blood (approximately 1 ml) was collected in heparinized microcentrifuge tubes (10 μl). The plasma was separated by centrifugation (900 × g) and stored at −80°C for determination of the halofantrine concentration by HPLC. The level of parasitemia was assessed on Giemsa-stained smears. At least 10 fields of about 100 cells were checked. The ratio of the parasitemia at each time after treatment to that before treatment was determined for each mouse. The animals were severely infected on day 5 (45% ± 15% parasitemia), and they died from day 7 postinfection if untreated.
(iv) Statistics.
All RBC counts and parasitemia levels are expressed as mean values ± standard deviations. The parasitemia data were analyzed by using the one-way analysis of variance test. Mean survival times were compared by using Student's t test, considering a probability of 5% to be significant.
Halofantrine pharmacokinetics. (i) Extraction of halofantrine and its metabolite from plasma samples.
Extraction and recovery assays were performed as previously described by Humberstone et al. (5). Plasma samples (0.4 ml) in polypropylene centrifuge tubes were spiked with 100 μl of internal standard (2 μg/ml in acetonitrile), and 1.0 ml of acetonitrile was added. The tubes were vortexed for 2 min to precipitate plasma proteins and centrifuged, and the supernatant was separated. In clean tubes, 4 ml of tert-butyl methyl ether and 1 ml of purified water containing 0.05% of orthophosphoric acid were added to the acetonitrile phase. The contents were vortexed for 2 min and separated by centrifugation for 10 min at 800 × g at 4°C. A 4-ml portion of the upper tert-butyl methyl ether layer was carefully removed and placed in a tube containing 50 μl of 0.005 M HCl in acetonitrile. The contents were evaporated to dryness under vacuum. The residue was reconstituted with 200 μl of acetonitrile, and 25 μl was injected onto the HPLC column.
(ii) HPLC analysis of halofantrine and its metabolite.
Concentrations of halofantrine and its main active metabolite, desbutylhalofantrine, in plasma were determined by HPLC under isocratic conditions by the method previously described by Mberu and Muhia (11). The HPLC system consisted of a Waters programmable pump (Waters 600 controller) and a Waters 717 WISP automatic injector (Waters-Millipore, Saint Quentin en Yvelines, France) connected to a Hypersil 5 octyldecyl silane 250- by 4.6-mm C18 60Δ 4-μm column (NOVA-PAK; Waters-Millipore) preceded by a Guard-Pak C18 end-capped 5-μm, 10- by 4.6-mm precolumn (Waters-Millipore). The column effluent was monitored with a variable-wavelength absorbance detector (Waters model 484) set at 254 nm. Data were analyzed on a Waters model 746 integrator. The mobile phase was acetonitrile-water (65:35, vol/vol) containing 1% triethylamine and adjusted to pH 4 with orthophosphoric acid. The flow rate was 1.5 ml/min, and all chromatograms were obtained at room temperature.
No interference between the internal standard, halofantrine, desbutylhalofantrine, and the sodium pentobarbital used in mouse anesthesia was detected in the chromatograms. Halofantrine, desbutylhalofantrine, and the internal standard were well resolved to baseline over the range of 10 to 3,000 ng used in this study, with retention times of 4.87, 3.10, and 5.84 min, respectively. The lowest measurable concentration of halofantrine and desbutylhalofantrine was 10 ng/ml from 0.4 ml of plasma, with a peak five times larger in area than the background. The recovery of the internal standard in plasma samples was 85% ± 10%.
(iii) Pharmacokinetic analysis.
In order to compare the different formulations, the mean area under the curve (AUC) for halofantrine and desbutylhalofantrine concentrations in plasma was calculated by the trapezoidal rule during the experimental period from 0.5 to 70 h (AUC0.5-70). Thus, the overestimated plasma clearance (CL) and the mean residence time (MRT) were calculated as follows: CL = dosei.v./AUC0.5-70, and MRT = 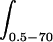 tCndtn/AUC0.5-70.
tCndtn/AUC0.5-70.
