

Abstract
The functional significance of the molecular swivel at the head-to-tail overlapping ends of contiguous tropomyosin (Tm) dimers in striated muscle is unknown. Contractile measurements were made in muscle fibers from transgenic (TG) mouse hearts that expressed a mutant α-Tm (TmH276N). We also reconstituted mouse cardiac troponin T (McTnT) N-terminal deletion mutants, McTnT1–44Δ and McTnT45–74Δ, into muscle fibers from TmH276N. For controls, we used the wild-type (WT) McTnT because altered effects could be correlated with the mutant forms of McTnT. TmH276N slowed crossbridge (XB) detachment rate (g) by 19%. McTnT1–44Δ attenuated Ca2+-activated maximal tension against TmWT (36%) and TmH276N (38%), but sped g only against TmH276N by 35%. The rate of tension redevelopment decreased (17%) only in McTnT1–44Δ + TmH276N fibers. McTnT45–74Δ attenuated tension (19%) and myofilament Ca2+ sensitivity (pCa50=5.93 vs. 6.00 in the control fibers) against TmH276N, but not against TmWT background. Thus, altered XB cycling kinetics decreased the fraction of strongly bound XBs in McTnT1–44Δ + TmH276N fibers, whereas diminished thin-filament cooperativity attenuated tension in McTnT45–74Δ + TmH276N fibers. In summary, our study is the first to show that the interplay between the N terminus of cTnT and the overlapping ends of contiguous Tm effectuates different states of Tm on the actin filament. —Mamidi, R., Michael, J. J., Muthuchamy, M., Chandra, M. Interplay between the overlapping ends of tropomyosin and the N terminus of cardiac troponin T affects tropomyosin states on actin.
Keywords: myofilament Ca2+ sensitivity, thin-filament activation, calcium-activated tension, molecular swivel, myofilament cooperativity
Cardinal to striated muscle contraction model is the activation- and deactivation-dependent cooperative movement of tropomyosin (Tm) on the actin-containing filaments (1–4). The thin-filament-based cooperative mechanisms are strongly dependent on two key contractile regulatory proteins of the thin filament: Tm and troponin (Tn). The coiled-coil helical dimers of Tm make weak electrostatic contacts with the actin filament (5, 6) and bind to each other in a head-to-tail manner to form a continuous flexible filamentous structure on the actin filament (7–9). The N terminus of cardiac troponin T (cTnT) is not only important for the head-to-tail polymerization of Tm (9, 10), but is also vital for creating a molecular swivel that includes an asymmetric 4-helix bundle at the overlapping junction of two contiguous Tm dimers (11).
It is now well established that the movement of Tm to discrete locations on the actin filament is associated with diverse states of Tm during activation and deactivation of the thin filament (2, 12–14). Although the crystal structure of Tm and chicken fast skeletal TnT suggests a strong link between the molecular swivel structure at the overlapping ends of Tm and the facilitation of Tm movement on the actin filament, the N terminus of cTnT poses new challenges to our understanding of Tm structure and function. This is because cTnT contains an extended N terminus rich in acidic amino acids (15) and because its role in modulating different states of Tm has not been studied before. The physiological role of the N terminus of cTnT takes on new significance because, in the fully relaxed state, the αα-Tm dimer of the cardiac muscle is confined to the outer domain of cardiac actin filaments, whereas the αα-, αβ-, or ββ-Tm dimer of the fast skeletal muscle is confined to the inner domain of fast skeletal muscle actin filaments (2). The location of Tm on the outer domain of actin corresponds to the blocked state of the McKillop-Geeves model (16), whereas the location of Tm on the inner domain of actin corresponds to the closed state of the McKillop-Geeves model. cTnT also imparts unique cardiac-specific effects on the cardiac thin filament because the N terminus of cTnT modulates the blocked- to closed-state transition of the thin filament via its effect on allosteric/cooperative mechanisms (17). Thus, in the relaxed state, cTnT has an intrinsic ability to stabilize the cardiac thin filament in the blocked state (17, 18), suggesting that cTnT may further increase the energy barrier between the blocked- to closed-state transition of the cardiac thin filament. Interestingly, in contrast to cTnT, fast skeletal muscle TnT significantly stabilizes the thin filament in the closed state (19). Inferences drawn from the above studies suggest that, in the fully relaxed state, the TnT moiety has a divergent effect on the location of Tm on the actin filament in cardiac and fast skeletal muscle. A direct consequence of this divergent effect of the TnT moiety is that the Tm filament is relatively more close to activation in the fast skeletal muscle than in the cardiac muscle. Collectively, these observations imply that the energy barrier between different states of Tm in cardiac and fast skeletal muscle is not the same (2); such differences have significant implications not only for the disparate effects of Tm/TnT isoforms on the kinetic rates, but also on the magnitude of thin-filament activation.
