

Abstract
Aggregation and accumulation of Aβ42 play an initiating role in Alzheimer's disease (AD); thus, selective lowering of Aβ42 by γ-secretase modulators (GSMs) remains a promising approach to AD therapy. Based on evidence suggesting that steroids may influence Aβ production, we screened 170 steroids at 10 μM for effects on Aβ42 secreted from human APP-overexpressing Chinese hamster ovary cells. Many acidic steroids lowered Aβ42, whereas many nonacidic steroids actually raised Aβ42. Studies on the more potent compounds showed that Aβ42-lowering steroids were bonafide GSMs and Aβ42-raising steroids were inverse GSMs. The most potent steroid GSM identified was 5β-cholanic acid (EC50=5.7 μM; its endogenous analog lithocholic acid was virtually equipotent), and the most potent inverse GSM identified was 4-androsten-3-one-17β-carboxylic acid ethyl ester (EC50=6.25 μM). In addition, we found that both estrogen and progesterone are weak inverse GSMs with further complex effects on APP processing. These data suggest that certain endogenous steroids may have the potential to act as GSMs and add to the evidence that cholesterol, cholesterol metabolites, and other steroids may play a role in modulating Aβ production and thus risk for AD. They also indicate that acidic steroids might serve as potential therapeutic leads for drug optimization/development.—Jung, J. I., Ladd, T. B., Kukar, T., Price, A. R., Moore, B. D., Koo, E. H., Golde, T. E., Felsenstein, K. M. Steroids as γ-secretase modulators.
Keywords: Alzheimer's disease, amyloid, Aβ, cholesterol metabolites
Accumulation of amyloid β (Aβ) in the brain is one of the hallmark pathological features of Alzheimer's disease (AD). Aβ is a normally secreted peptide proteolytically derived from the amyloid precursor protein (APP) through sequential cleavage by β- and γ-secretase, respectively (1). β-Secretase cleaves the ectodomain of APP, producing soluble APPβ (sAPPβ) and carboxyl-terminal fragment β (CTFβ). CTFβ is subsequently cleaved by γ-secretase, producing soluble Aβ and the APP intracellular domain (AICD). Aβ was originally described as an ∼4-kDa protein, as it exhibits extensive heterogeneity at both its amino and carboxyl termini when isolated from the AD or trisomy 21 brain (1–3). Under normal conditions, a diverse collection of Aβ peptides is also produced, but these peptides do not entirely account for the heterogeneity of Aβ deposited in the brain (4–7). Aβ1–40 is the major species normally produced, along with a number of minor species, including but not limited to Aβ1–37, Aβ38, Aβ39, and Aβ42 (1). In particular, Aβx–42 has been implicated as the pathogenic form in AD (8). In vitro experiments have demonstrated that Aβx–42 is more prone to aggregation than Aβx–40 and other shorter nonmutant Aβ peptides, and Aβx–42 is typically the earliest detectable form of Aβ deposited in the brain (3–5). A more direct causal role in AD comes from the study of genetic mutations in presenilin 1 (PS1), presenilin 2 (PS2), and APP that lead to either relative or absolute increases in Aβ42 (1). Transgenic modeling studies also show that Aβ42 is required for Aβ deposition in the mouse brain and that Aβ40 may actually protect from amyloid deposition (13, 14). In consideration of this evidence, selective lowering of Aβ42 appears to be a promising approach to prevent Aβ aggregation and ultimately Aβ deposition in the brain (9–14).
Small molecules that lower Aβ42 by shifting γ-secretase cleavage are generally referred to as γ-secretase modulators (GSMs). Since the initial finding that a subset of nonsteroidal anti-inflammatory drugs (NSAIDs) possess GSM activity, numerous compounds with GSM activity have been identified (15–21). In general, there are 2 recognized classes of GSMs: NSAID-like GSMs, which have a hydrophobic scaffold and have an absolute requirement for a carboxylic acid group for Aβ42 lowering; and nonacidic GSMs, including the diarylaminothiazoles and diarylureas (15, 16, 22, 23). Many nonacidic compounds structurally related to the NSAID-like GSMs raise Aβ42 and are referred to as inverse GSMs (iGSMs; ref. 20). GSMs from both classes have shown therapeutic efficacy in preclinical mouse models after chronic administration, with significant reduction in brain amyloid histopathology and behavioral deficit readouts having been demonstrated (15, 16, 19). As opposed to γ-secretase inhibitors (GSIs), which have inherent mechanism-based toxicity, GSMs are postulated to represent an intrinsically safe approach for AD therapy because they do not inhibit normal APP processing or processing of other γ-secretase substrates, e.g., Notch 1 (15–19). Instead, they alter γ-secretase cleavage and appear to have some degree of specificity, as they have stronger modulatory effects on APP and APLP2 then other γ-secretase substrates evaluated to date (21).
Nevertheless, while the preclinical potential of GSMs has been clearly demonstrated, it has proven challenging to identify a compound from the existing chemical leads that combines high potency with a high therapeutic safety window required for long-term chronic administration to treat AD (24). Therefore, there is a considerable need for the elaboration of the biology of novel chemical lead GSM structures with potential for improved safety as described herein. In this study, we have discovered a novel class of GSMs from the screening of a small library of ∼170 commercially available steroids for their potential to lower Aβ42. A subset of both naturally occurring and synthetic derivatives of cholesterol, which included all readily available compounds that contained a carboxylic acid as well as structurally related nonacidic steroids, was identified. This screen identified 40 steroids that raised Aβ42 and 41 steroids that lowered Aβ42. Focused studies on the most potent Aβ42-modulating steroids, 5β-cholanic acid (S529; ursocholanic acid) and 4-androsten-3-one-17β-carboxylic acid ethyl ester (S15), demonstrated that they were indeed bonafide GSM or inverse GSMs, respectively. Notably, the GSM identified in this study, S529, and its endogenous counterpart, 5β-cholanic acid-3β-ol (lithocholic acid), have the potential to interact with and regulate the EphA2 and FXR receptors (25–27). Finally, we further explored whether the steroid hormones estrogen and progesterone were also GSMs. Although estrogen has complex effects on APP processing, as previously reported (28), both estrogen and progesterone also appear to possess weak iGSM activity. Our results suggest that an endogenous Aβ42 modulatory system may exist and that its regulation or direct use of the steroids may represent a novel therapeutic approach to the treatment of AD.
MATERIALS AND METHODS
Compounds and cell culture
A collection of 170 diverse steroids, both naturally occurring and synthetic, was obtained from Steraloids (Newport, RI, USA) without preference to inherent biological properties; GSM-1 and Cmpd2 were synthesized by A. Fauq (Mayo Clinic Chemical Core, Jacksonville, FL, USA); and compound E was purchased from EMD Millipore (Billerica, MA, USA). All compounds were dissolved in dimethyl sulfoxide (DMSO) as a 30–60 mM stock. We attempted using a 45% solution of hydroxypropyl-β-cyclodextrin (HP-β-CD) as the vehicle since the compound(s) may precipitate from solution when DMSO is the vehicle and added to aqueous medium. Although HP-β-CD is broadly used for challenging pharmaceutical formulations, it was not able to enhance drug availability and in all likelihood lowered the available concentration, which resulted in no significant effects on the Aβ measurements when compared with vehicle (data not shown).