RESULTS
NC are a suitable carrier for halofantrine because the free base is very soluble in oil (>180 mg/ml) and has a favorable oil-water partition coefficient (log P = 6.5). A high drug concentration was obtained, and the drug was encapsulated with a yield of more than 95% (13). The mean diameter of the NC was around 180 nm as determined by quasielastic light scattering, and the populations were monodisperse. On the other hand, when the halofantrine solution was prepared by the protocol described in the literature (8), a 15-fold dilution of a 50-mg/ml organic solution in dextrose led to a suspension containing particles of 470 nm in diameter which aggregated on standing. Therefore, when the total dose to be administered allowed, a 5-mg/ml organic solution was prepared and was diluted 20 times a few minutes before injection to obtain a completely limpid preparation.
Four-day screening test.
The 4-day screening test was chosen for initial screening of the activity of the different halofantrine formulations administered by the i.v. route. Oral halofantrine was used as a control because this is the normal route of administration for this drug. The results are shown in Fig. 1 and Table 1. Untreated control mice were highly parasitized (about 50%) by day 4, with a low hematocrit, and they died on days 5 to 10. A similar profile of survival was seen for mice treated with unloaded PLA-POLOX NC (Fig. 1). Mice treated with unloaded PLA-PEG NC had a slightly extended mean survival time, with 40% of the mice living for more than 15 days postinfection, although the parasitemia and RBC levels were not significantly different from those of untreated controls (Table 1).
FIG. 1.

Survival of P. berghei-infected CD1 mice after treatment with halofantrine i.v. (four doses of 1 mg/ml) compared with controls in the 4-day test. Mice were treated from day 0 to day 3 after infection with one million parasitized RBC. Each group consisted of 10 mice. Controls with unloaded NC received 0.6 mg of polymer per mouse.
TABLE 1.
Treatment of P. berghei-infected CD1 mice with different formulations of halofantrine (4-day test)
| Treatment | Day 4
|
Day 7
|
MSTe (days) | ||
|---|---|---|---|---|---|
| % Parasitemiac | RBC (106/μl) | % Parasitemiac | RBC (106/μl) | ||
| Control | 52.6 ± 13.8 (31-71) | 2.5 ± 0.7 | 37.6 ± 6.1 (31-44) | 2.1 ± 0.6 | 7.1 (5-10) |
| Unloaded PLA-POLOX NC | NDg | ND | ND | ND | 6.6 (5-9) |
| Unloaded PLA-PEG 20-30 NC | 50.4 ± 17.0 (21-69) | 2.6 ± 0.8 | 31.1 ± 19.5 (27-52) | 2.0 ± 0.4 | 13.6 (7-26) |
| Halofantrine formulations | |||||
| i.v. solutiona | 0.5 ± 0.8 (0-2.0)d | 5.5 ± 1.1 | 1.1 ± 0.5 (0-1.5)d | 7.8 ± 1.1 | >60 (n = 10) |
| PLA-POLOX NCa | 0.5 ± 0.9 (0-2.5)d | 6.0 ± 1.3 | 0.9 ± 0.9 (0-2.5)d | 8.1 ± 1.0 | >60 (n = 8)f |
| PLA-PEG 20-30 NCa | 0.5 ± 0.5 (0-1.5)d | 4.9 ± 0.2 | 1.3 ± 1.2 (0-4)d | 7.0 ± 0.7 | >60 (n = 10) |
| Oral suspensionb | 0.4 ± 0.7 (0-1.6)d | 4.9 ± 0.7 | 0.3 ± 0.6 (0-1.6)d | 8.0 ± 0.7 | >60 (n = 10) |
Mice were treated i.v. with 1 mg/kg/day in a 4-day-test.
Mice were treated orally with 4 mg/kg/day in a 4-day-test.
Values are mean parasitemia ± standard deviations (95% confidence intervals are in parentheses).
Significantly different from controls in one-way analysis of variance (95% confidence intervals).
MST, mean survival time. The range or number of surviving mice is in parentheses.
One mouse each died on days 15 and 28.
ND, not determined.
