

Abstract
We examined the effects of a natural secondary bile acid, hyodeoxycholic acid (HDCA), on lipid metabolism and atherosclerosis in LDL receptor-null (LDLRKO) mice. Female LDLRKO mice were maintained on a Western diet for 8 wk and then divided into 2 groups that received chow, or chow + 1.25% HDCA, diets for 15 wk. We observed that mice fed the HDCA diet were leaner and exhibited a 37% (P<0.05) decrease in fasting plasma glucose level. HDCA supplementation significantly decreased atherosclerotic lesion size at the aortic root region, the entire aorta, and the innominate artery by 44% (P<0.0001), 48% (P<0.01), and 94% (P<0.01), respectively, as compared with the chow group. Plasma VLDL/IDL/LDL cholesterol levels were significantly decreased, by 61% (P<0.05), in the HDCA group as compared with the chow diet group. HDCA supplementation decreased intestinal cholesterol absorption by 76% (P<0.0001) as compared with the chow group. Furthermore, HDL isolated from the HDCA group exhibited significantly increased ability to mediate cholesterol efflux ex vivo as compared with HDL of the chow diet group. In addition, HDCA significantly increased the expression of genes involved in cholesterol efflux, such as Abca1, Abcg1, and Apoe, in a macrophage cell line. Thus, HDCA is a candidate for antiatherosclerotic drug therapy.—Shih, D. M., Shaposhnik, Z., Meng, Y., Rosales, M., Wang, X., Wu, J., Ratiner, B., Zadini, F., Zadini, G., Lusis, A. J. Hyodeoxycholic acid improves HDL function and inhibits atherosclerotic lesion formation in LDLR-knockout mice.
Keywords: bile acid, intestinal cholesterol absorption, low-density lipoprotein, high-density lipoprotein, reverse cholesterol transport
Atherosclerosis is initiated by the accumulation of low-density lipoprotein (LDL) in the intima of the artery, and elevated LDL cholesterol is the leading risk factor for cardiovascular disease (CVD) (1). The HMG-CoA reductase inhibitor statins have been widely used to lower plasma LDL cholesterol level (2) and reduce the incidence of major vascular events and cardiovascular death (3–7). In addition to statins, research over the past several decades has led to the development of a number of drugs that influence cholesterol homeostasis. For example, ezetimibe inhibits the action of Niemann-Pick C1-like 1 (NPC1L1), a protein on the intestinal brush border membrane that plays an important role in cholesterol absorption (8). Bile acid sequestrants, another class of cholesterol-lowering drugs, also inhibit cholesterol absorption, as well as bile acid reabsorption (9). High-density lipoprotein (HDL) cholesterol levels, on the other hand, are inversely correlated with disease risk, and HDL mediates reverse cholesterol transport (10) and exhibits antioxidative activities (1). Drug therapies that attempted to raise HDL cholesterol level (11, 12) or improve HDL functions (13) have been examined, but with limited success thus far.
In this study, we investigated the effects of a naturally occurring bile acid, hyodeoxycholic acid (HDCA), for its effects on atherosclerotic lesion development in LDL receptor-knockout (LDLRKO) mice, an animal model that closely resembles the human atherosclerotic condition. HDCA is a secondary hydrophilic bile acid formed in the small intestine by the gut flora (14). HDCA has been shown to prevent gallstone formation in the hamsters (15) and prairie dogs (16). Sehayek et al. (17) investigated the effects of HDCA on lipoprotein metabolism and atherosclerosis in mice and observed that it is atheroprotective. One of the mechanisms for the atheroprotective effects of HDCA likely involves its inhibitory effect on intestinal cholesterol absorption, which leads to substantially decreased plasma total cholesterol levels (17–19). In vitro studies have also demonstrated that HDCA activates G-protein-coupled bile acid receptor 1 [GPBAR1 (TGR5); ref. 20], liver X receptor (LXR; ref. 21), but not farnesoid X receptor (FXR; ref. 22). In this study, we demonstrated that HDCA supplementation significantly decreased atherosclerotic lesion formation in multiple vessels in the LDLRKO mice. Furthermore, we showed that HDCA not only inhibited intestinal cholesterol absorption but also exerted other antiatherogenic effects, including improving the ability of HDL to mediate cholesterol efflux from foam cells and increasing the expression of genes involved in cholesterol efflux in macrophages.
MATERIALS AND METHODS
Animals and study design
Female LDLRKO mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). For atherosclerosis studies, 8-wk-old female LDLRKO mice were fed a Western diet (21% fat, 0.15% cholesterol; TD.88137; Harlan Laboratories, Indianapolis, IN, USA) for 8 wk. One group of mice (baseline group) was euthanized at this time point for lesion measurement in the aortic root region and in the innominate artery. Atherosclerotic lesion in the whole aorta was not examined in the baseline group. The remaining mice were then divided into 2 groups and fed the following diets for another 15 wk before euthanasia: group 1, chow diet (5% fat, AIN-76A Rodent Diet; Research Diets, New Brunswick, NJ, USA); and group 2, chow diet + 1.25% (wt/wt) HDCA. For other studies, 8-wk-old female LDLRKO mice were fed a chow diet or chow diet + 1.25% HDCA for 3 wk before phenotype measurements. The HDCA used in the study was purchased from Alfa Aesar (B20506-22; Alfa Aesar, Ward Hill, MA, USA). Food consumption and body weight were recorded weekly. Animals were measured for total body fat mass and lean mass by magnetic resonance imaging (MRI) using Bruker Minispec (Bruker Corp., Billerica, MA, USA) with software from Eco Medical Systems (Houston, TX, USA) (23).
Lipid, total bile acids, HDCA assays, serum chemistry tests, gel filtration chromatography, dichlorofluorescein (DCF) assay, and immunoblotting
For plasma lipid and lipoprotein level determinations, mice were denied access to food for 16 h before bleeding. Total cholesterol, HDL cholesterol, free cholesterol, triglycerides, and free fatty acid levels were determined by enzymatic colorimetric assays (24). Phosphatidylcholine levels were assayed using an enzymatic colorimetric assay from Wako (Richmond, VA, USA). Plasma samples were fractionated by fast-performance liquid chromatography (FPLC) as described previously (25). Serum chemistry tests were performed by Pathology and Laboratory Medicine Services of the Department of Laboratory Animal Management at the University of California, Los Angeles. To determine the extent of lipid oxidation of HDL samples, 2 μg of HDL cholesterol in 175 μl phosphate-buffered saline (PBS) was added to each well of a 96-well plate, followed by 1 h incubation at 37°C. DCFH (5 μg) in 25 μl PBS was then added to each well, followed by an additional 1 h of incubation at 37°C. The DCF fluorescence intensity was then determined with a plate reader at an excitation wavelength of 485 nm and an emission wavelength of 530 nm, as described previously (26). For immunoblotting, FPLC fractions or HDL samples were fractionated by SDS-PAGE; transferred onto a nylon membrane; incubated with a rabbit antibody against mouse apolipoprotein A1 (apoA1), apo B-48/100, or apoE (Meridian Life Science, Memphis, TN, USA); washed; incubated with a secondary antibody; and detected using electrochemiluminescence (GE Healthcare Bio-Sciences, Piscataway, NJ, USA). Total bile acid levels were assayed using a kit from Diazyme Laboratories (Poway, CA, USA) according to the manufacturer's protocol. For determination of total bile acid levels in HDL, FPLC-isolated HDL was concentrated by using Amicon centrifugal filter units (EMD Millipore, Billerica, MA, USA). HDL samples carrying plasma-equivalent amount of HDL cholesterol were assayed together with plasma samples (the sources of the HDL preparation) for comparison of total bile acid level. Plasma HDCA levels were determined by Tandem Labs (Durham, NC, USA) using a LC/MS/MS method as described below. Standards were prepared in methanol:water (2:1) at HDCA concentrations of 1.00–1000 ng/ml. Samples and standards were extracted by protein precipitation using 100 μl sample or standard and addition of 400 μl methanol containing the internal standard d4-ursodeoxycholic acid (UDCA; 100 ng/ml). The samples were vortexed and centrifuged, and the supernatant (400 μl) was combined with water (400 μl). HDCA was separated on a Supelco Ascentis Express C-18 column (50×2.1 mm, 2.7 μm; Sigma-Aldrich, St. Louis, MO, USA) at a flow rate of 0.200 ml/min using 2 mobile phases: 10 mM ammonium acetate in water with 0.1% ammonium hydroxide (pH 9); and 10 mM ammonium acetate in methanol with 0.1% ammonium hydroxide. Elution was started with 50% B initially and held for 0.5 min, followed by a linear gradient to 80% B at 4 min and a second gradient to 95% B at 5 min. This composition was held for 2 min, followed by reequilibration to 50% B and a total run time of 8 min. Eluent was directly introduced into a SciexAPI5000 (AB Sciex, Framingham, MA, USA), and HDCA was detected by MS/MS monitoring m/z 391.1 in both Q1 and Q3 with a 40 eV collision voltage to lower background interference. The internal standard was detected at m/z 395.1. HDCA eluted at 4.26 min, and the internal standard was detected at 4.08 min. With the use of this system, HDCA was baseline separated from the isobaric species UDCA, deoxycholic acid (5.3 min), and chenodeoxycholic acid (5.1 min).
