

Abstract
Prostaglandin E2 (PGE2) is an important biological mediator involved in the defense against Mycobacterium tuberculosis (Mtb) infection. Previously, we reported that in macrophages (Mφs), infection with avirulent Mtb H37Ra resulted in inhibition of necrosis by an inhibitory effect on mitochondrial permeability transition via the PGE2 receptor EP2. However, human Mφs also express EP4, a PGE2 receptor functionally closely related to EP2 that also couples to stimulatory guanine nucleotide binding protein, but the functional differences between EP2 and EP4 in Mtb-infected Mφs have been unclear. EP4 antagonist addition to H37Ra-infected Mφs inhibited the expression of cyclooxygenase 2 (COX2) and microsomal prostaglandin E synthase-1 (mPGES-1), which are involved in PGE2 production. Moreover, H37Ra infection induced PGE2 production through the Toll-like receptor (TLR) 2/p38 mitogen-activated protein kinase (MAPK) signaling pathway. Induction of COX2 and mPGES-1 expression by TLR2 stimulation or Mtb infection was increased after additional stimulation with EP4 agonist. Hence, in Mtb-infected Mφs, PGE2 production induced by pathogen recognition receptors/p38 MAPK signaling is up-regulated by EP4-triggered signaling to maintain an effective PGE2 concentration.—Nishimura, T., Zhao, X., Gan, H., Koyasu, S., and Remold, H. G. The prostaglandin E2 receptor EP4 is integral to a positive feedback loop for prostaglandin E2 production in human macrophages infected with Mycobacterium tuberculosis.
Keywords: cyclooxygenase 2, Toll-like receptor 2, p38 mitogen-activated protein kinase
Tuberculosis is one of the most devastating infectious diseases globally. The World Health Organization Global Tuberculosis Report 2012 mentioned that the number of new cases of tuberculosis was 8,700,000 in 2011, and 1,400,000 deaths occurred due to tuberculosis (1). The success of Mycobacterium tuberculosis (Mtb) is closely related to its capacity to block the immune response of the host. Virulent Mtb inhibits apoptosis, a form of programmed cell death beneficial to the host because it is integral to killing intracellular bacilli and stimulation of adaptive immune responses. Virulent Mtb induces necrosis characterized by disruption and lysis of the macrophage (Mφ) plasma membrane, facilitating bacterial escape into the surrounding tissue to start a new cycle of infection and eventually dissemination from the lung to other tissues (2–4). In contrast, avirulent Mtb induces apoptosis, which leads to sequestering of the pathogen in apoptotic bodies and an augmented innate and clonal immune response.
The critical effector molecules that determine the death modality of the Mtb-infected Mφs were found to include prostaglandin E2 (PGE2), required for protection of the Mφs against Mtb-induced necrosis, and lipoxin A4 (LXA4), which blocks PGE2 production in the infected Mφs (5). PGE2 and LXA4 are predominantly elaborated by Mφs infected with avirulent or virulent Mtb, respectively. PGE2 is under certain conditions involved in the induction of apoptosis (protection) of the infected Mφs in vitro. In contrast, LXA4 was found to cause necrosis (spread of infection). As PGE2 and lipoxins are critically important as regulators of the death modality of Mφs, we became interested in the mechanism of PGE2 production in Mtb-infected human Mφs.
PGE2 belongs to a large group of lipids referred to as prostanoids, which are products of prostaglandin H2 (PGH2), the immediate product of cyclooxygenase 2 (COX2). PGE2 specifically interacts with 4 prostanoid receptors named EP1, EP2, EP3, and EP4, which are 7-transmembrane-spanning receptors that activate intracellular second messenger pathways by interacting with heterotrimeric guanine nucleotide binding (G) proteins, promoting intracellular pathways that either activate or inhibit inflammation (6, 7). EP1 regulates Ca2+ channel gating via Gαq, liberating Ca2+ from intracellular stores and elevating cytosolic Ca2+ concentration (8). EP3 mainly couples to Gαi and decreases intracellular cyclic adenosine monophosphate (cAMP) formation (9). EP2 and EP4, which are dominant PGE2 receptors in Mφs (10, 11), were initially characterized as coupling to Gαs and increased intracellular cAMP formation (12). EP2 is thought to trigger mainly a protein kinase A (PKA)-dependent pathway, whereas EP4 uses the phosphoinositide 3-kinase signaling pathway, as well as the PKA signaling pathway (13). There are also published differences between EP2 and EP4 in the affinity for PGE2 and in the desensitization to PGE2 (6). Several findings suggest that EP2 and EP4 are part of an activation loop for cAMP production, leading to increased COX2 expression and PGE2 production in mouse lung fibroblasts (14), human myeloid-derived suppressor cells (15), and in the mouse Mφ cell line RAW 264.7 (16). We reported earlier on a specific function of EP2 in Mtb-infected murine Mφs. Protection against mitochondrial permeability transition (MPT) resulting in necrosis of the Mtb-infected Mφs was dependent on triggering EP2, as Mφs from EP2−/− mice but not from EP1−/−, EP3−/−, and EP4 −/− mice became sensitive to MPT (5). However, the role of EP4 in Mtb-infected Mφs has been unclear. Here, we show by using the virulent Mtb strain H37Rv and the avirulent strain H37Ra for infection of human Mφs that EP4 is part of a positive feedback loop important for the regulation of PGE2 production. This finding could be of critical importance to maintain adequate PGE2 levels to restrict pulmonary Mtb growth in light of the short half-life of this eicosanoid in the tissue.
MATERIALS AND METHODS
Reagents and bacteria
Phospho-p38 mitogen-activated protein kinase (MAPK; Thr180/Tyr182) antibody, p38 MAPK antibody, glyceraldehyde 3-phosphate dehydrogenase (GAPDH; 14C10) rabbit mAb, and COX1 (D2G6) rabbit mAb were from Cell Signaling Technology (Beverly, MA, USA); p38 MAPK inhibitor SB202190 was from Enzo Life Sciences (Farmingdale, NY, USA). COX2 (murine) affinity-purified polyclonal antibody, microsomal prostaglandin E synthase-1 (mPGES-1) polyclonal antibody, cytosolic prostaglandin E synthase (cPGES; FL) polyclonal antibody, mPGES-2 polyclonal antibody, EP2 receptor polyclonal antibody, EP4 receptor (C-Term) polyclonal antibody, the EP1/EP3 agonist sulprostone, the EP2 antagonist AH6809, the EP4 antagonist AH23848, PGE2, and the EP2 agonist butaprost free acid (butaprost) were from Cayman Chemical (Ann Arbor, MI, USA). AH23848 has a pA2 value against PGE2 of 5.4 at EP4 (17). The EC50 for butaprost as determined by using human EP2 transfected CHO cells is 6.3 × 10−8 M (18). The EP4 agonist ONO-AE1-329 and the EP4 antagonist ONO-AE2-227 were provided by Ono Pharmaceuticals (Osaka, Japan). Pam3CSK4, FSL-1 (Pam2CGDPKHPKSF), PAb hTLR2 (polyclonal antibody to human TLR2), and PAb control (normal rat IgG control) were purchased from Invivogen (San Diego, CA, USA). Optimal concentrations for inhibitors, agonists, and antagonists were determined by assessing Mφ viability using a 3-(4,5-dimethylthiazol-2-vl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt life-death assay (CellTiter 96 Aqueous One Solution cell proliferation assay; Promega, Madison, WI, USA). The optimal concentration of ONO-AE1-329 was determined by dose-response experiments measuring cAMP accumulation in agonist-stimulated Mφs (Supplemental Fig. S1).
