

Abstract
Interspecies prion transmission often leads to stable changes in physical and biological features of prion strains, a phenomenon referred to as a strain mutation. It remains unknown whether changes in the replication environment in the absence of changes in PrP primary structure can be a source of strain mutations. To approach this question, RNA content was altered in the course of amplification of hamster strains in serial protein misfolding cyclic amplification (sPMCAb). On adaptation to an RNA-depleted environment and then readaptation to an environment containing RNA, strain 263K gave rise to a novel PrPSc conformation referred to as 263KR+, which is characterized by very low conformational stability, high sensitivity to proteolytic digestion, and a replication rate of 106-fold/PMCAb round, which exceeded that of 263K by almost 104-fold. A series of PMCAb experiments revealed that 263KR+ was lacking in brain-derived 263K material, but emerged de novo as a result of changes in RNA content. A similar transformation was also observed for strain Hyper, suggesting that this phenomenon was not limited to 263K. The current work demonstrates that dramatic PrPSc transformations can be induced by changes in the prion replication environment and without changes in PrP primary structure.—Gonzalez-Montalban, N., Jin Lee, Y., Makarava, N., Savtchenko, R., Baskakov, I. V. Changes in prion replication environment cause prion strain mutation.
Keywords: protein misfolding cyclic amplification, neurodegenerative diseases, protein misfolding, amyloid, conformational stability, proteinase K digestion
Misfolding and aggregation of the prion protein normal cellular isoform (PrPC) into a transmissible β-sheet-rich state, the PrP scrapie isoform (PrPSc), underlie a group of lethal neurodegenerative diseases of mammals (1). Multiple prion strains can be formed within the same amino acid sequence of the prion protein, giving rise to multiple strain-specific disease phenotypes (2–6). On cross-species prion transmission, stable changes in strain properties and disease phenotypes have frequently been observed, a phenomenon referred to as a strain mutation (7–11). Heritable changes in PrPSc conformation are believed to underlie the molecular mechanism behind strain mutations. It is generally assumed that prion strain mutations are primarily attributable to species-specific variations in the PrP primary sequence (11,12). It has been proposed that strain mutations occur due to lack of conformational compatibility between the strain-specific PrPSc structure of a donor species and the PrPC amino acid sequence of an acceptor species (12). Consistent with this hypothesis, molecular imaging studies revealed that the cross-β spine can switch its folding pattern within individual fibrils when the amyloid structure formed by hamster recombinant PrP was exposed to mouse recombinant PrP (13).
According to an emerging view, pools of PrPSc conformers within individual strains or isolates are intrinsically heterogeneous, presumably due to spontaneously occurring conformational mutations in PrPSc structure (14). Changes in the prion replication environment, including transmission between species, can give a selective advantage to minor PrPSc conformers, which otherwise remain undetected, leading to changes in PrPSc properties and/or disease phenotype (15–19). Consistent with this view, recent studies showed that drug-resistant prions can emerge quickly in cultured cells following treatment with prion inhibitors swainsonine or quinacrine (15,18). Reversible changes in the properties of protein misfolding cyclic amplification (PMCA)-derived vs. brain-derived PrPSc could be observed as a result of PrPSc amplification in serial PMCA (sPCMA; refs. 17, 20). These studies suggest that prion strains might indeed exhibit high levels of conformational plasticity and are subject to transformation when exposed to new replication environments.
While rare spontaneous strain mutations are believed to be one of the major sources of prion conformational diversity, the molecular origin and mechanisms underlying mutations are unknown. Can mutations occur in the absence of changes in PrP primary structure? What are the roles of cellular cofactors and the prion replication environment in strain mutations? Previous studies demonstrated that prions replicate with the assistance of other molecules (21). RNAs and polyanions were shown to form a favorable biochemical environment that catalyzes prion replication (3, 17, 21–23). Although RNA can serve as a catalyst, it does not appear to be an essential component of PrPSc particles (24, 25), nor is it important for defining prion strain-specific features (23). Recent studies revealed that lipids might serve as possible cellular cofactors that are important for generating prions with high infectivity titers and for defining strain-specific features (3, 6, 26).
The current study asked the question whether changes in prion replication environment and, specifically, RNA content in the absence of alteration in PrP primary structure lead to a stable change in PrPSc properties. We found that while adaptation of 263K or hyper (HY) PrPSc to an RNA-depleted environment in PMCA with beads (PMCAb) did not change their features, PrPSc of both strains underwent remarkable transformation on readaptation to an environment containing RNA. The 263K PrPSc readapted to the RNA-containing environment (referred to as 263KR+) displayed dramatically lower conformational stability and proteinase K (PK) resistance than that of 263K. Moreover, the PMCAb amplification rate for 263KR+ was found to be 3–4 orders of magnitude higher than that of 263K. Furthermore, the current study revealed that 263KR+ was lacking in the original brain-derived or PMCAb-derived 263K material, but emerged as a result of change in RNA content. The present work demonstrated that changes in the prion replication environment not only created conditions for selective amplification of minor PrPSc conformers, but could also give rise to a PrPSc transformation. In other words, change in replication environment plays an active role in generating prion conformational diversity even in the absence of changes in PrP primary structure.
MATERIALS AND METHODS
Ethics statement
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the U.S. National Institutes of Health (NIH). The protocol was approved by the Institutional Animal Care and Use Committee of the University of Maryland (assurance number A32000-01: permit number: 0312020).