We have recently demonstrated that two different N-terminal regions of cTnT show differential thin-filament activation under the wild-type (WT) α-Tm (TmWT) background and that these regions of cTnT exhibit divergent effects under the β-Tm background (20). In this study, we hypothesized that the intermolecular interplay between the N-terminal end of cTnT and the head-to-tail overlapping region of two contiguous Tms effectuates different states of Tm on the actin filament. Since different states of Tm on actin are strongly correlated to varied levels of thin-filament activation, altered function could be ascribed to different states of Tm. To test our hypothesis, we used cardiac muscle fibers from the transgenic (TG) mouse that expressed a mutant form of Tm (TmH276N) in the heart. The H276N mutation in Tm has been shown to destabilize the head-to-tail overlapping ends of Tm, leading to an attenuation of the rate of ventricular relaxation (21). Furthermore, we reconstituted the following mouse cTnT (McTnT) N-terminal deletion mutants into cardiac muscle fibers from TmH276N TG mice: McTnT 1–44 deletion (McTnT1–44Δ), which is known to attenuate Ca2+-activated maximal tension without altering crossbridge (XB) cycling kinetics (20); and McTnT 45–74 deletion (McTnT45–74Δ), which is known to attenuate thin-filament cooperativity without affecting Ca2+-activated maximal tension (20).
Simultaneous isometric tension and ATPase activity, myofilament Ca2+ sensitivity, and the rate of tension redevelopment were measured in detergent-skinned cardiac muscle fiber bundles. Our observations demonstrate that TmWT and TmH276N have divergent effects on how McTnT1–44Δ and McTnT45–74Δ attenuate Ca2+- activated maximal tension. Specifically, altered XB cycling kinetics attenuated Ca2+-activated maximal tension in McTnT1–44Δ + TmH276N reconstituted fibers, whereas the decreased thin-filament cooperativity attenuated Ca2+-activated maximal tension in McTnT45–74Δ + TmH276N reconstituted fibers. TmH276N decreased the rate of XB detachment, whereas TmH276N + McTnT1–44Δ increased the rate of XB detachment.
Our observations provide the first explicit evidence for the emergence of different states of Tm on the actin filament, engendered by a significant interplay between the N terminus of cTnT and the overlapping ends of two contiguous Tm dimers.
MATERIALS AND METHODS
Ethical approval and animal treatment protocols
The TG mice (FVB/N strain) expressing ∼82% of TmH276N in the myocardium were previously generated and well characterized (21). Nontransgenic (NTG) TmWT mice (FVB/N strain) containing the endogenous WT Tm in the myocardium were used as controls. All animals used in this study received adequate care and humane treatment according to the protocols approved by the Washington State University Institutional Animal Care and Use Committee.
Preparation of detergent-skinned cardiac muscle fiber bundles
Isoflurane was used to deeply anesthetize mice, and the depth of the anesthesia was evaluated by a lack of pedal withdrawal reflex (22, 23). Hearts were then quickly removed and transferred to an ice-cold high-relaxing (HR) solution containing 20 mM 2,3-butanedione monoxime (BDM), 50 mM N,N-bis-(2-hydroxyethyl)-2-amino-ethane-sulfonic acid (BES), 20 mM EGTA, 6.29 mM MgCl2, 6.09 mM Na2ATP, 30.83 mM potassium propionate, 10 mM sodium azide, 1.0 mM DTT, and 4 mM benzamidine-HCl. The pH of the solution was adjusted to 7.0 with KOH. pCa (pCa = −log [Ca2+]free) of the HR solution was 9.0. A fresh cocktail of protease inhibitors (5 μM bestatin, 2 μM E-64, 10 μM leupeptin, 1 μM pepstatin, and 200 μM PMSF) was added to the HR solution. Papillary muscle bundles, isolated from the left ventricles of the hearts, were further dissected into smaller fibers of ∼0.15 mm in width and 2 mm in length. Fibers were chemically skinned overnight at 4°C in HR solution containing 1% Triton X-100.
Expression and purification of recombinant mouse cardiac Tn subunits
Recombinant c-myc-tagged McTnT, McTnT1–44Δ, McTnT45–74Δ, mouse cardiac TnI (McTnI), and mouse cardiac TnC (McTnC) proteins, all cloned into pSBETa vector, were expressed in BL21*DE3 cells (Novagen, Madison, WI, USA) and purified as described earlier (20, 23). Purity of the protein fractions was confirmed using a 12.5% SDS gel. Pure protein fractions were then pooled and dialyzed extensively against deionized water containing 15 mM β-mercaptoethanol, lyophilized, and stored at −80°C.