Compounds were screened either in Chinese hamster ovary (CHO)-2B7 cells or in human neuroglioma (H4) cells stably expressing the APP695 wild-type (wt) protein, maintained as described previously (29). For compound testing, the cells were incubated for 16 h in the presence of the compound appropriately diluted into OptiMEM-reduced serum medium (Life Technologies, Carlsbad, CA, USA) containing 1% fetal bovine serum. The CellTiter 96 aqueous nonradioactive cell proliferation assay [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay; Promega, Madison, WI, USA] was used to determine toxicity according to the manufacturer's protocol (30). Unless otherwise noted, 1% DMSO was used as the vehicle control.
Antibodies and ELISAs
Monoclonal antibodies to Aβ were generated by the Mayo Clinic Immunology Core facilities (Jacksonville, FL, USA). Ab5 recognizes an epitope in the amino terminus of Aβ (Aβ1–16), recognizes both monomeric and aggregated Aβ, and is human specific. Ab13.1.1. was raised against Aβ35–40, is specific for Aβx–40, and exhibits minimal cross-reactivity with other Aβ peptides. Aβ2.1.3. was raised against Aβ35–42 and is specific to Aβx–42. The Aβ38 antibody (Ab38), supplied by P. Mehta (Institute of Basic Research, Staten Island, NY, USA), specifically recognizes Aβx–38 and shows no cross-reactivity with other Aβ peptides (data not shown). For cell-based screens, Aβ was captured from conditioned medium with either Ab5, Ab38, Ab13.1.1, or Ab2.1.3 (coated at 10–50 μg/ml in EC buffer: 5 mM NaH2PO4·H2O, 20 mM Na2HPO4, 400 mM NaCl, 2.5 mM EDTA, 151.5 μM BSA, 813 μM CHAPS, and 7.7 mM NaN3) on Immulon 4HBX Flat-Bottom Microfilter 96-well plates (Thermo Scientific, Waltham, MA, USA). Total Aβ level was determined by capture with Ab5 and detected with horseradish peroxidase (HRP)-conjugated 4G8 (a monoclonal antibody against Aβ17–24; Covance, Waltham, MA, USA) with the other Aβ peptides detected with HRP-conjugated Ab5. For the cell-free assay, HRP-conjugated 4G8 was used as the secondary detection antibody for all testing. Aβ standards (Bachem, King of Prussia, PA, USA) were prepared by dissolving in hexafluoroisopropanol (HFIP) at 1 mg/ml with sonication, dried under nitrogen, resuspended at 2 mg/ml HFIP, sonicated again, and dried under nitrogen. The resulting Aβ was resuspended in 0.01% ammonium hydroxide, portioned into aliquots in EC buffer, and frozen at −80°C. Following these steps, the Aβ is monomeric, as determined by size-exclusion chromatography (data not shown).
Mass spectrometry of Aβ
For matrix-assisted laser desorption/ionization time of flight (MALDI-TOF) mass spectrometry analyses of Aβ peptides, CHO 2B7 cells were treated with the various compounds, as described above. Secreted Aβ peptides were analyzed by essentially as described previously (5, 29). Briefly, Aβ1–x was immunoprecipitated using Ab5, with the peptide being eluted with trifluoroacetic acid:acetonitrile:water (1:20:20) saturated with α-cyano-4-hydroxycinnamic acid (Sigma, St. Louis, MO, USA). Samples were analyzed on a Voyager DE STR MALDI-TOF mass spectrometer (PE Biosystems, Foster City, CA, USA). For identification of Aβ species, the expected mass (m/z) is compared with the observed mass (m/z), italicized in parentheses: Aβ37. 4071.5 (4075.93); Aβ38, 4131.5 (4133.32); Aβ39, 4230.7 (4323.04); Aβ40, 4323.8 (4331.38); Aβ42, 4514.0 (4515.01).
Notch cleavage assay
DNA encoding AβD1-Q15-mNotch1V1711-E1809 fusion with a FLAG tag was synthesized and cloned into pET-21b (+) vector (Life Technologies). The protein was overexpressed in Escherichia coli BL21 and purified using a HiTrap Q-column (GE Life Sciences, Little Chalfont, UK) with NaCl elution gradient. Gel filtration and anti-FLAG M2 affinity column can be used if further purification is needed. CHAPSO solubilized γ-secretase was prepared as described from CHO S-1 cell line (31, 32). For in vitro assay, 25 μM substrate was incubated with CHAPSO solubilized γ-secretase (100 μg/ml protein concentration) in sodium citrate buffer (150 mM, with 1× complete protease inhibitor, pH 6.8; Roche, Indianapolis, IN, USA) for 2 h at 37°C in the presence or absence of test compounds. The final total volume was 120 μl. To capture the N-terminal product, 50 μl magnetic sheep-anti-mouse IgG beads (Life Technologies) was incubated with 4.5 μg Ab5 antibody for 30 min at room temperature with constant shaking. The beads were then washed with PBS and incubated with 60 μl in vitro assay mixture for 30 min. The C-terminal product was captured with anti-FLAG M2 magnetic beads (Sigma). Bound beads were washed 3 times with water. Samples were eluted with 10 μl 0.1% trifluoroacetic acid (Thermo Scientific) in water. Eluate (2 μl) was mixed with an equal volume of saturated α-cyano-4-hydroxycinnamic acid (ACCA) solution (Sigma) in 60% acetonitrile, 40% methanol. The sample mixture (1 μl) was loaded onto an ACCA-pretreated MSP 96 target plate and analyzed with a Bruker Microflex mass spectrometer (Bruker Daltonics, Billerica, MA, USA) for the cleavage product.
In vitro γ-secretase assays
Cell-free γ-secretase assays were performed as described previously (31, 32). The carbonate-extracted membranes derived from the H4 neuroglioma cells overexpressing APP695wt were incubated at 37°C for 2 h; Aβ levels were quantified by a panel of sandwich ELISAs; and AICD by immunoblotting, as described, with rabbit anti-APP-CT20; 1:1000 (Calbiochem, La Jolla, CA, USA), using an Odyssey scanner (LiCor Biosciences, Lincoln, NE, USA).
Measurement of sAPPα
CHO-2B7 cells were incubated with 17β-estradiol or progesterone at the indicated concentrations (10, 100, or 200 μM) for 16 h. The conditioned medium was collected, and the cells were harvested/lysed in RIPA buffer for immunoblotting for sAPPα and CTFα detection, respectively (28). Ab5 (1:500) was used for sAPPα detection, and anti-APP-CT-20 (1:1000) was used for CTFα detection. Both CTFα and sAPPα were normalized to actin following quantitation using Odyssey 3.0 software (LiCor Biosciences).
Statistical analysis
In vitro data were expressed and graphed as means ± se using GraphPad Prism 5 software (GraphPad, San Diego, CA, USA). Unless otherwise noted, analysis was either by Student's t test or by 1-way ANOVA followed by Dunnett's post hoc testing for group differences. MALDI-TOF MI calculations were analyzed using 2-way ANOVA with repeated measures. The level of significance was set at P < 0.05 in all tests.