Atherosclerosis lesion measurement and immunohistochemistry
Atherosclerotic lesion size and calcification at the aortic root region were determined as described previously (24, 27). En face analyses of lesions in the entire aorta were performed as described previously (28). Atherosclerotic lesions at the innominate artery were analyzed as described previously (29). Immunohistochemistry was performed, as described previously (30), using primary antibodies against CD68 (AbD Serotec, Raleigh, NC, USA) and α-actin (Epitomics, Burlingame, CA, USA) to determine macrophage and smooth muscle cell content, respectively, of the aortic root lesion.
Liver and fecal lipid extraction and fecal total bile acids assays
Liver or feces (50 mg) was homogenized in PBS and 0.88% KCl, respectively. The lipids in the homogenate were then extracted using the Folch method (31). The extracted lipids were dried down and resuspended in 1% Triton X-100 before lipid assays were performed as described above. To determine fecal total bile acid levels, 50 mg of dried fecal sample was homogenized in 0.4 ml 0.1 N NaOH, combined with 5 ml ethanol, and incubated at 80°C for 3 h. The samples were then centrifuged at 1500 g for 10 min. The supernatant was collected and neutralized by the addition of 7.4 μl of 0.1 N HCl per 100 μl supernatant. The neutralized supernatant (5 μl) was then assayed for total bile acids as described above.
Intestinal cholesterol absorption assay
Animals were placed in metabolic cages for 24 h, denied access to food for 4–6 h, and then given an intragastric bolus of 200 μl corn oil containing 1.67 μCi of [14C]-cholesterol (Perkin Elmer Wellesley, MA, USA; specific activity 50 mCi/mmol) and 0.67 μCi of [3H]β-sitostanol (American Radiolabeled Chemicals, St. Louis, MO, USA). The animals were returned to metabolic cages, and fecal samples were collected for 24 h. Collected fecal samples were dried by overnight incubation at 55°C. A sample of 100 mg dried feces was homogenized in 1.5 ml of 0.88% KCl, 7.2 ml of chloroform/methanol (2:1) was then added to extract lipid. After the 2 phases were separated by centrifugation, the lower organic phases were collected. To measure radioactivity, 0.1 ml of the organic phase were mixed with 7 ml of scintillation cocktail before counting. The following formula was used to calculate intestinal cholesterol absorption efficiency:
Cell culture and treatment conditions
RAW 264.7 cells, a mouse macrophage cell line, were cultured in growth medium containing DMEM (cat. no. 11995-065; Life Technologies, Grand Island, NY, USA) supplemented with 10% FBS (Hyclone, South Logan, UT, USA), 100 U/ml penicillin, and 100 μg/ml streptomycin. For treatment, RAW 264.7 cells were plated in 6-well plates (7.5×10−5 cells/well) in growth medium for 2 d. After being washed with PBS, the cells were incubated for 24 h in treatment medium (DMEM supplemented with 1% FBS (Hyclone), 100 U/ml penicillin, and 100 μg/ml streptomycin) containing various chemicals or dimethyl sulfoxide (DMSO) as the vehicle control. The cells were then washed with PBS before total RNA was isolated as described below.
Cholesterol efflux assay
RAW 264.7 cells were plated in 24-well plates (300,000 cells/well) and grown for 1 d. Cells in each well were then incubated with 1 ml of growth medium containing 25 μg/ml human acetylated LDL and 1 μCi 3H-cholesterol/ml for 48 h in a CO2 incubator. After being washed with PBS, the cells were then incubated with DMEM + 0.2% fatty acid-free bovine serum albumin (BSA; Sigma-Aldrich) overnight. After being washed with PBS, the cells were incubated with 0.5 ml of test samples (mouse HDL in DMEM+0.2% BSA) for 4 h at 37°C. Afterward, the supernatant was collected, and the cells were lysed with 0.1 N NaOH. Radioactivity that was associated with the supernatant and the cells, respectively, was then measured by liquid scintillation. The cholesterol efflux efficiency was calculated by the following formula:
RNA Isolation and quantitative RT-PCR analyses
Total RNA samples from tissues and cells were isolated using Trizol reagent (Life Technologies) according to manufacturer's protocol. The cDNA was synthesized using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). Quantitative PCR was performed using gene-specific primers (Table 1) and the Roche SYBR green master mix in a Roche Lightcycler 480 system (Roche, Basel, Switzerland). The mRNA levels of specific genes were normalized to the mRNA levels of the housekeeping gene RPL13a of the same sample.
Table 1.
Primers used for quantitative PCR analysis
| Primer | Sequence |
|---|---|
| Abca1-F | CTTCCCACATTTTTGCCTGG |
| Abca1-R | AAGGTTCCGTCCTACCAAGTC |
| Abcg1-F | CTGTCTGATGGCCGCTTTCT |
| Abcg1-R | CTGGACACGACCTCGTCCAC |
| Abcg5-F | GGTGTCCTGCATGTGTCCTA |
| Abcg5-R | ATTTGCCTGTCCCACTTCTG |
| Abcg8-F | AACCCTGCGGACTTCTACG |
| Abcg8-R | CTGCAAGAGACTGTGCCTTCT |
| Apoe-F | GACCCTGGAGGCTAAGGACT |
| Apoe-R | AGAGCCTTCATCTTCGCAAT |
| Asbt-F | GACTCGGGAACGATTGTGAT |
| Asbt-R | GGTTCAATGATCCAGGCACT |
| Bsep-F | ACAGAAGCAAAGGGTAGCCATC |
| Bsep-R | GGTAGCCATGTCCAGAAGCAG |
| Cyp7a1-F | CAACGGGTTGATTCCATACC |
| Cyp7a1-R | ATTTCCCCATCAGTTTGCAG |
| G6pase-F | GCTGAGTGGCTCAACCTTGT |
| G6pase-R | ATTCATGCACCCACCAAAAG |
| Hmgcr-F | GAATGCCTTGTGATTGGAGTTG |
| Hmgcr-R | ACACAGGCCGGGAAGAATG |
| Lxra-F | TGTGCGCTCAGCTCTTGT |
| Lxra-R | CCCTG GACATTACCAAGACAC |
| Lxrb-F | GCTCTGCCTACATCGTGGTC |
| Lxrb-R | CTCATGGCCCAGCATCTT |
| Mrp2-F | CTGAGTGCTTGGACCAGTGA |
| Mrp2-R | GTTAACAGCTGCCTGTGCAA |
| Npc1l1-F | GGTGCTGGCTGTGGGAGCTG |
| Npc1l1-R | GGTGCGGCCAATGTGAGCCT |
| Osta-F | GTCTCAAGTGATGAACTGCCA |
| Osta-R | TTGAGTGCTGAGTCCAGGTC |
| Ostb-F | TGACAAGCATGTTCCTCCTG |
| Ostb-R | TGCAGGTCTTCTGGTGTTTCT |
| Pepck-F | CAGCTGCTGCAGAACACAAGG |
| Pepck-R | GCTAACTGCTACAGCTAACGTG |
| Pparg1-F | GTGAACCACTGATATTCAGGACATTT |
| Pparg1-R | CCACAGAGCTGATTCCGAAGT |
| Rpl13a-F | CCCTCCACCCTATGACAAGA |
| Rpl13a-R | TTCTCCTCCAGAGTGGCTGT |
| Shp-F | GGAGCCTTGAGCTGGGTCCC |
| Shp-R | GAGGATTCGGGCCAGGCGG |
| Srb1-F | ATGGTGCCCTCCCTCATC |
| Srb1-R | ACAGGCTGCTCGGGTCTAT |
Statistical analysis
Student's t test was used for comparison of means.
RESULTS
HDCA supplementation significantly increased circulating HDCA levels and did not affect general health of the mice
The LDLRKO mice fed the HDCA diet were generally healthy, with a 10% lower body weight at the end of the 15 wk feeding, as compared with the chow diet group (Table 2). Fifteen weeks of HDCA supplementation also significantly decreased fat mass and increased lean mass in LDLRKO mice (Table 2). The serum chemistry data showed that HDCA supplementation did not raise the serum levels of any organ toxicity markers as compared with the chow group. In fact, circulating levels of several liver toxicity markers, such as aspartate aminotransferase (AST), direct bilirubin, total bilirubin, lactate dehydrogenase (LDH), and γ-glutamyltransferase (GGT), were significantly lower in mice fed the HDCA diet as compared with the chow diet (Table 3). The circulating HDCA levels of the chow and HDCA groups were 0.9 ± 0.4 and 42.4 ± 14.0 μM, respectively (chow vs. HDCA, P<0.0001).
Table 2.
