

Abstract
Sarcolipin (SLN) regulates muscle-based nonshivering thermogenesis and is up-regulated with high-fat feeding (HFF). To investigate whether other muscle-based thermogenic systems compensate for a lack of Sln and to firmly establish SLN as a mediator of diet-induced thermogenesis (DIT), we measured muscle and whole-body energy expenditure in chow- and high-fat-fed Sln−/− and wild-type (WT) mice. Following HFF, resting muscle metabolic rate (Vo2, μl/g/s) was increased similarly in WT (0.28±0.02 vs. 0.31±0.03) and Sln−/− (0.23±0.03 vs. 0.35±0.02) mice due to increased sympathetic nervous system activation in Sln−/− mice; however, whole-body metabolic rate (Vo2, ml/kg/h) was lower in Sln−/− compared with WT mice following HFF but only during periods when the mice were active in their cages (WT, 2894±87 vs. Sln−/−, 2708±61). Treatment with the β-adrenergic receptor (β-AR) antagonist propranolol during HFF completely prevented muscle-based DIT in Sln−/− mice; however, it had no effect in WT mice, resulting in greater differences in whole-body metabolic rate and diet-induced weight gain. Our results suggest that β-AR signaling partially compensates for a lack of SLN to activate muscle-based DIT, but SLN is the primary and more effective mediator.—Bombardier, E., Smith, I. C., Gamu, D., Fajardo, V. A., Vigna, C., Sayer, R. A., Gupta, S. C., Bal, N. C., Periasamy, M., Tupling, A. R. Sarcolipin trumps β-adrenergic receptor signaling as the favored mechanism for muscle-based diet-induced thermogenesis.
Keywords: calcium cycling, energy expenditure, skeletal muscle, obesity
simply stated, obesity results from a chronic imbalance between energy intake (i.e., feeding) and energy expenditure (i.e., metabolic rate). Behavioral and environmental factors leading to reduced activity levels and increased caloric intake are at the root of the problem; however, some individuals appear to increase energy expenditure in response to overfeeding and thus maintain a stable body weight. Diet-induced thermogenesis (DIT) is an increase in energy expenditure in response to caloric excess, which is thought to be regulated primarily by the sympathetic nervous system (SNS) through β-adrenergic receptor (β-AR) activation of mitochondrial uncoupling protein (UCP-1) in brown adipose tissue (BAT) (1, 2). However, given that BAT depots are minimal in larger mammals, this has prompted the search for alternative quantitatively important adaptive thermogenic pathways (3). Skeletal muscle has been speculated to play a role in DIT (4, 5), although a definitive mechanism for muscle-based DIT has proven to be elusive. Sarco(endo)plasmic reticulum Ca2+-ATPases (SERCA) pumps have been identified as major regulators of nonshivering thermogenesis (6, 7) and, therefore, could potentially contribute to muscle-based DIT.
Sarcolipin (SLN) is a small protein molecule that regulates the activity of SERCA pumps and is a key modulator of skeletal muscle relaxation (8). The first suggestion that SLN may have a thermogenic function was based on in vitro reconstitution experiments showing that SLN could uncouple ATP hydrolysis from Ca2+ transport by SERCA pumps (9) and increase the amount of heat released per molecule of ATP hydrolyzed (10). Promoting inefficient metabolism in muscle represents a potential treatment for obesity and its complications. We recently showed using Sln−/− mice that SLN functions to maintain core body temperature during acute cold challenge and that loss of Sln predisposes mice to diet-induced obesity (11), suggesting that SLN is an alternative thermogenic pathway that can mediate muscle-based DIT and metabolic inefficiency. The latter results were particularly impressive given the fact that our high-fat feeding (HFF) experiments were conducted below thermoneutrality of mice, where induction of compensatory alternative thermogenic pathways can mask an obesity phenotype in transgenic animals (1, 12). Interestingly, induction of the classical UCP-1 response occurred similarly in both wild-type (WT) and Sln−/− mice (11); however, it is not currently known whether other thermogenic systems exist within skeletal muscle that can be recruited to compensate for a lack of SLN and mediate muscle-based DIT. To answer this question and firmly establish SLN as a mediator of muscle-based DIT and metabolic inefficiency, we measured both muscle and whole-body energy expenditure in either chow or high-fat fed Sln−/− and WT mice.
MATERIALS AND METHODS
Sln−/− mice
The generation of Sln−/− mice has been described previously (13). Animals were housed in an environmentally controlled room with a standard 12:12-h light-dark cycle and allowed access to food and water ad libitum. Experiments were performed on littermate Sln−/− and WT males that were between the ages of 4–6 mo. All animal procedures were reviewed and approved by the Animal Care Committee of the University of Waterloo and are consistent with the guidelines established by the Canadian Council on Animal Care.