RESULTS
Identification of oxysterol-type GSMs
The effects of steroid treatment on Aβ42 levels were examined using a cell-based screen in CHO cells overexpressing the WT 695-aa isoform of APP (wtAPP695; CHO-2B7 cells) at an initial screening concentration of 10 μM (29). After 16 h treatment, secreted Aβ42 levels from the conditioned medium were measured by an Aβ42-specific sandwich ELISA (Supplemental Table S1). The 20 most effective Aβ42-lowering steroids and the 20 most effective inducers of Aβ42 were selected for additional analysis. To differentiate the compounds from general Aβ inhibitors or inducers and to categorize them as GSMs or iGSMs, the effect of the compounds on Aβ38 production, as well as total Aβ production, by specific ELISA assays was examined (data not shown). Aside from those compounds having no effect on generalized inhibition or induction, the results indicated that many of the steroids tested showed either GSM or iGSM activity with either increases or decreases in Aβ38, respectively, and little, if any, effect on total Aβ. As additional confirmation, GSM and iGSM activities were examined in a second cell line, H4 human neuroblastoma cells stably transfected with wtAPP695. The magnitude of the effect between the cells varied on a compound-by-compound basis, but it is difficult to make absolute comparisons due to differing pharmacokinetic variables; however, with few exceptions the overall rank of order of the compounds was essentially unchanged (data not shown). Interestingly, a number of the Aβ42-lowering steroids exist endogenously, including but not limited to 5β-cholanic acid-3β-ol (lithocholic acid) and 5-cholenic acid-3β-ol. The best representative Aβ42-lowering and Aβ42-raising steroids were selected for additional testing, including EC50 determination, in comparison to known GSMs (Fig. 1).
Figure 1.

γ-Secretase modulatory activity of select compounds in CHO-2B7 cells. A) Chemical structures of the various GSM compounds tested, along with their calculated EC50 for Aβ42 modulation determined in triplicate. GSM-1 (piperidine acetic acid–type), EC50 = 92.17 nM; Cmpd2 (piperazinyl pyrimidine type), EC50 = 44.81 nM; S529, EC50 = 5.68 μM; and 4-androsten-3-one-17β-carboxylic acid ethyl ester (S15), 50% increase at 6.25 μM. B–E) Concentration-response curves for Aβ42 (B), Aβ38 (C), Aβ40 (D) and total Aβ (E). Aβ levels from DMSO-treated cells (n=6) served as the control.
Representative GSM compounds include S529 and other various steroids, which comprise various hydroxy and keto derivatives, including various endogenous bile acids. Examples of the structure-activity relationship (SAR) are presented in Fig. 2. S529 acid is an acidic steroid with GSM activity, with a similar pharmacophore structure as the NSAID carboxylates, containing a lipophilic domain with a carboxylic acid group. Other representative compounds, showing iGSM activity, are the androstenedione-based compounds, including S15, which has a lipophilic domain and a modified carboxylic acid group containing an ethyl ester (Fig. 1A), were chosen for further study based on the magnitude and potency of their effects on Aβ42. For comparison, GSM-1, a piperidine acetic acid chemotype GSM, and compound 2 (Cmpd2), a piperazinyl pyrimidine nonacidic type GSM, were also utilized (22, 23). Dose-response curves are shown in Fig. 1B–E. S529 decreased Aβ42 up to 75% and raised Aβ38 >300% in a dose-dependent manner. At these concentrations, S529 was not toxic when evaluated by MTS assays (30). The EC50 of S529 for lowering Aβ42 is ∼5.7 μM in CHO-2B7 cells. The identified inverse modulator S15 selectively raised Aβ42 by 200% and decreased Aβ38 up to 20% at 25 μM in a dose-dependent manner. Cell toxicity was indicated for S15 at concentrations >50 μM. This pattern is similar to that of fenofibrate, a drug utilized to reduce cholesterol levels; however, it is a classic iGSM (20). The steroid modulators did not appreciably alter Aβ40 or total Aβ levels. For comparison, GSM-1 showed the same pattern of Aβ modulation by raising Aβ38 and lowering Aβ42, with no changes in Aβ40 or total Aβ. This phenotype is different from that of Cmpd2, which lowers both Aβ40 and Aβ42, with concomitant increases in Aβ37 and Aβ38. The EC50 for lowering Aβ42 for GSM-1 is 92.2 nM and for Cmpd2 is 44.8 nM. Cell toxicity was observed at concentrations >10 μM for GSM-1 and 2 μM for Cmpd2, respectively. Inhibition of the proteolytic processing of Notch and other γ-secretase substrates by the classical GSIs has severely limited their clinical development (15, 16). GSMs, by definition, do not show effects on Notch proteolytic processing. To further characterize the identified steroid class for their effects on γ-secretase-mediated Notch processing, the compounds were examined by proteolytic analysis of an Aβ-Notch fusion protein. Consistent with their profile as GSMs, no apparent alteration of Notch processing was detected as compared with the GSI LY-411,575 (Supplemental Fig. S1).
Figure 2.
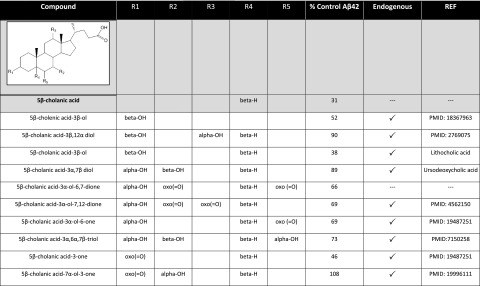
Markush structure of acidic steroid/sterol and associated structure-activity relationships. Effects of the various S529 derivatives, some of which are known endogenous steroidal metabolites (indicated by a checkmark) on Aβ42 production are summarized as compared to the DMSO vehicle-treated cells.
Aβ-specific immunoprecipitation and mass spectrometry (IP/MS) demonstrates that S529 shows a similar Aβ profile to known acidic-type GSMs
IP/MS peptide analysis was performed from conditioned media from cells treated, at the maximal effective concentration where no toxicity was observed, either with GSM-1, Cmpd2, S529, S15, or DMSO vehicle alone, respectively (Fig. 3A–E). From the vehicle-treated cells, Aβ40 was the major species detected, with minor amounts of Aβ37, Aβ38, Aβ39, and Aβ42 also detected. GSM-1 treatment altered the Aβ profile showing marked increases in Aβ38 and decreases in Aβ42 without changing Aβ40; Cmpd2 lowered both Aβ40 and Aβ42 peaks, with Aβ37 and Aβ38 increasing after treatment. S529 treatment showed a similar pattern to GSM-1, while S15 showed an increase in Aβ42 and modestly reduced Aβ38, consistent with iGSM. We performed quantification of the Aβ peptide spectra (see Supplemental Fig. S2A) using the standardized method by measuring the height of each Aβ peak (i.e., Aβ37, Aβ38, Aβ39, Aβ40, and Aβ42) between 3500 (m/z) and 5000 (m/z) where Aβ shifting occurs. The analyses show the ratio between each Aβ peak height to the total Aβ peak heights. Moreover, we have established a quantification method to selectively illustrate the magnitude of overall shifting in the Aβ profiles after compound treatment (Fig. 3F), by comparing the ratio of each peak to the sum of the total peaks and then calculating the difference between sum of the longer Aβ peptides (i.e., Aβ42) and that of the shorter Aβ peptides (i.e., Aβ37, Aβ38, and Aβ39). The result was subsequently normalized to the vehicle control. As this value represents the modulation induced by either a GSM or an iGSM, it is referred to as the modulation index (MI). The MI following each treatment is shown in Fig. 3F. All of the GSMs show a negative MI, whereas an iGSM shows a positive MI. Cmpd2, which alters both Aβ42 and Aβ40, demonstrated a more significant MI (−0.79) than GSM-1 (−0.26) or S529 (−0.06), which alter only Aβ42.