Body weight, composition, and daily food consumption data of LDLRKO mice fed various diets
| Diet | n | Initial BW (g) | BW after WD (g) | Final BW, age 31 wk (g) | Final fat mass (% BW) | Final lean mass (%BW) | Food consumption (g/mouse/d) |
|---|---|---|---|---|---|---|---|
| Chow | 9 | 17.5 ± 0.8 | 25.0 ± 2.5 | 27.1 ± 2.4 | 22.6 ± 1.5 | 77.4 ± 1.5 | 2.5 ± 0.3 |
| HDCA | 14 | 18.1 ± 1.1 | 25.2 ± 2.5 | 24.2 ± 1.5** | 19.1 ± 0.8* | 80.9 ± 0.8* | 2.3 ± 0.3 |
Values are means ± sd of each group. Initial body weight (BW) was measured at 8 wk of age. BW after Western diet (WD) was measured at 16 wk of age after the mice were placed on a WD for 8 wk. Final BW and composition (fat mass and lean mass percentages) were measured at euthanasia, at 31 wk of age, after the mice received 8 wk of WD, followed by 15 wk feeding of chow diet or chow diet + 1.25% HDCA (HDCA). Average food consumption data were obtained from wk 2 to 14 during the period of chow diet or HDCA diet feeding.
P = 0.05,
P = 0.01 vs. chow.
Table 3.
Serum chemistry test data of LDLRKO mice fed various diets for 15 wk
| Diet | n | AST (U/L) | ALT (U/L) | DBILI (mg/dl) | TBILI (mg/dl) | BUN (mg/dl) | CK (U/L) | LDH (U/L) | GGT (U/L) | ALP (U/L) | CREA (mg/dl) | TPROT (g/dl) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Chow | 4 | 149 ± 62 | 48 ± 28 | 1.2 ± 0.2 | 1.1 ± 0.2 | 19 ± 1 | 156 ± 112 | 249 ± 65 | 30 ± 8 | 102 ± 11 | 0.3 ± 0 | 6.4 ± 0.2 |
| HDCA | 4 | 67 ± 4* | 27 ± 4 | 0.3 ± 0.02* | 0.4 ± 0.1* | 24 ± 5 | 72 ± 30 | 150 ± 18* | 6 ± 1* | 114 ± 12 | 0.3 ± 0.1 | 6.3 ± 0.3 |
Values are means ± sd of each group. AST, aspartate aminotransferase; ALT, alanine transaminase; DBILI, direct bilirubin; TBILI, total bilirubin; BUN, blood urea nitrogen; CK, creatine kinase; LDH, lactate dehydrogenase; GGT, γ-glutamyltransferase; ALP, alkaline phosphatase; CREA, creatinine; TPROT, total protein.
P < 0.05 vs. chow.
HDCA inhibited atherosclerotic lesion formation in LDLRKO at multiple sites
In our feeding protocol, we fed the LDLRKO mice 8 wk of Western diet first to augment atherosclerotic lesion formation. After 8 wk of Western diet, 1 group of mice (baseline group) was euthanized to examine lesion formation at this time point. The remaining mice were then divided into 2 groups that received either the chow diet or chow diet + 1.25% HDCA for another 15 wk before lesion determination. The aim of this feeding protocol was to determine whether chow or chow + HDCA diets could regress or simply slow the progression of lesion formation from baseline. A previous study (32) with a similar feeding protocol of 3 mo Western diet feeding (baseline) followed by 3 mo chow diet feeding in male LDLRKO mice demonstrated a 35% decrease in lesion size in the baseline + chow diet group as compared with the baseline group. In another study (33), LDLRKO mice were fed a high-fat (21%), high-cholesterol (1.25%) diet for 6 mo (baseline), followed by 6 mo of chow diet feeding. The researchers reported a 68% increase in aortic lesion size in the 6-mo high-fat + 6-mo chow diet group as compared with the baseline group (33). In the present study, for the baseline group, the mean atherosclerotic lesion size at the aortic root region was 76,600 ± 5300 μm2/section. There was no calcification associated with any atherosclerotic lesion in the aortic root region of these mice, and there was no detectable lesion in the innominate artery of the baseline group (data not shown). After 15 wk of chow or chow + HDCA feeding, in the aortic root region, we did not observe lesion regression in either the chow group (561,200±42,300 μm2/section) or the chow + HDCA group (316,900±16,600 μm2/section) as compared with the baseline group (76,600±5,300 μm2/section). However, the chow + HDCA group exhibited significantly decreased atherosclerotic lesion size at the aortic root region, in the entire aorta, and in the innominate artery by 44, 48, and 94%, respectively, as compared with the chow group (Fig. 1A–C). Interestingly, in the HDCA group, there was a significant decrease in the inflammatory component of atherosclerotic lesions at the aortic root, as assessed by the relative area of macrophage-specific protein CD68 staining, as compared with the chow group (23 vs. 29% of total lesion area; P<0.05; Fig. 1D). The smooth muscle cell content of lesions, as assessed by the relative area of α-actin staining, showed a similar trend as macrophage content shown in Fig. 1D but was not statistically significant between the HDCA and chow groups (20 vs. 25% of total lesion area; P=0.07; Fig. 1E). In addition, we observed significantly less incidence of lesion calcification in the aortic root region of the HDCA group as compared with the chow group mice (62 vs. 91%; χ2 test, P=0.0003).
Figure 1.
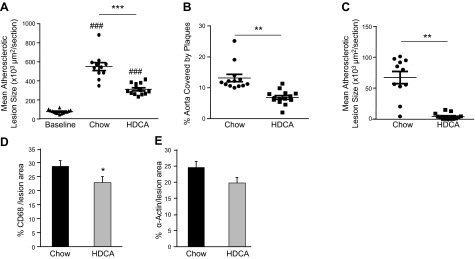
Atherosclerotic lesion size and composition of LDLRKO mice fed with various diets. After 8 wk of Western diet feeding (baseline), mice were switched to chow diet or chow diet supplemented with HDCA for another 15 wk. A–C) Atherosclerotic lesion size measurements at the aortic root region (A), in the entire aorta (B), and in the innominate artery region (C). Lines and error bars indicate mean ± se lesion area of each group. D, E) Relative macrophage content (D) and smooth muscle cell content (E) in the atherosclerotic lesions of the aortic root region were determined by immunohistochemistry using antibodies against CD68 and α-actin, respectively; 4–6 mice/group with 3–5 lesion sections/mouse were used in the study. *P < 0.05, **P < 0.01, ***P < 0.0001; ###P < 0.0001 vs. baseline group.
HDCA improved plasma lipoprotein profiles and decreased plasma glucose level
To begin to understand the underlying causes for decreased lesion formation in the HDCA group, we first determined plasma lipoprotein and lipid levels of LDLRKO mice that received 8 wk of Western diet (baseline group) and the other 2 groups that subsequently received 15 wk of the chow diet or chow diet + 1.25% HDCA. The chow and the chow + HDCA groups exhibited a 50% (P<0.05) and 81% (P<0.05) significant decrease in plasma very low density lipoprotein (VLDL)/intermediate-density lipoprotein (IDL)/LDL cholesterol levels, respectively, as compared with the Western diet baseline group (Table 4). Furthermore, animals that received HDCA exhibited s 51% (P<0.05) and 61% (P<0.05) decrease in plasma total and VLDL/IDL/LDL cholesterol levels, respectively, and a 28% (P<0.05) increase in triglyceride level as compared with the chow group (Table 4). Plasma HDL cholesterol was increased by 11% in the HDCA group as compared with the chow group (P=0.06). Plasma glucose level in the HDCA group was decreased by 37% (P<0.05) as compared with the chow group. Finally, plasma total bile acid levels were increased by 94% (P<0.05) in the HDCA group as compared with the chow group (Table 4). In another 3-wk feeding study, we observed that HDCA supplementation significantly decreased plasma total and VLDL/IDL/LDL cholesterol levels by 43% (P<0.05) and 51% (P<0.05), respectively, as compared with the chow diet group (Table 5). There was a significant 28% (P<0.05) increase in HDL cholesterol in the HDCA as compared with the chow group (Table 5). However, there was no significant difference in plasma triglyceride levels between the HDCA and chow diet groups after 3 wk feeding (Table 5). Pooled plasma samples from the 3-wk feeding experiment were fractionated by FPLC to examine lipoprotein profiles. As shown in Fig. 2A, the HDCA group exhibited significantly lower VLDL/IDL/LDL cholesterol levels and elevated HDL cholesterol as compared with the chow group. Phospholipid contents of the FPLC fractions (Fig. 2B) follow a similar trend as the cholesterol data. Therefore, as compared with the chow group, the HDCA group exhibited decreased phospholipid levels in the VLDL/IDL/LDL fractions and increased phospholipid levels in the HDL fractions. As compared with the chow group, the HDCA group exhibited lower triglyceride levels in the VLDL fractions, but an elevated triglyceride level in the IDL/LDL fraction (Fig. 2C). Interestingly, the lipid composition of VLDL and IDL/LDL isolated from the HDCA and chow groups was substantially different. As shown in Table 6, the VLDL of the HDCA group was relatively rich in triglyceride and poor in cholesterol as compared with that of the chow group. Similarly, the IDL/LDL from the HDCA group was relatively rich in triglyceride and poor in cholesterol as compared with that of the chow group (Table 6). We observed decreased apoB-48, apoB-100, and apoE levels in the VLDL fractions of the HDCA group as compared with the chow group (Fig. 2D). On the other hand, similar apoB-48, apoB-100, and apoE levels were observed in the LDL fractions of the chow and HDCA groups (Fig. 2D).
