

Abstract
We have previously shown that deletion of CuZnSOD in mice (Sod1−/− mice) leads to accelerated loss of muscle mass and contractile force during aging. To dissect the relative roles of skeletal muscle and motor neurons in this process, we used a Cre-Lox targeted approach to establish a skeletal muscle-specific Sod1-knockout (mKO) mouse to determine whether muscle-specific CuZnSOD deletion is sufficient to cause muscle atrophy. Surprisingly, mKO mice maintain muscle masses at or above those of wild-type control mice up to 18 mo of age. In contrast, maximum isometric specific force measured in gastrocnemius muscle is significantly reduced in the mKO mice. We found no detectable increases in global measures of oxidative stress or ROS production, no reduction in mitochondrial ATP production, and no induction of adaptive stress responses in muscle from mKO mice. However, Akt-mTOR signaling is elevated and the number of muscle fibers with centrally located nuclei is increased in skeletal muscle from mKO mice, which suggests elevated regenerative pathways. Our data demonstrate that lack of CuZnSOD restricted to skeletal muscle does not lead to muscle atrophy but does cause muscle weakness in adult mice and suggest loss of CuZnSOD may potentiate muscle regenerative pathways.—Zhang, Y., Davis, C., Sakellariou, G.K., Shi, Y., Kayani, A.C., Pulliam, D., Bhattacharya, A., Richardson, A., Jackson, M.J., McArdle, A., Brooks, S.V., Van Remmen, H. CuZnSOD gene deletion targeted to skeletal muscle leads to loss of contractile force but does not cause muscle atrophy in adult mice.
Keywords: oxidative stress, regeneration, central nuclei
Age-related muscle atrophy (sarcopenia) is a major contributor to frailty and loss of independence in the elderly (1). The problem is universal and leads to a significant vulnerability that opposes healthy aging. Yet, to date, the mechanisms underlying the muscle loss during aging remain to be defined. Oxidative stress and damage have been suggested to be among the factors contributing to the initiation and progression of muscle atrophy that occurs during aging (2). Consistent with a role of oxidative stress as a contributor to sarcopenia, studies from our group have found that loss of the superoxide scavenger copper zinc superoxide dismutase (CuZnSOD) in mice (Sod1−/− mice) leads to an accelerated and progressive loss of muscle during aging that mimics the loss that occurs in wild-type (WT) control mice past median life span and in older humans (3–7). Thus, the Sod1−/− mice are a powerful tool to study potential mechanisms of age-related muscle atrophy and to uncover potential targets for intervention for preventing sarcopenia in humans.
Mice lacking CuZnSOD have an increase in oxidative stress, as measured by elevated levels of oxidative damage in lipid, DNA, and protein in all tissues and in plasma (6–8), associated with a number of physiological declines, including an accelerated loss of skeletal muscle, hearing loss, and increased incidence of hepatocellular carcinoma (7–9). Hind-limb muscle mass in Sod1−/− mice is significantly reduced compared to age-matched WT control mice from as early as 3 mo of age and continues to decrease with age (7). In addition, many features of the muscles of Sod1−/− mice resemble those observed in old WT control mice, notably widespread degeneration of neuromuscular junctions (NMJs), loss of innervation, and loss of contractile force (10). Induction of contraction using direct muscle stimulation of muscle tissue, bypassing the NMJ, partially rescues the deficit in force, which indicates a loss of functional innervation in the Sod1−/− mice (4). These results suggest that loss of innervation is a critical part of the muscle atrophy phenotype exhibited by the Sod1−/− mice, but the importance of oxidative stress in motor nerves or in muscle to initiate these changes remains unclear.
To directly test whether changes initiated by lack of CuZnSOD within the muscle tissue alone can lead to muscle atrophy, we generated a tissue-specific Sod1-knockout mouse model targeting skeletal muscle Sod1 utilizing Cre recombinase expression driven by the human α skeletal actin promoter. If oxidative stress within the muscle itself is a primary initiator of muscle atrophy, we expected that loss of CuZnSOD specifically from muscle fibers would result in loss of muscle mass. Here, we report the effects of the muscle-specific deletion of CuZnSOD on muscle mass, morphology, function, and adaptive responses.
MATERIALS AND METHODS
Generation of muscle-specific Sod1-knockout (mKO) mice
The conditional Sod1-knockout (Sod1flox/flox) mice were generated using a mouse Sod1 genomic DNA that has 2 LoxP sites in the introns flanking exons 3 and 4 of Sod1, with a neomycin cassette for selection (see Fig. 1A), on a C57BL6 background, by the Transgenic Animal Model Core at the University of Michigan. The incorporation of the conditional Sod1 construct was confirmed by Southern blot analysis. The Sod1flox/flox mice were bred with a mouse strain that expresses muscle-specific actin α1 (Acta1)-Cre recombinase [B6.Cg-Tg(Acta1-cre)79Jme/J; the Jackson Laboratory, Bar Harbor, ME, USA] to generate mKO mice.
Figure 1.
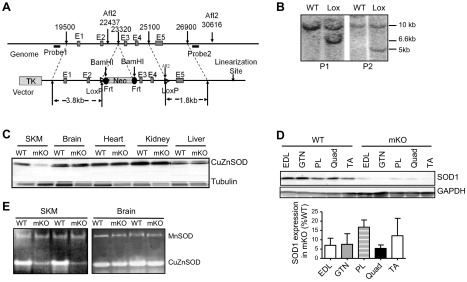
Generation of mKO mice. A) Schematic diagram of the Sod1 genomic construct and the recombination events in the generation of Sod1flox/flox mice. E1–E5, Sod1 exon 1 to 5; probe 1 and probe 2, DNA probes for Southern blot analysis to confirm the recombination between genomic DNA and the Cre-Lox/Sod1 construct; TK, thymidine kinase; Neo, neomycin resistance; Frt, flipase recognition target; LoxP, recombination site recognized by Cre recombinase. B) Southern blot analysis of the integration of Cre-Lox/Sod1 constructs into mouse genome. C) Western blot analysis of Sod1 expression in various tissues of mKO mice. D) Western blot analysis of Sod1 expression in multiple skeletal muscles in mKO mice. E) Enzymatic activity analysis of CuZnSOD.
All mice were fed ad libitum a standard NIH-31 chow and maintained under barrier conditions in microisolator cages on a 12-h light-dark cycle. For tissue collection, animals were euthanized by CO2 inhalation, followed by cervical dislocation. The tissues were immediately excised and placed on ice. After collection, all tissues were snap-frozen and stored at −80°C. All procedures were approved by the Institutional Animal Care and Use Committee at the University of Texas Health Science Center at San Antonio and the Audie L. Murphy Veterans Hospital (San Antonio, TX, USA).