Figure 3.

Pattern of Aβ isoforms after MALDI-TOF analysis from conditioned medium from GSM-treated cells. A–E) Effect of various GSM treatments on Aβ isoforms with representative Aβ spectra from 2–3 experiments with 3 replicates/experiment: DMSO (A), GSM-1 (B), Cmpd2 (C), S529 (D), and S15 (E). Identity of the Aβ peptide is noted above the appropriate peak and stacked bar graph, demonstrating the ratio of each peak height to the sum of all the peaks. F) Modulation index (MI) reflecting overall shifting in each Aβ profile induced by the treatments was calculated. As an example, the MI is determined by comparison of the DMSO-treated group to the drug-treated group such that the peak height for the longer Aβ species, Aβ42, is 0.08, and the sum of the peaks for Aβ37, Aβ38, and Aβ39 is 0.17, with the difference being 0.09. With the use of the same calculation, the difference between the longer and shorter Aβ peptides for GSM-1 is 0.35. This value is normalized with the value from DMSO treatment, resulting in MI = −0.26. Negative value indicates GSM activity; positive value indicates iGSM activity. **P < 0.01, ***P < 0.001.
In vitro γ-secretase activity study shows direct modulatory effects of select steroids
To ascertain whether the apparent shift in γ-secretase cleavage on APP is due to direct modulation by the select steroids, broken cell in vitro γ-secretase activity assays were performed. It has been previously demonstrated that direct γ-secretase activity can be measured using a cell-free membrane preparation with APP or β-secretase-derived C-terminal fragment C99 as substrate (32). The carbonate-extracted membranes from H4 APP695wt cells are incubated in the presence of the GSM compounds and the select steroids. Figure 4A shows the results from Aβ42, Aβ40, and total Aβ ELISAs. S529, as well as GSM-1 and Cmpd2, lowered Aβ42 levels, while S15 raised Aβ42 levels. As in the cell-based assay, Cmpd2 lowered Aβ40, as well as Aβ42. Furthermore, overall γ-secretase activity was assessed by examination of the production of the AICD by immunoblot. AICD fragments, the intracellular fragments remaining after γ-secretase cleavage, were detected by an APP C-terminal antibody (anti-CT-20; Fig. 4B). Unlike the γ-secretase inhibitor (compound E)-treated membranes, the steroidal compounds and the known GSMs had no discernible effects on the production of AICD (Fig. 4B) nor the precise position ε-cleavage site that generates an AICD fragment of 49 or 50 aa in length (Fig. 4C). These observations are consistent with those described elsewhere (33).
Figure 4.

In vitro γ-secretase activity assay shows a direct association of each GSM and iGSM in APP processing. A) Graph shows that each treatment was capable of modulating γ-secretase cleavage activity in a cell-free assay using carbonate-extracted membranes from H4-APP695wt-overexpressing cells. Control (Ctrl) refers to the net activity of membranes incubated for 2 h with DMSO (T2) after the subtraction of the basal γ-secretase activity (T0). To measure basal γ-secretase activity, samples were treated with compound E, an irreversible pan-γ-secretase inhibitor, at the starting point T0 of membrane incubation (61). Each sample is then normalized to the control. Each group is normalized to percentage of control (n=2); graph is representative of 4 assays. *P<0.05, ***P<0.001; unpaired, 2-tailed Student's t test. B) AICD immunoblot, along with its quantitation is shown after each drug treatment. AICD is markedly reduced or not detected after compound E treatment. GSM-1, Cmpd2, S529, and S15 treatments generated AICD with no significant quantitative changes. *P < 0.05; 1-way ANOVA with Dunnett's multiple comparison test. C) With the use of bicine urea gels, AICD fragments can be separated into C49 and C50 after each drug treatment (62). No significant changes are detected in either the level or position of ε-cleavage or after GSM-1, Cmpd 2, S15, or S529 treatment consistent with modulatory activity (33). MW, molecular weight.
Steroid hormones demonstrate complex effects on Aβ production
Since the steroid hormones estrogen and progesterone have been implicated as protective molecules for AD, we explored the possibility of whether they may play a role as GSMs. Utilizing the cell-based Aβ assay, we performed end-specific ELISA analysis from the conditioned medium after each treatment. 17β-Estradiol, the most common form of estrogen, and progesterone raised Aβ42 levels and lowered Aβ38 levels, characteristic of an iGSM (Fig. 5A, B). IP/MS analysis of the treated conditioned medium was consistent with their signature as iGSMs (Fig. 5C–F and Supplemental Fig. S2B). In vitro γ-secretase assay confirms that their effects for raising Aβ42 are also through direct modulation of γ-secretase (Supplemental Fig. S3). They possess a positive MI value, consistent with inverse modulation (Fig. 5G). It has been suggested that 17β-estradiol has a role as an α-secretase activator (28); therefore, we measured sAPPα levels from the medium, and CTFα level from the cell lysates, which were treated with 17β-estradiol or progesterone (Fig. 6). Modest increases with respect to sAPPα levels were detected, whereas CTFα fragments were significantly increased at 100 and 200 μM with 17β-estradiol treatment (Fig. 6A, C). No statistically significant changes were detected for sAPPα and CTFα levels after progesterone treatment (Fig. 6B, D). MTS assay demonstrated that at the highest concentration (200 μM) 17β-estradiol did not appear detrimental to the cells, while progesterone appeared to be toxic at these levels (data not shown). The physiological relevance of the Aβ42 increases that were detected is questionable, but this observation raises the potential of these types of molecules to act as iGSMs.
Figure 5.

Estrogen and progesterone show inverse GSM activity. A, B) Dose-response curves of 17β-estradiol (A) and progesterone (B) in CHO-2B7 cells show varying effects on Aβ42, Aβ38, and total Aβ levels. iGSM response of 17β-estradiol is weak but notable at 200 μM (note that the total Aβ increase at 50 μM is not typical for these studies). Effects of progesterone >50 μM (grayed area) are rejected due to cellular toxicity. C–F) IP/MS analyses of 17β-estradiol (D) and progesterone (E, F) in CHO-2B7 APP-overexpressing cells. DMSO (C) is used as the control to compare Aβ spectra of estrogen and progesterone-treated group (n=3). Species of Aβ peptides are marked above the each peak. G) MI for 17β-estradiol and progesterone. ***P < 0.001; 1-way ANOVA with Dunnett's multiple comparison test.
Figure 6.

α-Secretase assay was performed for 17β-estradiol and progesterone. Immunoblots show minimal change in sAPPα and increase in CTFα in 17β-estradiol-treated cells (A) while sAPPα and CTFα levels did not change in progesterone-treated cells (C). Each blot was quantified (B, D). DMSO served as the control; results are representative of 2–3 repeats of 3 individual experiments. *P < 0.05, **P < 0.01; 1-way ANOVA with Dunnett's multiple comparison test.