Oral treatment of 4 mg/kg/day for 4 consecutive days was able to reduce parasitemia to very low levels on days 4 and 7 and to cure all of the mice (Table 1). The RBC levels was also restored, with an overshoot at day 7, due to a erythropoietic response to the earlier hemolysis. By the i.v. route, four doses of 1 mg/kg as a solution or as PLA-PEG NC cured 100% of the mice, whereas with PLA-POLOX NC, 80% were cured (Fig. 1). For all three preparations, the parasitemia at days 4 and 7 was substantially reduced and the hematocrit was restored (Table 1).
Dose-response study.
For the dose-response study, a different experimental protocol was chosen in an attempt to distinguish between the different i.v. formulations. It also represents a better model for the treatment of severe malaria in humans because the infection was already established when the drug was administered. NC formulations containing up to 100 mg of halofantrine/kg (with a constant polymer dose of 6 mg/kg) were administered with no adverse effects. On the other hand, injection of 100 mg of halofantrine/kg caused immediate death of the treated mice.
The mean survival times of the different experimental groups and the evolution of the parasitemia are shown in Table 2. A single i.v. dose of 1 mg of halofantrine per kg as a solution or as PLA-PEG NC was unable to arrest the infection or to prolong the survival of the mice compared with the untreated control. Similarly, unloaded PLA-POLOX NC had no effect on the course of the disease; unloaded PLA-PEG NC were not tested in this protocol. On the other hand, 1 mg of halofantrine/kg in PLA-POLOX cured 80% of the mice. A similar survival was observed with 3 mg of this formulation/kg, but in this case the parasitemia declined more rapidly. All other formulations tested (solutions of 3 to 30 mg/kg, PLA-POLOX NC at 10 to 100 mg/kg, and PLA-PEG NC at 3 to 100 mg/kg) produced 100% cures. The parasitemia declined in an approximately dose-dependent manner and had disappeared completely at day 32 in all surviving mice. At doses of 3 mg/kg and above, the parasitemia declined more rapidly with the NC formulations than with the solution. At day 5, the parasitemia was slightly lower with PLA-POLOX NC than with PLA-PEG NC at all doses except 100 mg/kg. However, at days 10 and 14, this tendency was reversed, except at 1 mg/ml, which was ineffective in the form of PLA-PEG NC.
TABLE 2.
Antimalarial activities of different formulations in the P berghei-infected mouse model after a single i.v. dose
| Formulation of halofantrine | Dose (mg/kg) | MST (days)b | No. of survivors (n = 10) | Mean parasitemia (%) ± SDa on day:
|
|||
|---|---|---|---|---|---|---|---|
| 5 | 10 | 14 | 32 | ||||
| i.v. solution (8) | 1 | 18 (13-24) | 0 | 35.4 ± 10.5 | 43.0 ± 2.6 | 31.8 ± 4.1 | |
| 3 | >60e | 10 | 16.0 ± 12.5 | 27.5 ± 15.8 | 23.0 ± 13.6 | 0 | |
| 10 | >60e | 10 | 12.8 ± 8.2 | 7.3 ± 0.9 | 7.5 ± 6.0 | 0 | |
| 30 | >60e | 10 | 13.3 ± 7.3 | 2.4 ± 1.4 | 2.5 ± 4.6 | 0 | |
| 100f | |||||||
| PLA-POLOX NC | 1 | >60c,e | 8 | 30.5 ± 6.7 | 42.5 ± 8.0 | 22.8 ± 4.9 | 0 |
| 3 | >60d,e | 8 | 6.3 ± 2.2 | 16.3 ± 0.8 | 17.8 ± 3.9 | 0 | |
| 10 | >60e | 10 | 4.7 ± 2.7 | 2.3 ± 1.3 | 4.1 ± 1.3 | 0 | |
| 30 | >60e | 10 | 5.6 ± 3.6 | 1.8 ± 2.2 | 2.4 ± 1.8 | 0 | |
| 100 | >60e | 10 | 7.5 ± 1.0 | 1.6 ± 1.0 | 0 | 0 | |
| PLA-PEG NC | 1 | 9e (6-12) | 0 | 33.8 ± 7.5 | 45.1 ± 1.5 | ||
| 3 | >60e | 10 | 7.7 ± 2.6 | 11.5 ± 3.5 | 12.5 ± 6.1 | 0 | |
| 10 | >60e | 10 | 5.6 ± 3.7 | 7.4 ± 1.5 | 1.6 ± 1.0 | 0 | |
| 30 | >60e | 10 | 7.8 ± 3.9 | 2.5 ± 2.0 | 0.8 ± 0.5 | 0 | |
| 100 | >60e | 10 | 7.1 ± 2.9 | 1.7 ± 1.4 | 0.3 ± 0.1 | 0 | |
| Unloaded PLA NC | 7 (5-10) | 0 | 32.6 ± 9.2 | ||||
| Untreated control | 11 (6-24) | 0 | 34.7 ± 6.1 | 28.5 ± 7.8 | 33.5 ± 12.0 | ||
The value on day 3 was 15.3 ± 7.9.