SOD enzymatic activity assay
The activities of CuZnSOD and manganese SOD (MnSOD) were measured using native gels, with negative staining, as described previously (11).
Mitochondrial function assays
Mitochondrial reactive oxygen species (ROS) production was measured indirectly as hydrogen peroxide (H2O2) release from intact isolated mitochondria using Amplex Red (Molecular Probes, Eugene, OR, USA), as described previously (12). Horseradish peroxidase (HRP; 1 U/ml) and superoxide dismutase (SOD; 30 U/ml; Sigma, St. Louis, MO, USA) were added to convert all superoxide into H2O2. We monitored Amplex Red oxidation by H2O2 every 2 s for ∼10 min at an excitation of 545 nm and an emission of 590 nm using a Fluoroskan-FL Ascent Type 374 multiwell plate reader (Labsystems, Helsinki, Finland). We performed all assays at 37°C, in 125 mM KCl, 10 mM HEPES, 5 mM MgCl2, and 2 mM K2HPO4 (pH 7.44), with 50–20 μg of mitochondrial protein per 100 μl of reaction buffer. Mitochondrial ATP production was measured in isolated muscle mitochondria as described previously (13). The respiratory substrates GM (2.5 mM glutamate and 2.5 mM malate) and SR (5 mM succinate and 0.5 μM rotenone) were added as indicated in Results.
Isolation of single mature skeletal muscle fibers
Single muscle fibers were isolated from the flexor digitorum brevis (FDB) muscles of mice as described elsewhere (14). Briefly, FDB muscles were incubated for 1.5 h at 37°C in 0.4% (w/v) sterile type I collagenase in Eagle's minimum essential medium (MEM) containing 2 mM glutamine, 50 IU penicillin, 50 μg/ml streptomycin, and 10% fetal bovine serum (FBS; Invitrogen Ltd, Paisley, UK). Muscles were agitated every 20 min to improve digestion of the connective tissue. Single fibers were released by gentle trituration with a wide-bore pipette and washed 3 times in MEM containing 10% FBS. Fibers were plated onto precooled 35-mm cell culture dishes precoated with Matrigel (BD Biosciences, Oxford, UK) and allowed to attach for 45 min before adding 2 ml of MEM containing 10% FBS. Fibers were incubated for 16 h at 37°C in 5% CO2. Fibers prepared and cultured in this manner are viable for up to 6 d in culture (15) but were routinely used at 16 h following isolation. Experiments were performed only on fibers that exhibited clear striations along the sarcolemma (e.g., see Fig. 4C).
Figure 4.

ROS generation in isolated mitochondria and muscle fibers from mKO mice. A, B) H2O2 (A) and ATP production (B) from isolated skeletal muscle mitochondria (n=5/genotype). C) Confocal images of a single isolated fiber from the FDB muscle after 16 h in culture under bright field (i), fluorescent image following loading with CM-DCFH DA (ii), and merged image of i and ii (iii); original view, ×60. Scale bar = 30 μm). B) Rate of change in DCF fluorescence from resting FDB fibers of WT and mKO mice over the 30-min time course (n=4/genotype). Solid bars, WT mice; shaded bars, mKO mice.
Use of 5-(and 6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (CM-DCFH DA) to monitor changes in ROS production in single isolated fibers
To monitor changes in ROS production, single FDB muscle fibers were loaded by incubation with 10 μM CM-DCFH DA (Molecular Probes, Eugene, OR, USA) in 2 ml Dulbecco's phosphate-buffered saline (D-PBS) for 30 min at 37°C as described previously (15, 16). Cells were washed twice with D-PBS and were maintained in MEM without Phenol Red during the experimental period. Intracellular ROS, particularly H2O2, convert CM-DCFH to its fluorescent derivate, CM-DCF (see Fig. 4C).
Fluorescent microscopy imaging
The imaging system consisted of a C1 confocal microscope (Nikon Instruments Europe BV, Surrey, UK) equipped with a 488-nm excitation argon laser, 515/30 emission filter set for the detection of DCF fluorescence. Using a ×60 objective, fluorescence images were captured and analyzed with the EZC1 3.9 (12-bit) acquisition software (Nikon). All experiments were performed at 25°C.
Transmission electron microscopy (TEM)
Gastrocnemius muscles were freshly dissected and fixed in 2% paraformaldehyde and 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer and then postfixed with 1% osmium tetroxide followed by 1% uranyl acetate. The blocks were dehydrated through a graded series of ethanol washes and embedded in resin. Blocks were cut in ultrathin (90 nm) sections on a Reichert Ultracut UCT (Reichert, Vienna, Austria), stained with uranyl acetate followed by lead citrate, and viewed on a Jeol 1230 EX transmission electron microscope (Jeol Ltd., Akishima, Japan) at 80 kV.
Lipid peroxidation assay
Lipid peroxidation was measured in mouse tissues by determining the levels of F2-isoprostanes by methods we have previously described and initially described by Morrow et al. (17, 18). The levels of F2-isoprostanes were measured using gas chromatography-mass spectrometry (GC-MS). The levels of F2-isoprostanes are expressed as nanograms F2-isoprostanes per gram tissue.
Glutathione and protein thiol measurements
The automated glutathione recycling method described by Anderson (19) was used to assess the total glutathione content of muscle samples, using a 96-well plate reader (Powerwave X340; BioTek Instruments Inc., Winooski, VT, USA). The protein thiol content of samples was analyzed by the method of Di Monte et al. (20) adapted for use on a 96-well plate reader.
Analyses of the 3-nitrotyrosine (3-NT) content of muscle carbonic anhydrase III
Total cellular protein was isolated, and 100 μg was separated by SDS-PAGE, followed by Western blotting as described below. The content of 3-NT was analyzed by using a mouse monoclonal antibody (Cayman Chemical Co., Ann Arbor, MI, USA), and the bands were visualized and densitometric quantification of the carbonic anhydrase III band was undertaken using a Chemi-Doc XRS System (Bio-Rad Laboratories Ltd., Hemel Hempstead, UK) (3).
Western blotting of muscle proteins
For assessment of specific proteins in muscle, 50 μg of total protein was applied to a 10–15% polyacrylamide gel with a 4% stacking gel (National Diagnostics Ltd, Atlanta, GA, USA). The separated proteins were transferred onto nitrocellulose membranes by Western blotting. Membranes were probed using antibodies against CuZnSOD (Stressgen Inc., Victoria, BC, Canada); HSC70, HSP60, and HSP25, (Enzo Life Sciences, Ltd., Exeter, UK), pAkt(Ser308), Akt, pS6(Ser235/236), and S6 (Cell Signaling, Danvers, MA, USA); GAPDH, and tubulin (Abcam PLC, Cambridge, UK) as described previously (3). HRP-conjugated anti-mouse or -rabbit IgG (Cell Signaling) or anti-rat (Sigma) was used as secondary antibody. Peroxidase activity was detected using an ECL kit (Amersham International, Cardiff, UK), and band intensities were analyzed using Quantity One software (Bio-Rad).