Table 4.
Plasma lipid and glucose levels of LDLRKO mice fed Western diet for 8 wk or Western diet (8 wk) followed by chow or chow + 1.25% HDCA diets for 15 wk
| Diet | n | TG | TC | HDL | VLDL/IDL/LDL | UC | FFA | Glc | BAa |
|---|---|---|---|---|---|---|---|---|---|
| Western | 4 | 185 ± 96 | 1333 ± 282 | 76 ± 10 | 1258 ± 291 | 387 ± 94 | 65 ± 14 | 148 ± 12 | ND |
| Chow | 12 | 113 ± 33# | 722 ± 155# | 98 ± 16# | 623 ± 152# | 184 ± 46# | 44 ± 6# | 212 ± 51 | 22 ± 7 |
| HDCA | 14 | 145 ± 36* | 352 ± 31*,# | 109 ± 13# | 243 ± 26*,# | 95 ± 7*,# | 43 ± 9# | 133 ± 48* | 42 ± 15* |
Values are means ± sd of each group. For all data except bile acids, units are milligrams per deciliter; for bile acids, micromolar. TG, trigylceride; TC, total cholesterol; HDL, high-density lipoprotein cholesterol; VLDL/IDL/LDL, very low density lipoprotein/intermediate-density lipoprotein/low-density lipoprotein cholesterol; UC, unesterified cholesterol; FFA, free fatty acid; Glc, glucose; BA, bile acid; ND, not determined.
Data were obtained from a separate study with a 3-wk diet-feeding protocol.
P < 0.05 vs. chow;
P < 0.05 vs. Western diet.
Table 5.
Plasma lipid and glucose levels of LDLRKO mice fed chow or chow + 1.25% HDCA diets for 3 wk
| Diet | n | TG | TC | HDL | VLDL/IDL/LDL | UC | FFA | Glc |
|---|---|---|---|---|---|---|---|---|
| Chow | 8 | 176 ± 51 | 680 ± 143 | 70 ± 16 | 610 ± 153 | 169 ± 38 | 59 ± 16 | 165 ± 59 |
| HDCA | 8 | 157 ± 56 | 389 ± 69* | 90 ± 11* | 300 ± 67* | 95 ± 20* | 59 ± 12 | 121 ± 27 |
Values are means ± sd (mg/dl) of each group. TG, trigylceride; TC, total cholesterol; HDL, high-density lipoprotein cholesterol; VLDL/IDL/LDL, very low density lipoprotein/intermediate-density lipoprotein/low-density lipoprotein cholesterol; UC, unesterified cholesterol; FFA, free fatty acid; Glc, glucose.
P < 0.05 vs. chow.
Figure 2.
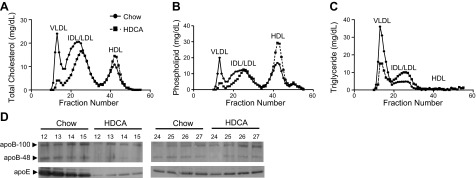
Plasma lipoprotein analysis of LDLRKO mice that received various diets. Pooled plasma samples from 5 mice/diet group that received chow or chow + HDCA diets for 3 wk were used for FPLC analysis. A–C) FPLC fractions were used to quantify total cholesterol (A), phospholipid (B), and triglyceride (C) concentrations. See Table 6 for lipid composition of VLDL and IDL/LDL from diet groups. D) The apoB-48, apoB-100, and apoE contents of the VLDL fractions (lanes 12–15) and LDL fractions (lanes 24–27) collected from FPLC were examined by immunoblotting as described in Materials and Methods.
Table 6.
Lipid composition of VLDL and IDL/LDL from chow and chow + HDCA diet groups as isolated by FPLC
| Diet and lipoprotein | Cholesterol (%) | Phospholipid (%) | Triglyceride (%) |
|---|---|---|---|
| Chow-VLDL | 32 | 20 | 48 |
| HDCA-VLDL | 19 | 16 | 65 |
| Chow-IDL/LDL | 54 | 33 | 13 |
| HDCA-IDL/LDL | 41 | 31 | 28 |
See Fig. 2A–C. Values are shown as individual lipid weight/total lipid weight (%).
HDCA supplementation decreased intestinal cholesterol absorption efficiency and increased daily cholesterol excretion through fecal output
Intestinal cholesterol absorption efficiency was determined using radiolabeled cholesterol and β-sitostanol. We observed a significant 76% decrease in intestinal cholesterol absorption efficiency in the HDCA group as compared with the chow group (Fig. 3A). We found that mice from the HDCA group exhibited a 91% increase in daily fecal cholesterol excretion as compared with the chow group (Fig. 3B). Triglyceride content in the fecal samples of the HDCA group was significantly higher as compared with the chow group (Fig. 3B). However, the amount of triglycerides present in the feces of the HDCA group accounted for only 0.4% of the triglycerides ingested by the mice. Therefore, HDCA did not have a substantial effect on triglyceride digestion and absorption in the intestine. The fecal samples of the HDCA group contained high levels of bile acids (Fig. 3B), which was most likely contributed by the excess HDCA, a bile acid, present in the diet.
Figure 3.
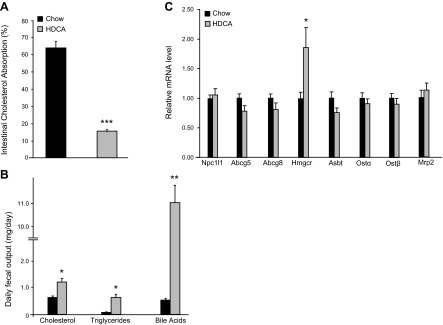
Intestinal cholesterol absorption, fecal lipid content, and gene expression in the small intestine of LDLRKO mice that received 3 wk feeding of chow diet or chow diet supplemented with HDCA. A) Intestinal cholesterol absorption rate. B) Daily fecal excretion of cholesterol, triglycerides, and bile acids. C) Small intestine expression of genes involved in cholesterol and bile acid transport of LDLRKO mice fed the chow or chow + HDCA diet. *P < 0.05, **P < 0.01, ***P < 0.0001 vs. chow diet group.
We then examined expression of genes involved in cholesterol and bile acid transport in the small intestine of these mice by qRT-PCR. The HDCA group exhibited no significant difference in the expression of genes involved in cholesterol absorption and excretion, such as Npc1l1, ATP-binding cassette subfamily G member 5 (Abcg5), and ATP-binding cassette subfamily G member 8 (Abcg8) (Fig. 3C). Expression levels of genes involved in bile acid transport, such as apical sodium-dependent bile salt transporter (Asbt), organic solute transporter α (Ostα), organic solute transporter β (Ostβ), and multidrug resistance protein 2 (Μrp2), were similar between chow and HDCA groups as well (Fig. 3C). Interestingly, the expression of 3-hydroxy-3-methylglutaryl-CoA reductase (Hmgcr), a gene involved in cholesterol synthesis, was significantly increased, by ∼100%, in the HDCA group as compared with the chow group, suggesting a lower intracellular cholesterol level in the enterocytes of the HDCA group, most likely due to the reduced intestinal cholesterol absorption.
Effects of HDCA on liver lipid content, hepatic gene expression, and bile composition
HDCA significantly decreased liver total cholesterol and unesterified cholesterol levels by ∼20% but did not change liver triglycerides or cholesterol ester levels as compared with the chow group (Fig. 4A). Bile composition analysis revealed that HDCA significantly increased total bile acids levels by 92% in the bile samples collected from the gallbladder as compared with those of the chow group (Fig. 4B), presumably due to increased hepatic excretion of HDCA. There were no significant differences in cholesterol or phophatidylcholine (PC) levels between the bile samples of HDCA and chow groups (Fig. 4B).
Figure 4.
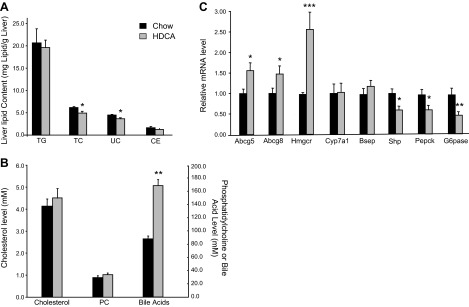
Liver lipid content, bile composition, and liver gene expression in the LDLRKO mice that received various diets for 3 wk. A) Liver lipid content was determined as described in Materials and Methods. B) Cholesterol, phophatdylcholine (PC), and total bile acids concentrations of bile collected from the gallbladder were examined. C) Hepatic expression of genes involved in cholesterol and bile acids homeostasis and gluconeogenesis was examined. *P < 0.05, **P < 0.01, ***P < 0.0001 vs. chow diet group.