Oxygen consumption in isolated intact soleus muscles
Resting oxygen consumption (Vo2) of isolated intact soleus muscles from WT and Sln−/− mice was measured at 30°C using the TIOX tissue bath system (Hugo Sachs Electronik–Harvard Apparatus, March-Hugstetten, Germany). The O2 partial pressure of a bubble-free Ringer solution (in mM: 121 NaCl, 5 KCl, 1.8 CaCl2, 0.5 MgCl2, 0.4 NaH2PO4, 24 NaHCO3, 5.5 glucose, and 0.1 EDTA, pH 7.3), which was preheated to 30°C and aerated with 95% oxygen and 5% carbon dioxide, was measured by a Clark O2 electrode (model 1302; Warner Instruments Corp., Hamden, CT, USA) connected to a polarographic circuit, the output of which voltage is directly proportional to the O2- partial pressure. The muscle length was adjusted to achieve optimal length (Lo) for twitch force production, and then the muscle was given 10 min to equilibrate inside the chamber before initiating data collection. The decrease in Po2 of the Ringer solution was recorded every 4 s in the presence of a resting muscle at Lo for 30 min. The muscle Vo2 was calculated by multiplying the measured drop in Po2 with time by the solubility of oxygen in Ringer solution at 30°C and the chamber volume (12.7 ml). The solubility of oxygen at 30°C was calculated to be 0.001203 M/atm. In reality, the TIOX system is not completely closed to the atmosphere, and the electrode consumes oxygen to perform measurements, resulting in loss of oxygen out of the solution even with no muscle mounted inside the chamber. Therefore, to account for the oxygen loss, blank trials were done at the beginning and at the end of daily data collection. A blank trial measures the rate of oxygen loss from the empty chamber (i.e., no muscle). The average daily rate of oxygen loss is then subtracted from the oxygen loss in the presence of the muscle to give the muscle Vo2. The average rate of oxygen loss (μl/s) over the course of the experiment was found to be 0.00708 ± 0.00054 with a coefficient of variation of 7.2%. Muscle Vo2 is reported relative to muscle wet weight (μl/g/s), and volumes are adjusted to standard temperature and pressure (1.0 atmosphere, 23°C).
Resting whole-body metabolic parameters
Daily average whole-body Vo2 (ml/kg/h), food consumption (g/d), and cage activity (movement counts) were measured in WT and Sln−/− mice using a comprehensive laboratory animal monitoring system (CLAMS; Oxymax series; Columbus Instruments, Columbus, OH, USA). CLAMS studies began after 2 h of acclimation to the metabolic chamber, and Vo2 data were collected at 26-min intervals over a 72-h period under a consistent environmental temperature (22°C). X- and Z-activity sensors allowed for monitoring of ambulatory activity (when 2 adjacent x-axis beams were broken in succession) and total cage activity. O2 consumption was averaged over a 24-h period, as well as divided into inactive metabolic rate (readings with an ambulatory activity count ≤2/26 min) and active metabolic rate (readings with an ambulatory activity count >2/26 min). During these studies, mice had free access to food and water.
Treadmill exercise
WT and Sln−/− mice were assessed for submaximal and maximal aerobic capacity (Vo2max) by running them on an enclosed motorized treadmill that was connected to the CLAMS. Submaximal tests were performed at 2 different speeds for 10 min, with sampling every 30 s. The mice exercised at a running speed of 8 m/min for 10 min, followed by 10 min at 16 m/min. On a separate occasion, Vo2max was determined using a progressive exercise test where the mice started running at 7 m/min for 90 s and then the speed was increased by 3 m/min every 90 s until a plateau in Vo2 was reached or the mouse was incapable of continuing.
High-fat-diet (HFD)-induced obesity
WT mice and Sln−/− littermates were individually housed and fed either a standard chow diet (Teklad 22/5 Rodent Diet; Harlan-Teklad, Madison, WI, USA; 3.62 kcal/g, 5% kcal from fat) or an HFD (TD.88137; Harlan-Teklad; 4.5 kcal/g, 42% kcal from fat) ad libitum for a period of 8 wk. Body weight was monitored weekly and was expressed as weight gained in grams per mouse. Before and following the 8-wk diet period and before tissue and serum collection, whole-body Vo2 was assessed using the CLAMS, and glucose tolerance tests were performed on unfed mice (12 h food withrawal) where a total of 5–10 μl of blood was drawn from the tail and assayed for glucose using a blood glucose meter (Accu-Chek Aviva; Roche Diagnostics, Mississauga, ON, Canada) at 0, 30, 60, and 120 min following an intraperitoneal injection of 10% d-glucose at a dose of 1 g/kg body weight. Tissue and serum were collected from anesthetized (0.65 mg somnitol/kg body weight) chow- and high-fat-fed mice that were denied access to food for 4 h. Blood was collected from the left ventricle (700 μl) and spun down at 5000 g for 8 min. The resulting serum was collected and stored at −80°C until analysis. Soleus and intrascapular BAT were excised from the mice, cleaned of extraneous and connective tissues, weighed, and then stored at −80°C for Western blotting analyses. The epididymal and retroperitoneal fat pads were also removed and weighed. Adiposity was determined from the weights of the epididymal and retroperitoneal fat pad and calculated as an adiposity index, defined as 100 × (sum of fat pad weights)/body weight (ref. 14).
Propranolol study
Propranolol (2.5 mg/d, 90-d time-release pellet; Innovative Research of America, Sarasota, FL, USA) supplementation was administered via subcutaneous pellet implantation to WT and Sln−/− mice that were fed an HFD for 8 wk as described above.