Rota-rod performance
Rota-rod performance was measured as described by Muller et al. (7). The initial speed of the rod was set to 2 rpm with a linear acceleration of 0.1 rpm/s. Falls from the rod were detected by an infrared sensor. Each mouse performed 4 trials/d on 3 consecutive days, with 20 min rest between trials.
Treadmill test
Treadmill running test was conducted using a 6-lane rodent treadmill (Exer-6; Columbus Instruments, Columbus, OH, USA) modified from methods previously described (13). Briefly, mice were acclimated for 3 d prior to test (0 m/min for 10 min, followed by 5 m/min for 10 min, then 10 m/min for 10 min). On the day of testing, mice were run at a 15° incline initially at 12 m/min, increasing the speed in increments of 3 m/min every 5 min. Exhaustion was determined by a failure to engage the treadmill despite the presence of a mild shock and physical prodding. Cumulative running distance and total running time were recorded.
Muscle function
Mice were anesthetized with intraperitoneal injections of tribromoethanol (400 mg/kg) supplemented to maintain adequate anesthesia throughout the procedure. Gastrocnemius (GTN) muscle contractile properties were measured in situ as described by Larkin et al. (4). In anesthetized mice, the whole GTN muscle was isolated, and the distal tendon was severed and secured to the lever arm of a servomotor (model 305B; Aurora Scientific, Aurora, ON, Canada). The muscle was activated by stimulation of the tibial nerve using a bipolar platinum wire electrode. Stimulation voltage and muscle length (Lo) were adjusted to give a maximum isometric twitch. At Lo, 300-ms trains of stimuli were applied at increasing frequency until the maximum isometric tetanic force (Po) was achieved. In some cases, this procedure was repeated using direct muscle stimulation rather than nerve stimulation via a cuff electrode around the proximal and distal ends of the muscle. For fatigue tests, muscles were stimulated with 100-Hz trains of 0.5 s duration, once each 5 s for 15 min. Muscles were subsequently removed, and deeply anesthetized mice were euthanized by administration of a pneumothorax. Muscles were blotted and weighed, and muscle fiber length (Lf) was calculated by multiplying Lo by 0.45 (4). Total fiber cross-sectional area (CSA) was calculated by dividing the muscle mass (mg) by the product of Lf (mm) and density (1.06 g/cm2) for calculation of specific Po (N/cm2).
Muscle fiber counts and areas and identification of fiber types
Muscles that had been coated in tissue-freezing medium (Triangle Biomedical Sciences, Durham, NC, USA) and rapidly frozen in isopentane cooled by dry ice were cryosectioned at −20°C through the midbelly with a thickness of 12 μm, and fluorescent immunohistochemical (IHC) staining was initiated the same day. Frozen sections were rinsed with PBS and permeabilized in 0.2% Triton X-100 in PBS for 5 min. Sections were incubated overnight in blocking solution (Vector M.O.M. Mouse Ig Blocking Reagent; Vector Laboratories, Burlingame, CA, USA) in a sealed chamber at 4°C and then soaked in M.O.M. Dilutent (Vector Laboratories) for 5 min. Fibers were identified by staining for myosin heavy chain and laminin to visualize fiber outlines, as described previously (4), using antibodies for type 1 myosin (mouse IgM; A4.840; Alexis Biochemicals, Lausen, Switzerland), type 2 myosin (mouse IgG; MS-1236-S; Thermo Fisher Scientific, Waltham, MA, USA), and laminin (rabbit IgG; L9393; Sigma). Secondary AlexaFluor antibodies, all from Invitrogen, were goat α mouse IgM (A-21426), goat α mouse IgG (A-21120), and goat α rabbit IgG (A-11008). Regenerating fibers were identified by staining muscle sections for a developmental isoform of myosin, embryonic myosin heavy chain (eMHC; F1.652; Developmental Studies Hybridoma Bank, Iowa City, IA, USA), and AlexFluor 555 goat anti-mouse IgG1 (Invitrogen) secondary antibody. Wheat germ agglutinin (WGA) lectin AlexaFluor 488 (Invitrogen) was used to identify extracellular matrix. Nuclei were identified using DAPI. Positive controls for regenerating fibers were obtained by injecting GTN muscles of control mice with 1.2% barium chloride to induce degeneration and regeneration. Cross sections were analyzed in a blinded procedure using CyteSeer 2.0.4 (Vala Sciences Inc., San Diego, CA, USA) for fiber CSAs and for fiber number, including the number of fibers expressing eMHC and the number of fibers with central nuclei.
Measurement of dry weight of skeletal muscle
Extensor digitorum longus (EDL), GTN, soleus, tibialis anterior (TA), quadriceps (Quad), and plantaris (PL) muscles were removed from 6-mo-old mice. Wet weight was measured for each muscle, followed by drying the tissue in an oven overnight (24 h). Dry weight was then measured. The ratio between dry and wet weight was calculated and compared by Student's t test between WT and mKO mice. In addition, multiple muscles from 16- to 18-mo-old mKO and WT mice were isolated, trimmed, and weighed to determine whether the CuZnSOD deficiency similarly affected a variety of skeletal muscles.
Statistical analysis
All data are expressed as means ± sem. The difference between WT and mKO mice was analyzed by Student's t test or 1-way ANOVA with post hoc test. A value of P < 0.05 (P<0.05) was considered statistically significant.
RESULTS
Generation of mKO mice
Sod1flox/flox mice were generated using a construct with 2 LoxP sites flanking exon 3 and 4, as described in Materials and Methods and shown in Fig. 1A. The recombination between the Sod1 construct and the WT allele was confirmed by Southern blot analysis using DNA probes corresponding to the 5′ and 3′ ends of the construct (P1 and P2). Positive clones display a 6.8-kb band for P1 with BamHI digestion and 5.5-kb band for P2 with AflII digestion (Fig. 1B). Crossing the Sod1flox/flox mice with mice expressing a skeletal muscle-specific Cre recombinase, Acta1-Cre, produced mice with skeletal muscle-specific deletion of Sod1 gene (designated mKO), which resulted in a >90% decrease of CuZnSOD protein in skeletal muscle of mKO mice, as confirmed by Western blot analysis, but no change in other tissues, including brain, heart, kidney, and liver (Fig. 1C). The 85–95% reduction, compared with WT levels, in CuZnSOD expression was consistent for a wide variety of muscles, including EDL, TA, Quad, PL, and GTN muscles (Fig. 1D). Skeletal muscle-specific deletion of CuZnSOD was further confirmed by enzymatic activity assay (Fig. 1E), which showed < 10% enzymatic activity in mKO mice compared with that measured in WT mice in skeletal muscle only. Moreover, despite the dramatic reduction of CuZnSOD activity in skeletal muscle of mKO mice, the activity of MnSOD, the mitochondrial SOD, is not up-regulated in mKO mice (Fig. 1D). Similarly, protein levels of catalase MnSOD and extracellular SOD are not up-regulated in muscle from mKO mice (Supplemental Fig. S1).