PMCAb
Normal brain homogenate (NBH; 10%) from healthy hamsters was prepared as described previously (27) and used as a substrate for PMCAb (28). NBH (10%) in conversion buffer was used as the substrate in PMCAb reactions. To produce RNA-depleted NBH, 50 μl of 10 mg/ml RNase A (R4875; Sigma-Aldrich, St. Louis, MO, USA) was added to 5 ml of 10% NBH to a final RNase concentration of 100 μg/ml. To prepare mock-digested NBH, 50 μl of RNA-free water was added to 5 ml of 10% NBH. Both mixtures were incubated at 37°C for 1 h under gentle rotation, and then total RNA was purified and analyzed using gel electrophoresis. The lack of RNA in RNase-treated NBH was confirmed by agarose gel.
To prepare seeds, 10% scrapie brain homogenates in PBS were serially diluted 102- to 104-fold in conversion buffer, as indicated, and 10 μl of the dilution was used to seed amplification in 90 μl of fresh substrate. Samples in 0.2-ml thin-wall PCR tubes (14230205; Fisher Scientific, Hampton, NH, USA) supplemented with 3 Teflon beads (McMaster-Carr, Los Angeles, CA, USA) were placed in a floating rack inside a Misonix S-4000 microplate horn (Misonix, Inc., Farmingdale, NY, USA) filled with 350 ml of water. Two coils of rubber tubing attached to a circulating water bath were installed to maintain 37°C inside the sonicator chamber. The standard sonication program consisted of 30 s sonication pulses delivered at 50% power efficiency applied every 30 min during a 24-h period, each round consisting of 48 cycles.
PK digestion
To analyze PMCAb products, 10 μl of each sample was supplemented with 2.5 μl SDS and 2.5 μl PK to a final concentration of SDS and PK of 0.25% and 50 μg/ml, respectively, followed by incubation at 37°C for 1 h. Digestion was terminated by addition of SDS-sample buffer and boiling for 10 min. Samples were loaded onto NuPAGE 12% BisTris gels, transferred to PVDF membrane, and stained with 3F4 antibody for detecting PrPSc.
To analyze scrapie brain homogenate (BH), an aliquot of 10% BH was mixed with an equal volume of 4% sarcosyl in PBS, supplemented with 50 mM Tris (pH 7.5), and digested with 50 μg/ml PK for 1 h at 37°C with 1000 rpm shaking (Eppendorf Thermomixer; Eppendorf, Hauppauge, NY, USA). The digestion was terminated by addition of SDS-sample buffer and boiled for 10 min and loaded onto NuPAGE 12% BisTris gels. After transfer to PVDF membrane, PrPSc was detected with 3F4 antibody.
PK resistance assay
To analyze resistance as a function of PK concentration, 10% scrapie BHs prepared by sonication in PBS (see above) were diluted to 1% using 1% NBH from healthy hamsters in PBS (pH 7.5), 0.15 M NaCl, 1% Triton, and 0.25% SDS. For sPMCAb-derived products, samples were only supplemented with 0.25% SDS. Lyophilized PK (P6556; Sigma-Aldrich) was dissolved in the PK buffer containing 20 mM Tris (pH 7.5) and 1 mM CaCl2, and the same buffer was used to prepare serial dilutions of PK. Each aliquot of scrapie BH or sPMCAb product was supplemented with the same volume, but increasing concentrations of PK, with a final concentration ranging from 0 to 2.5 mg/ml. After incubation at 37°C for 1 h, the reaction was stopped by addition of SDS-sample buffer and boiling the samples for 10 min. Samples were loaded onto NuPAGE 12% BisTris gels, transferred to PVDF membrane, and detected with 3F4 antibody.
Conformational stability assay
Scrapie BHs (10%) were diluted 10 times with conversion buffer, then supplemented with an equal volume of guanidine hydrochloride (GdnHCl) solution in PBS to a final concentration of GdnHCl ranging from 0.4 to 4 M, and incubated at room temperature for 1 h. Next, 9 vol of 2% sarcosyl in PBS was added to all samples, followed by 1 h incubation at room temperature, and then the samples were treated with 20 μg/ml PK for 1 h at 37°C with shaking. The digestion was stopped by 2 mM PMSF, and the proteins were precipitated in 4 vol of ice-cold acetone, incubated overnight at −20°C, and subsequently centrifuged 30 min at 16,000 g. Pellets were dried for 30 min, resuspended in 1× SDS-sample buffer, loaded into NuPAGE 12% bisTris gels, then transferred to PVDF membrane, and stained with 3F4 antibody.
Bioassay
Weanling Golden Syrian hamsters (all males) were inoculated intracerebrally under 2% O2/4 MAC isoflurane anesthesia. Each hamster received 50 μl of sPMCAb reaction products diluted 10-fold in PBS. After inoculation, hamsters were observed daily for disease using a blinded scoring protocol. Hamsters without any signs of clinical disease were euthanized at 453 d postinoculation.
PrP immunostaining
Formalin-fixed brains were divided at the midline in halves and processed for immunohistochemistry. Brain halves were treated with 96% formic acid prior to embedding in paraffin. For detection of disease-associated PrP, brain slices were subjected to hydrated autoclaving for 30 min at 121°C, followed by treatment with 96% formic acid for 5 min and staining using mouse monoclonal anti-PrP antibody 3F4 (1:1000, Covance, Berkeley, CA, USA) and StableDAB Peroxidase Substrate (KPL, Gaithersburg, MD, USA). Hematoxylin (Sigma-Aldrich) was used for counterstaining. Brain halves from uninoculated age-matched animals were used as controls. Images were taken by Nikon Eclips 90i (Nikon Instruments Inc., Melville, NY, USA) and analyzed by WCIF ImageJ software (NIH, Bethesda, MD, USA).