HDCA significantly increased hepatic expression of genes involved in cholesterol homeostasis, such as Abcg5, Abcg8, and Hmgcr by 56, 47, and 163%, respectively (Fig. 4C). The expression of genes involved in bile acid homeostasis, such as cytochrome P450, family 7, subfamily A, polypeptide 1 (Cyp7a1) and bile salt export pump (Bsep), was not changed by HDCA supplementation (Fig. 4C), whereas the expression of Shp, a target gene of Fxr and a transcription repressor, was significantly decreased by HDCA (Fig. 4C). Interestingly, the expression of genes involved in gluconeogenesis, such as phosphoenolpyruvate carboxykinase (Pepck) and glucose-6-phosphatase (G6pase), was significantly decreased by 38 and 52%, respectively, in the HDCA group as compared with the chow group (Fig. 4C).
HDCA supplementation improved HDL function as measured by a cholesterol efflux assay
HDL was isolated by FPLC from pooled plasma samples collected from LDLRKO mice fed the chow or HDCA diets. Western blot analysis of the HDL samples showed no difference in apoA1 levels between the HDCA and chow groups (Fig. 5A). The levels of total bile acid carried by HDL from the chow and HDCA groups were 3.6 and 4.4 μM, respectively (P<0.05; Fig. 5B), corresponding to 13 and 11% of the total bile acid present in plasma. When normalized by HDL cholesterol concentration, the chow and HDCA HDL samples contain 5.2 nmol bile acid/mg HDL cholesterol, and 4.9 nmol bile acid/mg HDL cholesterol (P=0.24), respectively. Interestingly, HDL isolated from the HDCA group carried significantly less amounts of lipid oxidation products, as determined by the DCF assay (Fig. 5C). We then examined cholesterol efflux ability of these HDLs. Using an HDL dose of 7.5 μg cholesterol/ml, we observed significant 45% increases in cholesterol efflux ability in the HDCA group as compared with the chow group (Fig. 5D). At an HDL dose of 15 μg cholesterol/ml, the HDCA group exhibited 49% increases in cholesterol efflux ability as compared with the chow group (Fig. 5D). Therefore, our data suggest improved HDL function in mice that received HDCA in the diet as compared with the chow group.
Figure 5.
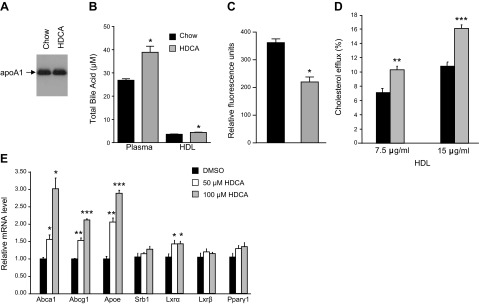
Examination of HDL properties and cholesterol efflux ability of HDL isolated from LDLRKO mice that received various diets, and the effect of HDCA treatment on expression of genes involved in cholesterol efflux in RAW 264.7 cells. A–D) HDL samples were isolated from pooled plasma of mice fed the chow or chow + HDCA diets by FPLC and used in immunoblotting using an antibody against mouse apoA1 (A), determination of total bile acid levels in HDL and plasma (B), examination of extent of lipid oxidation by the DCF assay (C), and cholesterol efflux assay as described in Materials and Methods (D). *P < 0.05, **P < 0.01, ***P < 0.0001 vs. chow diet group. E) Gene expression analysis of RAW 264.7 cells treated with DMSO (vehicle) or 50 or 100 μM of HDCA for 24 h. *P < 0.05, **P < 0.01, ***P < 0.0001 vs. DMSO group.
HDCA increased the expression of genes involved in cholesterol efflux in RAW 264.7 cells
We examined whether HDCA influences the expression of genes involved in cholesterol efflux in a macrophage cell line, RAW 264.7. The concentrations of HDCA used in treating the RAW cells in our study, 50 and 100 μM, were close to the circulating HDCA levels of LDLRKO mice that received the 1.25% HDCA supplementation in diet (mean 42.4 μM, range 31–66 μM). HDCA treatment significantly increased the expression of ATP-binding cassette subfamily A member 1 (Abca1), ATP-binding cassette subfamily G member 1 (Abcg1), and apolipoprotein E (Apoe) in RAW cells in a dose-response way. Thus, at a dose of 50 μM HDCA, the expression of Abca1, Abcg1, and Apoe was significantly increased by 57, 54, and 106%, respectively, as compared with the vehicle-treated group (Fig. 5E). Furthermore, at a dose of 100 μM HDCA, the expression of Abca1, Abcg1, and Apoe was significantly increased by 201, 112, and 189%, respectively, as compared with the vehicle-treated group (Fig. 5E). Expression of Srb1 was similar between the vehicle-treated and HDCA-treated groups (Fig. 5E). Expression of the nuclear receptor Lxrα, but not Lxrβ or peroxisome proliferator-activated receptor γ1 (Pparγ1), was modestly increased by HDCA treatment by 36%, at both the 50 and 100 μM doses, as compared with the control group (Fig. 5E).
DISCUSSION
We investigated the effects of HDCA supplementation on lipid metabolism and atherosclerotic lesion development in LDLRKO mice. Substantial decreases (44–94%) of atherosclerotic lesion size in the aortic root, the entire aorta, and the innominate artery were observed in mice supplemented with 1.25% HDCA in diet for 15 wk as compared with the chow group (Fig. 1A–C). In the aortic root region, the atherosclerotic lesions of the HDCA group also exhibited a significant decrease in macrophage content as compared with the chow group (Fig. 1D), probably due to decreased circulating IDL/LDL cholesterol level and improved HDL function in the HDCA group. We also observed a significant decrease in the incidence of calcification associated with the lesions of the aortic root region in the HDCA group as compared with the chow group. We found that HDCA supplementation resulted in a >60% decrease in plasma VLDL/IDL/LDL cholesterol level (Table 4). The main cause of decreased plasma VLDL/IDL/LDL cholesterol in the HDCA group was likely due to a 76% reduction in intestinal cholesterol absorption efficiency (Fig. 3A) as compared with the chow group. Furthermore, HDL isolated from the HDCA group exhibited significantly increased ability to mediate cholesterol efflux ex vivo as compared with HDL of the chow diet group (Fig. 5D). In cell culture studies, we observed that HDCA-treated macrophage cells, RAW 264.7, exhibited increased expression of genes involved in cholesterol efflux, including Abca1, Abcg1, and apoe, as compared with the vehicle-treated control (Fig. 5E). Therefore, HDCA appears to exhibit multiple antiatherogenic functions.
Previously, Sehayek et al. (17) showed that HDCA suppressed atherosclerosis formation at the aortic root by decreasing plasma cholesterol levels in male LDLRKO mice. The current study, by using female LDLRKO mice, confirmed the findings of Sehayek et al. that HDCA decreased lesion formation, intestinal cholesterol absorption, and plasma cholesterol levels in mice. Furthermore, our study provided several additional novel findings. First, HDCA supplementation decreased atherosclerosis formation at multiple sites: the aortic root, whole aorta, and innominate artery. The decrease in lesion size caused by HDCA supplementation was more dramatic at the innominate artery (94% decrease) than at the aortic root or the entire aorta (44 and 48% decrease, respectively). A previous study also observed major reduction of atherosclerosis in fractalkine-deficient mice as compared with the wild-type mice at the innominate artery, instead of the aortic root (34). The cause of the differential atherosclerosis development at various sites is likely due to the complex interaction between the specific flow conditions at these different anatomical locations and systemic risk factors such as hypercholesterolemia, genetics, and gender (35). Second, the current study examined lesion composition and demonstrated that HDCA supplementation significantly decreased macrophage content and the incidence of calcification in lesions, indicating HDCA treatment significantly decreased inflammatory component of the lesion and slowed lesion progression. Third, we demonstrated that HDCA significantly improved the ability of HDL in mediating cholesterol efflux from cholesterol-loaded macrophages. Fourth, HDCA significantly increased the expression of genes involved in cholesterol efflux, such as Abca1, Abcg1, and Apoe, in a macrophage cell line. Thus, in addition to inhibition of cholesterol absorption, this study uncovered 2 additional mechanisms, improving HDL function and induction of cholesterol efflux genes in macrophages, which may explain the antiatherogenic properties of HDCA.