Serum chemistry
Serum concentrations of leptin were measured using an enzyme-immunoassay (EIA; Alpco Diagnostics, Salem, NH, USA), and assay kits were used for nonesterified fatty acids (NEFAs; NEFA C, Wako Chemicals, Neuss, Germany) and low-density lipoprotein (LDL) cholesterol (total serum cholesterol/HDL-ADVANCE ASSAY; Diagnostic Chemicals, Charlottetown, PEI, Canada). Norepinephrine (NE) and epinephrine were determined using high-performance liquid chromatography and electrochemical detection as described previously (15).
Western blot analysis
Expression of SLN (custom-made antibody, 1:3000 dilution; ref. 16) and UCP-1 (PA1-3883, 1in 500; Thermo Fisher Scientific, Rockford, IL USA) were determined using standard Western blotting techniques. The tissue homogenates containing equal amount of protein were separated using standard SDS-PAGE (16% Tris-tricine gel for SLN and 7.5% acrylamide gels for UCPs). The proteins were transferred to a 0.2-μm nitrocellulose membrane for SLN or polyvinylidene difluoride membranes for UCP-1, and, after blocking with 5% skim milk containing 0.05% Tween 20 for 60 min at room temperature, the membranes were incubated with the appropriate primary antibody. After being washed in Tris-buffered saline with 0.05% Tween, the membranes were then treated for 30 or 60 min with horseradish-conjugated secondary antibody. Signals were detected with an enhanced chemiluminescence reagent (GE Healthcare, Baie d'Urfe, QC, Canada) using a bioimaging system, and densitometric analysis was performed using the GeneSnap software (Syngene, Frederick, MD, USA).
Statistics
Analyses were performed using Prism (GraphPad Software, La Jolla, CA, USA). Data are presented as means ± sem. Comparisons were by Student's t test or ANOVA followed by a Tukey's post hoc test. Values of P < 0.05 were considered significant.
RESULTS
Sln−/− mice develop greater obesity, hyperlipidemia, and glucose intolerance than WT mice following HFF
Sln−/− mice develop greater obesity and glucose intolerance than WT mice when fed an HFD (45% kcal from fat) for 12 wk (11). In this study, mice were fed a similar HFD (42% kcal from fat) for 8 wk, which also resulted in greater weight gain (P<0.05; Fig. 1A), adiposity (P<0.05; Fig. 1B), higher levels of circulating leptin (P<0.05; Fig. 1C), NEFAs (P<0.05; Fig. 1D), and LDL cholesterol (P<0.05; Fig. 1E), and more severe glucose intolerance (Fig. 1F) in Sln−/− mice compared with WT mice.
Figure 1.

Sln−/− mice develop greater obesity, hyperlipidemia, and glucose intolerance than WT mice following HFF. A) Increase in body weight during 8 wk of HFF. Values are means ± sem (n=16 or 17). *P < 0.05. B) Adiposity index [100 × (sum of fat pad weights)/body weight] of WT and Sln−/− mice that were fed either chow or HFD. Values are means ± sem (n=11–12 for chow, 16–17 for HFD). *P < 0.0001 vs. chow; #P < 0.05 vs. WT. C) Serum leptin concentration in WT and Sln−/− mice that were fed either chow or HFD. *P < 0.0001 vs. chow; #P < 0.05 vs. WT. D) NEFA concentration in WT and Sln−/− mice that were fed either chow or HFD. *P < 0.05 vs. chow; #P < 0.05 vs. WT. E) LDL in WT and Sln−/− mice that were red either chow or HFD. *P < 0.0001 vs. chow; #P < 0.05 vs. WT. F) Glucose tolerance tests were repeated within the same mice before (chow group) and after (HFD group) consuming an HFD. Values are means ± sem (n=14 or 16; *P<0.01 vs. chow, #P<0.01 vs. WT).
Effects of HFF on SLN protein expression and both muscle and whole-body energy expenditure
Comparisons between HFD- and control chow-fed mice show that SLN in the soleus muscle of WT mice was elevated ∼3-fold in response to the HFD (P<0.001; Fig. 2). To directly determine whether SLN mediates muscle-based DIT and whether the obesity of Sln−/− mice was due to impaired muscle-based DIT, measurements of resting muscle and whole-body metabolic rate were compared between chow- and high-fat-fed mice. Unexpectedly, resting muscle Vo2 was increased by HFF similarly in Sln−/− and WT mice (HFD > chow, P<0.05; Fig. 3A); however, following HFF, average daily whole-body Vo2 was significantly lower in Sln−/− mice compared with WT mice (Table 1, P<0.05). Notably, as shown in Table 1, average daily food intake and both total and ambulatory cage activity during HFF were similar between Sln−/− and WT mice. Average whole-body Vo2 and ambulatory cage activity data collected at 26-min intervals over 24 h are presented in Fig. 3B, showing one full dark (8:00 AM to 8:00 PM) and light (8:00 PM to 8:00 AM) cycle. Since activity patterns for individual mice vary over 24 h, the average group dual beam counts per 26 min were always >10. However, on average, mice spent ∼4 h/d being inactive (≤2 dual beam counts/reading; i.e., per 26 min interval; WT, 9.4±2 vs. Sln−/−, 9.3±1 readings), with ∼75% of the inactive time spent during the light cycle (WT, 7.0±1 vs. Sln−/−, 7.1±1 readings). Interestingly, when we analyzed whole-body Vo2 during active and inactive periods separately, we found that the differences between Sln−/− and WT mice were only significant during active periods when the mice were ambulating (≥3 dual beam counts/26 min) around their cages (P<0.05; Fig. 3C). We next assessed the energy requirements during submaximal treadmill exercise of chow-fed animals and found lower Vo2 at both 8 and 16 m/min in Sln−/− mice compared with WT (P<0.05; Fig. 3D). There were no differences in Vo2max between WT and Sln−/− mice (Fig. 3E).