RESULTS
Adaptation of 263K to PMCAb in RNA-depleted brain homogenate
Consistent with the previous studies (21, 29, 30), amplification of brain-derived 263K in PMCAb was found to be RNA dependent (Fig. 1A). To test whether 263K could adapt to replicate in the absence of RNA, PMCAb reactions were seeded with 100-fold diluted 263K brain material, and serial reactions were carried out using RNA-depleted BH (Fig. 1B, C). Relatively steady amplification was observed in nine out of ten independent sPMCAb reactions (data not shown). Typically, at 10-fold dilutions between serial rounds the signal intensity declined slowly in sPMCAb (Fig. 1C). Previously, the amplification rate of 263K in PMCAb in NBH was found to be ∼300-fold/round (30). Therefore, this result indicates that in the absence of RNA the amplification rate of 263K was ∼30-fold lower than in the presence of RNA. To maintain amplification, after 9 sPMCAb rounds the dilution fold between rounds was reduced from 1:10 to 1:5 (Fig. 1B, C). The 263K form that was adapted to RNA-depleted BH will be referred to as 263KR−.
Figure 1.
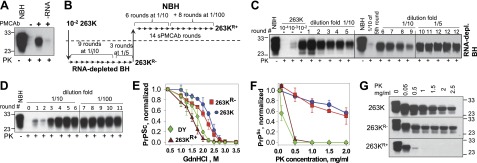
263K transformation on changes in replication environment. A) RNA dependency of 263K amplification. 104-fold diluted 263K brain material subjected to amplification in PMCAb conducted in NBH or RNA-depleted NBH. B) Experimental design for testing adaptation of 263K to RNA-depleted NBH and then readaptation to NBH. C) Adaptation of 263K to RNA-depleted NBH. 102-fold diluted 263K brain material was subjected to 9 sPMCAb rounds conducted at 10-fold dilution between rounds and then to 3 rounds at 5-fold dilution between rounds. End products of round 12 for 3 independent sPMCAb reactions are shown and referred to as 263KR−. sPMCAb reactions were conducted in RNA-depleted NBH. Undigested 10% NBH and serial dilution of 263K brain material are shown as references. D) Readaptation of 263KR− to NBH. 263KR− was subjected to 6 sPMCAb rounds with 10-fold dilution between rounds and then to an additional 8 rounds (5 of which are shown) with 100-fold dilution between rounds. sPMCAb reactions were conducted using NBH. 263KR− readapted to NBH is referred to as 263KR+. E) Conformational stability profiles for brain-derived 263K (blue circle), 263KR− (red square), 263KR+ (brown triangle), and brain-derived Drowsy (DY; green diamond). Means ± sd for 3 independent denaturation experiments are shown. F, G) Analysis of PK resistance. F) PK-resistance profiles for brain-derived 263K (blue circle), 263KR− (red square), 263KR+ (brown triangle), and brain-derived DY (green diamond). The data were normalized relative to the intensity of PK-resistant products at 0.05 mg/ml PK. Means ± sd for 3 independent PK-digestion experiments are shown. G) Representative Western blots of 263K, 263KR−, and 263KR+ treated with increasing concentrations of PK as indicated. For conformational stability and PK resistance assays, products of the 12th PMCAb round in RNA-depleted BH were used to assay 263KR−, and products of the 14th PMCAb round in NBH were used to assay 263KR+. All Western blots were stained with 3F4 antibody.
Adaptation of 263KR− to an environment with RNA resulted in transformation of strain-specific biochemical features
To test whether 263KR− could be readapted to replicate in an environment containing RNA, the products of the 12th sPMCAb round conducted in RNA-depleted BH were used to seed sPMCAb reactions in NBH (Fig. 1B). Three independent experiments on readaptation were performed. In each experiment, 263KR− displayed weak amplification in the first 2 or 3 serial rounds (Fig. 1D). After the second or third round, the amplification yield significantly increased and remained steady in subsequent rounds even when the dilution rate between rounds was increased from 10- to 100-fold (Fig. 1D). 263KR− readapted to the environment with RNA will be referred to as 263KR+.
To test whether the physical properties of 263K changed on its adaptation to the RNA-depleted environment and then readaptation to the environment containing RNA, conformational stability, PK-resistance and amplification rates were analyzed for 263K, 263KR−, and 263KR+. The conformational stability and PK-resistance profiles were found to be very similar for 263KR− and 263K (Fig. 1E–G). Surprisingly, 263KR+ displayed substantially lower conformational stability and much lower PK resistance than 263K or 263KR− (Fig. 1E–G). In fact, 263KR+ conformational stability was found to be even lower than that of Drowsy (DY), which is known as the least stable hamster strain (Fig. 1E and refs. 31, 32).
For comparing amplification rates, a set of sPMCAb reactions with dilution folds between serial rounds starting from 1:10 and higher were performed (Fig. 2). The amplification rate is defined operationally as the highest dilution between sPMCAb rounds at which amplification was still capable of compensating for the effect of dilution. Consistent with previous results, 263K showed an amplification rate between 100- and 300-fold/round (Fig. 2A). 263KR− amplification rate was less than 30-fold/round (Fig. 2B). Unexpectedly, 263KR+ showed astonishingly high amplification rate of ∼106-fold/round, which exceeded that of 263K by almost 4 orders of magnitude (Fig. 2C). To make sure that such an unusually high rate was not due to cross-contamination during sample handling (33), the dilution folds that exceeded a limiting dilution (1014- and 1020-fold) were used between serial rounds. No PK resistant products were detected in these experiments (Fig. 2C). Similar to amplification of the 263K, amplification of 263KR+ was found to be RNA dependent. In RNA-depleted brain homogenate, 263KR+ amplification rate dropped to 10- to 100-fold/round (Fig. 2D).
Figure 2.