This study chose a feeding protocol of 8 wk feeding of Western diet (baseline) followed by 15 wk feeding of chow diet with or without 1.25% HDCA originally intended to determine whether there would be lesion size reduction from baseline after the chow or chow + HDCA diet feeding. A previous study (32) with a similar feeding protocol in male LDLRKO mice demonstrated a 35% decrease in lesion size in the 3 mo Western diet + 3 mo chow diet group as compared with the baseline (3 mo Western diet) group. However, we observed substantial lesion size increases in both chow and chow + HDCA groups as compared with the baseline group (Fig. 1A). One explanation for continued lesion size increase after switching to a low-fat chow diet is the fact that we only observed a 46% decrease in plasma total cholesterol level when compare the chow (722 mg/dl) to the baseline data (1333 mg/dl). A previous study (36) using a similar chow diet (AIN-76) as the one used in this study reported a plasma total cholesterol level of 633 mg/dl in LDLRKO mice, similar to our chow diet value (722 mg/dl). These values are substantially higher than other chow diet total cholesterol values reported by other studies, ∼300 mg/dl (32, 33). Therefore, at least part of the reason we observed substantial lesion progression in the chow group as compared with the baseline group is probably due to the relatively modest decrease in plasma total cholesterol (46%) seen in the chow group as compared with the baseline group (Western diet; Table 4).
The significant decrease in intestinal cholesterol absorption observed in the HDCA group as compared with the chow group (Fig. 3A) was likely due to the inhibitory effect of HDCA on intestinal cholesterol absorption as previously demonstrated (19). An in vitro study showed that hydrophilic bile acids such as HDCA exhibit significantly lower capability in promoting mixed micelle formation than hydrophobic bile acids (37). It was postulated that hydrophilic bile acids such as HDCA and UDCA may interfere with the formation of mixed micelle containing cholesterol in the intestinal lumen and, therefore, decrease the absorption of cholesterol by enterocytes (19). In fact, UDCA has been used to treat or prevent cholesterol gallstone disease (38, 39). Furthermore, UDCA is sometimes used to treat cerebrotendinous xanthomatosis (CTX), a lipid storage disease caused by mutation in the CYP27A1 gene. However, chenodeoxycholic acid is considered more effective than UDCA in treating CTX (40–42).
In our study, we observed decreased total cholesterol and unesterified cholesterol levels in the livers of the HDCA group as compared with the chow group (Fig. 4A). This probably led to significantly lower levels of circulating hepatotoxicity markers, such as AST and bilirubin, in the HDCA group as compared with the chow group (Table 3). A previous study found that hepatic accumulation of unesterified cholesterol was associated with increased circulating levels of alanine aminotransferase (ALT) and AST in the NPC1L1-knockout mice as compared with the wild-type mice (43).
In the current study, plasma VLDL cholesterol, phospholipid, and triglyceride levels were all substantially decreased in the HDCA group as compared with the chow group (Fig. 2A–C). This is probably due to decreased VLDL particle numbers, since substantially lower levels of apoB-48 and apoB-100 were observed in the VLDL fractions of the HDCA group as compared with the chow group (Fig. 2D). Furthermore, the VLDL lipid composition of the HDCA group was substantially different from that of the chow group, with relatively lower cholesterol and higher triglyceride content in the former as compared with the latter (Table 6). This is probably caused by the decreased intestinal cholesterol absorption and decreased liver cholesterol level in the HDCA group. We postulate that as a result of the low availability of cholesterol in the liver, more triglyceride is packaged into the VLDL of the HDCA group as compared with the chow group. The lipid composition of IDL/LDL is also different between the HDCA and chow groups, with the former carrying relatively lower cholesterol and higher triglyceride content as compared with the latter (Table 6). Again, this is probably due to the altered lipid composition of the VLDL in the HDCA group. Interestingly, we observed a pronounced decrease in cholesterol and phospholipid levels in the IDL fractions of the HDCA group as compared with the chow group (Fig. 2A, B). Since IDL is known to promote atherosclerosis in both humans (44) and mice (45), the IDL cholesterol reducing effect of HDCA may be an important factor in its protection against atherosclerosis. Our data on the effect of HDCA supplementation on plasma triglyceride levels were inconclusive. On one hand, after feeding of 8 wk Western diet + 15 wk HDCA diet, we observed a significant 28% increase in plasma triglyceride levels as compared with the 8 wk Western diet + 15 wk chow group (Table 4). In contrast, we did not observe any significant difference in plasma triglyceride levels between the chow and HDCA groups after 3 wk of feeding (Table 5). Therefore, the exposure to Western diet (0 vs. 8 wk), the age of mice (11 vs. 31 wk), and the duration of the HDCA supplementation (3 vs. 15 wk) probably all influenced whether HDCA has an effect on plasma triglyceride levels.
Our data showed that HDL isolated from mice fed the HDCA diet had the same level of apoA1, decreased level of lipid oxidation products, and increased capability to mediate cholesterol efflux from the macrophage cell line RAW 264.7 cells, as compared with that of the chow group. A recent publication demonstrated that the capacity of HDL to mediate cholesterol efflux from macrophages had a strong inverse correlation with both carotid intima-media thickness and the likelihood of coronary artery disease in a human population, independently of the HDL cholesterol level (46). Thus, the ability of HDCA to improve cholesterol efflux capacity of HDL may be one of the mechanisms responsible for the atheroprotective effects of HDCA. Oxidative modification of HDL and apoAI has been shown to impair the ability of HDL to mediate cholesterol efflux (47–49). Decreased lipid oxidation in HDL also correlates with improved reverse cholesterol transport ability of HDL in cell culture (50). In the present study, the extent of apoAI oxidation of HDL isolated from the chow and HDCA group was not determined. However, the lower extent of lipid oxidation observed in the HDCA HDL as compared the chow HDL (Fig. 5C) may contribute to improved cholesterol efflux ability of the former. In addition, although the total bile acid level per HDL is similar for the HDCA and chow groups, presumably, the HDL isolated from the former is enriched with HDCA as compared with the latter. This qualitative difference in bile acid composition may also contribute to the improved cholesterol efflux capability of the HDCA HDL.
We observed that HDCA treatment increased the mRNA levels of LXR target genes, including Abca1, Abcg1, and Apoe (51), in a mouse macrophage cell line (Fig. 5E). Previously, both free and taurine-conjugated HDCA have been shown to activate LXRα, with ED50 of 17 and 3 μM, respectively (21), in cell culture. Free and taurine-conjugated HDCA also activate LXRβ, less effectively, with ED50 of 55 and 11 μM, respectively (21). Our data suggest that at least part of the atheroprotective effect of HDCA could be mediated through activation of LXR in macrophages, leading to increased expression of LXR target genes, Abca1, Abcg1, and Apoe, all of which promote cholesterol efflux.
Bile acids have been shown to activate the G-protein-coupled receptor TGR5, leading to increased energy expenditure, decreased obesity, and improved insulin sensitivity (52, 53). HDCA has been shown to activate TGR5 (20) but not FXR (22) in vitro. We found that after 15 wk of HDCA supplementation, the mice were less obese (Table 2) and exhibited significantly lower fasting glucose levels as compared with the chow diet group (Table 4). Expressions of gluconeogenesis genes, Pepck and G6pase, whose expression are known to be decreased by both TGR5 (53) and SHP through the activation of FXR (54), were significantly lower in the HDCA group as compared with the chow group (Fig. 4C). Since Shp mRNA levels were significantly lower in the livers of the HDCA group than in those of the control group, it is unlikely that the decreased expression of Pepck and G6pase in the HDCA-treated mice was due to the action of FXR and SHP. More likely, this is due to the activation of TGR5 by HDCA. In agreement, hepatic expression of Cyp7a1 and Bsep, 2 genes known to be down- and up-regulated by FXR, respectively, was not altered by HDCA supplementation (Fig. 4C), suggesting HDCA did not activate FXR in the mouse liver. In the small intestine, HDCA did not significantly increase or decrease the expression of FXR target genes Ostα, Ostβ, and Mrp2, either (Fig. 3C). Therefore, our data suggest that HDCA is an agonist for TGR5 but not FXR in vivo. Our data do not support HDCA as an antagonist for FXR.
In summary, our study demonstrates that HDCA influences cholesterol and glucose homeostasis in LDLRKO mice. HDCA supplementation inhibited intestinal cholesterol absorption, lowered plasma VLDL/IDL/LDL cholesterol levels, improved HDL function, and decreased obesity and plasma glucose levels in mice. The glucose-lowering and obesity-preventing effects of HDCA are most likely due to the activation of TGR5. Furthermore, HDCA not only significantly decreased the atherosclerotic lesion size but also decreased the contribution of inflammatory components, macrophages, and incidence of calcification within lesions. Our findings suggest that HDCA is an attractive candidate for the treatment of obesity, diabetes, and atherosclerosis.
Acknowledgments
The authors thank Lawrence Castellani, Sarada Charugundla, Yu-Rong Xia, Carmen Volpe, and Zhiqiang Zhou for excellent technical support.
The authors thank Balbir Brar for critical reading of the manuscript.
This study was supported in part by AtheroNova, Inc., and by U.S. National Heart, Lung, and Blood Institute grants 1R01-HL-094322-04 (to A.J.L.) and 2-P01-HL-030568-26A1 (to D.M.S.).