MST, mean survival time. The range is in parentheses.
Two mice died on day 14.
Two mice died on day 12.
Significantly different from controls by Student's t test (95% confidence intervals).
Correlation between pharmacokinetics of halofantrine and efficacy.
In the pharmacokinetic study, we chose a noncurative dose of 1 mg/kg in a severe malaria model so as to monitor the evolution of parasitemia and to be able to determine the relationship between the pharmacokinetics and the pharmacological effect (22).
Figure 2A shows that both NC formulations were able to modify the clearance profile of halofantrine in plasma compared with the solution and that this difference was maintained for a long period (up to 70 h for PLA-PEG NC). The AUC for halofantrine-loaded NC was increased more than sixfold compared with the solution (Table 3). Figure 2B shows that there was little difference in the clearance profiles of the principal metabolite, desbutylhalofantrine. However, a higher concentration in plasma was observed with PLA-POLOX NC at short times (up to 24 h) and with PLA-PEG NC at longer times (after 40 h).
FIG. 2.

Plasma clearance profiles of halofantrine and its main metabolite and evolution of parasitemia after i.v. administration of 1 mg/kg as a solution, conventional NC (PLA-POLOX NC), or PEG-grafted NC (PLA-PEG NC) to P. berghei-infected mice on day 5 after infection. Results are means ± standard deviations for 6 to 10 mice. (A) Plasma clearance profiles of halofantrine. (B) Plasma clearance profiles of desbutylhalofantrine. (C) Evolution of parasitemia. Results are expressed as a fraction of the parasitemia measured just before treatment in the same mouse. In the control (untreated) group, four of eight mice died between 28 and 48 h and one more mouse died between 48 and 71 h.
TABLE 3.
Pharmacokinetic parameters of i.v. halofantrine in different formulations
| Formulationa | Halofantrine
|
Desbutylhalofantrine
|
|||
|---|---|---|---|---|---|
| AUC0.5-70 (μg · h/ml) | CL (ml/h) | MRT0.5-70 (h) | AUC0.5-70 (μg · h/ml) | MRT0.5-70 (h) | |
| Solution | 1.91 | 13.09 | 24.4 | 11.7 | 34.4 |
| PLA-POLOX NC | 12.00 | 2.08 | 16.8 | 15.0 | 33.1 |
| PLA-PEG NC | 11.80 | 2.11 | 22.4 | 15.5 | 41.3 |
The halofantrine concentration in NC was 0.125 mg/ml, and that in isotonic solution was 0.25 mg/ml. The total dose was 1 mg/kg.
The efficacy of halofantrine was monitored at the same time as its pharmacokinetics. Figure 2C shows the evolution of parasitemia in the different groups. The results are presented as a fraction of the parasitemia measured in the mice just before treatment (45% ± 15%). Untreated control mice maintained a high level of parasitemia and began to die 7 days after infection, i.e., the equivalent of 48 h after drug administration. The evolution of parasitemia in the treated mice is consistent with the pharmacokinetic profiles. Parasitemia started to decline earlier in mice treated with PLA NC and PLA-PEG NC than in those treated with the solution, which reduced parasitemia after 48 h only. Furthermore, the effect of PLA NC could be seen slightly earlier than that of PLA-PEG NC (P < 0.05 between 2 and 16 h) (Fig. 2C). However, at longer times, PLA-PEG NC showed a slight advantage over PLA NC (P < 0.05 at 48 and 70 h). On the other hand, a dramatic and significant difference was observed between both NC formulations and halofantrine solution (P < 0.05 at 28, 48, and 70 h for PLA-PEG NC and at 2, 8, 28, and 48 h for PLA NC).