Reconstitution of recombinant mouse cardiac Tn subunits into detergent-skinned mouse cardiac muscle fibers
Reconstitution of recombinant Tn subunits into chemically skinned muscle fibers was performed as described earlier (22, 23). cmyc-tagged WT McTnT (McTnTWT) protein was used as the control. The cmyc-tag was added only to McTnTWT, but not to McTnT1–44Δ and McTnT45–74Δ proteins. This is because the extent of incorporation of TnT mutants was easily visualized based on differential mobility of the deletion mutants when compared to the endogenous TnT (Fig. 1). We have previously shown that the c-myc tag at the N terminus of McTnTWT does not affect the normal cardiac function (24, 25). Replacement of endogenous Tn subunits was performed using an extraction solution containing McTnT (McTnTWT, McTnT1–44Δ, or McTnT45–74Δ; 0.9 mg/ml, w/v) and McTnI (1.0 mg/ml, w/v), according to a protocol described previously (23). The McTnT-McTnI-treated fibers were then incubated with McTnC (3.0 mg/ml, w/v) overnight at 4°C. NTG TmWT mouse cardiac fibers reconstituted with McTnTWT + McTnI + McTnC are referred to as McTnTWT fibers; NTG TmWT mouse cardiac fibers reconstituted with McTnT1–44Δ + McTnI + McTnC are referred to as McTnT1–44Δ fibers; and NTG TmWT mouse cardiac fibers reconstituted with McTnT45–74Δ + McTnI + McTnC are referred to as McTnT45–74Δ fibers. TG TmH276N mouse cardiac muscle fibers reconstituted with McTnTWT + McTnI + McTnC are referred to as McTnTWT + TmH276N fibers; TG TmH276N mouse cardiac muscle fibers reconstituted with McTnT1–44Δ + McTnI + McTnC are referred to as McTnT1–44Δ + TmH276N fibers; and TG TmH276N mouse cardiac muscle fibers reconstituted with McTnT45–74Δ + McTnI + McTnC are referred to as McTnT45–74Δ + TmH276N fibers.
Figure 1.
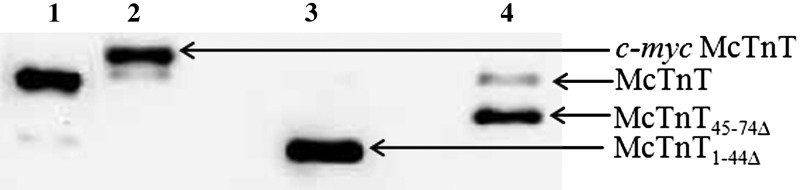
Western blot analysis of the reconstituted fibers to estimate the level of incorporation of McTnT proteins. Reconstituted fibers containing TmWT background were solubilized using 2% SDS (26). Solubilized fiber samples were run on a 10% SDS gel and were then transferred onto a PVDF membrane for Western blot analysis. Lane 1 shows purified recombinant McTnT protein. Lanes 2–4 show samples from McTnTWT, McTnT1–44Δ, and McTnT45–74Δ reconstituted fiber samples, respectively.
For SDS-PAGE analysis, reconstituted muscle fibers were solubilized and resuspended in 2% SDS solution (10 μl/fiber) as described previously (26). SDS-solubilized fibers were mixed with an equal volume of protein loading dye that contained 125 mM Tris-HCl (pH 6.8), 20% glycerol, 2% SDS, 0.01% bromphenol blue, and 50 mM β-mercaptoethanol. Equal quantities of protein (∼2 μg of total protein) from solubilized samples were loaded and separated by SDS-PAGE (4% acrylamide stacking gel and 10% acrylamide separating gel). Proteins were transferred onto a PVDF membrane for Western blot analysis using a Trans-Blot Turbo Transfer System (Bio-Rad Laboratories Inc., Hercules, CA, USA). The incorporation of McTnTWT, McTnT1–44Δ, and McTnT45–74Δ was assessed using a monoclonal anti-TnT primary antibody (clone JLT-12; Sigma-Aldrich, St. Louis, MO, USA), followed by a HRP-labeled anti-mouse secondary antibody (Amersham Biosciences, Pittsburgh, PA, USA). Densitometric scanning of Western blots was done using ImageJ software (U.S. National Institutes of Health, Bethesda, MD, USA; obtained at http://rsbweb.nih.gov/ij/).
Measurement of steady-state isometric tension and ATPase activity in reconstituted cardiac muscle fibers
T-shaped aluminum clips were used to attach the muscle fiber between a motor arm (322C; Aurora Scientific Inc., Aurora, ON, Canada) and a force transducer (AE 801; Sensor One Technologies Corp., Sausalito, CA, USA). The sarcomere length (SL) of the muscle fibers was set to 2.3 μm under relaxing conditions (pCa 9.0) using a laser light diffraction pattern (22, 27, 28). After 2 cycles of maximal activation and relaxation, the SL was readjusted if needed. The muscle fiber was then immersed in a constantly stirred chamber containing Ca2+ solutions of concentrations ranging from pCa 4.3 to 9.0. The composition of various pCa solutions was calculated using methods described earlier (29). The composition of the maximal Ca2+ activation solution (pCa 4.3) was 31 mM potassium propionate, 5.95 mM Na2ATP, 6.61 mM MgCl2, 10 mM EGTA, 10.11 mM CaCl2, 50 mM BES, 5 mM sodium azide, and 10 mM phosphoenolpyruvate (PEP). The composition of the relaxing solution (pCa 9.0) was 51.14 mM potassium propionate, 5.83 mM Na2ATP, 6.87 mM MgCl2, 10 mM EGTA, 0.024 mM CaCl2, 50 mM BES, 5 mM NaN3, and 10 mM PEP. The activation and relaxing solutions also contained 0.5 mg/ml pyruvate kinase (500 U/mg), 0.05 mg/ml lactate dehydrogenase (870 U/mg), 20 μM diadenosine pentaphosphate, 10 μM oligomycin, and a cocktail of protease inhibitors. The pH of the Ca2+ solutions was adjusted to 7.0 using KOH, and the ionic strength was 180 mM. Isometric steady-state tensions were digitally recorded on a computer.