Footnotes
- Abca1
- ATP-binding cassette subfamily A member 1
- Abcg1
- ATP-binding cassette subfamily G member 1
- Abcg5
- ATP-binding cassette subfamily G member 5
- Abcg8
- ATP-binding cassette subfamily G member 8
- apoA1
- apolipoprotein A1
- Apoe
- apolipoprotein E
- Asbt
- apical sodium-dependent bile salt transporter
- ALP
- alkaline phosphatase
- ALT
- alanine aminotransferase
- AST
- aspartate aminotransferase
- BSA
- bovine serum albumin
- Bsep
- bile salt export pump
- BUN
- blood urea nitrogen
- CE
- cholesterol ester
- CK
- creatine kinase
- CREA
- creatinine
- CVD
- cardiovascular disease
- Cyp7a1
- cytochrome P450, family 7, subfamily A, polypeptide 1
- DBILI
- direct bilirubin
- DCF
- dichlorofluorescein
- DMSO
- dimethyl sulfoxide
- FPLC
- fast-performance liquid chromatography
- FXR
- farnesoid X receptor
- G6pase
- glucose-6-phosphatase
- GGT
- γ-glutamyltransferase
- Hmgcr
- 3-hydroxy-3-methylglutaryl-CoA reductase
- IDL
- intermediate-density lipoprotein
- HDCA
- hyodeoxycholic acid
- HDL
- high-density lipoprotein
- LDH
- lactate dehydrogenase
- LDL
- low-density lipoprotein
- LDLRKO
- low-density lipoprotein receptor knockout
- LXR
- liver X receptor
- Mrp2
- multidrug resistance protein 2
- NPC1L1
- Niemann-Pick C1-like 1
- Ostα
- organic solute transporter α
- Ostβ
- organic solute transporter β
- PBS
- phosphate-buffered saline
- PC
- phophatidylcholine
- Pepck
- phosphoenolpyruvate carboxykinase
- PPARγ1
- peroxisome proliferator-activated receptor γ1
- TBILI
- total bilirubin
- TC
- total cholesterol
- TG
- triglyceride
- TGR5
- G-protein-coupled bile acid receptor 1 (GPBAR1)
- TPROT
- total protein
- UC
- unesterified cholesterol
- UDCA
- ursodeoxycholic acid
- VLDL
- very low density lipoprotein
REFERENCES
- 1. Lusis A. J. (2000) Atherosclerosis. Nature 407, 233–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Grundy S. M. (1988) HMG-CoA reductase inhibitors for treatment of hypercholesterolemia. N. Engl. J. Med. 319, 24–33 [DOI] [PubMed] [Google Scholar]
- 3. Scandinavian Simvastatin Survival Study Group (1994) Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet 344, 1383–1389 [PubMed] [Google Scholar]
- 4. Shepherd J., Cobbe S. M., Ford I., Isles C. G., Lorimer A. R., MacFarlane P. W., McKillop J. H., Packard C. J. (1995) Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. West of Scotland Coronary Prevention Study Group. N. Engl. J. Med. 333, 1301–1307 [DOI] [PubMed] [Google Scholar]
- 5. The LIPID Study Group (1998) Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. N. Engl. J. Med. 339, 1349–1357 [DOI] [PubMed] [Google Scholar]
- 6. Downs J. R., Clearfield M., Weis S., Whitney E., Shapiro D. R., Beere P. A., Langendorfer A., Stein E. A., Kruyer W., Gotto A. M., Jr. (1998) Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA 279, 1615–1622 [DOI] [PubMed] [Google Scholar]
- 7. Baigent C., Blackwell L., Emberson J., Holland L. E., Reith C., Bhala N., Peto R. E., Barnes H., Keech A., Simes J., Collins R. (2010) Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 376, 1670–1681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Davidson M. H. (2011) Therapies targeting exogenous cholesterol uptake: new insights and controversies. Curr. Atheroscler. Rep. 13, 95–100 [DOI] [PubMed] [Google Scholar]
- 9. Couture P., Lamarche B. (2013) Ezetimibe and bile acid sequestrants: impact on lipoprotein metabolism and beyond. Curr. Opin. Lipidol. 24, 227–232 [DOI] [PubMed] [Google Scholar]
- 10. Rader D. J. (2006) Molecular regulation of HDL metabolism and function: implications for novel therapies. J. Clin. Invest. 116, 3090–3100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Digby J. E., Ruparelia N., Choudhury R. P. (2012) Niacin in cardiovascular disease: recent preclinical and clinical developments. Arterioscler. Thromb. Vasc. Biol. 32, 582–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Barter P., Rye K. A. (2011) Cholesteryl ester transfer protein inhibition to reduce cardiovascular risk: where are we now? Trends Pharmacol. Sci. 32, 694–699 [DOI] [PubMed] [Google Scholar]
- 13. Watson C. E., Weissbach N., Kjems L., Ayalasomayajula S., Zhang Y., Chang I., Navab M., Hama S., Hough G., Reddy T., Soffer D., Rader D. J., Fogelman A. M., Schecter A. (2011) Treatment of patients with cardiovascular disease with L-4F, an apo-A1 mimetic, did not improve select biomarkers of HDL function. J. Lipid Res. 52, 361–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Eyssen H. J., De Pauw G., Van Eldere J. (1999) Formation of hyodeoxycholic acid from muricholic acid and hyocholic acid by an unidentified gram-positive rod termed HDCA-1 isolated from rat intestinal microflora. Appl. Environ. Microbiol. 65, 3158–3163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Singhal A. K., Cohen B. I., Finver-Sadowsky J., McSherry C. K., Mosbach E. H. (1984) Role of hydrophilic bile acids and of sterols on cholelithiasis in the hamster. J. Lipid Res. 25, 564–570 [PubMed] [Google Scholar]
- 16. Singhal A. K., Cohen B. I., Mosbach E. H., Une M., Stenger R. J., McSherry C. K., May-Donath P., Palaia T. (1984) Prevention of cholesterol-induced gallstones by hyodeoxycholic acid in the prairie dog. J. Lipid Res. 25, 539–549 [PubMed] [Google Scholar]
- 17. Sehayek E., Ono J. G., Duncan E. M., Batta A. K., Salen G., Shefer S., Neguyen L. B., Yang K., Lipkin M., Breslow J. L. (2001) Hyodeoxycholic acid efficiently suppresses atherosclerosis formation and plasma cholesterol levels in mice. J. Lipid Res. 42, 1250–1256 [PubMed] [Google Scholar]
- 18. Cohen-Solal C., Parquet M., Ferezou J., Serougne C., Lutton C. (1995) Effects of hyodeoxycholic acid and alpha-hyocholic acid, two 6 alpha-hydroxylated bile acids, on cholesterol and bile acid metabolism in the hamster. Biochim. Biophys. Acta 1257, 189–197 [DOI] [PubMed] [Google Scholar]
- 19. Wang D. Q., Tazuma S., Cohen D. E., Carey M. C. (2003) Feeding natural hydrophilic bile acids inhibits intestinal cholesterol absorption: studies in the gallstone-susceptible mouse. Am. J. Physiol. Gastrointest. Liver Physiol. 285, G494–G502 [DOI] [PubMed] [Google Scholar]
- 20. Sato H., Macchiarulo A., Thomas C., Gioiello A., Une M, Hofmann A. F., Saladin R., Schoonjans K., Pellicciari R., Auwerx J. (2008) Novel potent and selective bile acid derivatives as TGR5 agonists: biological screening, structure-activity relationships, and molecular modeling studies. J. Med. Chem. 51, 1831–1841 [DOI] [PubMed] [Google Scholar]
- 21. Song C., Hiipakka R. A., Liao S. (2000) Selective activation of liver X receptor alpha by 6alpha-hydroxy bile acids and analogs. Steroids 65, 423–427 [DOI] [PubMed] [Google Scholar]
- 22. Makishima M., Lu T. T., Xie W., Whitfield G. K., Domoto H., Evans R. M., Haussler M. R., Mangelsdorf D. J. (2002) Vitamin D receptor as an intestinal bile acid sensor. Science 296, 1313–1316 [DOI] [PubMed] [Google Scholar]
- 23. Taicher G. Z., Tinsley F. C., Reiderman A., Heiman M. L. (2003) Quantitative magnetic resonance (QMR) method for bone and whole-body-composition analysis. Anal. Bioanal. Chem. 377, 990–1002 [DOI] [PubMed] [Google Scholar]
- 24. Mehrabian M., Qiao J. H., Hyman R., Ruddle D., Laughton C., Lusis A. J. (1993) Influence of the apoA-II gene locus on HDL levels and fatty streak development in mice. Arterioscler. Thromb. 13, 1–10 [DOI] [PubMed] [Google Scholar]
- 25. Hedrick C. C., Castellani L. W., Warden C. H, Puppione D. L., Lusis A. J. (1993) Influence of mouse apolipoprotein A-II on plasma lipoproteins in transgenic mice. J. Biol. Chem. 268, 20676–20682 [PubMed] [Google Scholar]
- 26. Navab M., Hama S. Y., Hough G. P., Subbanagounder G., Reddy S. T., Fogelman A. M. (2001) A cell-free assay for detecting HDL that is dysfunctional in preventing the formation of or inactivating oxidized phospholipids. J. Lipid Res. 42, 1308–1317 [PubMed] [Google Scholar]