The Mtb strains H37Rv and H37Ra (American Type Culture Collection, Manassas, VA, USA) were portioned into aliquots in small tubes after culturing and stored at −80°C. Prior to infection, the frozen stock was thawed at room temperature. For bacterial counts, 100-μl aliquots were serially diluted 10-fold with 0.02% Tween 80 in PBS and plated on 7H11 agar plates (Thermo Fisher Scientific Remel Products, Lenexa, KS, USA). At 28 d after plating, the number of colonies was evaluated.
Cells and culture
As described before (19), human Mφs were obtained by culturing human mononuclear cells from buffy coat preparations (Research Blood Components, Boston, MA, USA) for 9 or 10 d in Iscove's modified Dulbecco's medium (IMDM) containing 10% (v/v) human AB serum (Gemini Bio-Products, Sacramento, CA, USA) at a density of 2.0 × 106 cells/2 ml/well in 6-well cluster plates (Corning, Corning, New York, USA). The cells were cultured in the absence of serum in IMDM 12 h prior to infection of Mtb or treatment.
Adherent Mφs were cultured with Mtb at MOI 10:1 and were washed after 4 h with Hank's balanced salt solution (HBSS) to remove nonadherent bacilli, and fresh IMDM was added. In all experiments with inhibitors, agonists, antagonists and antibodies, Mφs were preincubated for 45 min with the respective components.
Assay of PGE2 and cAMP production
PGE2 concentrations in Mφ medium and cAMP concentrations in Mφ lysates were determined using Prostaglandin E2 Express EIA kit and cyclic AMP EIA kit (Cayman Chemical, Ann Arbor, MI, USA), respectively, according to the manufacturer's instructions.
Immunoblot analysis
Human Mφs were infected with Mtb and lysed with RIPA Buffer (Boston BioProducts, Ashland, MA, USA) containing 1% protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO, USA) and 1% phosphatase inhibitor cocktail II (Boston BioProducts). Protein was measured with the bicinchoninic acid assay. Cell lysate batches from each sample containing equal amounts of protein were fractionated using 10, 12, or 13% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), and proteins were transferred to polyvinylidene fluoride membrane at 4°C. The membrane was blocked for 1 h with 2% ECL advance blocking reagent (GE Healthcare, Piscataway, NJ, USA) in TBS/0.01% Tween 20 at room temperature and was then probed with the appropriate primary antibodies. After washing, blots were probed with peroxidase-conjugated goat anti-rabbit IgG (H+L) (Jackson ImmunoResearch, West Grove, PA, USA). ECL advance solutions A and B (GE Healthcare, Piscataway, NJ, USA) were added, and blots were exposed to HyBlot CL (Denville Scientific, Metuchen, NJ, USA). Signal intensities were quantified by using ImageJ software (U.S. National Institutes of Health, Bethesda, MD, USA).
In vitro cell death assays
Necrosis of Mφs was evaluated by determining the number of plasma membrane damaged cells by using Live/Dead fixable dead-cell stain kits (Life Technologies, Carlsbad, CA, USA), according to the specification of the manufacturer. This assay is based on the reaction of a fluorescent reactive dye with cellular amines. On necrosis, the dye permeates the damaged membranes of necrotic cells and reacts with free amines both in the interior and on the cell surface, resulting in intense fluorescent staining. In non-necrotic cells, the dye only reacts with cell-surface amines, resulting in relatively dim staining. The number of necrotic cells was counted by flow cytometry using a FACS canto2 (BD Bioscience, Sparks, MD, USA).
Measurement of Mtb uptake by human Mφs
As described before (20), to measure Mtb uptake, Mφs were inoculated with H37Ra at an MOI of 1:1 for 4 h. After washing, the cells were lysed by the addition of 250 μl/well of 0.2% SDS, and SDS was neutralized with 750 μl of 5% FBS. Cell lysates (100 μl) were serially diluted 10-fold and were plated on 7H11 agar plates. Colonies were counted after the plates had been incubated for 28 d at 37°C.
RNA isolation and quantitative real-time RT-PCR
RNA from Mφs was isolated and purified using the RNeasy mini kit (Qiagen, Hilden, Germany) and was transcribed into cDNA with the QuantiTect reverse transcription kit (Qiagen, Hilden, Germany). The cDNA was denatured for 10 min at 95°C (21–24). Specific DNA fragments for EP2, EP4, COX2, and mPGES-1 were amplified by PCR with a Max3000p Stratagene Cycler (Agilent Technology, Santa Clara, CA, USA) for 40 cycles with 30 s at 95°C, 60 s at 55°C (EP2), 60°C (EP4 and COX2) or 65°C (mPGES-1), and 60 s at 72°C. The oligonucleotide primers used for EP2 were 5′-TGAAGTTGCAGGCGAGCA-3′ (forward) and 5′-GACCGCTTACCTGCAGCTGTAC-3′ (reverse), for EP4 were 5′-CATCTGCTCCATCCCGCT-3′ (forward) and 5′-GGATGGCCTGCAAATCTGG-3′ (reverse), for COX2 were 5′-GCTGGAACATGGAATTACCCA-3′ (forward) and 5′-CTTTCTGTACTGCGGGTGGAA-3′ (reverse), for mPGES-1 were 5′-AGTATTGCAGGAGCGACC-3′ (forward) and 5′-CCAGAAAGGAGTAGACGAAGC-3′ (reverse), for GAPDH were 5′-TGCACCACCAACTGCTTAGC-3′ (forward) and 5′-GGCATGGACTGTGGTCATGAG-3′ (reverse). The relative gene expression of EP2, EP4, COX2, and mPGES-1 was determined by the ΔΔCT method comparing expression in uninfected and infected Mφs. GAPDH was used as a reference gene.
Statistics
Results are expressed as means ± sem. Unpaired t test was used to analyze the change of EP2 and EP4 expression between uninfected and infected Mφs. Comparisons among ≥3 experimental groups were performed with ANOVA followed by Bonferroni-corrected post hoc test. A value of P < 0.05 was considered statistically significant. All analysis was performed by using GraphPad Prism 5 (GraphPad Software, La Jolla, CA, USA).
RESULTS
EP4 expression is elevated in Mtb-infected Mφs
To determine whether EP2 and EP4 differ in their capacity to protect against Mtb-induced Mφ death, we treated Mtb-infected Mφs with EP2 and EP4 agonists to evaluate their effects on the induction of necrosis. Treatment of H37Rv-infected primary human Mφs with butaprost, an EP2 agonist, as well as with PGE2 augmented protection against necrosis (Fig. 1) consistent with our previous findings in Mφs from EP2−/− mice (5). Treatment with EP4 agonist had no effect on induction of necrosis. These experiments suggested that EP2 but not EP4 is directly involved in protective mechanisms against Mtb and raised the question of whether infection with H37Ra or with H37Rv alters their expression at the mRNA and protein level. The mRNA expression of EP2 and EP4 was significantly increased at 24 h after Mtb infection (Fig. 2A). Similarly, the expression of EP2 and EP4 protein was slightly increased (Fig. 2B). As Gα-coupled PGE2 receptors induce COX2 expression (14–16), we investigated whether EP4 is involved in the regulation of PGE2 production.