Steady-state isometric ATPase activity was measured according to a protocol described earlier (22, 30). Briefly, near-UV light was passed through the muscle chamber, split (50:50) via a beam splitter, and detected at 340 nm (sensitive to changes in NADH) and 400 nm (insensitive to changes in NADH). ATPase activity was measured using an enzyme-linked assay. ATP regeneration from ADP was coupled to the breakdown of PEP to pyruvate and ATP catalyzed by pyruvate kinase, which was linked to the synthesis of lactate catalyzed by lactate dehydrogenase. The breakdown of NADH during the synthesis of lactate was proportional to the ATP consumption and was measured by changes in UV absorbance at 340 nm. The signal for NADH was calibrated by multiple injections of 250 pmol of ADP. The tension cost was estimated from the slope of the relationship between steady-state isometric tension and ATPase activity, as described earlier (23, 27).
Measurement of rate of tension redevelopment (ktr) in reconstituted cardiac muscle fibers
Measurement of ktr in maximally activated (pCa 4.3) muscle fiber was according to a modified protocol originally described by Brenner and Eisenberg (31). Once the muscle fiber reached a steady-state isometric force, the motor arm was commanded to slacken the muscle fiber by 10% of the muscle length, using a high-speed length-control device (Aurora Scientific). After a brief shortening period of 25 ms, the motor arm was commanded to rapidly (within 0.5 ms) swing past the original set point by a 10% stretch in muscle length. The stretch was applied to ensure that any remaining XBs were mechanically detached, and the residual force was not greater than ∼10% of the initial steady-state isometric force. ktr was determined by fitting the following monoexponential equation to the force redevelopment:
where F is the force at time t, Fss is the steady-state isometric force, and Fres is the residual force from which the redevelopment of force occurs.
Data analysis
We analyzed the data using a 2-way analysis of variance (ANOVA). One factor in this analysis was McTnT variant (McTnTWT or McTnT1–44Δ or McTnT45–74Δ), and the other was Tm variant (TmWT or TmH276N). Thus, we used a 2-way ANOVA to test the hypothesis that the effect of the McTnT variant on a given contractile parameter depended on the type of Tm variant (interaction effect). When the interaction effect was significant, it showed that the effects of McTnTWT, McTnT1–44Δ, or McTnT45–74Δ were dissimilar against TmWT and TmH276N. When the interaction effect was not significant, we interpreted the main effects due to McTnT variants and Tm variants. Post hoc tests (multiple pairwise comparisons) were made using uncorrected Fisher's LSD method to test the effects of McTnT variants or Tm variants on the measured contractile parameters. The Hill equation was fitted to normalized pCa-tension data to estimate pCa50 (−log [Ca2+]free required for half-maximal activation) and Hill coefficient (nH) values. Data presented here for TmWT fibers are from our recently published study (20). Values are reported as means ± se. The criterion for statistical significance was set at P < 0.05. Asterisks in the figures and table represent statistical significance using post hoc comparisons.
RESULTS
Generation of McTnT1–44Δ and McTnT45–74Δ deletion mutants
Using McTnT1–44Δ and McTnT45–74Δ proteins, we have recently deciphered the cardiac-specific functional role of two distinct regions within the N-terminal end of McTnT: region 1–44 of McTnT, which is rich in negatively charged residues; and region 45–74 of McTnT, which is unique to the cardiac isoform of TnT (for further details, see Mamidi et al., ref. 20). McTnT1–44Δ attenuated Ca2+-activated maximal tension, whereas McTnT45–74Δ attenuated thin-filament cooperativity, without effecting Ca2+-maximal tension (20). Because of their divergent actions on the cardiac thin filament and their strategic location near the head-to-tail overlapping ends of two contiguous Tms, we hypothesized that the N-terminal end of cTnT would alter cTnT-Tm interactions in the overlapping region of two contiguous Tms to bring about different states of Tm on the actin filament. Therefore, McTnT1–44Δ and McTnT45–74Δ were ideal for investigating how these mutants divergently modified the function of cardiac muscle fibers from TG mouse hearts that expressed TmH276N; the H276N mutation in Tm is known to destabilize the head-to-tail overlapping ends of Tms, an effect that attenuates the rate of ventricular relaxation (21).