Figure 2.
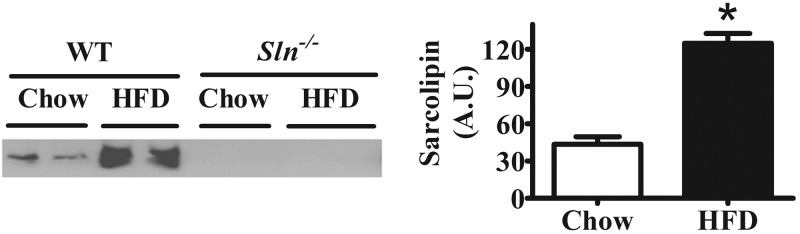
Effects of HFF on SLN protein expression in soleus. Western blot analysis of SLN content in soleus muscle from WT and Sln−/− mice that were fed either chow diet or HFD. Left panel: representative Western blot. Right panel: mean densitometry data. A.U., arbitrary units. Values are means ± sem (n=2), *P < 0.001).
Figure 3.

Effects of HFF on resting muscle Vo2 and whole-body Vo2 measured under sedentary housing conditions and during submaximal treadmill exercise in Sln−/− and WT mice. A) Resting Vo2 of isolated soleus muscles from WT and Sln−/− mice that were fed either chow diet or HFD. Values are means ± sem (n=9–10). *P < 0.05, HFD > chow. B) Whole-body Vo2 and ambulatory activity (dual beam activity counts) collected during 26-min intervals over 24 h (1 full dark and light cycle) from WT and Sln−/− mice. Values are means (error bars are not displayed for ease of presentation; n=16–17). C) Whole-body Vo2 measurements of WT and Sln−/− mice were separated by inactive (readings with a total activity count ≤2) and active time periods. Values are means ± sem (n=16–17). *P < 0.05. D, E) Submaximal steady state Vo2 (D) and Vo2max (E) measured in WT and Sln−/− mice during treadmill exercise. Values are means ± sem (n=18). *P < 0.05.
Table 1.
Daily whole-body Vo2, food intake, and physical activity in HFF WT and Sln−/− mice
| Parameter | WT | Sln−/− |
|---|---|---|
| Daily Vo2 (ml O2/kg body mass/h) | 2708 ± 71 | 2553 ± 57* |
| Food intake (g/d) | 3.65 ± 0.2 | 3.21 ± 0.2 |
| Total activity (counts/d) | 10,150 ± 822 | 8984 ± 563 |
| Dual beam activity (counts/d) | 3253 ± 393 | 2674 ± 255 |
Values are means ± sem (n=16–17).
P < 0.05.
SNS activation is higher but UCP-1 content and BAT mass are not different in Sln−/− mice compared with WT mice following HFF
We next wanted to investigate potential mechanisms for muscle-based DIT in Sln−/− mice. We measured plasma concentrations of catecholamines as an index of SNS activity, which we thought might be activated in response to HFF to a greater extent in Sln−/− mice. Plasma NE (Fig. 4A) and epinephrine (Fig. 4B) levels were not different between chow-fed groups and were elevated (P<0.01) by HFF only in the Sln−/− mice by ∼2-fold. Despite increased catecholamines in HFD-fed Sln−/− mice, HFF caused similar increases in UCP-1 content (HFD>chow, P<0.05; Fig. 5A) and BAT mass (HFD>chow, P<0.0001; Fig. 5B) in Sln−/− and WT mice.
Figure 4.
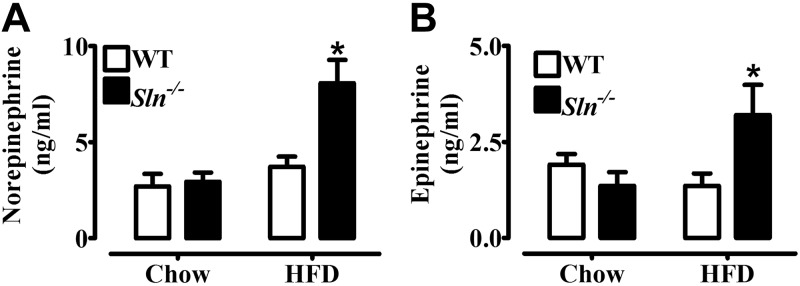
SNS activation is higher in Sln−/− mice compared with WT mice following HFF. Fasting plasma norepinephrine (A) and epinephrine concentrations (B) from WT and Sln−/− mice that were fed either chow or HFD. Values are means ± sem (n=10). *P < 0.01.