Figure 1.

Effect of EP2 and EP4 stimulation on Mφ necrosis induced by H37Rv. H37Rv (MOI 10:1)-infected human Mφs were treated with PGE2 (1 or 10 μM), butaprost (EP2 agonist; 1 or 10 μM) or ONO-AE1-329 (EP4 agonist; 1 or 10 μM). The number of cells with damaged plasma membrane indicating necrosis was measured at 48 h after infection (n=3; values are expressed as means ± sem). *P < 0.05 vs. Mφs infected with H37Rv only.
Figure 2.

EP2 and EP4 expression in Mtb-infected Mφs. A) At 24 h after infection with H37Ra and H37Rv (MOI 10:1), relative gene expression of EP2 and EP4 was analyzed by quantitative real-time RT-PCR using GAPDH as a reference gene (n=3; mean ± sem). *P < 0.05. B) EP2 and EP4 protein expression measured by Western blot analysis (left panel). GAPDH was used as a loading control (n=3). The intensity of the EP2 and EP4 protein was normalized to that of GAPDH and presented as a fold change relative to uninfected control cells. Values are expressed as means ± sem of 3 independent experiments (right panel).
Mtb-infected Mφs induce COX2 and mPGES-1, resulting in PGE2 production
In PGE2 synthesis, arachidonic acid (AA) originating from the nuclear and plasma membrane is processed to PGH2 by COX1 or COX2, which is then converted into PGE2 by cPGES, mPGES-1, or mPGES-2 (7, 25). COX1, cPGES, and mPGES-2 are constitutively expressed and play a role in basal PGE2 production. COX2 and mPGES-1 are functionally coupled (26), and their expression is induced by a number of stimulants, resulting in PGE2 production at sites of stress and inflammation (7). However, COX1 and COX2 expression is induced in inflammatory joints of mice in an arthritis model (27), and mPGES-2 is induced to high levels in cancer cells (25).
Avirulent H37Ra induced higher expression of COX2 in host Mφs, which correlated with higher PGE2 production in Mφs infected with H37Ra (5). To determine how PGE2 production is regulated in Mtb-infected Mφs, we evaluated the expression of COX1, COX2, cPGES, mPGES-1, and mPGES-2 in Mφs infected with avirulent H37Ra and virulent H37Rv. H37Ra and, to a lesser extent, H37Rv infection induced expression of COX2 protein at 4 and 12 h and expression of mPGES-1 at 24 and 48 h after infection. COX2 expression was restricted to early time points in Mφs infected with virulent H37Rv with almost no COX2 present at 24 and 48 h (Fig. 3). In contrast, COX1, cPGES and mPGES-2 expression was not affected by infection of Mφs with either virulent or avirulent Mtb. These findings indicate that COX2 and mPGES-1 are required for PGE2-production in Mtb-infected Mφ. In contrast to H37Rv-infected Mφs, COX2 protein is expressed up to 48 h after infection in Mφs infected with H37Ra, correlating with higher levels of PGE2 produced by H37Ra-infected Mφs.
Figure 3.

Top panel: time-dependent protein expression of COX1, COX2, cPGES, mPGES-2, and mPGES-1 in H37Ra- and H37Rv-infected (MOI 10:1) Mφs evaluated by Western blot analysis (n=3). Bottom panels: intensity of the COX2 protein (left panel) and mPGES-1 protein (right panel) was normalized to that of GAPDH and presented as a fold change relative to H37Ra-infected Mφs at 48 h after infection. Values are expressed as means ± sem of 3 independent experiments.
EP4 triggering and p38 MAPH activation in Mtb-infected Mφs induce PGE2 production
To better characterize the role of EP4 in PGE2 production, we examined the effect of H37Ra infection on Mφs pretreated with the EP4 antagonist AH23848. AH23848 inhibited COX2 and mPGES-1 expression at the mRNA and protein level (Fig. 4A, B) and inhibited PGE2 production in a dose-dependent manner (Fig. 4C). The other EP4 antagonist, ONO-AE2-227, as well as AH23848, inhibited COX2 and mPGES-1 expression (Fig. 4A). In H37Ra-infected Mφs, treatment with the EP2 antagonist AH6809 or the EP1/3 agonist sulprostone (negative control) had no effect on the expression of COX2 or of mPGES-1 (Fig. 4A). As PGE2 also affects phagocytosis of Mφs (28–30), we tested Mtb uptake in EP4 antagonist-reated and untreated Mφs. There was no significant difference in Mtb uptake between EP4 antagonist-treated and untreated Mφs (Supplemental Fig. S2), suggesting that the EP4 signaling pathway predominantly modulates PGE2 production in Mtb-infected Mφs downstream of Mtb uptake.
Figure 4.
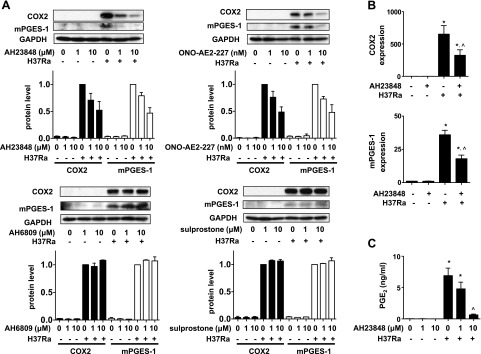
Regulation of COX2 and mPGES-1 expression and PGE2 production in H37Ra-infected human Mφs by EP4. Mφs were treated with the EP2 antagonist AH6809 and the EP1/EP3 agonist sulprostone (1 or 10 μM), the EP4 antagonists AH23848 and ONO-AE2-227 (1 or 10 μM and 1 or 10 nM, respectively), and inoculated with H37Ra (MOI 10:1). A) Top blot panels: Western blots of COX2 and mPGES-1 of H37Ra-infected Mφs untreated and treated with EP4 antagonists at 24 h after infection (n=3). Top graph panels: quantization of COX 2 and mPGES-1 protein. Intensity of the COX2 protein (left panel) and mPGES-1 protein (right panel) was normalized to that of GAPDH and is presented as a fold change relative to Mφs infected with H37Ra only. Bottom blot panels: Western blots of COX2 and mPGES-1 protein in H37Ra-infected Mφs untreated and treated with AH6809 or sulprostone at 24 h after infection (n=3). Bottom graph panels: quantization of COX2 and mPGES-1 protein. In all experiments, values are expressed as means ± sem of 3 independent experiments. B) Relative gene expression of COX2 and mPGES-1 in AH23848 (10 μM)-treated and untreated human Mφs infected with H37Ra (MOI 10:1) at 24 h after infection (n=3; means±sem). *P < 0.05 vs. uninfected Mφs; ^P < 0.05 vs. Mφs infected with H37Ra alone. C) PGE2 production measured by ELISA at 48 h after infection (n=3; means± sem). *P < 0.05 vs. uninfected Mφs; ^P < 0.05 vs. Mφs infected with H37Ra.