Changes of muscle mass and morphology in mKO mice
Because muscle loss occurs at early ages in the constitutive Sod1−/− mice, we asked whether muscle-specific loss of CuZnSOD would have the same effect. As shown in Fig. 2A, no obvious difference is apparent in muscle morphology between WT and mKO mice. Both the absolute mass and mass relative to body mass ratio for GTN muscle were greater at 6 mo of age for mKO compared with control mice (Fig. 2B). The elevation in muscle mass was not restricted to the GTN muscle but was observed in a variety of muscles (Supplemental Table S2). To probe the basis for the higher GTN muscle masses in mKO mice, we measured the numbers of fibers in cross sections of GTN muscles and determined the average muscle fiber CSA. Although a trend showed an increase in the number of fibers in muscles of mKO mice, the increase did not reach statistical significance (Fig. 2B), and no difference was found in the average fiber CSA between age-matched mKO mice and WT mice (Fig. 2B).
Figure 2.

Muscle morphology in mKO mice. A) Hind-limb muscles from mKO and WT mice (10 mo old). B) Muscle mass expressed in milligrams (top left panel) and as a percentage of body mass (top right panel); number of muscle fibers in a CSA (bottom left panel), and average fiber CSAs for GTN muscles from control mice (solid bars) and mKO mice (shaded bars) at 6–8 mo of age (n=5). *P < 0.05 vs. WT control. C) Details of muscle fiber morphology in representative histological sections stained with hematoxylin and eosin. D) Electron micrographs of longitudinal sections of GTN muscles. E) Immunofluorescent images stained for embryonic myosin heavy chain (red); WGA lectin to visualize extracellular matrix (green), and DAPI to visualize nuclei (blue) in GTN muscles from WT control mice (top left panel), WT mice treated with BaCl2 for 7 d (top right panel), and mKO mice (bottom panels). Black arrows indicate the normal peripheral localization of myonuclei; yellow arrows mark the centrally located nuclei that are frequently seen in mKO muscles.
Histological analysis of GTN muscles revealed that a substantial number of muscle fibers in mKO mice contained centrally located nuclei, while fibers with centrally located nuclei were rarely detectable in WT mice (Fig. 2C). Electron microscopy further revealed altered structure, with muscle fibers in mKO mice showing multiple nuclei in the cytoplasm within a single section (Fig. 2D). In many cases, the central nuclei are localized to small sections of individual myofibers (Fig. 2D). Despite these abnormalities, the overall level of central nucleation remained constant throughout life at ∼26% of fibers in muscles of mKO mice compared with only 2% of fibers in WT mice (Supplemental Fig. S2). To explore whether the presence of central nuclei is indicative of underlying regeneration in the muscles of mKO mice, GTN muscle sections were stained for eMHC (Fig. 2E). Consistent with the lack of central nuclei in WT control muscles, no eMHC-positive fibers were observed in WT mice. In muscles of mKO mice, some isolated regions displayed fibers that both were positive for eMHC and contained central nuclei, but numerous fibers were also present that showed a persistence of the central nuclei without eMHC expression. Overall, this spotty eMHC expression resulted in only 1.1 ± 0.5% of the fibers in the whole GTN cross section that were positive for eMHC (Supplemental Fig. S2). Although more widespread, similar staining patterns were observed in control muscles 7 d following injection of BaCl2, which suggests transient expression of eMHC in regenerating fibers that is resolved while central nuclei remain. In total, these histological changes are consistent with ongoing, but focal, degeneration-regeneration in the muscle from mKO mice.
Muscle function is reduced in mKO mice
Sod1−/− mice show significant reductions in force-generating capacity associated with the loss of muscle mass. Therefore, we were interested to determine whether muscle-specific deletion of Sod1 gene would be sufficient to alter muscle function in the mKO mice. To test this, we measured maximum isometric force generated by GTN muscles in situ in response to nerve stimulation. As shown in Fig. 3A, maximum isometric specific force of GTN muscle was reduced by 20% in mKO mice compared with WT mice. To determine whether the weakness of muscles of mKO mice was due to NMJ dysfunction, contractions were also elicited using direct stimulation of the muscle tissue. Under these circumstances, when the NMJ was bypassed, no change in force was observed for muscles of control or mKO mice (Fig. 3B) compared to force determined by nerve stimulation, indicating no functional denervation in those muscles, as previously observed (4) and confirmed here (Fig. 3B) for Sod1−/− mice.
Figure 3.

Muscle function in mKO mice. A) Maximum isometric specific force (N/cm2), normalized by total fiber CSA, for GTN muscles from control mice (solid bars) and mKO mice (shaded bars) at 6–8 mo of age (n=5). *P < 0.05 vs. WT control. B) Force generated during direct muscle stimulation and nerve stimulation for GTN muscles of control (solid black), mKO (shaded bars), and Sod1−/− (open bars) mice. C) Rota-rod performance in control mice and mKO mice at 6–8 mo of age was measured for 3 consecutive days, with 4 trials/d. Data represent average ± se time (s) that mice remain on the rotating rod before falling off. D) Treadmill performance test. Data from the treadmill performance test show no difference in the duration of time that control (WT, n=6) and mKO (n=5) mice were able to run. E) Force production during a series of repeated isometric contractions, expressed as a percentage of the initial force. Lines represent the average response of 4 muscles form 6- to 8-mo-old mice. Although mKO mice showed a tendency to lose force faster, the data are not significantly different between WT and mKO mice.
To assess the potential effects of the defect in muscle contractility on overall neuromuscular function, we measured rota-rod performance and endurance using treadmill running. Despite the weakness observed for isolated whole GTN, the mKO mice showed no difference in the rota-rod test; i.e., the latency to fall (length of time the mice remain on the rotating rod) was not significantly different between mKO and WT mice (Fig. 3C). Moreover, no impairment was found in treadmill running performance in mKO compared to WT mice (Fig. 3D). Consistent with the similarity in the abilities of mKO and control mice to run, fatigability of isolated GTN muscles as measured by the relative decline in force during repeated isometric contractions was also not different between mKO and WT control groups (Fig. 3E). These findings indicate that the lack of CuZnSOD in the muscle contributes to functional deficits that appear to be localized to skeletal muscle.