Figure 5.

Effects of HFF on UCP-1 protein expression in BAT and intrascapular BAT weight in Sln−/− and WT mice. A) Western blot analysis of UCP-1 content in BAT from WT and Sln−/− mice that were fed either chow diet or HFD. Top panel: representative Western blot. Bottom panel: mean densitometry data. Values are means ± sem (n=9 for chow, 13–15 for HFD). *P < 0.05, HFD > chow. B) Intrascapular BAT weight from WT and Sln−/− mice that were fed either chow diet or HFD. Values are means ± sem (n=11–12 for chow, 16–17 for HFD). *P < 0.0001, HFD > chow.
Propranolol treatment decreases muscle-based DIT in Sln−/− but not WT mice, resulting in even greater genotype differences in diet-induced weight gain
To determine whether augmented SNS activation of β-AR signaling could explain muscle-based DIT in Sln−/− mice, we treated mice with propranolol to block β-AR signaling during the HFD, and measurements of muscle and whole-body Vo2 were performed and compared with untreated control animals that consumed the same HFD. Propranolol treatment completely prevented muscle-based DIT in Sln−/− mice (P<0.05) but had no effects in WT mice (Fig. 6A). In keeping with these results, propranolol treatment resulted in even greater genotype differences in daily whole-body Vo2 following HFF, with Vo2 being 13% lower in Sln−/− mice compared with WT mice (P<0.05; Fig. 6B). Notably, both active and inactive whole-body Vo2 were significantly lower in Sln−/− mice that were treated with propranolol and fed an HFD compared with their WT counterparts (P<0.05; Fig. 6C). Given that propranolol inhibited muscle-based DIT in Sln−/− mice but not WT mice, the genotype differences in weight gained over 8 wk of HFF were even greater with propranolol treatment (Sln−/−>WT, P<0.05; Fig. 6D).
Figure 6.

Effects of propranolol treatment on diet-induced thermogenesis and weight gain in Sln−/− and WT mice. A) Resting VO2 of isolated soleus muscles from WT and Sln−/− mice that were treated with propranolol and fed an HFD (HFD+P, n=10) compared with nontreated mice that were fed either chow or HFD (data from Fig. 3A, n=9–10). Values are means ± sem. *P < 0.05 vs. WT HFD + P and Sln−/− HFD. B) Daily (24-h average) whole-body Vo2 of propranolol-treated WT and Sln−/− mice after consuming an HFD for 8 wk. Values are means ± sem (n=10). *P < 0.05. C) Whole-body Vo2 measurements of propranolol-treated WT and Sln−/− mice were separated by inactive (readings with a total activity count ≤2) and active time periods. Values are means ± sem (n=10). *P < 0.05. D) Total weight gained (g) over 8 wk while consuming an HFD in control (untreated) and propranolol-treated WT and Sln−/− mice. Values are means ± sem (n=10; Sln−/−>WT, P<0.05).
DISCUSSION
In this study, we investigated the interplay between SLN and another major thermogenic pathway, β-AR signaling, in the process of muscle-based DIT. SLN is a regulator of muscle-based nonshivering thermogenesis that is up-regulated in response to HFF and protects against diet-induced obesity in mice (11). Here, we show in response to HFF that resting muscle metabolic rate is increased similarly in Sln−/− and WT mice due to increased SNS activation in Sln−/− mice; however, whole-body metabolic rate was lower in Sln−/− mice following HFF, but only when the mice were ambulatory in their cages. Treatment with the β-AR antagonist propranolol during HFF completely prevented muscle-based DIT in Sln−/− mice; however, it had no effect in WT mice, resulting in greater differences in whole-body metabolic rate during both active and inactive periods and weight gain following HFF. UCP-1 plays a major role in adaptive thermogenesis and is believed by some to be the only adaptive thermogenic mechanism protective against diet-induced obesity (1, 5). However, our data support the view that several alternative thermogenic pathways exist and that inactivation of one major pathway forces the induction of compensatory alternative pathways (3, 12).