DISCUSSION
These studies have identified the acidic steroid as a novel class of GSM, demonstrating that endogenous GSMs, resulting from the normal metabolic oxidation of cholesterol, may exist, and show that select steroid hormones may alter Aβ production in a highly complex fashion. Although many acidic steroids selectively act as GSMs, closely related nonacidic analogs, including ester derivatives, either have no effect or manifest iGSM activity. Of the steroids tested, S529 was the most potent GSM in the collection, while S15 was the most potent iGSM among the nonacidic steroids. Analysis of the activities of these GSMs using cell-free γ-secretase assays shows that S529 and S15 were able to directly interact with the γ-secretase/substrate complex and influence Aβ42 levels. S529 demonstrates an Aβ-altering pattern similar to the carboxylate containing compound GSM-1, which selectively lowers Aβ42 while concomitantly raising Aβ38 levels without affecting total Aβ. This pattern is distinct from the nonacidic Cmpd2, which lowers both Aβ40 and Aβ42 and raises Aβ37 and Aβ38 without affecting total Aβ.
It is not fully understood what contributes to the differential activities of acidic and nonacidic GSMs. There are most certainly differences in binding interactions either with the APP substrate, the PS1 γ-secretase enzyme, or a ternary complex of both (34, 35). Given the evidence for a pore- or channel-like active site for γ-secretase substrate cleavage and the requirement of high lipophilicity for high potency, it is likely that all GSMs modulate processivity of γ-secretase cleavage through complex interactions with substrate, presenilin, and also membrane lipids. The binding sites and binding modes may vary with each class and each compound, respectively (36–41). The original low-to-moderate potency GSMs, derived from carboxylate containing NSAIDs, are suggested to bind directly to APP (36, 37). However; the optimized, high-potency carboxylic acid GSM-1 is reported to bind the PS1 N-terminal fragment (NTF) in the γ-secretase complex (38). Nonacidic GSMs have been shown to bind to γ-secretase components rather than the substrate (38–41). Overall, the evidence suggests a primary GSM binding interaction with PS1 and a weaker binding interaction with substrate, which is nonetheless highly significant for overall potency due to the chelate effect. One may postulate that the essential carboxylate group of acidic GSMs forms a strong ionic bond at a lysine residue at a binding site that is not occupied by nonacids. Cholesterol has been shown to bind APP CTFβ at a site that overlaps with the site affected as the acidic GSM binding site on APP; we and others have found that lysine residue 624, which is presumed to delineate the APP ectodomain from its transmembrane domain, dramatically alters γ-secretase processivity without altering the initial ε-cleavage (42). Thus, one might speculate that acidic steroids can bind to APP CTFβ in a manner similar to cholesterol but also form an ionic bond to the positively charged lysine, resulting in enhanced processivity and thus γ-secretase modulation. Additional studies will be needed to assess this possible mechanism of action. Our study supports the general concept that a lipophilic anchor, represented by the steroid-backbone, with a carboxylate side chain, is required for Aβ42-lowering activity (18). The mechanism for iGSM activity has not been defined; however, the shift in the activity profile can be attributed to the elimination of the aforementioned lysine binding interaction. It is predicted that they act on the same site as GSMs (20, 21).
This study adds to the growing body of evidence that cholesterol and cholesterol metabolites, including bile acids, may play a role in modulating Aβ production. Although the exact mechanisms showing their effects on Aβ production are not fully defined, it is apparent that many of them have effects on APP secretases. For example, there is a positive correlation between cholesterol and Aβ production. Cholesterol has been reported to negatively regulate α-secretase, whereas β- and γ-secretase activities are positively regulated by cholesterol (43–46). Considering that cholesterol directly influences γ-secretase activity and cholesterol colocalizes within the lipid rafts with APP and γ-secretase, cholesterol could alter physical conformation of γ-secretase to change the site or rate of cleavage of APP (46–49). Likewise, since many cholesterol metabolites found in the brain (including some bile acids) also show a correlation with Aβ production (49–51), perhaps they function as endogenous Aβ regulators through modulation of γ-, β-, and α- secretase activity, as well as trafficking of APP and its associated secretases. It is conceivable that, like cholesterol, a bile acid may be able to bind the same site proposed by Beel and colleagues (52, 53) but is further capable of influencing the processivity of the γ-secretase activity. Therefore, potentially increasing levels of these acidic steroids or analog compounds therapeutically may serve as a novel approach to modulating Aβ42 production with respect to AD prevention and treatment. Interestingly, acidic steroids, such as the closely related chenodeoxycholic acid, are already used therapeutically for the treatment of gallstones.
Beyond the investigation of the select steroidal and cholesterol metabolites, we also explored steroid hormones as potential Aβ-altering molecules. Although they are frequently reported to play a protective role in AD, it remains unknown whether estrogen or progesterone have direct effects on Aβ production (54–60). Our results show that 17β-estradiol and progesterone can potentially raise Aβ42 levels; a characteristic of iGSMs. 17β-Estradiol appears to have additional effects on APP processing by increasing α-secretase activity. We confirmed that 17β-estradiol lowered total Aβ levels in concentration-dependent manner and increased CTFα, the cleavage product of α-secretase. This is a finding consistent with previous reports (28, 57). Although progesterone did not increase α-secretase cleavage products, it is implicated as a neuroprotective molecule and is associated with a Aβ clearance mechanism by activating Aβ-degrading enzymes (60). These observations suggest that the roles of 17β-estradiol and progesterone in altering Aβ metabolism are complex and may include iGSM activities. The biological relevance of these observations is questionable considering absolute values for these steroid hormones under normal conditions. However, it does imply that similar molecules with greater intrinsic iGSM activity may exist endogenously that may have relevance to the disease state.
These studies have identified many acidic steroids as GSMs and nonacidic steroids as iGSMS, further linking steroid metabolism to APP processing. Although unproven, these data suggest that the naturally occurring steroids may have the potential to act as endogenous GSMs and add to the evidence that cholesterol, cholesterol metabolites, and other steroids may play a role in modulating Aβ production and thus risk for AD (63). They also indicate that S529 and other steroids might serve as a potential lead for drug optimization and development. Along a similar line, a triterpene derivative of Actaea racemosa (black cohosh) has been characterized as a GSM (64, 65). (It is of note that the black cohosh GSM profile is distinct from those previously defined GSMs, suggesting that it potentially has complicated effects on the γ-secretase processivity.) However, triterpenes are precursors to steroids in both plants and animals, suggesting that this entire steroidal class of molecules may represent a largely untapped potential for drug development and may serve as a viable alternative for AD therapy.
Supplementary Material
Acknowledgments
These studies were supported by funds from U.S. National Institutes of Health grant AG-20206 and the Coins for Alzheimer's Research Trust, supported by Rotarians in the southeastern United States.
The authors are extremely grateful to Dr. Rong Wang (Department of Genetics and Genomic Sciences, Mount Sinai School of Medicine, New York, NY, USA) for the use of the PE Biosystems Voyager DE STR MALDI-TOF mass spectrometer.