DISCUSSION
No overt signs of toxicity or abnormal behavior were observed when halofantrine NC were injected i.v. into mice, and the maximum tolerated dose was higher than that of halofantrine solubilized in dimethylacetamide-PEG. This is in accordance with observations of the reduced cardiotoxicity of halofantrine in NC in rats (10), although electrocardiograms were not performed in the present study. Furthermore, NC were stable for 10 months at 4°C, while the solution had to be prepared extemporaneously due to rapid precipitation of the drug. This instability could explain the severe local irritation experienced when this formulation was administered to human patients (8).
A severely infected mouse model was used to determine the pharmacokinetics of halofantrine administered as the three different formulations. It is clear that entrapment in NC changes the pharmacokinetic profile (Fig. 2A). Halofantrine delivered as a solution is removed rapidly from the circulation, in accordance with its rapid uptake by tissues (7, 8). On the other hand, NC would be able to release the drug progressively in the blood compartment and to reduce its uptake by the tissues. It was not possible to calculate a volume of distribution for the different formulations because of the difficulty of extrapolating the AUC to infinity. The pharmacokinetics of the principal metabolite, desbutylhalofantrine, which has potency similar to that of the parent drug, varied less between the formulations (Fig. 2B). This may be because although the NC-associated drug was taken up more gradually into tissues, most was accumulated in the liver, the principal site of metabolism by cytochrome P450 (1).
Despite the different biodistribution profiles of unloaded PLA-PEG and PLA-POLOX NC (15), there was very little difference between the pharmacokinetic parameters of halofantrine encapsulated in these formulations. This could be a consequence of performing the experiments with heavily parasitized mice as opposed to healthy animals. The involvement of the liver and spleen in clearing senescent and parasitized RBC in infected animals might saturate the mononuclear phagocyte system, thus affecting NC pharmacokinetics and reducing the difference between PLA-POLOX NC and PLA-PEG NC pharmacokinetics (6). Another parameter which is affected by malaria infection is liver blood flow, which is often a limiting factor in drug elimination.
Furthermore, the halofantrine measured in the plasma included both that still associated with the NC and drug which had been released. For a highly lipophilic compound like halofantrine, release will be strongly dependent on partitioning between the oily core and suitable acceptor molecules, usually proteins (9). Low- and high-density lipoproteins are the main halofantrine-binding proteins in plasma (3). Although it has also been shown that in individuals suffering from malaria the lipoprotein level in the blood may be reduced (12), a large part of the drug found in the plasma at later incubation times may be associated with lipoproteins rather than NC. It should also be noted that in this study the drug was measured in plasma, not whole blood, so the amount associated with RBC, its site of action, was not taken into account. For this reason, it was interesting to compare the pharmacokinetic profiles with the efficacy against the malaria parasites.
There was in fact a close correlation between plasma halofantrine concentrations and the pharmacological effect (reduction of parasitemia) (Fig. 2C). PLA-PEG NC were able to reduce the parasitemia more rapidly than PLA-POLOX NC. This was also shown in the dose-response study reported in Table 2. This may be due to the more rapid availability of drug from these NC, since in vitro experiments have indicated that release is retarded in the presence of PEG (13). On the other hand, the decrease in parasitemia was more sustained for the PLA-PEG formulation, correlating with a higher plasma halofantrine concentration at 70 h and the fact that these NC might be expected to remain in the circulation longer. The slower release of halofantrine from PLA-PEG NC may also explain why they were more effective in the survival study (Fig. 1). These NC also had some effect in prolonging survival without associated drug. This phenomenon is difficult to explain but may be due to an association of PEG with the RBC membrane, preventing reinfection.