Analysis of amplification rate. A series of sPMCAb reactions were seeded with sPMCAb-derived 263K (A), 263KR− (B), or 263KR+ (C), and carried out using 10-, 30-, 100-, 300-, 103-, 104-, 105-, 106-, 1014-, or 1020-fold dilutions between rounds, as indicated. D) Analysis of RNA dependency of 263KR+ amplification. sPMCAb-derived 263KR+ material was diluted 10-, 100-, 1000-, 2000-, or 10,000-fold, as indicated, into NBH or RNA-depleted BH and subjected to one PMCAb round. Western blots in all panels were stained with 3F4 antibody.
What is the origin of 263KR+?
The results on transformation of the 263K physical features on adaptation to RNA-depleted and then to RNA-containing NBH could be explained by two alternative hypotheses: 263KR+ was present in brain-derived 263K in very minor amounts and was selectively amplified as a result of changes in the replication environment; or 263KR+ was not present in brain-derived 263K, but emerged de novo as a result of a 263K mutation caused by alteration of the replication environment. To distinguish between these two possibilities, we exploited the fact that 263KR+ amplification rate exceeded significantly that of brain-derived 263K. If 263KR+ was present as a small subfraction in 263K material, we should be able to selectively amplify it in sPMCAb reactions conducted with high dilution folds between serial rounds. In sPMCAb reactions conducted with the dilution fold 1:2000, brain-derived 263K decayed quickly without any sign of 263KR+ (Fig. 3A). This experiment was repeated multiple times using brain-derived material from several animals or sPMCAb-derived 263K material that was serially propagated in NBH. No signs of 263KR+ were observed in 263K (Fig. 3B).
Figure 3.

263KR+ was absent in brain-derived or sPMCAb-derived 263K. A) sPMCAb reactions seeded with 104-fold diluted 263K brain material (top panel) or 104-fold diluted 263K brain material mixed with 2000-fold diluted sPMCAb-derived 263KR+ (bottom panel). 2000-fold dilution between serial rounds were used in both reactions. B) Brain-derived 263K material was subjected to 10 sPMCAb rounds in NBH, then sPMCAb reactions were seeded with sPMCAb-derived 263K (upper panel) or 263KR+ (lower panel). 1000- or 2000-fold dilutions between serial rounds were used as indicated. Miniscule amounts of 263KR+ seeds were mixed with sPMCAb-derived 263K seeds at ratios 1:1000 or 1:2000 (top panel, lanes 5–7 and 11–13, respectively) to test whether sPMCAb-derived 263K interferes with 263KR+ amplification. C) Brain-derived 263K material was subjected to 24 sPMCAb rounds in NBH (not shown) and then to 4 additional rounds using 100-, 1000-, or 10,000-fold dilution factor between rounds. Western blots were stained with 3F4 antibody.
To make sure that the negative results were not due to inhibition of 263KR+ amplification (if such exists) by excessive amounts of 263K, miniscule amounts of 263KR+ were mixed with brain-derived or sPMCAb-derived 263K, and the mixtures were used to seed sPMCAb. In contrast to the fast decay of the signal in serial reactions seeded with 263K alone, steady amplification was observed in reactions seeded with 263KR+:263K mixtures (Fig. 3A, B). To test whether 263KR+ would emerge during prolonged amplification in sPMCAb in the absence of changes in replication environment, brain-derived 263K was subjected to 24 sPMCAb rounds in the absence of changes in RNA content. Then the products of 24th sPMCAb round were tested for the presence of 263KR+ using the experimental format with high dilution folds between sPMCAb rounds. The sPMCAb-derived material showed steady amplification at 100-fold dilutions, a decrease in yield yet relatively steady amplification at 1000-fold dilutions, and quick decay at 10,000-fold dilution (Fig. 3C). While the amplification capacity of sPMCAb-derived 263K appeared to be slightly higher than that of brain-derived 263K, it was approximately three orders of magnitude lower than that expected for 263KR+. A modest increase in amplification capacity was consistent with the previous study, where serial amplification of brain-derived material in sPMCAb was found to improve amplification rate (17). Nevertheless, these results confirmed that the changes in replication environment were essential for a transformation of 263K PrPSc.
To rule out the possibility that 263KR+ was the result of self-formation during sPMCAb in the absence of RNA, three independent non-seeded sPMCAb reactions, each consisting of 26 rounds, were performed in RNA-depleted NBH (Supplemental Fig. S1). All reactions were negative dismissing self-formation de novo as the cause for the appearance of 263KR+. In summary, these results support the hypothesis that 263KR+ was not present in the original brain-derived 263K material, but emerged from 263K due to a change in replication environment.