Western blot analysis to estimate the level of incorporation of exogenously added Tn subunits into detergent-skinned mouse cardiac fiber bundles
In our reconstitution protocol, the exogenously added Tn complex consisted of McTnT (McTnTWT, McTnT1–44Δ, or McTnT45–74Δ), McTnI, and McTnC proteins. We have previously demonstrated that the entire endogenous Tn complex is replaced when a vast excess of exogenously added cTnT competes with the endogenous cTnT (32). Thus, the level of McTnTWT or cTnT mutant incorporation can be used as a measure of the level of replacement of the endogenous Tn complex. The differential migration pattern of c-myc tagged McTnT, when compared to the endogenous cTnT, enabled us to assess the incorporation of McTnTWT. The incorporation of McTnT1–44Δ or McTnT45–74Δ was assessed based on the differences in the gel migration pattern when compared to the endogenous cTnT. SDS-solubilized fiber samples were run on a 10% SDS gel and were transferred onto a PVDF membrane for Western blot analysis. ImageJ software was used for precise quantification of the optical intensities of the protein bands. The total optical band intensity (i.e., the total amount of cTnT in a single lane) was assumed to be the sum of the optical band intensities of endogenous McTnT and the incorporated McTnT (McTnTWT, McTnT1–44Δ, or McTnT45–74Δ) bands. The level of incorporation was then calculated by dividing the optical band intensity of the exogenously added McTnT with the total band intensity. The level of protein incorporation was as follows: 89% for McTnTWT (Fig. 1, lane 2), 100% for McTnT1–44Δ (Fig. 1, lane 3), and 83% for McTnT45–74Δ (Fig. 1, lane 4). The level of replacement of endogenous McTnT in the reconstituted TmH276N fiber groups was similar to that of the reconstituted TmWT fiber groups.
Effect of McTnT deletion mutants on Ca2+-activated maximal tension in TmWT and TmH276N fibers
Because the H276N mutation in Tm is known to destabilize the head-to-tail overlapping ends of Tms (21), we wanted to test our hypothesis that the effects of McTnT1–44Δ and McTnT45–74Δ on thin filaments would be altered by TmH276N. First, we compared the effects of McTnT1–44Δ and McTnT45–74Δ on Ca2+-activated maximal tension by measuring tension in reconstituted fibers at pCa 4.3. Two-way ANOVA revealed a significant interaction effect (P<0.05), suggesting that the destabilization of the C-terminal ends of Tm (TmH276N) modified the effect of McTnT deletion mutants on Ca2+-activated maximal tension (Fig. 2A). To determine the factor that was responsible for the significant interaction effect, we carried out subsequent post hoc tests using pairwise multiple comparisons (Fischer's LSD). Post hoc tests revealed that McTnT45–74Δ, but not McTnT1–44Δ, had differential effects on maximal tension depending on the type of Tm present. For example, McTnT45–74Δ did not alter maximal tension in TmWT fibers, but significantly decreased maximal tension by ∼19% in TmH276N fibers (Fig. 2B). Thus, the differential effects of McTnT45–74Δ on maximal tension gave rise to a significant Tm-cTnT interaction effect (Fig. 2A). On the other hand, McTnT1–44Δ significantly decreased maximal tension by ∼36% in TmWT fibers and by ∼38% in TmH276N fibers (Fig. 2B). Thus, McTnT1–44Δ attenuated Ca2+-activated maximal tension regardless of the Tm variant (TmWT or TmH276N). These data demonstrate that the destabilized ends of TmH276N modified the effects of McTnT45–74Δ on maximal tension, but it had no effect on McTnT1–44Δ.
Figure 2.

Effect of McTnT deletion mutants on Ca2+-activated maximal tension in TmWT and TmH276N fibers. Ca2+-activated maximal tension was measured at pCa 4.3. A) Two-way ANOVA revealed a significant Tm-cTnT interaction effect (P<0.05), indicating that the differences between the effects of McTnT1–44Δ and McTnT45–74Δ were dissimilar against TmWT and TmH276N backgrounds. B) Subsequent post hoc multiple pairwise comparisons (Fischer's LSD) revealed that differential effect of McTnT45–74Δ on tension against TmWT and TmH276N was responsible for the interaction effect. Specifically, McTnT45–74Δ decreased maximal tension in TmH276N fibers by ∼19% (P<0.01), but not in TmWT fibers. On the other hand, McTnT1–44Δ decreased tension against TmWT (∼36%) and TmH276N (∼38%) uniformly. At least 7 determinations were done for each group. Values are reported as means ± sem. **P < 0.01; ***P < 0.0001.
Effect of McTnT deletion mutants on Ca2+-activated maximal ATPase activity in TmWT and TmH276N fibers
As described above, TmH276N had disparate effects on Ca2+-activated maximal tension of McTnT1–44Δ and McTnT45–74Δ reconstituted fibers. Therefore, we sought to determine whether Ca2+-activated maximal ATPase activity of McTnT deletion mutants was also differentially affected by TmH276N mutation. As described previously, the rate of ATP consumption and tension were measured simultaneously (23, 27). We compared the ATPase activity of different groups of reconstituted fibers at pCa 4.3. Two-way ANOVA revealed no significant interaction effect (P=0.12); however, we observed a trend that was similar to that of maximal tension (Fig. 3A). Significant main effects (P<0.001), due to McTnT deletion mutants and Tm variants, on Ca2+-activated maximal ATPase activity were observed. Post hoc multiple comparisons revealed that the maximal ATPase activity decreased by ∼26% in McTnT1–44Δ + TmWT fibers, when compared to McTnTWT + TmWT fibers; and by ∼21% in McTnT1–44Δ + TmH276N fibers, when compared to McTnTWT + TmH276N fibers (Fig. 3B). On the other hand, McTnT45–74Δ did not affect Ca2+-activated maximal ATPase activity in TmWT and TmH276N fibers. However, McTnT45–74Δ decreased maximal ATPase activity by ∼10% (P=0.14) in TmH276N fibers, but this decrease was not significant because McTnTWT + TmH276N also intrinsically decreased Ca2+-activated maximal ATPase activity by ∼10% (P=0.058) when compared to McTnTWT + TmWT fibers (Table 1). Thus, although not statistically significant, Ca2+-activated maximal ATPase activity of McTnT45–74Δ + TmH276N fibers showed a trend (Fig. 3) similar to that observed for maximal tension of McTnT45–74Δ + TmH276N fibers (Fig. 2).