We recently identified SLN as a novel regulator of muscle-based thermogenesis in mammals, showing that Sln−/− mice have severely reduced cold tolerance and develop greater obesity and glucose intolerance than WT mice when fed an HFD (45% kcal from fat) for 12 wk (11). In this study, mice were fed a similar HFD (42% kcal from fat) for 8 wk, and, as expected, Sln−/− mice gained more weight and developed greater adiposity, hyperlipidemia, and more severe glucose intolerance than WT mice. The pronounced increase in SLN content in soleus following HFF [3- to 5-fold in our earlier study (11) and ∼3-fold here) suggests that SLN is recruited to activate muscle-based DIT. To directly determine whether SLN mediates muscle-based DIT and whether the obesity of Sln−/− mice was due to impaired muscle-based DIT, measurements of resting muscle and whole-body metabolic rate were compared between chow- and high-fat fed mice. Our unexpected results showing that resting muscle Vo2 was increased by HFF similarly in Sln−/− and WT mice indicate that SLN is not required for muscle-based DIT. Nevertheless, following HFF, average daily whole-body Vo2 was significantly lower in Sln−/− mice compared with WT mice, which can explain the greater diet-induced obesity in Sln−/− mice, since food intake and cage activity during HFF were similar between Sln−/− and WT mice. The apparent discrepancy between the muscle and whole-body results could be explained by our separate analyses of whole-body Vo2 during active and inactive time periods, where we found that the differences between Sln−/− and WT mice were only significant during active periods when the mice were ambulatory in their cages. If we increase the lower limit of ambulatory activity counts per 26 min from 3 to 25, then the differences in active Vo2 between WT and Sln−/−mice are even greater (∼6 vs. 10%, data not shown). These results indicate that SLN-mediated DIT and SLN-independent DIT in WT and Sln−/− mice, respectively, are both effective at increasing resting muscle metabolic rate, but only SLN-mediated DIT can operate across a range of metabolic rates (i.e., during physical activity) and thus have a greater effect on daily whole-body energy expenditure to decrease obesity. This is consistent with the fact that skeletal muscle accounts for ∼90% of the increased energy requirements associated with exercise (17), of which 20–50% is due to Ca2+ pumping by SERCA pumps (18, 19). To test our hypothesis further, mice performed treadmill exercise at 2 constant submaximal work rates (8 and 16 m/min), and whole-body Vo2 was assessed. As expected, the energy requirements during submaximal treadmill exercise were lower in Sln−/− mice, which can be explained by the fact that in WT muscle, SLN continues to interact with SERCA pumps in the presence of Ca2+ (11), which would promote metabolic inefficiency during physical activity or exercise. There were no differences in Vo2max between WT and Sln−/− mice, indicating that oxidative capacity of skeletal muscle was likely not affected by Sln ablation. Collectively, the exercise Vo2 data suggest that Sln−/− mice have greater reserve than WT mice, which is supported by our observation that, on average, Sln−/− mice could achieve a significantly greater running speed during Vo2max testing than WT mice (data not shown).
We next wanted to investigate potential mechanisms for muscle-based DIT in Sln−/− mice. Several “energy-wasting” or uncoupling reactions exist in skeletal muscle (4, 17), but, to date, evidence that any of these reactions are involved in adaptive thermogenesis is either lacking or equivocal. We measured plasma concentrations of catecholamines as an index of SNS activity, which we thought might be activated in response to HFF to a greater extent in Sln−/− mice. Plasma NE and epinephrine levels were elevated after HFD, but only in the Sln−/− mice by ∼2-fold, whereas there were no differences between chow-fed WT and Sln−/− mice. These data suggest that Sln ablation forces greater activation of adaptive adrenergic thermogenesis, an adaptation that is apparently insufficient to fully compensate for the loss of Sln and prevent greater diet-induced obesity and glucose intolerance in Sln−/− mice. These findings are striking because even though HFF caused similar increases in UCP-1 content and BAT mass in Sln−/− and WT mice, BAT activation should be higher in Sln−/− mice with 2-fold higher plasma NE.
Skeletal muscle is a target of β-AR signaling (20), but the current evidence for SNS-mediated thermogenesis in skeletal muscle is not very strong (21). To determine whether augmented SNS activation of β-AR signaling could explain muscle-based DIT in Sln−/− mice, we treated mice with propranolol to block β-AR signaling during the HFD, and measurements of muscle and whole-body VO2 were performed and compared with untreated control animals that consumed the same HFD. Propranolol treatment completely prevented muscle-based DIT in Sln−/− mice but had no effects in WT mice, indicating that β-AR signaling does not contribute to SLN-mediated muscle-based DIT in WT mice, but in the absence of SLN, it is the dominant mechanism for muscle-based DIT. In keeping with these results, propranolol treatment resulted in even greater genotype differences in daily whole-body Vo2 following HFF, with Vo2 being 13% lower in Sln−/− mice compared with WT mice. More importantly, in contrast to HFF without propranalol, where whole-body Vo2 of Sln−/− mice was only lower than WT mice during active periods, both active and inactive whole-body Vo2 were significantly lower in Sln−/− mice after HFF with propranalol when compared to their WT counterparts. This would be expected, based on the genotype differences in resting muscle Vo2 observed following HFF given that skeletal muscle accounts for ∼30% of basal metabolic rate in rodents (17). The fact that genotype differences in weight gained over 8 wk of HFF were even greater with propranolol treatment indicates that adrenergic thermogenesis could at least partially compensate for a lack of SLN and therefore was somewhat effective at reducing diet-induced obesity in Sln−/− mice.
Our results show that SLN is the dominant thermogenic mechanism in skeletal muscle that can increase metabolic inefficiency across a range of metabolic rates, presumably by uncoupling Ca2+ transport from ATP hydrolysis by SERCA pumps (9, 10). Induction of this thermogenic pathway in muscle by HFF reduces reliance on the SNS, which is activated to a greater extent in the absence of SLN and only contributes to muscle-based DIT under those conditions. However, our results indicate that adrenergic thermogenesis can only partially compensate for a lack of SLN, since SLN-knockout mice develop greater diet-induced obesity and glucose intolerance. Chronic SNS overactivity may be a causal link between obesity and accompanying type 2 diabetes and hypertension (22) and is a major cause of heart failure (23). Our findings raise the possibility that failure to activate SLN in response to overfeeding may predispose individuals to metabolic syndrome, type 2 diabetes, and cardiovascular disease. These results may also help to explain why some obese individuals are metabolically healthy whereas others develop type 2 diabetes and cardiovascular disease (24). Promoting inefficient metabolism in muscle represents a potential treatment for obesity and its complications. For example, skeletal muscle-specific overexpression of mitochondrial uncoupling proteins UCP-1 and UCP-3 increases metabolic rate and resistance to diet-induced obesity in mice (25–27). Targeting SR Ca2+ cycling as another potential site in muscle to promote inefficient metabolism to prevent and/or treat obesity is a novel and potentially promising approach.