Bioassay of 263KR− and 263KR+
To test whether 263KR+ exhibited a disease phenotype different from 263K, sPMCAb reaction products were tested in animal bioassay. Syrian hamsters were inoculated intracerebrally with the following sPMCAb products: sPMCAb-derived 263K (products of 10th sPMCAb round), 263KR− (products of 12th sPMCAb round in RNA-depleted NBH), intermediate 263KR+ (products of 6th sPMCAb round in NBH), and endpoint 263KR+ (products of 14th sPMCAb round in NBHs). sPMCAb-derived 263K was used as a reference. The amount of PK-resistant material was very similar in all inoculums (Supplemental Fig. S2A). Animals inoculated with 263KR− showed the same incubation time to disease and clinical signs as those inoculated with sPMCAb-derived 263K (Table 1). Moreover, brain-derived 263K and 263KR− material was found to have very similar if not identical conformational stability in the GdnHCl denaturation assay (Supplemental Fig. S2B). This result is consistent with the previous study (23) and suggests that adaptation of 263K to RNA-depleted BH did not noticeably change the strain-specific disease phenotype. While animals inoculated with an intermediate 263KR+ preparation showed longer incubation time to disease than those inoculated with sPMCAb-derived 263K, the clinical signs and PrPSc deposition pattern were identical in these two groups (Table 1 and Supplemental Fig. S3). Immunostaining revealed PrPSc deposition typical for 263K including diffuse immunoreactivity in the granule cell layer and deep nuclei regions of cerebellum, small plaques and diffuse deposits in layers of the cerebral cortex and small plaques in subventricular zones (Supplemental Fig. S3). Surprisingly, animals inoculated with the endpoint 263KR+ preparation did not develop any clinical sings for up to 453 d postinoculation, when they were euthanized (Table 1). PrP immunostaining did not detect any disease-associated PrP in brains of animals from the endpoint 263KR+ or age-matched control groups (Supplemental Fig. S3). No PK-resistant PrP was found in the group inoculated with the endpoint 263KR+ using Western blot (Supplemental Fig. S2C). Moreover, no signal was detected on Western blot in this group on treatment with very low concentrations of PK (Supplemental Fig. S2C) similar to those used for detecting PK-sensitive PrPSc (34). However, small amounts of PrPSc were detected using sPMCAb in 2 of 8 animals from this group (Supplemental Fig. S2D). In the PK-resistance assay, PrPSc from 263KR+-inoculated animals was found to be substantially more sensitive to PK than 263K (Supplemental Fig. S2E). Moreover, PrPSc from 263KR+-inoculated animals could be steadily amplified in sPMCAb at high dilution folds between rounds, which is a characteristic feature of 263KR+ but not 263K (Supplemental Fig S2F). These results confirmed that the minor amount of PrPSc found in 263KR+-inoculated animals displayed 263KR+- but not 263K-specific features.
Table 1.
Incubation time to disease for PMCAb-derived 263K, 263KR− and 263KR+
| Inoculum | Incubation time (d) | Attack rate |
|---|---|---|
| sPMCAb-derived 263K (10th round) | 95 ± 3 | 8/8 |
| 263KR−(12th round) | 91 ± 1 | 8/8 |
| 263KR+(6th round) | 120 ± 6 | 7/7 |
| 263KR+(14th round) | >453a | 0/8 |
PMCAb products were diluted 10-fold prior to inoculation. Incubation times are means +sd. Attack rate is judged by clinical signs and positive Western blot.
Animals were euthanized at 453 d postinoculation.
Phenomenon of PrPSc transformation is not specific to 263K
To test whether the phenomenon of PrPSc transformation in response to changes in cofactor environment is a general one, a series of sPMCAb experiments similar to those outlined in Fig. 1B was performed using HY strain. HY showed steady amplification in sPMCAb reactions conducted in RNA-depleted NBH (Fig. 4A). However, as opposed to 263K, after 6 sPMCAb rounds the amplification rate of HY increased to such an extent that the dilution fold between rounds could be raised from 1:10 to 1:100 without loss of signal (Fig. 4A). In sPMCAb reactions seeded with HYR− and conducted in the presence of RNA, the amplification rate was stable at 1:100 dilution between rounds (Fig. 4B). The PK digestion assay carried out at increasing concentrations of PK revealed that HYR− was similar to that of PMCAb-derived HY, whereas HYR+ was substantially more sensitive to PK (Fig. 4C). Using the experimental format presented in Fig. 2, the amplification rates of HY, HYR−, and HYR+ were compared (Fig. 4D). The amplification rate of HYR+ was found to be similar to that of 263KR+, but 3–4 orders of magnitude higher than those of HY or HYR− (Fig. 4D). In a manner similar to 263K, HY showed a dramatic transformation of its physical properties after its adaptation to RNA-depleted NBH and then readaptation to NBH. This result suggests that the phenomenon is not limited to 263K.
Figure 4.
HY transformation on changes in replication environment. A) Adaptation of HY to RNA-depleted NBH. 102-fold diluted HY brain material was subjected to 11 sPMCAb rounds conducted at 10-dilution fold between rounds. The products of the 8th round were used to seed sPMCAb reactions at 100-dilution fold between rounds. End products of the 11th round are referred to as HYR−. sPMCAb were conducted in RNA-depleted NBH. Undigested 10% NBH is shown as a reference. B) Readaptation HYR− to NBH. HYR− was subjected to 10 sPMCAb rounds at 100-fold dilution between rounds. sPMCAb were conducted using NBH. HYR− readapted to NBH is referred to as HYR+. C) Analysis of PK resistance. Western blots of sPMCAb-derived HY, HYR− and HYR+ treated with increasing concentrations of PK as indicated. D) Analysis of amplification rates. A series of sPMCAb reactions were seeded with 104-fold diluted HY brain material (top panel), n-fold diluted HYR− or HYR+ (middle panels), and HYR+ or 263KR+ (bottom panel) and conducted using 30-, 100-, 300-, 103-, 104-, 106-, or 107-fold dilutions between rounds, as indicated, where n equals the dilution fold between rounds. All Western blots were stained with 3F4 antibody.
DISCUSSION
The current studies demonstrated that strain-specific PrPSc properties can be transformed dramatically by changing the prion replication environment, a phenomenon that provides important new insight into the mechanism underlying prion strain mutation. We found that simple depletion of RNA from PMCAb reactions did not change PrPSc properties. In fact, 263KR− and 263K had very similar if not identical characteristics with respect to their physical features and infectivity. Surprisingly, readaptation of 263KR− to a PMCAb environment containing RNA led to the emergence of new PrPSc conformers referred to as 263KR+. Remarkably, 263KR+ was not found in brain-derived 263K, nor was it present in sPMCAb reactions conducted within the same environment. Instead, reversible changes in RNA content of sPMCAb reactions were found to be essential to give rise to 263KR+. The fact that 263KR+ failed to replicate in animals strongly supports the idea that 263KR+ was lacking in 263K brain material, but emerged in vitro.