- 27. Qiao J. H., Xie P. Z., Fishbein M. C, Kreuzer J., Drake T. A., Demer L. L., Lusis A. J. (1994) Pathology of atheromatous lesions in inbred and genetically engineered mice. Genetic determination of arterial calcification. Arterioscler. Thromb. 14, 1480–1497 [DOI] [PubMed] [Google Scholar]
- 28. Shih D. M., Xia Y. R., Wang X. P., Miller E., Castellani L. W., Subbanagounder G., Cheroutre H., Faull K. F., Berliner J. A., Witztum J. L., Lusis A. J. (2000) Combined serum paraoxonase knockout/apolipoprotein E knockout mice exhibit increased lipoprotein oxidation and atherosclerosis. J. Biol. Chem. 275, 17527–17535 [DOI] [PubMed] [Google Scholar]
- 29. Bennett B. J., Wang S. S., Wang X., Wu X., Lusis A. J. (2009) Genetic regulation of atherosclerotic plaque size and morphology in the innominate artery of hyperlipidemic mice. Arterioscler. Thromb. Vasc. Biol. 29, 348–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Orozco L. D., Kapturczak M. H., Barajas B., Wang X., Weinstein M. M., Wong J., Deshane J., Bolisetty S., Shaposhnik Z., Shih D. M., Agarwal A., Lusis A. J., Araujo J. A. (2007) Heme oxygenase-1 expression in macrophages plays a beneficial role in atherosclerosis. Circ. Res. 100, 1703–1711 [DOI] [PubMed] [Google Scholar]
- 31. Folch J., Lees M., Sloane Stanley G. H. (1957) A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 226, 497–509 [PubMed] [Google Scholar]
- 32. Ramachandran S., Penumetcha M., Merchant N. K., Santanam N., Rong R., Parthasarathy S. (2005) Exercise reduces preexisting atherosclerotic lesions in LDL receptor knock out mice. Atherosclerosis 178, 33–38 [DOI] [PubMed] [Google Scholar]
- 33. Tsimikas S., Shortal B. P., Witztum J. L., Palinski W. (2000) In vivo uptake of radiolabeled MDA2, an oxidation-specific monoclonal antibody, provides an accurate measure of atherosclerotic lesions rich in oxidized LDL and is highly sensitive to their regression. Arterioscler. Thromb. Vasc. Biol. 20, 689–697 [DOI] [PubMed] [Google Scholar]
- 34. Teupser D., Pavlides S., Tan M., Gutierrez-Ramos J. C., Kolbeck R., Breslow J. L. (2004) Major reduction of atherosclerosis in fractalkine (CX3CL1)-deficient mice is at the brachiocephalic artery, not the aortic root. Proc. Natl. Acad. Sci. U. S. A. 101, 17795–17800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. VanderLaan P. A., Reardon C. A., Getz G. S. (2004) Site specificity of atherosclerosis: site-selective responses to atherosclerotic modulators. Arterioscler. Thromb. Vasc. Biol. 24, 12–22 [DOI] [PubMed] [Google Scholar]
- 36. Teupser D., Persky A. D, and Breslow J. L. (2003) Induction of atherosclerosis by low-fat, semisynthetic diets in LDL receptor-deficient C57BL/6J and FVB/NJ mice: comparison of lesions of the aortic root, brachiocephalic artery, and whole aorta (en face measurement). Arterioscler. Thromb. Vasc. Biol. 23, 1907–1913 [DOI] [PubMed] [Google Scholar]
- 37. Van de Heijning B. J., Stolk M. F., van Erpecum K. J., Renooij W., Groen A. K., vanBerge-Henegouwen G. P. (1994) Bile salt-induced cholesterol crystal formation from model bile vesicles: a time course study. J. Lipid Res. 35, 1002–1011 [PubMed] [Google Scholar]
- 38. Bachrach W. H., Hofmann A. F. (1982) Ursodeoxycholic acid in the treatment of cholesterol cholelithiasis. Part I. Dig. Dis. Sci. 27, 737–761 [DOI] [PubMed] [Google Scholar]
- 39. Uy M. C., Talingdan-Te M. C., Espinosa W. Z., Daez M. L., Ong J. P. (2008) Ursodeoxycholic acid in the prevention of gallstone formation after bariatric surgery: a meta-analysis. Obes. Surg. 18, 1532–1538 [DOI] [PubMed] [Google Scholar]
- 40. Batta A. K., Salen G., Tint G. S. (2004) Hydrophilic 7 beta-hydroxy bile acids, lovastatin, and cholestyramine are ineffective in the treatment of cerebrotendinous xanthomatosis. Metabolism 53, 556–562 [DOI] [PubMed] [Google Scholar]
- 41. Broughton G., 2nd. (1994) Chenodeoxycholate: the bile acid. The drug. A review. Am. J. Med. Sci. 307, 54–63 [DOI] [PubMed] [Google Scholar]
- 42. Koopman B. J., Wolthers B. G., van der Molen J. C., G. T, Nagel G. T., Waterreus R. J., Oosterhuis H. J. (1984) Capillary gas chromatographic determinations of urinary bile acids and bile alcohols in CTX patients proving the ineffectivity of ursodeoxycholic acid treatment. Clin. Chim. Acta 142, 103–111 [DOI] [PubMed] [Google Scholar]
- 43. Beltroy E. P., Liu B., Dietschy J. M., Turley S. D. (2007) Lysosomal unesterified cholesterol content correlates with liver cell death in murine Niemann-Pick type C disease. J. Lipid Res. 48, 869–881 [DOI] [PubMed] [Google Scholar]
- 44. Krauss R. M., Lindgren F. T., Williams P. T., Kelsey S. F., Brensike J., Vranizan K., Detre K. M., Levy R. I. (1987) Intermediate-density lipoproteins and progression of coronary artery disease in hypercholesterolaemic men. Lancet 2, 62–66 [DOI] [PubMed] [Google Scholar]
- 45. Plump A. S., Smith J. D., Hayek T., Aalto-Setälä K., Walsh A., Verstuyft J. G., Rubin E. M., Breslow J. L. (1992) Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell 71, 343–353 [DOI] [PubMed] [Google Scholar]
- 46. Khera A. V., Cuchel M., de la Llera-Moya M., Rodrigues A., Burke M. F., Jafri K., French B. C., Phillips J. A., Mucksavage M. L., Wilensky R. L., Mohler E. R., Rothblat G. H., Rader D. J. (2011) Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N. Engl. J. Med. 364, 127–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nagano Y., Arai H., Kita T. (1991) High density lipoprotein loses its effect to stimulate efflux of cholesterol from foam cells after oxidative modification. Proc. Natl. Acad. Sci. U. S. A. 88, 6457–6461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shao B., Pennathur S., Pagani I., Oda M. N., Witztum J. L., Oram J. F., Heinecke J. W. (2010) Modifying apolipoprotein A-I by malondialdehyde, but not by an array of other reactive carbonyls, blocks cholesterol efflux by the ABCA1 pathway. J. Biol. Chem. 285, 18473–18484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zheng L., Nukuna B., Brennan M. L., Sun M., Goormastic M., Settle M., Schmitt D., Fu X., Thomson L., Fox P. L., Ischiropoulos H., Smith J. D., Kinter M., Hazen S. L. (2004) Apolipoprotein A-I is a selective target for myeloperoxidase-catalyzed oxidation and functional impairment in subjects with cardiovascular disease. J. Clin. Invest. 114, 529–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Navab M., Ananthramaiah G. M., Reddy S. T., Van Lenten B. J., Ansell B. J., Fonarow G. C., Vahabzadeh K., Hama S., Hough G., Kamranpour N., Berliner J. A., Lusis A. J., Fogelman A. M. (2004) The oxidation hypothesis of atherogenesis: the role of oxidized phospholipids and HDL. J. Lipid Res. 45, 993–1007 [DOI] [PubMed] [Google Scholar]
- 51. Calkin A. C., Tontonoz P. (2012) Transcriptional integration of metabolism by the nuclear sterol-activated receptors LXR and FXR. Nat. Rev. Mol. Cell Biol. 13, 213–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Watanabe M., Houten S. M., Mataki C., Christoffolete M. A., Kim B. W., Sato H., Messaddeq N., Harney J. W., Ezaki O., Kodama T., Schoonjans K., Bianco A. C., Auwerx J. (2006) Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature 439, 484–489 [DOI] [PubMed] [Google Scholar]
- 53. Thomas C., Gioiello A., Noriega L., Strehle A., Oury J., Rizzo G., Macchiarulo A., Yamamoto H., Mataki C., Pruzanski M., Pellicciari R., Auwerx J., Schoonjans K. (2009) TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 10, 167–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Thomas C., Pellicciari R., Pruzanski M., Auwerx J., Schoonjans K. (2008) Targeting bile-acid signalling for metabolic diseases. Nat. Rev. Drug Discov. 7, 678–693 [DOI] [PubMed] [Google Scholar]