Mitochondrial function and ROS generation in skeletal muscle from mKO mice
Previous studies from our group have reported significant alterations in mitochondrial function in skeletal muscle from Sod1−/− mice, including increased ROS generation and reduced ATP production (7). To determine whether muscle-specific loss of the Sod1 gene affects mitochondrial function, we measured ROS generation and ATP production in mitochondria isolated from skeletal muscle of mKO and WT mice. As shown in Fig. 4A, B, we found no change in mitochondrial ROS or ATP production in muscle from mKO mice compared to muscle from control mice, suggesting that mitochondrial function is not compromised in mKO mice. In addition, the level of ROS generation in intact muscle fibers as measured by the rate of change in CM-DCF fluorescence is not different between mKO mice and control mice under resting conditions (Fig. 4C, D).
Loss of CuZnSOD in mKO mice does not affect the redox status in skeletal muscle
CuZnSOD is the primary scavenger of superoxide anion in the cytosolic compartment of the cell. Thus, loss of CuZnSOD would be predicted to have significant effects on oxidative stress and redox balance. Indeed, we and others found significant increases in oxidative damage (lipid, protein, and DNA oxidation) and altered redox markers (reduced glutathione peroxidase activity and increased NFκB activity) in the constitutive Sod1−/− mice (5, 7, 8). To determine the effect of reduced superoxide scavenging potential in response to CuZnSOD gene deletion in the mKO mice, we measured markers of antioxidant status in GTN muscles. The levels of total/oxidized glutathione and protein thiols, indicators of global antioxidant status, were measured. As shown in Fig. 5A, the levels of total glutathione, oxidized glutathione, and total protein thiols were similar in muscles of mKO and WT mice, indicating no change in the overall redox status in mKO mice. Protein nitration, measured by the level of 3-NT in carbonic anhydrase III, was also similar in skeletal muscle from mKO mice and WT mice (Fig. 5B). Similarly, we measured the level of F2-isoprostanes, an indicator of lipid oxidation, and found no difference between mKO mice and WT mice (Fig. 5C).
Figure 5.
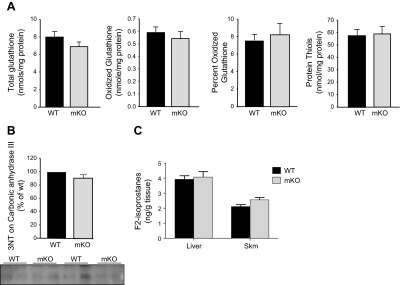
Oxidative damage is not elevated in skeletal muscle of mKO mice. A) Glutathione oxidation and total protein thiol contents in skeletal muscle. B) Nitration of carbonic anhydrase III in skeletal muscle. C) Lipid peroxidation, measured as F2-isoprostanes in skeletal muscle.
Cell signaling and stress responses in skeletal muscle of mKO mice
The changes in muscle fiber morphology and muscle function in the mKO mice implicate potential alterations in some important cellular processes in response to the loss of Sod1 gene. Because the histological analysis suggested a potential increase in newly regenerated fibers, we hypothesized that Akt–mammalian target of rapamycin (mTOR) signaling, a central regulatory pathway for cell growth and proliferation, would be affected by the loss of Sod1 in mKO mice (21). As shown in Fig. 6A, the Akt-mTOR activity was elevated in skeletal muscle of mKO mice, as measured by the phosphorylation of Akt (Ser308) and ribosome subunit S6 (a downstream target of mTOR signaling). Akt-mTOR signaling was not affected in mKO mice in tissues other than skeletal muscle (data not shown].
Figure 6.

Molecular signaling pathways in muscle from mKO mice (n=4/genotype). A) Akt-mTOR signaling. B) NFκB signaling, measured as the nuclear translocation of NFκB p65 subunit. C) Expression of heat-shock proteins. All data are expressed as the percentage of WT control mice. *P < 0.05 vs. WT.
We had previously shown an elevated response in heat-shock and NFκB signaling pathways in Sod1−/− mice (5). We hypothesized that the expression of genes involved in the adaptive response to stress in skeletal muscle may be altered in the mKO mice. As shown in Fig. 6C, the amount of nuclear p65 in skeletal muscle is similar between mKO mice and WT mice, suggesting no up-regulation of NFκB signaling in muscles of mKO mice. Similarly, the protein expression of Hsc70, Hsp60, and Hsp25 remains the same in the muscle of mKO mice as WT mice, indicating the loss of Sod1 gene in skeletal muscle did not activate the heat-shock response (Fig. 6B).
Maintenance of greater muscle mass into late life in mKO mice
Lastly, we tested whether overall muscle structure and function changed dramatically with aging in mKO mice. GTN muscle mass and isometric force generation was measured in very young (3–4 mo), adult (6–8 mo), and middle-aged (16–17 mo) mKO and control mice. We found no loss of muscle mass in the mKO mice throughout the period studied (Fig. 7A). In fact, the mass of the GTN muscles increases with age for mKO mice and remains elevated compared with muscles of control mice out to at least 16–17 mo of age, the oldest age studied thus far. In addition, one might hypothesize that ongoing degeneration and regeneration in the muscles of mKO mice would lead to deterioration of force-generating capability. Although muscles of mKO mice were weaker than those of control mice at all ages studied, force did not decline for either group for the duration of the present study (Fig. 7B).
Figure 7.

Age-related changes of GTN muscle mass and maximum isometric force in mKO and WT mice. A) GTN muscle mass. B) Maximum isometric specific force. Data are presented as means ± sem; n = 5. *P < 0.05 vs. WT.
DISCUSSION
Oxidative stress has long been implicated as a significant contributing factor to age-related loss of muscle mass. A causal connection is supported by our previous work showing elevated levels of oxidative stress and accelerated muscle atrophy in mice lacking the superoxide scavenging enzyme CuZnSOD (Sod1−/− mice). The characteristics of the muscle loss in Sod1−/− mice closely mimics the changes that occur in human sarcopenia (22, 23), as evidenced by the observations in these mice of degeneration of neuromuscular junctions, loss of innervation, and fiber degeneration in the muscle itself (5, 6). Although these findings indicate that changes in both motor neurons and skeletal muscle are associated with the pathophysiology of sarcopenia, the relative role of each of these tissues or the different cell types involved in the initiation or progression of sarcopenia has not been clearly defined. Our aim was to address the question of whether the process of age-associated muscle atrophy is due to autonomous effects in the muscle itself, as opposed to events in the motor neuron that influence the skeletal muscle to degenerate. Specifically, we asked whether the loss of CuZnSOD restricted to muscle fibers would have an effect on atrophy similar to that seen in the constitutive whole-body knockout mouse, in which muscles and neurons as well as other tissues and cell types are affected. Our approach was to generate a mouse model in which the Sod1 gene was deleted specifically in skeletal muscle using a muscle specific Cre-lox construct to address the tissue specific effect of ROS on the development of muscle atrophy (24). Using this model, we investigated the effect of a muscle specific Sod1 gene deletion (mKO) on muscle structure and function. The major findings of this study are the following. First, in contrast to the muscle atrophy observed in Sod1−/− mice as early as 3 mo of age, mKO mice displayed maintenance or even slightly elevated muscle mass out to 17 mo of age. Second, similar to our previous findings in Sod1−/− mice, muscles from mKO mice showed functional deficits measured as reduced isometric specific force of isolated GTN muscles, although these reductions in muscle function were not as severe as those observed for Sod1−/− mice. Finally, a novel and unexpected result was that the muscle-specific deficiency of CuZnSOD resulted in the appearance of a degenerative-regenerative phenotype, with many fibers in the muscles of mKO mice exhibiting centrally located nuclei, as well as evidence for fibers expressing eMHC through the life span of the mice.