In the absence of selective pressure imposed by cellular clearance, the newly emerged PrPSc conformer was very sensitive to proteolytic digestion. While 263KR+ displayed a very fast replication rate in PMCAb, this conformer did not appear to be competent in the cellular environment of an animal brain. The failure of 263KR+ to induce disease in animals was presumably due to its very low conformational stability and high susceptibility to proteolytic clearance. In fact, 263KR+ was found to be less stable than even the least stable hamster-adapted strain Drowsy. Alternatively, the failure of 263KR+ to cause diseases might not be due to its fast clearance, but rather due to very poor replication in the environment of an animal brain despite its fast replication in PMCAb. A third possibility is that a change in replication environment induced a transformation of 263K into a self-propagating yet intrinsically innocuous state that lacked a molecular feature essential for prion infectivity. The differences between these three possibilities could be semantic, because it is not clear what physical characteristics that 263KR+ lacks are essential for infecting an animal. Nevertheless, this study emphasizes the critical role of host selection in evolution of self-replicating PrPSc states and illustrate that selection criteria for PrPSc replication in vitro and in vivo are different (17). Moreover, careful comparison of 263K and 263KR+ should identify those physical features that are important for prion infectivity.
Observation of clinical disease with prolonged incubation time in the group inoculated with the intermediate 263KR+ products can be attributed to small amounts of 263K/263KR− in this preparation. Indeed, the PrP deposition pattern in animals inoculated with intermediate 263KR+ products was the same as in animals inoculated with 263K. These results were consistent with the idea that 263KR+ emerged de novo during sPMCAb and was initially present at very low concentrations. As a result, multiple sPMCAb rounds were necessary to fully replace 263K/263KR− with 263KR+.
Detection of small amounts of 263KR+ in animal brains 453 d postinoculation suggests that either a small fraction of the 263KR+ inoculum was able to survive clearance or that 263KR+ was able to replicate in brains at a rate that did not compensate for the rate of clearance. It would be interesting to determine next whether 263KR+ can adapt to replicate in animal brains on its serial transmission. Previous studies revealed that stabilization of a prion strain of synthetic origin required multiple serial passages even when transmission was performed within the same host species (35, 36).
What are the origins of strain mutations and what causes strains to mutate? Previous studies postulated that prion strains or isolates are intrinsically heterogeneous; the heterogeneity arises due to spontaneously occurring mutations of PrPSc structure; under constant replication environment, selective pressure gives advantages to those PrPSc conformers that fit best in the particular environment; and on changes in replication environment, minor conformers that fit best to replicate in a new environment could be selected (14, 15). Indeed, the fact that natural prion isolates are intrinsically heterogeneous has been acknowledged for decades (2, 8, 9, 12, 37, 38). Like prion isolates of natural origin, synthetic prions produced by recombinant PrP fibrils were found to be conformationally diverse and subject to strain evolution and adaptation even when transmitted within the same host (5, 35, 39–42). Ghaemmaghami et al. (16, 36) showed that on serial passages, synthetic strains were prone to conformational transformation and competitive selection, and that the outcome of the selection was controlled by replication environment. Notably, these studies revealed that alteration in replication environment of cultured cells provided selective advantage to different synthetic strains replicating within the same PrP primary structure. Using cultured cells treated with swainsonine, Li et al. (15) showed that alteration of the cellular replication environment due to inhibition of complex N-linked glycans led to a selective amplification of minor PrPSc conformers in cultured cells. Moreover, selective amplification of minor PrPSc isoforms was shown to be accompanied by a gradual change in cell tropism. However, in experiments on cultured cells, the minor conformers were believed to be present in the original brain-derived material. The present work showed that changes in replication environment not only create conditions for selective amplification of minor PrPSc conformers, but could also give rise to a new self-replicating state. Indeed, we showed that 263KR+ did not exist in brain-derived or PMCAb-derived PrPSc populations, but emerged de novo on readaptation of 263KR− to the PMCAb environment containing RNAs. Moreover, in experiments in cultured cells and swainsonine, where the inhibition of glycosylation changed the glycosylation status of PrPC, it remained unclear whether selective amplification of new substrains was due to changes in the cellular environment or due to modifications in PrPC primary structure. Changes in PrPC glycosylation status was recently shown to give a selective advantage to different PrPSc states (19). In the current study the new PrPSc conformers emerged in the absence of treatments that modify PrPC primary structure. It is tempting to speculate that the alteration in the PMCAb environment gave rise to multiple new PrPSc conformers, but that only those conformers that had the highest PMCAb amplification rates were selected.
In previous studies, RNA was shown to serve a catalytic role in PrPSc replication (21,23,30). Indeed, previously RNA was found to increase amplification rates for diverse prion strains (23,30), whereas neither in the previous nor current studies was removal of RNAs able to cause significant or permanent alterations in strain phenotype (23, 24). Noteworthy, recent studies by Deleault and coworkers (6, 26) illustrated that cellular lipids might play the role of strain-specifying cofactors.
How do changes in RNA content lead to emergence of new PrPSc conformers? One can speculate that RNA might control fidelity of prion replication. RNA depletion might create favorable conditions for generating conformationally diverse PrPSc populations. In the absence of RNA, conformational mutations might occur at a higher rate as a result of deformed templating (5,43). Moreover, in an RNA-depleted environment the catalytic role of RNA might be compensated in part by other cellular polyanions creating diverse replication environments and boosting conformational diversity of the PrPSc pool. However, in the absence of RNA, the newly emerged conformers represented only a minor subfraction of the PrPSc pool and remained undetected. Supplementing RNA back to PMCAb reactions might have given selective advantages to those conformers that exhibited the highest PMCAb amplification rate. After multiple sPMCAb rounds, 263KR+ fully replaced the original strain. It remains to be tested in future studies whether this mechanism is correct. Nevertheless, considering that similar results were observed using HY, the observed phenomenon of dramatic strain transformation is unlikely to be 263K-specific. In summary, the current work revealed that changes in prion replication environment can be a source of PrPSc transformation even in the absence of changes in PrP primary structure.