On the basis of previous findings (31–33), we tested whether p38 MAPK-dependent pathways are involved in PGE2 production induced in Mtb-infected human Mφs. H37Ra infection induced p38 MAPK phosphorylation starting at 10 min after infection, which culminated at 60 min and decreased at later time points (Fig. 5A). To determine whether p38 MAPK activation regulates PGE2 production, we evaluated PGE2 production in H37Ra-infected Mφs treated with the p38 MAPK inhibitor SB202190. SB202190 inhibited COX2 and mPGES-1 expression and PGE2 production in H37Ra-infected Mφs (Fig. 5B, C), indicating that p38 MAPK signaling is required for PGE2 production. In addition, we determined whether EP4 triggering directly activates p38 MAPK. The addition of EP4 agonist to human Mφs did not induce p38 MAPK phosphorylation (Fig. 6A), and at 30 min and 4 h after H37Ra addition, there was no change of p38 MAPK phosphorylation between EP4 antagonist-treated and untreated Mφs (Fig. 6B). Thus, p38 MAPK signaling and EP4 signaling are independently required for optimal PGE2 production in Mtb-infected Mφs , and p38 phosphorylation is not directly induced by EP4 triggering.
Figure 5.

Regulation of COX2 and mPGES-1 expression and PGE2 production by p38 MAPK in H37Ra-infected human Mφs. A) Western blot of time-dependent p38 MAPK phosphorylation in H37Ra infection (MOI 10:1). Ratio of P-p38 MAPK to p38 MAPK is indicated for every time point (n=3). B) Left panel: Western blot of COX2 and mPGES-1 in Mφs treated with the p38 MAPK inhibitor SB202190 (1 or 10 μM) 24 h after H37Ra infection (MOI 10:1). Right panel: quantization of COX2 and mPGES-1 protein. Intensity of the COX2 protein (left) and mPGES-1 protein (right) was normalized to that of GAPDH and presented as fold change relative to Mφs infected with H37Ra only. Values are expressed as means ± sem of 3 independent experiments. C) PGE2 production by H37Ra (MOI 10:1)-infected Mφs treated with SB202190 (10 μM) measured by ELISA at 24 h after infection (n=3; means±sem). *P < 0.05 vs. uninfected Mφs; ^P < 0.05 vs. Mφs infected with H37Ra only.
Figure 6.

Role of EP4 in the activation of the p38 MAPK signaling in H37Ra-infected human Mφs. A) Western blot of p38 MAPK phosphorylation in Mφs stimulated with the EP4 agonist ONO-AE1-329 (10 μM). Mφ lysates treated with DMSO (final concentration: 0.1%) only and with the TLR1/2 agonist Pam3CSK4 (10 μg/ml) were used as negative and positive controls, respectively (n=4). B) Western blot of p38 MAPK phosphorylation of H37Ra (MOI 10:1)-infected Mφs treated with and without the EP4 antagonist AH23848 (10 μM) at 30 min and 240 min after infection (n=4).
Mtb activates a TLR2/p38 MAPK signaling pathway, leading to PGE2 production
Although Toll-like receptor (TLR) 4, TLR9, and NOD2 were found to play a role in the recognition of Mtb (34), TLR2 in association with TLR1/TLR6 is the predominant Toll-like receptor recognizing mycobacterial determinants in the initiation of immune responses (35, 36). We therefore focused on the role of TLR2 in the production of PGE2.
To determine whether TLR2 stimulation induces COX2 and mPGES-1 expression through the p38 MAPK and EP4 signaling pathways in primary human Mφs, we first evaluated COX2 and mPGES-1 expression in TLR2 agonist-treated Mφs. Treatment with the TLR2 agonists Pam3CSK4 (TLR1/2 agonist) and FSL-1 (TLR2/6 agonist)-induced p38 MAPK phosphorylation and the expression of COX2 and mPGES-1 (Fig. 7A, B). Moreover, these TLR2-induced changes were blocked by the p38 MAPK inhibitor SB202190 and the EP4 antagonist AH23848 (Fig. 7C). Similar results were obtained using Mtb-infected Mφs (Figs. 4A and 5B).
Figure 7.
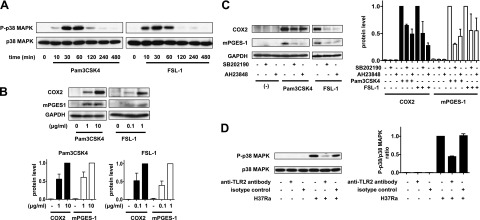
TLR2 stimulation of human Mφs induces COX2 and mPGES-1 expression through the p38 MAPK and EP4 signaling pathways. A) Western blot of p38 MAPK phosphorylation at different time points in Mφs treated with TLR1/2 agonist Pam3CSK4 (10 μg/ml) or TLR2/6 agonist FSL-1 (1 μg/ml; n=3). B) Top panel: Western blot of COX2 and mPGES-1 in Mφs treated with Pam3CSK4 (1 or 10 μg/ml) or FSL1 (0.1 or 1 μg/ml) at 24 h after treatment (n=3). Bottom panels: quantization of COX2 protein and mPGES-1 protein. Intensity of the COX2 protein (left panel) and mPGES-1 protein (right panel) was normalized to that of GAPDH and presented as fold change relative to Mφs treated with Pam3CSK4 (10 μg/ml) or FSL-1 (1 μg/ml). Values are expressed as means ± sem of 3 independent experiments. C) Left panel: Western blot of COX2 and mPGES-1 in Mφs treated with the p38 MAPK inhibitor SB202190 (10 μM) or the EP4 antagonist AH23848 (10 μM) and the TLR2 agonists Pam3CSK4 (10 μg/ml) and FSL-1 (1 μg/ml) at 24 h after treatment (n=3). Right panels: quantization of COX2 and mPGES-1 protein expression from Western blot analysis on left side. The intensity of the COX2 protein (left panel) and mPGES-1 protein (right panel) was normalized to that of GAPDH and presented as fold change relative to TLR2 agonists treated Mφs. Values are means ± sem of 3 independent experiments. D) Left panel: Western blot of p38 MAPK phosphorylation at 30 min in H37Ra (MOI 10:1)-infected Mφs treated with an anti-TLR2 antibody (30 μg/ml), which neutralizes TLR2 activity (n=3). Right panel: ratios of P-p38 MAPK to p38 MAPK, shown as fold change relative to Mφs infected with H37Ra only. Values are means ± sem of 3 independent experiments.
To determine whether triggering of TLR2 is associated with p38 MAPK phosphorylation in Mtb-infected Mφs, we investigated the effect of blocking TLR2 signaling. p38 MAPK phosphorylation was inhibited by blocking TLR2 using a specific antibody (Fig. 7D). Thus, Mtb activates TLR2/p38 MAPK and EP4 signaling pathways, resulting in PGE2 production.
Mtb triggering of EP4 enhances PGE2 production induced by the TLR2/p38 MAPK signaling pathway
These findings suggested that PGE2 production in Mtb-infected Mφ depends on two different steps. We hypothesized that the first step consists of triggering pathogen recognition receptors (PRRs) by Mtb, which induces COX2 and mPGES-1 expression through p38 MAPK phosphorylation, resulting in PGE2 production. In the second event, PGE2 produced by the first step triggers EP4 and augments PGE2 production in a p38 MAPK-independent manner.