Figure 3.

Effect of McTnT deletion mutants on Ca2+-activated maximal ATPase activity in TmWT and TmH276N fibers. Ca2+-activated maximal ATPase was measured at pCa 4.3. A) Two-way ANOVA revealed no significant Tm-cTnT interaction effect (P=0.12) on maximal ATPase activity because the trend was similar in different groups. B) Subsequent post hoc multiple comparisons revealed that McTnT1–44Δ significantly decreased Ca2+-activated maximal ATPase activity by ∼26% (P<0.0001) in TmWT and ∼21% (P<0.01) inTmH276N fibers. At least 7 determinations were done for each group. Values are reported as means ± sem. **P < 0.01; ***P < 0.0001.
Table 1.
Steady-state contractile parameters of NTG (TmWT) and TG (TmH276N) cardiac muscle fibers reconstituted with the WT Tn complex
| Parameter | McTnTWT + McTnI + McTnC reconstituted into NTG TmWT cardiac muscle fibers | McTnTWT + McTnI + McTnC reconstituted into TG TmH276N cardiac muscle fibers |
|---|---|---|
| Tension (mN·mm−2) | 49.75 ± 0.94 | 53.19 ± 1.82 |
| ATPase (pmol·mm−3·s−1) | 304.38 ± 9.36 | 274.53 ± 7.42 |
| nH tension | 2.62 ± 0.13 | 2.94 ± 0.26 |
| pCa50 tension | 5.65 ± 0.01 | 5.63 ± 0.01 |
| Tension cost (pmol·mN−1·mm−1·s−1) | 6.39 ± 0.18 | 5.17 ± 0.15** |
| ktr (s−1) | 12.87 ± 0.46 | 13.38 ± 0.57 |
Isometric steady-state tension and ATPase activity were measured simultaneously, as described previously (22, 27). The Hill equation was fitted to the pCa-tension relationships to derive nH- and pCa50-tension values. Tension cost was estimated as described previously (23). ktr was measured according to a protocol described previously (31). At least 7 determinations were done for each group. Values are reported as means ± sem
P < 0.01 vs. TmWT.
Effect of McTnT deletion mutants on the rate constant of XB detachment (g) in TmWT and TmH276N fibers
The H276N mutation in Tm decreased the rate of ventricular relaxation (−dP/dt) in TG mouse hearts (21). Our hypothesis was that the destabilized ends of Tms would alter actin structure and thus modify XB cycling kinetics. One way to determine the effect of Tm variants on XB cycling kinetics is to estimate the tension cost from simultaneous measurements of tension and ATPase activity (30). We estimated tension cost values as described previously (23, 27). In brief, the relationship between steady-state isometric tension and the corresponding ATPase activity was determined at various levels of Ca2+ activation (22, 30). The slope of ATPase-tension plot is a measure of the amount of ATP consumed for a given amount of tension (tension cost). Because the ratio of ATPase (fg/f+g) to tension (f/f+g) is proportional to g, tension cost is a measure of the rate constant of XB detachment (33). Two-way ANOVA revealed no significant interaction effect (P=0.065), but showed a significant main effect of McTnT deletion mutants (P<0.001) and Tm variants (P<0.05) on tension cost (Fig. 4A). Subsequent post hoc multiple comparisons revealed that tension cost increased significantly by ∼35%, only in McTnT1–44Δ + TmH276N fibers (Fig. 4B). Thus, our data demonstrated that the rate of XB detachment increased only when both mutants McTnT1–44Δ and TmH276N worked together, whereas TmH276N alone slowed XB detachment kinetics (Table 1). A closer look at our data indicates that an increase in tension cost in McTnT1–44Δ + TmH276N fibers (when compared to McTnTWT + TmH276N fibers; Fig. 4B) was due to the observation that TmH276N attenuated tension cost when compared to TmWT background; specifically, tension cost decreased by ∼19% in McTnTWT + TmH276N fibers when compared to McTnTWT + TmWT fibers (Table 1). Furthermore, multiple pairwise comparisons also revealed that tension cost of McTnT1–44Δ + TmH276N fibers was significantly different from that of McTnT45–74Δ + TmH276N fibers. Collectively, these data demonstrate that the effect on XB detachment kinetics is not only influenced by the structural integrity of the overlapping ends of Tms, but is also modulated by the N-terminal end of cTnT.