Our observation that muscle atrophy was not observed in mKO mice supports the conclusion that compromised detoxification of superoxide anion within muscle fibers per se does not represent a primary factor initiating age-associated sarcopenia. The lack of CuZnSOD would be predicted to lead to an increase in superoxide anion and/or peroxynitrite, oxidative damage, and altered redox status, as we found in tissues from the Sod1−/− mice. Surprisingly, we found no evidence of increased oxidative damage to lipid or protein in the mKO mice. Moreover, GSH/GSSG ratios in muscle were unchanged in response to CuZnSOD deficiency and skeletal muscle mitochondrial ROS generation, and ROS levels were not elevated in isolated muscle fibers of the mKO mice. The seemingly minimal effect of the lack of CuZnSOD on oxidative damage in skeletal muscle fibers is surprising due to the observations that oxidative damage is dramatically elevated in many tissues, including muscle, in Sod1−/− mice even at very young ages (7). It is possible that the lack of CuZnSOD in skeletal muscle is not critical except under conditions of stress. In future studies, we will measure oxidative damage post exercise in vivo or after induced contraction in situ. Furthermore, although we find no overt oxidative damage in muscles of mKO compared with WT mice, oxidative modification of specific proteins or small localized transient changes in redox homeostasis may occur (25). In addition, subtle changes of ROS due to the loss of the Sod1 gene could result in the changes observed in molecular signaling and physical function (26).
The finding that a reduction in antioxidant scavenging potential in muscle was not sufficient to drive muscle atrophy is consistent with previous findings from our group showing that deletion of mitochondrial SOD, MnSOD, specifically in type IIB muscle fibers, increased oxidative stress but did not reduce muscle mass (13). Despite the lack of major alterations in oxidative stress in muscles of mKO mice, our findings indicate that the muscle atrophy observed during aging of Sod1−/− mice is not due primarily to deficiency of CuZnSOD in skeletal muscle, suggesting that a failure in redox homeostasis in motor neurons may be the primary initiating factor.
While muscle-specific deficiency of CuZnSOD does not recapitulate the accelerated muscle atrophy observed in Sod1−/− mice, muscles of both Sod1−/− (4) and mKO (present study) mice demonstrate muscle weakness, as measured by deficits in maximum specific isometric force. The observation that direct stimulation of the muscles of Sod1−/− mice consistently increased force production over that generated using nerve stimulation (4) indicates that the muscles of Sod1−/− mice contain a population of fibers that can produce force but that fails to contract in response to nerve stimulation, possibly due to faulty transmission at the neuromuscular junction. In contrast, direct muscle stimulation failed to rescue any of the weakness of GTN muscles in mKO mice, indicating that the weakness of these muscles is intrinsic to the muscle fibers. Direct effects of oxidants on the myofibrillar apparatus that influence force generation have been reported (27, 28). Oxidative modifications of specific contractile proteins may disrupt actin-myosin interactions such that the number of myosin cross-bridges strongly bound to actin is reduced, thereby reducing force-generating capacity of individual muscle fibers (29). The likelihood that the reduced specific force of muscles in mKO mice is due to oxidation of specific myofibrillar proteins appears low, however, based on the observations that the contractile proteins within muscle fibers of Sod1−/− mice do not suffer irreversible damage as a result of chronic exposure to high oxidative stress (30).
The presence of a population of regenerating fibers with reduced force-generating capability could also account for the weakness observed for the muscles of mKO mice. Force production by developmental isoforms of myosin is lower than that of adult isoforms (31, 32). Thus, the presence of fibers expressing eMHC is expected to contribute to weakness at the whole-muscle level. While fibers expressing eMHC produce less force than mature fibers, the small percentage of fibers expressing eMHC at any given time is insufficient to explain any significant portion of the whole muscle weakness. In WT mice, nuclei are typically restricted to the periphery of mature healthy fibers. Centrally located nuclei are associated with development and in rodent muscles represent a hallmark of muscle degeneration-regeneration (33), and the persistence of a sizeable number of fibers with central nuclei in muscles of mKO mice suggests that incomplete maturation of fibers cannot be ruled out as a contributor to the weakness. The presence of fibers with central nuclei in the mKO mice is also consistent with the possibility that Sod1 deficiency may result in increased vulnerability of muscle fibers to damage such that fibers are injured during normal activities that rarely damage WT muscles. Finally, additional histological abnormalities were observed in the muscles of mKO mice, such as cellular infiltrate and increased extracellular matrix and the presence of noncontractile material could also reduce specific force, but the extent of the changes were minor and not likely important contributors to whole-muscle weakness.
The greater muscle mass observed for mKO compared with control WT mice indicates increased fiber number, greater individual fiber sizes, the accumulation of nonmuscle cells or extracellular tissue, or some combination of these possibilities. Although the size and complex architecture of the GTN muscle preclude definitive conclusions regarding fiber numbers and CSAs based on histological analysis, our findings provide no support for increased fiber areas, i.e., hypertrophy. The lack of evidence for fiber hypertrophy is somewhat surprising in light of our observation of increased levels in muscles of mKO mice of phosphorylated Akt and S6 (34), but Akt-mTOR signaling affects numerous cellular functions (21). Among the processes regulated by Akt is cell proliferation. Therefore, based on the appearance of fibers with central nuclei, we speculate an increased satellite cell activation and proliferation in muscles of mKO may be underlying the signaling changes detected here. Note that the previous studies suggest that Acta-Cre is likely not expressed in satellite cells (35, 36), so we expect no direct impact on oxidative stress in this cell population and no direct impact on regenerative capacity. We might further speculate that the persistence of central nuclei reflects a defect in late-stage regeneration or muscle fiber maturation, such as the migration of nuclei to the periphery of the myofibers. Our finding of a trend toward an increase in mKO mice of the number of fibers in GTN muscles warrants further study to determine whether muscle-specific deficiency of Sod1 affects muscle development or may result in postnatal hyperplasia; i.e., de novo generation of new muscle fibers. An increase in the number of fibers appearing in a cross section could also be a result of fiber branching that would also contribute to the reduction in specific force (37).