The authors also thank Dr. Yong Ran for technical support in performing the notch inhibition assay. The authors are also grateful to Gideon Shapiro and Kristi Ayers for critical reading of the manuscript.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- Aβ
- amyloid β
- AD
- Alzheimer's disease
- AICD
- amyloid precursor protein intracellular domain
- APP
- amyloid precursor protein
- CHO
- Chinese hamster ovary
- Cmpd2
- compound 2
- CTFβ
- carboxyl-terminal fragment β
- DMSO
- dimethyl sulfoxide
- GSI
- γ-secretase inhibitor
- GSM
- γ-secretase modulator
- HRP
- horseradish peroxidase
- iGSM
- inverse γ-secretase modulator
- IP/MS
- immunoprecipitation and mass spectrometry
- MALDI-TOF
- matrix-assisted laser desorption/ionization time of flight
- MI
- modulation index
- MTS
- 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
- NSAID
- nonsteroidal anti-inflammatory drug
- PS1
- presenilin 1, S15, 4-androsten-3-one-17β-carboxylic acid ethyl ester
- S529
- 5β-cholanic acid
- sAPP
- soluble amyloid precursor protein
- wt
- wild type
REFERENCES
- 1. Golde T. E., Eckman C. B., Younkin S. G. (2000) Biochemical detection of Aβ isoforms: implications for pathogenesis, diagnosis, and treatment of Alzheimer's disease. Biochim. Biophys. Acta 1502, 172–187 [DOI] [PubMed] [Google Scholar]
- 2. Teller J. K., Russo C., DeBusk L. M., Angelini G., Zaccheo D., Dagna-Bricarelli F., Scartezzini P., Bertolini S., Mann D. M., Tabaton M., Gambetti P. (1996) Presence of soluble amyloid beta-peptide precedes amyloid plaque formation in Down's syndrome. Nat. Med. 2, 93–95 [DOI] [PubMed] [Google Scholar]
- 3. Russo C., Saido T. C., DeBusk L. M., Tabaton M., Gambetti P., Teller J. K. (1997) Heterogeneity of water-soluble amyloid β-peptide in Alzheimer's disease and Down's syndrome brains. FEBS Lett. 409, 411–416 [DOI] [PubMed] [Google Scholar]
- 4. Moore B., Chakrabarty P., Levites Y., Kukar T., Baine A.-M., Moroni T., Ladd T., Das P., Dickson D., Golde T. (2012) Overlapping profiles of Abeta peptides in the Alzheimer's disease and pathological aging brains. Alzheimers Res. Ther. 4, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang R., Sweeney D., Gandy S., Sisodia S. (1996) The profile of soluble amyloid beta protein in cultured cell media. Detection and quantification of amyloid beta protein and variants by immunoprecipitation-mass spectrometry. J. Biol. Chem. 271, 31894–31902 [DOI] [PubMed] [Google Scholar]
- 6. Seubert P., Vigo-Pelfrey C., Esch F., Lee M., Dovey H., Davis D., Sinha S., Schiossmacher M., Whaley J., Swindlehurst C., McCormack R., Wolfert R., Selkoe D., Lieberburg I., Schenk D. (1992) Isolation and quantification of soluble Alzheimer's β-peptide from biological fluids. Nature 359, 325–327 [DOI] [PubMed] [Google Scholar]
- 7. Suzuki N., Cheung T. T., Cai X. D., Odaka A., Otvos L., Eckman C., Golde T. E., Younkin S. G. (1994) An increased percentage of long amyloid beta protein secreted by familial amyloid beta protein precursor (beta APP717) mutants. Science 264, 1336–1340 [DOI] [PubMed] [Google Scholar]
- 8. Steven G, Y. The role of Aβ42 in Alzheimer's disease. J. Physiol. 92, 289–292 [Google Scholar]
- 9. Jarret J. T. (1993) The carboxy terminus of the β amyloid protein is critical for the seeding of amyloid formation: implication for the pathogenesis of Alzheimer's disease. Biochemistry 32, 4693–4697 [DOI] [PubMed] [Google Scholar]
- 10. Gravina S. A., Ho L., Eckman C. B., Long K. E., Otvos L., Younkin L. H., Suzuki N., Younkin S. G. (1995) Amyloid β protein (Aβ) in Alzheimer's disease brain. J. Biol. Chem. 270, 7013–7016 [DOI] [PubMed] [Google Scholar]
- 11. Iwatsubo T., Odaka A., Suzuki N., Mizusawa H., Nukina N., Ihara Y. (1994) Visualization of Aβ42(43) and Aβ40 in senile plaques with end-specific Aβ monoclonals: evidence that an initially deposited species is Aβ42(43). Neuron 13, 45–53 [DOI] [PubMed] [Google Scholar]
- 12. McGowan E., Pickford F., Kim J., Onstead L., Eriksen J., Yu C., Skipper L., Murphy M. P., Beard J., Das P., Jansen K., DeLucia M., Lin W.-L., Dolios G., Wang R., Eckman C. B., Dickson D. W., Hutton M., Hardy J., Golde T. (2005) Aβ42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron 47, 191–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim J., Onstead L., Randle S., Price R., Smithson L., Zwizinski C., Dickson D. W., Golde T., McGowan E. (2007) Aβ40 inhibits amyloid deposition in vivo. J. Neurosci. 27, 627–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang R., Wang B., He W., Zheng H. (2006) Wild-type presenilin 1 protects against alzheimer disease mutation-induced amyloid pathology. J. Biol. Chem. 281, 15330–15336 [DOI] [PubMed] [Google Scholar]
- 15. Weggen S., Eriksen J. L., Das P., Sagi S. A., Wang R., Pietrzik C. U., Findlay K. A., Smith T. E., Murphy M. P., Bulter T., Kang D. E., Marquez-Sterling N., Golde T. E., Koo E. H. (2001) A subset of NSAIDs lower amyloidogenic Aβ42 independently of cyclooxygenase activity. Nature 414, 212–216 [DOI] [PubMed] [Google Scholar]
- 16. Eriksen J. L., Sagi S. A., Smith T. E., Weggen S., Das P., McLendon D. C., Ozols V. V., Jessing K. W., Zavitz K. H., Koo E. H., Golde T. E. (2003) NSAIDs and enantiomers of flurbiprofen target γ-secretase and lower Aβ42 in vivo. J. Clin. Invest. 112, 440–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kukar T., Golde T. E. (2008) Possible mechanisms of action of NSAIDs and related compounds that modulate gamma-secretase cleavage. Curr. Top. Med. Chem. 8, 47–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zall A., Kieser D., Höttecke N., Naumann E. C., Thomaszewski B., Schneider K., Steinbacher D. T., Schubenel R., Masur S., Baumann K., Schmidt B. (2011) NSAID-derived γ-secretase modulation requires an acidic moiety on the carbazole scaffold. Bioorg. Med. Chem. 19, 4903–4909 [DOI] [PubMed] [Google Scholar]
- 19. Oehlrich D., Berthelot D. J. C., Gijsen H. J. M. (2010) γ-Secretase modulators as potential disease modifying anti-alzheimer's drugs. J. Med. Chem. 54, 669–698 [DOI] [PubMed] [Google Scholar]
- 20. Kukar T., Murphy M. P., Eriksen J. L., Sagi S. A., Weggen S., Smith T. E., Ladd T., Khan M. A., Kache R., Beard J., Dodson M., Merit S., Ozols V. V., Anastasiadis P. Z., Das P., Fauq A., Koo E. H., Golde T. E. (2005) Diverse compounds mimic Alzheimer disease-causing mutations by augmenting Aβ42 production. Nat. Med. 11, 545–550 [DOI] [PubMed] [Google Scholar]