Acknowledgments
The authors thank Marg Burnett for excellent technical assistance.
This work was supported in part by research grants from the Canadian Institutes of Health Research (CIHR; MOP 86618 and MOP 47296 to A.R.T.) and the U.S. National Institutes of Health (R01 HL-080551 to M.P.). E.B., I.C.S., and C.V. were supported by postgraduate scholarship doctoral awards from the Natural Sciences and Engineering Research Council of Canada. V.F. was supported by a doctoral award from CIHR. R.A.S. was supported by master's postgraduate scholarship awards from CIHR and from the Natural Sciences and Engineering Research Council of Canada. N.C.B. was supported by a postdoctoral fellowship from the American Physiological Society and the American Heart Association (10POST3360007).
Author contributions: A.R.T. and E.B. conceived of the study idea and designed the experiments; I.C.S. conducted the resting muscle Vo2 TIOX studies; E.B. and R.A.S. conducted the HFD feeding and treadmill exercise experiments, glucose tolerance tests, adiposity and serum chemistry analyses; E.B. and D.G. conducted the propranolol experiments; E.B., C.V., and R.A.S. conducted the whole-body Vo2 CLAMS measurements; V.A.F. conducted the Western blot analyses on UCP-1; S.C.G. conducted the Western blot analyses on SLN; M.P. generated the Sln−/− mouse model; A.R.T., E.B., I.C.S., V.A.F., S.C.G., and N.C.B. analyzed the data and assembled the figures; A.R.T., E.B., and D.G. wrote the article.
The authors declare no conflicts of interest.
Footnotes
- β-AR
- β-adrenergic receptor
- BAT
- brown adipose tissue
- CLAMS
- comprehensive laboratory animal monitoring system
- DIT
- diet-induced thermogenesis
- HFD
- high-fat diet
- HFF
- high-fat feeding
- LDL
- low-density lipoprotein
- NE
- norepinephrine
- NEFA
- nonesterified fatty acid
- SERCA
- sarco(endo)plasmic reticulum Ca2+ ATPase
- SLN
- sarcolipin
- SNS
- sympathetic nervous system
- UCP-1
- mitochondrial uncoupling protein
REFERENCES
- 1. Feldman H. M., Golozoubova V., Cannon B., Nedergaard J. (2009) UCP1 ablation induces obesity and abolishes diet-induced thermogenesis in mice exempt from thermal stress by living at thermoneutrality. Cell Metab. 9, 203–209 [DOI] [PubMed] [Google Scholar]
- 2. Bachman E. S., Dhillon H., Zhang C., Cinti S., Bianco A. C., Kobilka B. K., Lowell B. B. (2002) Beta-AR signaling required for diet-induced thermogenesis and obesity resistance. Science 297, 843–845 [DOI] [PubMed] [Google Scholar]
- 3. Kozak L. P. (2010) Brown fat and the myth of diet-induced thermogenesis. Cell Metab. 11, 263–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lowell B. B., Spiegelman B. M. (2000) Towards a molecular understanding of adaptive thermogenesis. Nature 404, 652–660 [DOI] [PubMed] [Google Scholar]
- 5. Tseng Y., Cypess A. M., Kahn C. R. (2010) Cellular bioenergetics as a target for obesity therapy. Nat. Rev. Drug. Discov. 9, 465–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Block B. A. (1994) Thermogenesis in muscle. Annu. Rev. Physiol. 56, 535–577 [DOI] [PubMed] [Google Scholar]
- 7. De Meis L. (2001) Role of the sarcoplasmic reticulum Ca2+-ATPase on heat production and thermogenesis. Biosci. Rep. 21, 113–137 [DOI] [PubMed] [Google Scholar]
- 8. Tupling A. R., Bombardier E., Gupta S. C., Hussain D., Vigna C., Bloemberg D., Quadrilatero J., Trivieri M. G., Babu G. J., Backx P. H., Periasamy M., MacLennan D. H., Gramolini A. O. (2011) Enhanced Ca2+ transport and muscle relaxation in skeletal muscle from sarcolipin-null mice. Am. J. Physiol. Cell Physiol. 301, C841–C849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Smith W. S., Broadbridge R., East J. M., Lee A. G. (2002) Sarcolipin uncouples hydrolysis of ATP from accumulation of Ca2+ by the Ca2+-ATPase of skeletal muscle sarcoplasmic reticulum. Biochem. J. 361, 277–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mall S., Broadbridge R., Harrison S. L., Gore M. G., Lee A. G., East J. M. (2006) The presence of sarcolipin results in increased heat production by Ca2+-ATPase. J. Biol. Chem. 281, 36597–36602 [DOI] [PubMed] [Google Scholar]