Both NC formulations provide a stable, well tolerated preparation of halofantrine, but they appear to have complementary properties. PLA-POLOX NC can provide a fast effect due to relatively rapid release of drug, whereas PLA-PEG NC provide a more sustained effect because of their continued presence in the circulation. A mixture of the two types of particle could be envisaged as a prolonged delivery system for i.v. administration of halofantrine. Of course, such a formulation would be destined for the treatment of severe malaria, in which parasites are sequestered within the tissues and in particular in cerebral capillaries. Although we have not tested our formulations in a model of cerebral malaria, we expect that the increased plasma halofantrine concentrations would improve its delivery to parasites sequestered in the tissues.
Acknowledgments
We thank Henri Vial and Anne Bonhoure (CNRS UMR 5539, Montpellier, France) for checking the activity against Plasmodium falciparum in vitro. We are grateful to Stéphanie Ronsse for her help with animal experiments and to SmithKline Beecham, Welwyn, United Kingdom, for supplying the internal standard, halofantrine, and desbutylhalofantrine.
V.C.F.M. received financial support from the Brazilian National Council of Scientific and Technological Development (CNPq). This work was supported by CNRS, France.
REFERENCES
- 1.Baune, B., V. Furlan, A. M. Taburet, and R. Farinotti. 1999. Effect of selected antimalarial drugs and inhibitors of cytochrome P-450 3A4 on halofantrine metabolism by human liver microsomes. Drug Metab. Dispos. 27:565-568. [PubMed] [Google Scholar]
- 2.Bryson, H. M., and K. I. Goa. 1992. Halofantrine. A review of its antimalarial activity, pharmacokinetic properties and therapeutical potential. Drugs 43:236-258. [DOI] [PubMed] [Google Scholar]
- 3.Cenni, B., J. Meyer, R. Brandt, and B. Betschart. 1995. The antimalarial drug halofantrine is bound mainly to low and high density lipoproteins in human serum. Br. J. Clin. Pharmacol. 39:519-526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fessi, H., F. Puisieux, J.-P. Devissaguet, N. Ammoury, and S. Benita. 1989. Nanocapsule formation by interfacial polymer deposition following solvent displacement. Int. J. Pharm. 55:R1-R4. [Google Scholar]
- 5.Humberstone, A. J., G. J. Currie, C. J. H. Porter, M. J. Scanlon, and W. N. Charman. 1995. A simplified liquid chromatography assay for the quantification of halofantrine and desbutylhalofantrine in plasma and identification of a degradation product of desbutylhalofantrine formed under alkaline conditions. J. Pharm. Biomed. Anal. 13:265-272. [DOI] [PubMed] [Google Scholar]
- 6.Illum, L., N. W. Thomas, and S. S. Davis. 1986. Effect of a selected suppression of the reticuloendothelial system on the distribution of model carrier particles. J. Pharm. Sci. 75:16-21. [DOI] [PubMed] [Google Scholar]
- 7.Karbwang, J., and K. N. Bangchang. 1994. Clinical pharmacokinetics of halofantrine. Clin. Pharmacokin. 27:104-119. [DOI] [PubMed] [Google Scholar]
- 8.Krishna, S., F. ter Kuile, W. Supanaranond, S. Pukrittayakamee, P. Teja-Isavadharm, D. Kyle, and N. J. White. 1993. Pharmacokinetics, efficacy and toxicity of parenteral halofantrine in uncomplicated malaria. Br. J. Clin. Pharmacol. 36:585-591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Legrand, P., G. Barratt, V. C. F. Mosqueira, H. Fessi and J.-P. Devissaguet. 1999. Polymeric nanocapsules as drug delivery systems: a review. S. T. P. Pharma Sci. 9:411-418. [Google Scholar]
- 10.Leite, E. A., A. G. Guimarães, H. N. Guimarães, and V. C. F. Mosqueira. 2001. Entrapment of antimalarial drug in biodegradable nanocapsules reduces side effect of heart damage. In M. Pinotti, L. Wykrota, and L. Poletto (ed.), Proceedings of the II Latin American Congress of Artificial Organs and Biomaterials, reference BRMG046. [CD-ROM.]