- 21. Sagi S. A., Lessard C. B., Winden K. D., Maruyama H., Koo J. C., Weggen S., Kukar T. L., Golde T. E., Koo E. H. (2011) Substrate sequence influences γ-secretase modulator activity, role of the transmembrane domain of the amyloid precursor protein. J. Biol. Chem. 286, 39794–39803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hall A., Elliott R. L., Giblin G. M., Hussain I., Musgrave J., Naylor A., Sasse R., Smith B. (2010) Piperidine-derived gamma-secretase modulators. Bioorg. Med. Chem. Lett. 20, 1306–1311 [DOI] [PubMed] [Google Scholar]
- 23. Rivkin A., Ahearn S. P., Chichetti S. M., Kim Y. R., Li C., Rosenau A., Kattar S. D., Jung J., Shah S., Hughes B. L., Crispino J. L., Middleton R. E., Szewczak A. A., Munoz B., Shearman M. S. (2010) Piperazinyl pyrimidine derivatives as potent γ-secretase modulators. Bioorg. Med. Chem. Lett. 20, 1269–1271 [DOI] [PubMed] [Google Scholar]
- 24. Gijsen H. J., Mercken M. (2012) γ-Secretase modulators: can we combine potency with safety? Int. J. Alzheimers Dis. 2012, 295207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tognolini M., Incerti M., Hassan-Mohamed I., Giorgio C., Russo S., Bruni R., Lelli B., Bracci L., Noberini R., Pasquale E. B., Barocelli E., Vicini P., Mor M., Lodola A. (2012) Structure-activity relationships and mechanism of action of Eph-ephrin antagonists: interaction of cholanic acid with the EphA2 receptor. Chem. Med. Chem. 7, 1071–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fukuchi J., Song C., Dai Q., Hiipakka R. A., Liao S. (2005) 5β-Cholane activators of the farnesol X receptor. J. Steroid Biochem. Mol. Biol. 94, 311–318 [DOI] [PubMed] [Google Scholar]
- 27. Suzuki T., Tamehiro N., Sato Y., Kobayashi T., Ishii-Watabe A., Shinozaki Y., Nishimaki-Mogami T., Hashimoto T., Asakawa Y., Inoue K., Ohno Y., Yamaguchi T., Kawanishi T. (2008) The novel compounds that activate farnesoid X receptor: the diversity of their effects on gene expression. J. Pharm. Sci. 107, 285–294 [DOI] [PubMed] [Google Scholar]
- 28. Amtul Z., Wang L., Westaway D., Rozmahel R. F. (2010) Neuroprotective mechanism conferred by 17beta-estradiol on the biochemical basis of Alzheimer's disease. Neuroscience 169, 781–786 [DOI] [PubMed] [Google Scholar]
- 29. Murphy M. P., Uljon S. N., Fraser P. E., Fauq A., Lookingbill H. A., Findlay K. A., Smith T. E., Lewis P. A., McLendon D. C., Wang R., Golde T. E. (2000) Presenilin 1 regulates pharmacologically distinct γ-secretase activities: implications for the role of presenilin in γ-secretase cleavage. J. Biol. Chem. 275, 26277–26284 [DOI] [PubMed] [Google Scholar]
- 30. Barltrop J. A., Owen T. C., Cory A. H., Cory J. G. (1991) 5-(3-carboxymethoxyphenyl)-2-(4,5-dimethylthiazolyl)-3-(4-sulfophenyl)tetrazolium, inner salt (MTS) and related analogs of 3-(4,5-dimethylthiazolyl)-2,5-diphenyltetrazolium bromide (MTT) reducing to purple water-soluble formazans as cell-viability indicators. Bioorg. Med. Chem. Lett. 1, 611–614 [Google Scholar]
- 31. Fraering P. C., Ye W., Strub J. M., Dolios G., LaVoie M. J., Ostaszewski B. L., van Dorsselaer A., Wang R., Selkoe D. J., Wolfe M. S. (2004) Purification and characterization of the human gamma-secretase complex. Biochemistry 43, 9774–9789 [DOI] [PubMed] [Google Scholar]
- 32. McLendon C., Xin T., Ziani-Cherif C., Murphy M. P., Findlay K. A., Lewis P. A., Pinnix I., Sambamurti K., Wang R., Fauq A., Golde T. E. (2000) Cell-free assays for γ-secretase activity. FASEB J. 14, 2383–2386 [DOI] [PubMed] [Google Scholar]
- 33. Lessard C., Tyan S.-H., Maruyama H., Cottrell, Suresh S., Golde T., Koo E. (2012) γ-Secretase modulators do not alter ε-cleavage of APP in modulating Aβ42 levels. Soc. Neurosci. Ann. Meet. 750.27, E66 [Google Scholar]
- 34. Li X., Dang S., Yan C., Gong X., Wang J., Shi Y. (2013) Structure of a presenilin family intramembrane aspartate protease. Nature 493, 56–61 [DOI] [PubMed] [Google Scholar]
- 35. De Strooper B., Iwatsubo T., Wolfe M. S. (2012) Presenilins and γ-secretase: structure, function, and role in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2, a006304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kukar T. L., Ladd T. B., Bann M. A., Fraering P. C., Narlawar R., Maharvi G. M., Healy B., Chapman R., Welzel A. T., Price R. W., Moore B., Rangachari V., Cusack B., Eriksen J., Jansen-West K., Verbeeck C., Yager D., Eckman C., Ye W., Sagi S., Cottrell B. A., Torpey J., Rosenberry T. L., Fauq A., Wolfe M. S., Schmidt B., Walsh D. M., Koo E. H., Golde T. E. (2008) Substrate-targeting [ggr]-secretase modulators. Nature 453, 925–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Richter L., Munter L.-M., Ness J., Hildebrand P. W., Dasari M., Unterreitmeier S., Bulic B., Beyermann M., Gust R., Reif B., Weggen S., Langosch D., Multhaup G. (2010) Amyloid beta 42 peptide (Aβ42)-lowering compounds directly bind to Aβ and interfere with amyloid precursor protein (APP) transmembrane dimerization. Proc. Natl. Acad. Sci. U. S. A. 107, 14597–14602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ebke A., Luebbers T., Fukumori A., Shirotani K., Haass C., Baumann K., Steiner H. (2011) Novel γ-secretase enzyme modulators directly target presenilin protein. J. Biol. Chem. 286, 37181–37186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kounnas M. Z., Danks A. M., Cheng S., Tyree C., Ackerman E., Zhang X., Ahn K., Nguyen P., Comer D., Mao L., Yu C., Pleynet D., Digregorio P. J., Velicelebi G., Stauderman K. A., Comer W. T., Mobley W. C., Li Y.-M., Sisodia S. S., Tanzi R. E., Wagner S. L. (2010) Modulation of γ-secretase reduces β-amyloid deposition in a transgenic mouse model of alzheimer's disease. Neuron 67, 769–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ohki Y., Higo T., Uemura K., Shimada N., Osawa S., Berezovska O., Yokoshima S., Fukuyama T., Tomita T., Iwatsubo T. (2011) Phenylpiperidine-type γ-secretase modulators target the transmembrane domain 1 of presenilin 1. EMBO J. 30, 4815–4824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Crump C. J., Fish B. A., Castro S. V., Chau D.-M., Gertsik N., Ahn K., Stiff C., Pozdnyakov N., Bales K. R., Johnson D. S., Li Y.-M. (2011) Piperidine acetic acid based γ-secretase modulators directly bind to presenilin-1. ACS Chem. Neurosci. 2, 705–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kukar T. L., Ladd T. B., Robertson P., Pintchovski S. A., Moore B., Bann M. A., Ren Z., Jansen-West K., Malphrus K., Eggert S., Maruyama H., Cottrell B. A., Das P., Basi G. S., Koo E. H., Golde T. E. (2011) Lysine 624 of the amyloid precursor protein (app) is a critical determinant of amyloid β peptide length. J. Biol. Chem. 286, 39804–39812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bodovitz S., Klein W. L. (1996) Cholesterol modulates secretase cleavage of amyloid precursor protein. J. Biol. Chem. 271, 4436–4440 [DOI] [PubMed] [Google Scholar]