Supplementary Material
Acknowledgments
The authors thank Robert Rohwer for assistance in conducting bioassays and Pamela Wright for editing the manuscript.
This work was supported by U.S. National Institutes of Health grants NS045585 and NS074998 to I.V.B.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- BH
- brain homogenate
- GdnHCl
- guanidine hydrochloride
- HY
- hyper
- NBH
- normal brain homogenate
- PK
- proteinase K
- PMCA
- protein misfolding cyclic amplification
- PMCAb
- protein misfolding cyclic amplification with beads
- PrP
- prion protein
- PrPC
- prion protein normal cellular isoform
- PrPSc
- prion protein scrapie isoform
- sPMCAb
- serial protein misfolding cyclic amplification
- sPMCAb
- serial protein misfolding cyclic amplification with beads
REFERENCES
- 1. Prusiner S. B. (1998) Prions. Proc. Natl. Acad. Sci. U. S. A. 95, 13363–13383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bessen R. A., Marsh R. F. (1992) Identification of two biologically distinct strains of transmissible mink encephalopathy in hamsters. J. Gen. Virol. 73, 329–334 [DOI] [PubMed] [Google Scholar]
- 3. Wang F., Wang X., Yuan C. G., Ma J. (2010) Generating a prion bacterially expressed recombinant prion protein. Science 327, 1132–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Makarava N., Kovacs G. G., Bocharova O. V., Savtchenko R., Alexeeva I., Budka H., Rohwer R. G., Baskakov I. V. (2010) Recombinant prion protein induces a new transmissible prion disease in wild type animals. Acta Neuropathol. 119, 177–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Makarava N., Kovacs G. G., Savtchenko R., Alexeeva I., Ostapchenko V. G., Budka H., Rohwer R. G., Baskakov I. V. (2012) A new mechanism for transmissible prion diseases. J. Neurosci. 32, 7345–7355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Deleault N. R., Walsh D. J., Piro J. R., Wang F., Wang X., Ma J., Rees J. R., Supattapone S. (2012) Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions. Proc. Acad. Natl. Sci. U. S. A. 109, E1938–E1946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kimberlin R. H., Cole S., Walker C. A. (1987) Temporary and permanent modifications to a single strain of mouse scrapie on transmission to rats and hamsters. J. Gen. Virol. 68, 1875–1881 [DOI] [PubMed] [Google Scholar]
- 8. Bruce M. E., Dickinson A. G. (1987) Biological evidence that the scrapie agent has an independent genome. J. Gen. Virol. 68, 79–89 [DOI] [PubMed] [Google Scholar]
- 9. Kimberlin R. H., Walker C. A., Fraser H. (1989) The genomic identity of different strains of mouse scrapie is expressed in hamsters and preserved on reisolation in mice. J. Gen. Virol. 70, 2017–2025 [DOI] [PubMed] [Google Scholar]
- 10. Bartz J. C., Bessen R. A., McKenzie D., Marsh R. F., Aiken J. M. (2000) Adaptation and selection of prion protein strain conformations following interspecies transmission of transmissible mink encephalopathy. J. Virol. 74 5542–5547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Peretz D., Williamson R. A., Legname G., Matsunaga Y., Vergara J., Burton D., DeArmond S., Prusiner S., Scott M. R. (2002) A change in the conformation of prions accompanies the emergence of a new prion strain. Neuron 34, 921–932 [DOI] [PubMed] [Google Scholar]
- 12. Angers R. C., Kang H. E., Napier D., Browning S., Seward T., Mathiason C., Balachandran A., McKenzie D., Castilla J., Soto C., Jewell J., Graham C., Hoover E. A., Telling G. C. (2010) Prion strain mutation determined by prion protein conformational compatibility and primary structure. Science 328, 1154–1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Makarava N., Ostapchenko V. G., Savtchenko R., Baskakov I. V. (2009) Conformational switching within individual amyloid fibrils. J. Biol. Chem. 284, 14386–14395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Collinge J., Clarke A. R. (2007) A general model of prion strains and their pathogenicity. Science 318, 930–936 [DOI] [PubMed] [Google Scholar]
- 15. Li J., Browning S., Mahal S. P., Oelschlegel A. M., Weissmann C. (2010) Darwinian evolution of prions in cell culture. Science 327, 869–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ghaemmaghami S., Watts J. C., Nquyen H. O., Hayashi S., DeArmond S. J., Prusiner S. B. (2011) Conformational transformation and selection of synthetic prion strains. J. Mol. Biol. 413, 527–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gonzalez-Montalban N., Baskakov I. V. (2012) Assessment of strain-specific PrPSc elongation rates revealed a transformation of PrPSc properties during protein misfolding cyclic amplification. PLoS One 7, 0041210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ghaemmaghami S., Ahn M., Lessard P., Giles K., Legname G., DeArmond S. J., Prusiner S. B. (2009) Continuous quinacrine treatment results in the formation of drug-resistant prions. PLOS Pathog. 5, e1000673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Makarava N., Savtchenko R., Baskakov I. V. (2013) Selective amplification of classical and atypical prions using modified protein misfolding cyclic amplification. J. Biol. Chem. 288, 33–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Klingeborn M., Race B., Meade-White K. D., Chesebro B. (2011) Lower specific infectivity of protease-resistant prion protein generated in cell-free reactions. Proc. Acad. Natl. Sci. U. S. A. 108, E1244–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Deleault N. R., Lucassen R. W., Supattapone S. (2003) RNA molecules stimulate prion protein conversion. Nature 425 717–720 [DOI] [PubMed] [Google Scholar]