To determine whether TLR2 and EP4 triggering exerts a synergistic effect on Mtb-infected human Mφs, which results in optimal up-regulation of PGE2 production, we evaluated COX2 and mPGES-1 expression in Mφs infected with virulent H37Rv, which produce small amounts of PGE2 (5), after treatment with EP4 agonist ONO-AE1-329. Although there was little COX2 and mPGES-1 protein expression induced by EP4 stimulation with ONO-AE1-329 alone, EP4 stimulation in H37Rv-infected Mφs induced substantial COX2 and especially mPGES-1 protein expression (Fig. 8A). A similar result was obtained with Mφs activated by the TLR2 agonist Pam3CSK4 (Fig. 8B). COX2 and mPGES-1 gene transcription was also enhanced by the EP4 agonist ONO-AE1-329 in H37Rv-infected Mφs (Fig. 8C). The EP2 agonist butaprost had no effect. Finally, COX2 and mPGES-1 expression in H37Rv-infected Mφs activated with the EP4 agonist ONO-AE1-329 was significantly inhibited by the EP4 antagonist AH23848 (Fig. 8D). Cumulatively, these findings indicate that EP4 stimulation augments COX2 and mPGES-1 expression by the TLR2/p38 MAPK signaling pathway, resulting in increased PGE2 production. These findings support a model in which costimulation of EP4 and TLR2 is necessary to yield optimal production of PGE2 (Fig. 9).
Figure 8.
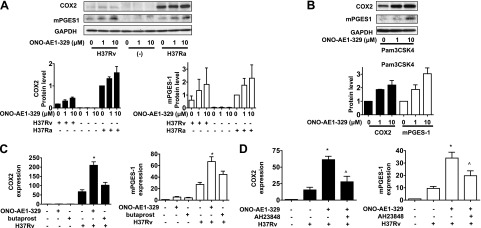
Effect of EP4 stimulation on COX2 and mPGES-1 expression in Mtb-infected Mφs. A) Top panel: Western blot of COX2 and mPGES-1 at 24 h after infection in H37Ra- and H37Rv (MOI 10:1)-infected Mφs treated with the EP4 agonist ONO-AE1-329 (1 or 10 μM; n=3). Bottom panels: quantization of COX2 and mPGES-1 from Western blot panel. Intensity of the COX2 and mPGES-1 protein was normalized to that of GAPDH and presented as fold change relative to only H37Ra-infected Mφs. Values are means of 3 independent experiments. B) Top panel: Western blot of COX2 and mPGES-1 in Mφs treated with the EP4 agonist ONO-AE1-329 (1 or 10 μM) and the TLR1/2 agonist Pam3CSK4 (10 μg/ml) at 24 h after treatment (n=3). Bottom panel: quantization of COX2 and mPGES-1 protein in Western blot analysis. Intensity of the COX2 protein and mPGES-1 protein was normalized to that of GAPDH and presented as fold change relative to Mφs treated with Pam3CSK4 only. Values are means ± sem of 3 independent experiments. C) Relative gene expression of COX2 and mPGES-1 in ONO-AE1-329 (10 μM)-treated H37Rv (MOI 10:1)-infected Mφs at 24 h after infection. (n=3; means±sem). EP2 agonist butaprost has a minimal effect. *P < 0.05 vs. Mφs infected with H37Rv only. D) Down-regulation of the relative gene expression of COX2 and mPGES-1 in ONO-AE1-329 (10 μM)-treated Mφs infected with H37Rv (MOI 10:1) by the EP4 antagonist AH23848 (10 μM; n=3; means±sem). *P < 0.05 vs. Mφs infected with H37Rv only; ^P < 0.05 vs. ONO-AE1-329-treated Mφs infected with H37Rv.
Figure 9.

Proposed mechanisms of PGE2 production induced by TLR2/p38 MAPK and EP4 stimulation, leading to optimal PGE2 production and protection against necrosis in Mtb-infected Mφs. PGE2 generated by TLR2 stimulation/p38 MAPK phosphorylation following Mtb infection (first step) triggers EP4 to produce increased amounts of PGE2 (second step). PGE2 blocked necrosis induced by Mtb infection via EP2.
DISCUSSION
The role of PGE2 in Mtb-infected Mφs is confusing because 4 functionally different PGE2 receptors exist, and many different cell types are able to produce PGE2, which might exert distinct effects on Mφs. Mφs are well-known producers of prostaglandins, including PGE2 (37). In THP-1 cells, dormant Mtb infection induces PGE2 production, which prevents the induction of necrosis (38). Similarly, avirulent H37Ra induces more PGE2 production than infection with virulent H37Rv, resulting in the inhibition of MPT and of necrosis (5). On the other hand, PGE2 inhibits efferocytosis, which increases clearance of Mtb (39). Thus, the mechanisms of PGE2 production in Mtb-infected Mφ and the role of PGE2 receptor EP4 are unclear.
Mtb-infected Mφs are known to induce COX2 expression (5, 33), and we demonstrate here that Mtb infection also induces mPGES-1 expression. In the LPS-treated murine Mφ cell line RAW 264.7, cAMP-response element binding protein (16), nuclear factor-κB, and early growth-response gene product 1 (26) enhance the transcriptional activation of both COX2 and mPGES-1. The induction of COX2 and mPGES-1 expression is coupled and mPGES-1 expression is generally delayed as compared to COX2 expression in several cell types (40, 41). Specifically, in Mφs, COX2 and mPGES-1 expression was induced at 4 and 24 h after Mtb infection, respectively. Of note, PGE2 production already started 4 h after addition of Mtb (5). These findings suggest that COX2 and constitutively expressed cPGES or mPGES-2 cooperate to produce PGE2 in the early phase of infection and that mPGES-1, as well as COX2, becomes an essential player in PGE2 production in the late phase of infection.
Recognition of mycobacterial components depends on TLR2, TLR4, TLR9, and TLR1/TLR6 that heterodimerize with TLR2 (34, 42). In our studies, TLR2 agonists induced p38 MAPK phosphorylation similar to Mφs infected with H37Ra. An inhibitory anti-TLR2 antibody blocked p38 MAPK phosphorylation, and a p38 MAPK inhibitor blocked the expression of COX2 and mPGES-1 in H37Ra-infected human Mφs. Our findings indicate that TLR2 is involved in p38 MAPK-dependent PGE2 production. Mtb-dependent release of TNF-α, which plays an important role in cell death of murine primary Mφs, dendritic cells (43), and murine RAW 264.7 cells (44), was also found to be TLR2 dependent. Furthermore, clinical studies demonstrate a role of TLR2 in Mtb infection. Several gene association studies demonstrate a correlation between single-nucleotide polymorphisms (SNPs) in TLR2 and tuberculosis (45, 46). These findings identify TLR2 genetic polymorphisms as important determinants of susceptibility to tuberculosis, which might regulate the production of lipid mediators and cytokines, such as PGE2 and TNF-α, which determine the pathophysiology of tuberculosis. Not only p38 MAPK, but also other MAPKs, might contribute to the first step of PGE2 production in Mtb-infected Mφs. Data using a phospho-MAPK array for Mtb-infected human Mφs indicated that p38 MAPK activation is stronger than that of other MAPKs (47). Interleukin-1β(IL-1β) might also be required for p38 MAPK-dependent PGE2 production of Mtb-infected Mφs. In human Mφs, H37Ra induces IL-1β production. In addition, PGE2 production is blocked by a caspase-1 inhibitor, which inactivates IL-1β (unpublished results). Cumulatively, we show that p38 MAPK is critical to initiate PGE2 production in Mtb-infected Mφ.