- 44. Kojro E., Gimpl G., Lammich S., März W., Fahrenholz F. (2001) Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the α-secretase ADAM 10 Proc. Natl. Acad. Sci. U. S. A. 98, 5815–5820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Simons M., Keller P., De Strooper B., Beyreuther K., Dotti C. G., Simons K. (1998) Cholesterol depletion inhibits the generation of β-amyloid in hippocampal neurons. Proc. Natl. Acad. Sci. U. S. A. 95, 6460–6464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wahrle S., Das P., Nyborg A. C., McLendon C., Shoji M., Kawarabayashi T., Younkin L. H., Younkin S. G., Golde T. E. (2002) Cholesterol-dependent γ-secretase activity in buoyant cholesterol-rich membrane microdomains. Neurobiol. Dis. 9, 11–23 [DOI] [PubMed] [Google Scholar]
- 47. Golde T. E., Eckman C. B. (2001) Cholesterol modulation as an emerging strategy for the treatment of Alzheimer's disease. Drug Disc. Today 6, 1049–1055 [DOI] [PubMed] [Google Scholar]
- 48. Bhattacharyya R., Kovacs D. M. (2010) ACAT inhibition and amyloid beta reduction. Biochim. Biophys. Acta 1801, 960–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Prasanthi J., Huls A., Thomasson S., Thompson A., Schommer E., Ghribi O. (2009) Differential effects of 24-hydroxycholesterol and 27-hydroxycholesterol on beta-amyloid precursor protein levels and processing in human neuroblastoma SH-SY5Y cells. Mol. Neurodegener. 4, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Brown J. (2004) Differential expression of cholesterol hydroxylases in Alzheimer's disease. J. Biol. Chem. 279, 34674–34681 [DOI] [PubMed] [Google Scholar]
- 51. Nunes A., Amaral J., Lo A., Fonseca M., Viana R. S., Callaerts-Vegh Z., D'Hooge R., Rodrigues C. P. (2012) TUDCA, a bile acid, attenuates amyloid precursor protein processing and amyloid-β deposition in APP/PS1 mice. Mol. Neurobiol. 45, 440–454 [DOI] [PubMed] [Google Scholar]
- 52. Beel A. J., Sakakura M., Barrett P. J., Sanders C. R. (2010) Direct binding of cholesterol to the amyloid precursor protein: an important interaction in lipid–Alzheimer's disease relationships? Biochim. Biophys. Acta 1801, 975–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Barrett P. J., Song Y., Van Horn W. D., Hustedt E. J., Schafer J. M., Hadziselimovic A., Beel A. J., Sanders C. R. (2012) The amyloid precursor protein has a flexible transmembrane domain and binds cholesterol. Science 336, 1168–1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yue X., Lu M., Lancaster T., Cao P., Honda S.-I., Staufenbiel M., Harada N., Zhong Z., Shen Y., Li R. (2005) Brain estrogen deficiency accelerates Aβ plaque formation in an Alzheimer's disease animal model. Proc. Natl. Acad. Sci. U. S. A. 102, 19198–19203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jaffe A. B., Toran-Allerand C. D., Greengard P., Gandy S. E. (1994) Estrogen regulates metabolism of Alzheimer amyloid beta precursor protein. J. Biol. Chem. 269, 13065–13068 [PubMed] [Google Scholar]
- 56. Chang D., Kwan J., Timiras P. S. (1997) Estrogens influence growth, maturation, and amyloid beta-peptide production in neuroblastoma cells and in a beta-APP transfected kidney 293 cell line. Adv. Exp. Med. Biol. 429, 261–271 [DOI] [PubMed] [Google Scholar]
- 57. Xu H., Gouras G. K., Greenfield J. P., Vincent B., Naslund J., Mazzarelli L., Fried G., Jovanovic J. N., Seeger M., Relkin N. R., Liao F., Checler F., Buxbaum J. D., Chait B. T., Thinakaran G., Sisodia S. S., Wang R., Greengard P., Gandy S. (1998) Estrogen reduces neuronal generation of Alzheimer beta-amyloid peptides. Nat. Med. 4, 447–451 [DOI] [PubMed] [Google Scholar]
- 58. Haskell S. G., Richardson E. D., Horwitz R. I. (1997) The effect of estrogen replacement therapy on cognitive function in women: a critical review of the literature. J. Clin. Epidemiol. 50, 1249–1264 [DOI] [PubMed] [Google Scholar]
- 59. Manthey D., Heck S., Engert S., Behl C. (2001) Estrogen induces a rapid secretion of amyloid β precursor protein via the mitogen-activated protein kinase pathway. Eur J. Biochem. 268, 4285–4291 [DOI] [PubMed] [Google Scholar]
- 60. Jayaraman A., Carroll J. C., Morgan T. E., Lin S., Zhao L., Arimoto J. M., Murphy M. P., Beckett T. L., Finch C. E., Brinton R. D., Pike C. J. (2012) 17β-Estradiol and progesterone regulate expression of β-amyloid clearance factors in primary neuron cultures and female rat brain. Endocrinology 153, 5467–5479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Seiffert D., Bradley J. D., Rominger C. M., Rominger D. H., Yang F., Meredith J. E., Wang Q., Roach A. H., Thompson L. A., Spitz S. M., Higaki J. N., Prakash S. R., Combs A. P., Copeland R. A., Arneric S. P., Hartig P. R., Robertson D. W., Cordell B., Stern A. M., Olson R. E., Zaczek R. (2000) Presenilin-1 and -2 are molecular targets for γ-secretase inhibitors. J. Biol. Chem. 275, 34086–34091 [DOI] [PubMed] [Google Scholar]
- 62. Klafki H.-W., Wiltfang J., Staufenbiel M. (1996) Electrophoretic separation of βA4 peptides (1–40) and (1–42). Anal. Biochem. 237, 24–29 [DOI] [PubMed] [Google Scholar]
- 63. Popp J., Lewczuk P., Kölsch H., Meichsner S., Maier W., Kornhuber J., Jessen F., Lütjohann D. (2012) Cholesterol metabolism is associated with soluble amyloid precursor protein production in Alzheimer's disease. Neurochem. 123, 310–316 [DOI] [PubMed] [Google Scholar]
- 64. Hubbs J. L., Fuller N. O., Austin W. F., Shen R., Ruicaho R., Creaser S. P., McKee T. D., Loureiro R. M. B., Tate B., Xia W., Ives J., Bronk B. S. (2012) Optimization of a natural product-based class of γ-secretase modulators. J. Med. Chem. 55, 9270–9282 [DOI] [PubMed] [Google Scholar]
- 65. Loureiro R. M., Dumin J. A., McKee T. D., Austin W. F., Fuller N. O., Hubbs J. L., Shen R., Jonker J., Ives J., Bronk BS., Tate B. (2013) Efficacy of SPI-1865, a novel gamma-secretase modulator, in multiple rodent models. Alzheimers Res. Ther. 5, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.