- 22. Deleault N. R., Harris B. T., Rees J. R., Supattapone S. (2007) Formation of native prions from minimal components in vitro. Proc. Acad. Natl. Sci. U. S. A. 104, 9741–9746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Saa P., Sferrazza G. F., Ottenberg G., Oelschlegel A. M., Dorsey K., Lasmezas C. I. (2012) Strain-specific role of RNAs in prion replication. J. Virol. 86, 10494–10504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Piro J. R., Harris B. T., Supattapone S. (2011) In situ photodegradation of incorporated polyanion does not alter prion infectivity. PLOS Pathog. 7, e1002001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Safar J. G., Kellings K., Serban A., Groth D., Cleaver J. E., Prusiner S. B., Riesner D. (2005) Search for a prion-specific nucleic acid. J. Virol. 79, 10796–10806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Deleault N. R., Piro J. R., Walsh D. J., Wang F., Ma J., Geoghegan J. C., Supattapone S. (2012) Isolation of phosphatidylethanolamine as a solitary cofactor for prion formation in the absence of nucleic acids. Proc. Acad. Natl. Sci. U. S. A. 109, 8546–8551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Makarava N., Kovacs G. G., Savtchenko R., Alexeeva I., Budka H., Rohwer R. G., Baskakov I. V. (2011) Genesis of mammalian prions: from non-infectious amyloid fibrils to a transmissible prion disease. PLoS Pathog. 7, e1002419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gonzalez-Montalban N., Makarava N., Ostapchenko V. G., Savtchenko R., Alexeeva I., Rohwer R. G., Baskakov I. V. (2011) Highly efficient protein misfolding cyclic amplification. PLoS Pathog. 7, e1001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Deleault N. R., Kascsak R., Geoghegan J. C., Supattapone S. (2010) Species-dependent differences in cofactor utilization for formation of the protease-resistant prion protein in vitro. Biochemistry 49, 3928–3934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gonzalez-Montalban N., Makarava N., Savtchenko R., Baskakov I. V. (2011) Relationship between conformational stability and amplification efficiency of prions. Biochemistry 50, 7933–7940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Peretz D., Scott M., Groth D., Williamson A., Burton D., Cohen F. E., Prusiner S. B. (2001) Strain-specified relative conformational stability of the scrapie prion protein. Protein Sci. 10, 854–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ayers J. L., Schutt C. R., Shikiya R. A., Aguzzi A., Kincaid A. E., Bartz J. C. (2011) The strain-encoded relationship between PrP replication, stability and processing in neurons is predictive of the incubation period of disease. PLOS Pathog. 7, e1001317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cosseddu G. M., Nonno R., Vaccari G., Bucalossi C., Fernandez-Borges N., Di Bari M. A., Castilla J., Agrimi U. (2011) Ultra-efficient PrP(Sc) amplification highlights potentialities and pitfalls of PMCA technology. PLOS Pathog. 7, e1002370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sajnani G., Silva C. J., Ramos A., Pastrana M. A., Onisko B. C., Erickson M. L., Antaki E. M., Dynin I., Vazquez-Fernandez E., Sigurdson C. J., Carter J. M., Requena J. (2012) PK-sensitive PrPSc is infectious and shares basic structural features with PK-resistant PrPsc. PLoS Pathog. 8, e1002547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Makarava N., Kovacs G. G., Savtchenko R., Alexeeva I., Budka H., Rohwer R. G., Baskakov I. V. (2012) Stabilization of a prion strain of synthetic origin requires multiple serial passages. J. Biol. Chem. 287, 30205–30214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ghaemmaghami S., Colby D. W., Nquyen H. O., Hayashi S., Oehler A., DeArmond S., Prusiner S. B. (2013) Convergent replication of mouse synthetic prion strains. Am. J. Pathol. 182, 866–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kimberlin R. H., Walker C. A. (1978) Evidence that the transmission of one source of scrapie agent to hamsters involves separation of agent strains from a mixture. J. Gen. Virol. 39, 487–496 [DOI] [PubMed] [Google Scholar]
- 38. Giles K., Glidden D. V., Patel S., Korth C., Groth D., Lemus A., DeArmond S. J., Prusiner S. B. (2010) Human prion strain selection in transgenic mice. Ann. Neurol. 68, 151–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Legname G., Nguyen H. O. B., Baskakov I. V., Cohen F. E., DeArmond S. J., Prusiner S. B. (2005) Strain-specified characteristics of mouse synthetic prions. Proc. Natl. Aca. Sci. U. S. A. 102, 2168–2173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Legname G., Nguyen H. O. B., Peretz D., Cohen F. E., DeArmond S. J., Prusiner S. B. (2006) Continuum of prion protein structures enciphers a multitude of prion isolate-specified phenotypes. Proc. Acad. Natl. Sci. U. S. A. 103, 19105–19110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Colby D. W., Giles K., Legname G., Wille H., Baskakov I. V., DeArmond S. J., Prusiner S. B. (2009) Design and construction of diverse mammalian prion strains. Proc. Acad. Natl. Sci. U. S. A. 106, 20417–20422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Colby D. W., Wain R., Baskakov I. V., Legname G., Palmer C. G., Nguyen H. O., Lemus A., Cohen F. E., DeArmond S. J., Prusiner S. B. (2010) Protease-sensitive synthetic prions. PLoS Pathog. 6, e1000736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Makarava N., Baskakov I. V. (2012) Genesis of transmissible protein states vie deformed templating. Prion 6, 252–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.