We further demonstrate that in Mtb-infected Mφs, a signaling pathway initiated by TLR2/p38 MAPK up-regulates PGE2 production by the action of EP4-triggered autocrine signaling pathways. In human synovial fibroblasts, PGE2 regulates the level and stability of COX2 mRNA induced by IL-1β treatment (48). In human Mφs, the induction of COX2 and mPGES-1 expression by an EP4 agonist alone was weak, but increased after the addition of TLR2 agonists or by Mtb infection. EP4 stimulation might stabilize or enhance transcription of COX2 and mPGES-1 induced by the PRRs/p38 MAPK signaling pathway. A mechanism triggering PGE2 receptors in a PRRs/p38 MAPK-dependent fashion leading to enhancement of cellular functions (positive feedback) was previously described. In the mouse Mφ cell line RAW 264.7, PGE2 stimulated EP2 expression and cAMP production, inducing the expression of COX2 and mPGES-1 (16). On the other hand, EP4 is the dominant regulator of matrix metalloproteinase-1-dependent mPGES-1 expression in mouse Mφs when compared with EP2 (49). We could not see differences in the effect of EP2 and EP4 expression in primary human Mφs before and after Mtb infection. EP2 and EP4 have clearly different properties, such as different degrees of desensitization and affinity to PGE2 (50). Furthermore, the functions of EP2 are distinct from EP4 functions with regard to phagocytosis, efferocytosis, and cell death (28, 29). Our data indicate that the EP4-mediated positive feedback of COX2 and mPGES-1 expression, resulting in enhanced PGE2 production in Mtb-infected Mφs might increase the local levels of prostanoids, including PGE2 in the tissues, where it is needed to maintain an effective prostanoid concentration. Because of the short half-life of biologically active PGE2 (51), it is not surprising that large amounts of PGE2 have to be continuously produced by a mechanism that up-regulates its production in a PGE2-dependent manner.
In this study, we confirmed that avirulent H37Ra induces higher expression of COX2 than virulent H37Rv (5). Interestingly, EP4 stimulation increased COX2 and mPGES-1 expression in H37Rv-infected Mφs and in H37Ra-infected Mφs, indicating that there is no fundamental difference in the regulation of PGE2 production by the EP4 signaling pathway in H37Ra and H37Rv-infected human Mφs. On the other hand, EP4 agonist treatment did not inhibit necrosis of Rv-infected Mφs (Fig. 1), probably because the up-regulation of COX2 and mPGES-1 expression by EP4 agonist treatment could not induce a sufficiently high PGE2 production to protect against MPT. We speculate that virulent factors of H37Rv block the PRRs/p38 MAPK signaling pathway, resulting in low PGE2 production and induction of necrosis compared with H37Ra.
In summary, we report here that a positive feedback loop in PGE2 production in Mtb-infected human Mφs is dependent on EP4 signaling, which maintains a critical effective concentration of PGE2, an eicosanoid involved in the defense against Mtb.
Supplementary Material
Acknowledgments
The authors are grateful to Dr. Eileen Remold-O'Donnell for helpful discussions. H.G.R. and T.N. conceived of and designed the experiments, analyzed the data and wrote the article; T.N. did the experiments with assistance from H.G. and X.Z.; S.K. provided intellectual input.
The authors declare no conflicts of interest.
This work is funded by the U.S. National Institutes of Health (grant AI072143). T.N. is supported by Strategic Young Researcher Overseas Visits Program for Accelerating Brain Circulation (Japan Society of the Promotion of Science).
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- AA
- arachidonic acid
- cAMP
- cyclic adenosine monophosphate
- COX1
- cyclooxygenase 1
- COX2
- cyclooxygenase 2
- cPGES
- cytosolic prostaglandin E synthase
- G
- guanine nucleotide binding
- GAPDH
- glyceraldehyde 3-phosphate dehydrogenase
- IL-1β
- interleukin-1β
- IMDM
- Iscove's modified Dulbecco's medium
- LXA4
- lipoxin A4
- Mφ
- macrophage
- MAPK
- mitogen-activated protein kinase
- Mtb
- Mycobacterium tuberculosis
- mPGES-1
- microsomal prostaglandin E synthase-1
- mPGES-2
- microsomal prostaglandin E synthase-2
- MPT
- mitochondrial permeability transition
- PAGE
- polyacrylamide gel electrophoresis
- PGE2
- prostaglandin E2
- PGH2
- prostaglandin H2
- PKA
- protein kinase A
- PRR
- pathogen recognition receptor
- SDS
- sodium dodecyl sulfate
- TLR
- Toll-like receptor
REFERENCES
- 1. World Health Organization (2012) Global Tuberculosis Report 2012, World Health Organization, Geneva, Switzerland [Google Scholar]
- 2. Chen M., Gan H., Remold H. G. (2006) A mechanism of virulence: virulent Mycobacterium tuberculosis strain H37Rv, but not attenuated H37Ra, causes significant mitochondrial inner membrane disruption in macrophages leading to necrosis. J. Immunol. 176, 3707–3716 [DOI] [PubMed] [Google Scholar]
- 3. Park J. S., Tamayo M. H., Gonzalez-Juarrero M., Orme I. M., Ordway D. J. (2006) Virulent clinical isolates of Mycobacterium tuberculosis grow rapidly and induce cellular necrosis but minimal apoptosis in murine macrophages. J. Leukoc. Biol. 79, 80–86 [DOI] [PubMed] [Google Scholar]
- 4. Sohn H., Lee K. S., Kim S. Y., Shin D. M., Shin S. J., Jo E. K., Park J. K., Kim H. J. (2009) Induction of cell death in human macrophages by a highly virulent Korean isolate of Mycobacterium tuberculosis and the virulent strain H37Rv. Scand. J. Immunol. 69, 43–50 [DOI] [PubMed] [Google Scholar]
- 5. Chen M., Divangahi M., Gan H., Shin D. S., Hong S., Lee D. M., Serhan C. N., Behar S. M., Remold H. G. (2008) Lipid mediators in innate immunity against tuberculosis: opposing roles of PGE2 and LXA4 in the induction of macrophage death. J. Exp. Med. 205, 2791–2801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sugimoto Y., Narumiya S. (2007) Prostaglandin E receptors. J. Biol. Chem. 282, 11613–11617 [DOI] [PubMed] [Google Scholar]
- 7. Legler D. F., Bruckner M., Uetz-von Allmen E., Krause P. (2010) Prostaglandin E2 at new glance: novel insights in functional diversity offer therapeutic chances. Int. J. Biochem. Cell Biol. 42, 198–201 [DOI] [PubMed] [Google Scholar]