In summary, our results clearly show that the loss of CuZnSOD in the muscle alone is not sufficient to initiate muscle atrophy. These findings indicate that the premature age-associated NMJ degeneration and muscle fiber loss observed in CuZnSOD-deficient mice are likely to be initiated by alterations in redox homeostasis in the motor neurons or, perhaps, other cell types. While muscles of mKO mice do not display muscle atrophy, the lack of CuZnSOD specifically in muscle resulted in reduced force-generating capacity, as previously reported for muscles of CuZnSOD-deficient mice. A novel and unexpected result of the muscle-specific deletion of Sod1 is the presence of extensive central nucleation that may result from increased regenerative activity in the muscles of mKO mice. Alternatively, Sod1 may play a role in signaling for the migration of nuclei to the periphery of muscle fibers, a process for which the mechanisms are essentially unknown.
Supplementary Material
Acknowledgments
The authors thank Jose Gomez and Anne Erickson for technical assistance.
The work was supported by U.S. National Institute on Aging grant AG-020591. The F1.652 antibody was obtained from the Developmental Studies Hybridoma Bank, developed under the auspices of the U.S. National Institute of Child Health and Human Development and maintained by The University of Iowa, Department of Biology (Iowa City, IA, USA).
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- 3-NT
- 3-nitrotyrosine
- Acta1
- actin α1
- CM-DCFH DA
- 5-(and 6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate
- CSA
- cross-sectional area
- CuZnSOD
- copper zinc superoxide dismutase
- D-PBS
- Dulbecco's phosphate-buffered saline
- EDL
- extensor digitorum longus
- eMHC
- embryonic myosin heavy chain
- FBS
- fetal bovine serum
- FDB
- flexor digitorum brevis
- GTN
- gastrocnemius
- MEM
- minimum essential medium
- HRP
- horseradish peroxidase
- mKO
- muscle-specific Sod1 knockout
- MnSOD
- manganese superoxide dismutase
- mTOR
- mammalian target of rapamycin
- NMJ
- neuromuscular junction
- PL
- plantaris
- Quad
- quadriceps
- ROS
- reactive oxygen species
- SOD
- superoxide dismutase
- Sod1−/−Flox
- conditional Sod1 knockout
- TA
- tibialis anterior
- WGA
- wheat germ agglutinin
REFERENCES
- 1. Marcell T. J. (2003) Sarcopenia: causes, consequences, and preventions. J. Gerontol. A Biol. Sci. Med. Sci. 58, M911–M16 [DOI] [PubMed] [Google Scholar]
- 2. Barker T., Traber M. G. (2007) From animals to humans: evidence linking oxidative stress as a causative factor in muscle atrophy. J. Physiol. 583, 421–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sakellariou G. K., Pye D., Vasilaki A., Zibrik L., Palomero J., Kabayo T., McArdle F., Van Remmen H., Richardson A., Tidball J. G., McArdle A., Jackson M. J. (2011) Role of superoxide-nitric oxide interactions in the accelerated age-related loss of muscle mass in mice lacking Cu,Zn superoxide dismutase. Aging Cell 10, 749–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Larkin L. M., Davis C. S., Sims-Robinson C., Kostrominova T. Y., Remmen H. V., Richardson A., Feldman E. L., Brooks S. V. (2011) Skeletal muscle weakness due to deficiency of CuZn-superoxide dismutase is associated with loss of functional innervation. Am. J. Physiol. Regul. Integ. Comp. Physiol. 301, R1400–R407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vasilaki A., van der Meulen J. H., Larkin L., Harrison D. C., Pearson T., Van Remmen H., Richardson A., Brooks S. V., Jackson M. J., McArdle A. (2010) The age-related failure of adaptive responses to contractile activity in skeletal muscle is mimicked in young mice by deletion of Cu,Zn superoxide dismutase. Aging Cell 9, 979–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jang Y. C., Lustgarten M. S., Liu Y., Muller F. L., Bhattacharya A., Liang H., Salmon A. B., Brooks S. V., Larkin L., Hayworth C. R., Richardson A., Van Remmen H. (2010) Increased superoxide in vivo accelerates age-associated muscle atrophy through mitochondrial dysfunction and neuromuscular junction degeneration. FASEB J. 24, 1376–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Muller F. L., Song W., Liu Y., Chaudhuri A., Pieke-Dahl S., Strong R., Huang T. T., Epstein C. J., Roberts L. J., 2nd, Csete M., Faulkner J. A., Van Remmen H. (2006) Absence of CuZn superoxide dismutase leads to elevated oxidative stress and acceleration of age-dependent skeletal muscle atrophy. Free Radic. Biol. Med. 40, 1993–2004 [DOI] [PubMed] [Google Scholar]
- 8. Elchuri S., Oberley T. D., Qi W., Eisenstein R. S., Jackson Roberts L., Van Remmen H., Epstein C. J., Huang T. T. (2005) CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene 24, 367–380 [DOI] [PubMed] [Google Scholar]
- 9. McFadden S. L., Ding D., Burkard R. F., Jiang H., Reaume A. G., Flood D. G., Salvi R. J. (1999) Cu/Zn SOD deficiency potentiates hearing loss and cochlear pathology in aged 129,CD-1 mice. J. Comp. Neurol. 413, 101–112 [PubMed] [Google Scholar]
- 10. Zong Y., Zhang B., Gu S., Lee K., Zhou J., Yao G., Figueiredo D., Perry K., Mei L., Jin R. (2012) Structural basis of agrin-LRP4-MuSK signaling. Genes Dev. 26, 247–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Van Remmen H., Salvador C., Yang H., Huang T. T., Epstein C. J., Richardson A. (1999) Characterization of the antioxidant status of the heterozygous manganese superoxide dismutase knockout mouse. Arch. Biochem. Biophys. 363, 91–97 [DOI] [PubMed] [Google Scholar]
- 12. Muller F. L., Liu Y., Jernigan A., Borchelt D., Richardson A., Van Remmen H. (2008) MnSOD deficiency has a differential effect on disease progression in two different ALS mutant mouse models. Muscle Nerve 38, 1173–1183 [DOI] [PubMed] [Google Scholar]