Figure 4.

Effect of McTnT deletion mutants on tension cost in TmWT and TmH276N fibers. The relationship between steady-state isometric tension and the corresponding ATPase activity was determined at various levels of Ca2+ activation (22, 30). Tension cost value was derived from the slope of tension-ATPase relationships. A) Two-way ANOVA revealed no significant Tm-cTnT interaction effect (P=0.064) on tension cost. B) Subsequent post hoc tests revealed that McTnT1–44Δ significantly increased tension cost by ∼35% in TmH276N fibers, because the mean average value of the McTnT1–44Δ + TmH276N group was significantly higher than the mean average value of the McTnTWT + TmH276N group. At least 6 determinations were done for each group. Values are reported as means ± sem. ***P < 0.001.
Effect of McTnT deletion mutants on XB turnover rate, ktr, in TmWT and TmH276N fibers
Because TmH276N had a slowing effect on the rate of XB detachment and McTnT1–44Δ sped the rate of XB detachment in the presence of TmH276N, we wanted to determine whether such changes in XB cycling kinetics had any effect on XB turnover rate. Therefore, we assessed XB turnover rate by estimating the rate of tension redevelopment ktr in maximally activated muscle fibers reconstituted with McTnT deletion mutants against TmWT and TmH276N backgrounds. Two-way ANOVA revealed a significant interaction effect (P<0.01), suggesting that TmH276N influenced the effect of McTnT deletions on ktr (Fig. 5A). Post hoc multiple pairwise comparisons revealed that ktr decreased significantly by ∼17% only in McTnT1–44Δ + TmH276N fibers (Fig. 5B). Collectively, these observations demonstrate that ktr decreased only when both mutants (McTnT1–44Δ and TmH276N) worked together. Our observations demonstrate that attenuation of XB turnover rate is not only mediated by the structural integrity of the overlapping ends of Tm, but is also affected by the N-terminal end of cTnT. McTnT45–74Δ had no effect on ktr, regardless of the type of Tm present (Fig. 5).
Figure 5.

Effect of McTnT deletion mutants on the rate of tension redevelopment in TmWT and TmH276N fibers. Rate of tension development ktr was measured as described previously (31). A) Two-way ANOVA revealed a significant interaction effect (P<0. 01), indicating that the differences between the effects of McTnT1–44Δ and McTnT45–74Δ on ktr were dissimilar against TmWT and TmH276N backgrounds. B) Post hoc multiple pairwise comparisons revealed that McTnT1–44Δ significantly decreased ktr (by ∼17%), only in TmH276N fibers. At least 7 determinations were done for each group. Values are reported as mean ± sem. *P < 0.05.
Effect of McTnT deletion mutants on myofilament cooperativity, nH, and Ca2+sensitivity, pCa50, in TmWT and TmH276N fibers
Our data show that neither McTnT45–74Δ nor TmH276N had any effect on Ca2+-activated maximal tension. This is indicated by the observation that Ca2+-activated maximal tension of McTnT45–74Δ + TmWT fibers was not different when compared to McTnTWT + TmWT fibers (Fig. 2B). Furthermore, Ca2+-activated maximal tension of McTnTWT + TmH276N fibers was not different when compared to McTnTWT + TmWT fibers (Table 1). However, Ca2+-activated maximal tension decreased significantly in McTnT45–74Δ + TmH276N fibers (Fig. 2B). Because McTnT45–74Δ attenuates thin-filament cooperativity (20) and TmH276N decreases the rate of XB detachment (Table 1) when present together, the interplay between these two mutants may affect the XB-regulatory unit (RU; Tm-Tn)-mediated thin-filament cooperativity and myofilament Ca2+ sensitivity (34). Normalized tension values were plotted against a range of pCa to estimate the pCa-tension relationships in reconstituted fibers (Fig. 6). The Hill equation was fitted to normalized pCa-tension data to estimate nH (which is a measure of myofilament cooperativity) and pCa50 (an indicator of myofilament Ca2+ sensitivity) values (Fig. 7A, C, respectively). Two-way ANOVA revealed no significant interaction effect (Fig. 7A), but showed a significant main effect (P<0.05) of McTnT deletion mutants on nH. Post hoc multiple pairwise comparisons revealed that nH decreased significantly (P<0.001) by ∼27% in McTnT45–74Δ + TmWT fibers and by ∼26% in McTnT45–74Δ + TmH276N fibers (Fig. 7B). On the other hand, McTnT1–44Δ did not affect nH, regardless of the type of Tm present in the fiber.
Figure 6.

Effect of McTnT deletion mutants on the pCa-tension relationship in TmWT and TmH276N fibers. Ca2+-activated tension was measured at the SL of 2.3 μm. Normalized tension values were plotted against a range of pCa to derive the pCa-tension relationships. Normalized pCa-tension relationships were fitted using the Hill equation; the fitted curves are shown as connecting lines. A) Effect of McTnT deletion mutants on the pCa-tension relationships in TmWT fibers. B) Effect of McTnT deletion mutants on the pCa-tension relationships in TmH276N fibers. At least 7 determinations were done for each group. Values are reported as means ± sem.