- 8. Tabata H., Tanaka S., Sugimoto Y., Kanki H., Kaneko S., Ichikawa A. (2002) Possible coupling of prostaglandin E receptor EP(1) to TRP5 expressed in Xenopus laevis oocytes. Biochem. Biophys. Res. Commun. 298, 398–402 [DOI] [PubMed] [Google Scholar]
- 9. Namba T., Sugimoto Y., Negishi M., Irie A., Ushikubi F., Kakizuka A., Ito S., Ichikawa A., Narumiya S. (1993) Alternative splicing of C-terminal tail of prostaglandin E receptor subtype EP3 determines G-protein specificity. Nature 365, 166–170 [DOI] [PubMed] [Google Scholar]
- 10. Arakawa T., Laneuville O., Miller C. A., Lakkides K. M., Wingerd B. A., DeWitt D. L., Smith W. L. (1996) Prostanoid receptors of murine NIH 3T3 and RAW 264.7 cells. Structure and expression of the murine prostaglandin EP4 receptor gene. J. Biol. Chem. 271, 29569–29575 [DOI] [PubMed] [Google Scholar]
- 11. Katsuyama M., Ikegami R., Karahashi H., Amano F., Sugimoto Y., Ichikawa A. (1998) Characterization of the LPS-stimulated expression of EP2 and EP4 prostaglandin E receptors in mouse macrophage-like cell line, J774.1. Biochem. Biophys. Res. Commun. 251, 727–731 [DOI] [PubMed] [Google Scholar]
- 12. Regan J. W. (2003) EP2 and EP4 prostanoid receptor signaling. Life Sci. 74, 143–153 [DOI] [PubMed] [Google Scholar]
- 13. Fujino H., West K. A., Regan J. W. (2002) Phosphorylation of glycogen synthase kinase-3 and stimulation of T-cell factor signaling following activation of EP2 and EP4 prostanoid receptors by prostaglandin E2. J. Biol. Chem. 277, 2614–2619 [DOI] [PubMed] [Google Scholar]
- 14. Vichai V., Suyarnsesthakorn C., Pittayakhajonwut D., Sriklung K., Kirtikara K. (2005) Positive feedback regulation of COX-2 expression by prostaglandin metabolites. Inflamm. Res. 54, 163–172 [DOI] [PubMed] [Google Scholar]
- 15. Obermajer N., Muthuswamy R., Lesnock J., Edwards R. P., Kalinski P. (2011) Positive feedback between PGE2 and COX2 redirects the differentiation of human dendritic cells toward stable myeloid-derived suppressor cells. Blood 118, 5498–5505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Diaz-Munoz M. D., Osma-Garcia I. C., Fresno M., Iniguez M. A. (2012) Involvement of PGE2 and the cAMP signalling pathway in the up-regulation of COX-2 and mPGES-1 expression in LPS-activated macrophages. Biochem. J. 443, 451–461 [DOI] [PubMed] [Google Scholar]
- 17. Coleman R. A., Grix S. P., Head S. A., Louttit J. B., Mallett A., Sheldrick R. L. (1994) A novel inhibitory prostanoid receptor in piglet saphenous vein. Prostaglandins 47, 151–168 [DOI] [PubMed] [Google Scholar]
- 18. Wilson R. J., Rhodes S. A., Wood R. L., Shield V. J., Noel L. S., Gray D. W., Giles H. (2004) Functional pharmacology of human prostanoid EP2 and EP4 receptors. Eur. J. Pharmacol. 501, 49–58 [DOI] [PubMed] [Google Scholar]
- 19. Gan H., Lee J., Ren F., Chen M., Kornfeld H., Remold H. G. (2008) Mycobacterium tuberculosis blocks crosslinking of annexin-1 and apoptotic envelope formation on infected macrophages to maintain virulence. Nat. Immunol. 9, 1189–1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gan H., He X., Duan L., Mirabile-Levens E., Kornfeld H., Remold H. G. (2005) Enhancement of antimycobacterial activity of macrophages by stabilization of inner mitochondrial membrane potential. J. Infect. Dis. 191, 1292–1300 [DOI] [PubMed] [Google Scholar]
- 21. Shoji M., Tanabe N., Mitsui N., Tanaka H., Suzuki N., Takeichi O., Sugaya A., Maeno M. (2006) Lipopolysaccharide stimulates the production of prostaglandin E2 and the receptor Ep4 in osteoblasts. Life Sci. 78, 2012–2018 [DOI] [PubMed] [Google Scholar]
- 22. Ferronato S., Lira M. G., Olivato S., Scuro A., Veraldi G. F., Romanelli M. G., Patuzzo C., Malerba G., Pignatti P. F., Mazzucco S. (2011) Upregulated expression of Toll-like receptor 4 in peripheral blood of ischaemic stroke patients correlates with cyclooxygenase 2 expression. Eur. J. Vasc. Endovasc. Surg. 41, 358–363 [DOI] [PubMed] [Google Scholar]
- 23. Hiraiwa H., Sakai T., Mitsuyama H., Hamada T., Yamamoto R., Omachi T., Ohno Y., Nakashima M., Ishiguro N. (2011) Inflammatory effect of advanced glycation end products on human meniscal cells from osteoarthritic knees. Inflamm. Res. 60, 1039–1048 [DOI] [PubMed] [Google Scholar]
- 24. Zaslona Z., Serezani C. H., Okunishi K., Aronoff D. M., Peters-Golden M. (2012) Prostaglandin E2 restrains macrophage maturation via E prostanoid receptor 2/protein kinase A signaling. Blood 119, 2358–2367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Park J. Y., Pillinger M. H., Abramson S. B. (2006) Prostaglandin E2 synthesis and secretion: the role of PGE2 synthases. Clin. Immunol. 119, 229–240 [DOI] [PubMed] [Google Scholar]
- 26. Diaz-Munoz M. D., Osma-Garcia I. C., Cacheiro-Llaguno C., Fresno M., Iniguez M. A. (2010) Coordinated up-regulation of cyclooxygenase-2 and microsomal prostaglandin E synthase 1 transcription by nuclear factor-κB and early growth response-1 in macrophages. Cell. Signal. 22, 1427–1436 [DOI] [PubMed] [Google Scholar]
- 27. Chen M., Boilard E., Nigrovic P. A., Clark P., Xu D., Fitzgerald G. A., Audoly L. P., Lee D. M. (2008) Predominance of cyclooxygenase 1 over cyclooxygenase 2 in the generation of proinflammatory prostaglandins in autoantibody-driven K/BxN serum-transfer arthritis. Arthritis Rheum. 58, 1354–1365 [DOI] [PubMed] [Google Scholar]
- 28. Aronoff D. M., Canetti C., Peters-Golden M. (2004) Prostaglandin E2 inhibits alveolar macrophage phagocytosis through an E-prostanoid 2 receptor-mediated increase in intracellular cyclic AMP. J. Immunol. 173, 559–565 [DOI] [PubMed] [Google Scholar]
- 29. Medeiros A. I., Serezani C. H., Lee S. P., Peters-Golden M. (2009) Efferocytosis impairs pulmonary macrophage and lung antibacterial function via PGE2/EP2 signaling. J. Exp. Med. 206, 61–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Serezani C. H., Kane S., Medeiros A. I., Cornett A. M., Kim S. H., Marques M. M., Lee S. P., Lewis C., Bourdonnay E., Ballinger M. N., White E. S., Peters-Golden M. (2012) PTEN directly activates the actin depolymerization factor cofilin-1 during PGE2-mediated inhibition of phagocytosis of fungi. Sci. Signal. 5, ra12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Guan Z., Baier L. D., Morrison A. R. (1997) p38 mitogen-activated protein kinase down-regulates nitric oxide and up-regulates prostaglandin E2 biosynthesis stimulated by interleukin-1β. J. Biol. Chem. 272, 8083–8089 [DOI] [PubMed] [Google Scholar]