- 11. Bal N. C., Maurya S. K., Sopariwala D. H., Sahoo S. K., Gupta S. C., Shaikh S. A., Pant M., Rowland L. A., Bombardier E., Goonasekera S. A., Tupling A. R., Molkentin J. D., Periasamy M. (2012) Sarcolipin is a newly identified regulator of muscle-based thermogenesis in mammals. Nat. Med. 18, 1575–1579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Anunciado-Koza R., Ukropec J., Koza R. A., Kozak L. P. (2008) Inactivation of UCP1 and the glycerol phosphate cycle synergistically increases energy expenditure to resist diet-induced obesity. J. Biol. Chem. 283, 27688–27697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Babu G. J., Bhupathy P., Timofeyev V., Petrashevskaya N. N., Reiser P. J., Chiamvimonvat N., Periasamy M. (2007) Ablation of sarcolipin enhances sarcoplasmic reticulum calcium transport and atrial contractility. Proc. Natl. Acad. Sci. U. S. A. 104, 17867–17872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Taylor B. A., Phillips S. J. (1996) Detection of obesity QTLs on mouse chromosomes 1 and 7 by selective DNA pooling. Genomics 34, 389–398 [DOI] [PubMed] [Google Scholar]
- 15. Green H. J., Jones S., Ball-Burnett M., Fraser I. (1991) Early adaptations in blood substrates, metabolites, and hormones to prolonged exercise training in man. Can. J. Physiol. Pharmacol. 69, 1222–1229 [DOI] [PubMed] [Google Scholar]
- 16. Babu G. J., Bhupathy P., Carnes C. A., Billman G. E., Periasamy M. (2007) Differential expression of sarcolipin protein during muscle development and cardiac pathophysiology. J. Mol. Cell. Cardiol. 42, 215–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rolfe D. F. S., Brown G. C. (1997) Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol. Rev. 77, 731–758 [DOI] [PubMed] [Google Scholar]
- 18. Szentesi P., Zaremba R., van Mechelen W., Stienen G. J. (2001) ATP utilization for calcium uptake and force production in different types of human skeletal muscle fibres. J. Physiol. 531, 393–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Barclay C. J., Woledge R. C., Curtin N. A. (2007) Energy turnover for Ca2+ cycling in skeletal muscle. J. Muscle Res. Cell Motil. 28, 259–274 [DOI] [PubMed] [Google Scholar]
- 20. Roatta S., Farina D. (2010) Sympathetic actions on the skeletal muscle. Exerc. Sport Sci. Rev. 38, 31–35 [DOI] [PubMed] [Google Scholar]
- 21. Dulloo A. G. (2002) Biomedicine. A sympathetic defense against obesity. Science 297, 780–781 [DOI] [PubMed] [Google Scholar]
- 22. Masuo K., Rakugi H., Ogihara T., Esler M. D., Lambert G. W. (2010) Cardiovascular and renal complications of type 2 diabetes in obesity: role of sympathetic nerve activity and insulin resistance. Curr. Diabetes Rev. 6, 58–67 [DOI] [PubMed] [Google Scholar]
- 23. Lehnart S. E., Wehrens X. H., Kushnir A., Marks A. R. (2004) Cardiac ryanodine receptor function and regulation in heart disease. Ann. N. Y. Acad. Sci. 1015, 144–159 [DOI] [PubMed] [Google Scholar]
- 24. Primeau V., Coderre L., Karelis A. D., Brochu M., Lavoie M. E., Messier V., Sladek R., Rabasa-Lhoret R. (2011) Characterizing the profile of obese patients who are metabolically healthy. Int. J. Obes. (Lond.) 35, 971–981 [DOI] [PubMed] [Google Scholar]
- 25. Li B., Nolte L. A., Ju J.-S., Han D. H., Coleman T., Holloszy J. O., Semenkovich C. F. (2000) Skeletal muscle respiratory uncoupling prevents diet-induced obesity and insulin resistance in mice. Nat. Med. 6, 1115–1120 [DOI] [PubMed] [Google Scholar]
- 26. Clapham J. C., Arch J. R. S., Chapman H., Haynes A., Lister C., Moore G. B. T., Piercy V., Carter S. A., Lehner I., Smith S. A., Beeley L. J., Godden R. J., Herrity N., Skehel M., Changani K. K., Hockings P. D., Reid D. G., Squires S. M., Hatcher J., Trail B., Latcham J., Rastan S., Harper A. J., Cadenas S., Buckingham J. A., Brand M. D., Abuin A. (2000) Mice overexpressing human uncoupling protein-3 in skeletal muscle are hyperphagic and lean. Nature 406, 415–418 [DOI] [PubMed] [Google Scholar]
- 27. Son C., Hosoda K., Ishihara K., Bevilacqua L., Masuzaki H., Fushiki T., Harper M. E., Nakao K. (2004) Reduction of diet-induced obesity in transgenic mice overexpressing uncoupling protein 3 in skeletal muscle. Diabetologia 47, 47–54 [DOI] [PubMed] [Google Scholar]