- 11.Mberu, E. K., and D. K. Muhia. 1992. Measurement of halofantrine and its major metabolite desbutylhalofantrine in plasma and blood by high-performance liquid chromatography: a new methodology. J. Chrom. Biom. Appl. 581:156-160. [DOI] [PubMed] [Google Scholar]
- 12.Mohanty, S., S. K. Mishra, B. S. Das, S. K. Satpathy, D. Mohanty, J. K. Patnaik, and T. K. Bose. 1992. Altered plasma lipid pattern in falciparum malaria. Ann. Trop. Med. Parasitol. 86:601-606. [DOI] [PubMed] [Google Scholar]
- 13.Mosqueira, V. C. F., P. Legrand, R. Gref, and G. Barratt. 1999. In-vitro release kinetic studies of PEG-modified nanocapsules and nanospheres loaded with a lipophilic drug: halofantrine base. Proc. Int. Symp. Control. Rel. Bioact. Mater. 26:1074-1075. [Google Scholar]
- 14.Mosqueira, V. C. F., P. Legrand, R. Gref, B. Heurtault, M. Appel, and G. Barratt. 1999. Interactions between a macrophage cell line (J774 A1) and surface-modified poly(d,l-lactide) nanocapsules bearing poly(ethylene glycol). J. Drug Target. 7:65-78. [DOI] [PubMed] [Google Scholar]
- 15.Mosqueira, V. C. F., P. Legrand, J.-L. Morgat, M. Vert, E. Mysiakine, R. Gref, J.-P. Devissaguet, and G. Barratt. 2001. Biodistribution of novel long-circulating PEG-grafted nanocapsules in mice: effects of PEG chain length and density. Pharm. Res. 18:1411-1419. [DOI] [PubMed] [Google Scholar]
- 16.Mosqueira, V. C. F., P. Legrand, A. Gulik, O. Bourdon, R. Gref, D. Labarre, and G. Barratt. 2001. Relationship between complement activation, cellular uptake and surface physicochemical aspects of novel PEG-modified nanocapsules. Biomaterials 22:2969-2979. [DOI] [PubMed] [Google Scholar]
- 17.Osdene, T. S., P. B. Russell, and L. Rane. 1967. 2,4,7-Triamino-6-ortho-substituted arylpteridines. A new series of potent antimalarial agents. J. Med. Chem. 10:431-436. [DOI] [PubMed] [Google Scholar]
- 18.Peters, W., L. Ze-Lin, B. L. Robinson, and D. C. Warhurst. 1986. The chemotherapy of rodent malaria, XL. Ann. Trop. Med. Parasitol. 80:483-489. [DOI] [PubMed] [Google Scholar]
- 19.Porter, C. J. H., S. A. Charman, and W. N. Charman. 1996. Lymphatic transport of halofantrine in the triple-cannulated anesthetized rat model: effect of lipid vehicle dispersion. J. Pharm. Sci. 85:351-356. [DOI] [PubMed] [Google Scholar]
- 20.Quellec, P., R. Gref, L. Perrin, E. Dellacherie, F. Sommer, Y. M. Verbavatz, and M. J. Alonso. 1998. Protein encapsulation within polyethylene glycol-coated nanospheres. I. Physicochemical characterization. J. Biomed. Mater. Res. 42:45-54. [DOI] [PubMed] [Google Scholar]
- 21.Whitty, C. J. M., and F. Sanderson. 1999. New therapies and changing patterns of treatment of malaria. Curr. Opin. Infect. Dis. 12:579-584. [DOI] [PubMed] [Google Scholar]
- 22.Winstanley, P. A., and W. M. Watkins. 1992. Pharmacology and parasitology: integrating experimental methods and approaches to falciparum malaria. Br. J. Clin. Pharmacol. 33:575-581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamaoka, T., Y. Tabata, and Y. Ikada. 1994. Distribution and tissue uptake of poly(ethylene glycol) with different molecular weights after intravenous administration to mice. J. Pharm. Sci. 83:601-606. [DOI] [PubMed] [Google Scholar]