- 32. Oikawa A., Kobayashi M., Okamatsu Y., Shinki T., Kamijo R., Yamamoto M., Hasegawa K. (2007) Mitogen-activated protein kinases mediate interleukin-1β-induced receptor activator of nuclear factor-κB ligand expression in human periodontal ligament cells. J. Periodontal Res. 42, 367–376 [DOI] [PubMed] [Google Scholar]
- 33. Bansal K., Narayana Y., Patil S. A., Balaji K. N. (2009) M. bovis BCG induced expression of COX-2 involves nitric oxide-dependent and -independent signaling pathways. J. Leukoc. Biol. 85, 804–816 [DOI] [PubMed] [Google Scholar]
- 34. Kleinnijenhuis J., Oosting M., Joosten L. A., Netea M. G., Van Crevel R. (2011) Innate immune recognition of Mycobacterium tuberculosis. Clin. Dev. Immunol. 2011:405310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stenger S., Modlin R. L. (2002) Control of Mycobacterium tuberculosis through mammalian Toll-like receptors. Curr. Opin. Immunol. 14, 452–457 [DOI] [PubMed] [Google Scholar]
- 36. Krutzik S. R., Modlin R. L. (2004) The role of Toll-like receptors in combating mycobacteria. Semin. Immunol. 16, 35–41 [DOI] [PubMed] [Google Scholar]
- 37. Matsumoto H., Naraba H., Murakami M., Kudo I., Yamaki K., Ueno A., Oh-ishi S. (1997) Concordant induction of prostaglandin E2 synthase with cyclooxygenase-2 leads to preferred production of prostaglandin E2 over thromboxane and prostaglandin D2 in lipopolysaccharide-stimulated rat peritoneal macrophages. Biochem. Biophys. Res. Commun. 230, 110–114 [DOI] [PubMed] [Google Scholar]
- 38. Iona E., Pardini M., Gagliardi M. C., Colone M., Stringaro A. R., Teloni R., Brunori L., Nisini R., Fattorini L., Giannoni F. (2012) Infection of human THP-1 cells with dormant Mycobacterium tuberculosis. Microbes Infect. 14, 959–967 [DOI] [PubMed] [Google Scholar]
- 39. Martin C. J., Booty M. G., Rosebrock T. R., Nunes-Alves C., Desjardins D. M., Keren I., Fortune S. M., Remold H. G., Behar S. M. (2012) Efferocytosis is an innate antibacterial mechanism. Cell Host Microbe 12, 289–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Inada M., Matsumoto C., Uematsu S., Akira S., Miyaura C. (2006) Membrane-bound prostaglandin E synthase-1-mediated prostaglandin E2 production by osteoblast plays a critical role in lipopolysaccharide-induced bone loss associated with inflammation. J. Immunol. 177, 1879–1885 [DOI] [PubMed] [Google Scholar]
- 41. Stichtenoth D. O., Thoren S., Bian H., Peters-Golden M., Jakobsson P. J., Crofford L. J. (2001) Microsomal prostaglandin E synthase is regulated by proinflammatory cytokines and glucocorticoids in primary rheumatoid synovial cells. J. Immunol. 167, 469–474 [DOI] [PubMed] [Google Scholar]
- 42. Quesniaux V., Fremond C., Jacobs M., Parida S., Nicolle D., Yeremeev V., Bihl F., Erard F., Botha T., Drennan M., Soler M. N., Le Bert M., Schnyder B., Ryffel B. (2004) Toll-like receptor pathways in the immune responses to mycobacteria. Microbes Infect. 6, 946–959 [DOI] [PubMed] [Google Scholar]
- 43. Drage M. G., Pecora N. D., Hise A. G., Febbraio M., Silverstein R. L., Golenbock D. T., Boom W. H., Harding C. V. (2009) TLR2 and its co-receptors determine responses of macrophages and dendritic cells to lipoproteins of Mycobacterium tuberculosis. Cell. Immunol. 258, 29–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Basu S., Pathak S. K., Banerjee A., Pathak S., Bhattacharyya A., Yang Z., Talarico S., Kundu M., Basu J. (2007) Execution of macrophage apoptosis by PE_PGRS33 of Mycobacterium tuberculosis is mediated by Toll-like receptor 2-dependent release of tumor necrosis factor-α. J. Biol. Chem. 282, 1039–1050 [DOI] [PubMed] [Google Scholar]
- 45. Thuong N. T., Hawn T. R., Thwaites G. E., Chau T. T., Lan N. T., Quy H. T., Hieu N. T., Aderem A., Hien T. T., Farrar J. J., Dunstan S. J. (2007) A polymorphism in human TLR2 is associated with increased susceptibility to tuberculous meningitis. Genes Immun. 8, 422–428 [DOI] [PubMed] [Google Scholar]
- 46. Caws M., Thwaites G., Dunstan S., Hawn T. R., Lan N. T., Thuong N. T., Stepniewska K., Huyen M. N., Bang N. D., Loc T. H., Gagneux S., van Soolingen D., Kremer K., van der Sande M., Small P., Anh P. T., Chinh N. T., Quy H. T., Duyen N. T., Tho D. Q., Hieu N. T., Torok E., Hien T. T., Dung N. H., Nhu N. T., Duy P. M., van Vinh Chau N., Farrar J. (2008) The influence of host and bacterial genotype on the development of disseminated disease with Mycobacterium tuberculosis. PLoS Pathog. 4, e1000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rand L., Green J. A., Saraiva L., Friedland J. S., Elkington P. T. (2009) Matrix metalloproteinase-1 is regulated in tuberculosis by a p38 MAPK-dependent, p-aminosalicylic acid-sensitive signaling cascade. J. Immunol. 182, 5865–5872 [DOI] [PubMed] [Google Scholar]
- 48. Faour W. H., He Y., He Q. W., de Ladurantaye M., Quintero M., Mancini A., Di Battista J. A. (2001) Prostaglandin E2 regulates the level and stability of cyclooxygenase-2 mRNA through activation of p38 mitogen-activated protein kinase in interleukin-1 β-treated human synovial fibroblasts. J. Biol. Chem. 276, 31720–31731 [DOI] [PubMed] [Google Scholar]
- 49. Khan K. M., Kothari P., Du B., Dannenberg A. J., Falcone D. J. (2012) Matrix metalloproteinase-dependent microsomal prostaglandin E synthase-1 expression in macrophages: role of TNF-α and the EP4 prostanoid receptor. J. Immunol. 188, 1970–1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sugimoto Y., Narumiya S., Ichikawa A. (2000) Distribution and function of prostanoid receptors: studies from knockout mice. Prog. Lipid Res. 39, 289–314 [DOI] [PubMed] [Google Scholar]
- 51. Forstermann U., Neufang B. (1983) Elimination from the circulation of cats of 6-keto-prostaglandin E1 compared with prostaglandins E2 and I2. J. Pharm. Pharmacol. 35, 724–728 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.