- 13. Lustgarten M. S., Jang Y. C., Liu Y., Muller F. L., Qi W., Steinhelper M., Brooks S. V., Larkin L., Shimizu T., Shirasawa T., McManus L. M., Bhattacharya A., Richardson A., Van Remmen H. (2009) Conditional knockout of Mn-SOD targeted to type IIB skeletal muscle fibers increases oxidative stress and is sufficient to alter aerobic exercise capacity. Am. J. Physiol. Cell Physiol. 297, C1520–C532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shefer G., Yablonka-Reuveni Z. (2005) Isolation and culture of skeletal muscle myofibers as a means to analyze satellite cells. Methods Mol. Biol. 290, 281–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Palomero J., Pye D., Kabayo T., Spiller D. G., Jackson M. J. (2008) In situ detection and measurement of intracellular reactive oxygen species in single isolated mature skeletal muscle fibers by real time fluorescence microscopy. Antioxid. Redox Signal.10, 1463–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McArdle A., Pattwell D., Vasilaki A., Griffiths R. D., Jackson M. J. (2001) Contractile activity-induced oxidative stress: cellular origin and adaptive responses. Amer. J. Physiol. Cell Physiol. 280, C621–627 [DOI] [PubMed] [Google Scholar]
- 17. Morrow J. D., Roberts L. J., 2nd, (1999) Mass spectrometric quantification of F2-isoprostanes in biological fluids and tissues as measure of oxidant stress. Methods Enzymol. 300, 3–12 [DOI] [PubMed] [Google Scholar]
- 18. Ward W. F., Qi W., Van Remmen H., Zackert W. E., Roberts L. J., 2nd, Richardson A. (2005) Effects of age and caloric restriction on lipid peroxidation: measurement of oxidative stress by F2-isoprostane levels. J. Gerontol. A Biol. Sci. Med. Sci. 60, 847–851 [DOI] [PubMed] [Google Scholar]
- 19. Anderson M. E. (1985) Determination of glutathione and glutathione disulfide in biological samples. Methods Enzymol. 113, 548–555 [DOI] [PubMed] [Google Scholar]
- 20. Di Monte D., Ross D., Bellomo G., Eklow L., Orrenius S. (1984) Alterations in intracellular thiol homeostasis during the metabolism of menadione by isolated rat hepatocytes. Arch. Biochem. Biophys. 235, 334–342 [DOI] [PubMed] [Google Scholar]
- 21. Weichhart T. (2012) Mammalian target of rapamycin: a signaling kinase for every aspect of cellular life. Methods Mol. Biol. 821, 1–14 [DOI] [PubMed] [Google Scholar]
- 22. Campbell M. J., McComas A. J., Petito F. (1973) Physiological changes in ageing muscles. J. Neurol. Neurosurg. Psychiat. 36, 174–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Doherty T. J., Vandervoort A. A., Brown W. F. (1993) Effects of ageing on the motor unit: a brief review. Can. J. Appl. Physiol. 18, 331–358 [DOI] [PubMed] [Google Scholar]
- 24. Fulle S., Protasi F., Di Tano G., Pietrangelo T., Beltramin A., Boncompagni S., Vecchiet L., Fano G. (2004) The contribution of reactive oxygen species to sarcopenia and muscle ageing. Exper. Gerontol. 39, 17–24 [DOI] [PubMed] [Google Scholar]
- 25. Wu R. F., Terada L. S. (2010) Focal oxidant and Ras signaling on the ER surface activates autophagy. Autophagy 6, 828–829 [DOI] [PubMed] [Google Scholar]
- 26. Martindale J. L., Holbrook N. J. (2002) Cellular response to oxidative stress: signaling for suicide and survival. J. Cell. Physiol. 192, 1–15 [DOI] [PubMed] [Google Scholar]
- 27. Plant D. R., Lynch G. S. (2001) Rigor force responses of permeabilized fibres from fast and slow skeletal muscles of aged rats. Clin. Exp. Pharmacol. Physiol. 28, 779–781 [DOI] [PubMed] [Google Scholar]
- 28. Andrade F. H., Reid M. B., Allen D. G., Westerblad H. (1998) Effect of hydrogen peroxide and dithiothreitol on contractile function of single skeletal muscle fibres from the mouse. J. Physiol. 509(Pt.2), 565–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lowe D. A., Surek J. T., Thomas D. D., Thompson L. V. (2001) Electron paramagnetic resonance reveals age-related myosin structural changes in rat skeletal muscle fibers. Amer. J. Physiol. Cell Physiol. 280, C540–547 [DOI] [PubMed] [Google Scholar]
- 30. Larkin L. M., Hanes M. C., Kayupov E., Claflin D. R., Faulkner J. A., Brooks S. V. (2012) Weakness of whole muscles in mice deficient in Cu, Zn superoxide dismutase is not explained by defects at the level of the contractile apparatus. [E-pub ahead of print] Age (Dordr.) 10.1007/s11357-012-9441-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Geiger P. C., Cody M. J., Macken R. L., Bayrd M. E., Fang Y. H., Sieck G. C. (2001) Mechanisms underlying increased force generation by rat diaphragm muscle fibers during development. J. Appl. Physiol. 90, 380–388 [DOI] [PubMed] [Google Scholar]
- 32. Johnson B. D., Wilson L. E., Zhan W. Z., Watchko J. F., Daood M. J., Sieck G. C. (1994) Contractile properties of the developing diaphragm correlate with myosin heavy chain phenotype. J. Appl. Physiol. 77, 481–487 [DOI] [PubMed] [Google Scholar]
- 33. Ciciliot S., Schiaffino S. (2010) Regeneration of mammalian skeletal muscle. Basic mechanisms and clinical implications. Curr. Pharm. Des. 16, 906–914 [DOI] [PubMed] [Google Scholar]
- 34. Leger B., Cartoni R., Praz M., Lamon S., Deriaz O., Crettenand A., Gobelet C., Rohmer P., Konzelmann M., Luthi F., Russell A. P. (2006) Akt signalling through GSK-3beta, mTOR and Foxo1 is involved in human skeletal muscle hypertrophy and atrophy. J. Physiol. 576, 923–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stern-Straeter J., Bonaterra G. A., Hormann K., Kinscherf R., Goessler U. R. (2009) Identification of valid reference genes during the differentiation of human myoblasts. BMC Mol. Biol. 10, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Baroffio A., Hamann M., Bernheim L., Bochaton-Piallat M. L., Gabbiani G., Bader C. R. (1996) Identification of self-renewing myoblasts in the progeny of single human muscle satellite cells. Differentiation 60, 47–57 [DOI] [PubMed] [Google Scholar]
- 37. Chan S., Head S. I., Morley J. W. (2007) Branched fibers in dystrophic mdx muscle are associated with a loss of force following lengthening contractions. Amer. J. Physiol. Cell Physiol. 293, C985–C992 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.