Figure 7.

Effect of McTnT deletion mutants on myofilament cooperativity, nH, and myofilament Ca2+ sensitivity, pCa50, in TmWT and TmH276N fibers. The Hill equation was fitted to the pCa-tension relationships to estimate nH and pCa50 values. A) Two-way ANOVA revealed no significant Tm-cTnT interaction effect (P=0.98) on nH. B) Subsequent post hoc multiple pairwise comparisons revealed a significant main effect (P<0.05) of McTnT deletion mutants on nH. Post hoc tests revealed that McTnT45–74Δ decreased nH significantly (P<0.001) in TmWT and TmH276N fibers, respectively. C) Two-way ANOVA revealed no significant Tm-cTnT interaction effect (P=0.63) on pCa50. D) Subsequent post hoc multiple pairwise comparisons revealed significant main effects of McTnT deletion mutants (P<0.0001) and Tm variants (P<0.05) on pCa50. Post hoc tests revealed that McTnT1–44Δ significantly decreased pCa50, while McTnT45–74Δ significantly increased pCa50 in TmWT and TmH276N fibers. At least 7 determinations were done for each group. Values are reported as means ± sem. **P < 0.01; ***P < 0.001.
pCa50 values estimated from different groups of fibers were compared to determine the effect of McTnT mutants and Tm variants on myofilament Ca2+ sensitivity. Two-way ANOVA revealed no significant interaction effect (Fig. 7C), but showed a significant main effect of McTnT deletion mutants (P<0.0001) and Tm variants (P<0.05) on myofilament Ca2+ sensitivity. Post hoc multiple pairwise comparisons revealed the following: McTnT1–44Δ significantly decreased pCa50, regardless of the type of Tm present; and McTnT45–74Δ significantly increased pCa50, regardless of the type of Tm present (Fig. 7D). Although McTnT45–74Δ had a general trend to increase pCa50, multiple pairwise comparisons indicated an interesting feature about it when a different Tm background was considered. For example, pCa50 of McTnT45–74Δ + TmH276N fibers (5.93) was significantly lower when compared to McTnT45–74Δ + TmWT fibers (6.00). Interestingly, Ca2+-activated maximal tension (Fig. 2B) of McTnT45–74Δ + TmH276N fibers (43 mN/mm2) was also significantly lower than that of McTnT45–74Δ + TmWT fibers (51 mN/mm2). Our observations demonstrate that McTnT45–74Δ desensitizes TmH276N fibers to Ca2+, leading to a significant attenuation of Ca2+-activated maximal tension.
DISCUSSION
The biological significance of the intermolecular interplay between the cardiac-specific N terminus of cTnT and the head-to-tail overlapping region of two contiguous Tms is unknown. In this study, we investigated how the functional effect of deletions in the N terminus of cTnT was modulated by a mutation in α-Tm (TmH276N) that destabilized the head-to-tail overlapping ends of Tms. McTnT1–44Δ and McTnT45–74Δ attenuated Ca2+-activated maximal tension in cardiac muscle fibers from TmH276N TG mouse hearts via independent mechanisms. Conversely, the structural integrity of the head-to-tail overlap of Tms also modulated the functional outcome of deletions in the N terminus of cTnT. We discuss our novel data in terms of a link between cTnT-Tm interactions in the 4-helix bundle of the Tm overlapping ends (11) and the emergence of different states of Tm on the actin filament.
Destabilization of the head-to-tail overlapping region by the H276N mutation in Tm slows XB detachment kinetics
As schematically represented in Fig. 8, the structural integrity of the head-to-tail overlap region of Tm is promoted by the splaying of the N terminus of Tm (residues 1–15), the binding of one of the chains from the C termini of adjacent Tms, and the binding of the N terminus of TnT to form a 4-helix bundle (11). As for the C terminus of Tm, residues 263, 266, 267, and 270 play crucial role in forming a bend in the helical structure of Tm that is essential for the formation of a molecular swivel, which contains the 4-helix bundle. Specifically, mutations at residues 267 and 270 decrease the affinity of TnT for Tm on actin (11). Moreover, the H276N mutation in Tm has also been shown to alter cardiac contractility by decreasing the stability of the C-terminal helix of Tm (21). Although the TG overexpression of TmH276N in the mouse heart had no effect on Ca2+-activated maximal tension, the rate of ventricular relaxation −dP/dt was significantly attenuated in isolated heart preparations, suggesting that the destabilized C-terminal ends of Tm affected contractile function (21). Our current observations corroborated these findings because detergent-skinned muscle fibers containing TmH276N showed a significant decrease in tension cost. Because tension cost is a measure of XB detachment rate constant (33), our observations suggest that the destabilizing effect of the H276N mutation on the overlapping ends of Tms slows XB detachment kinetics in cardiac muscle fibers containing TmH276N (Table 1). This slowing effect on XB detachment rate by H276N mutation may provide a molecular basis for the attenuation of the rate of ventricular relaxation observed in TG heart preparations (21).
Figure 8.
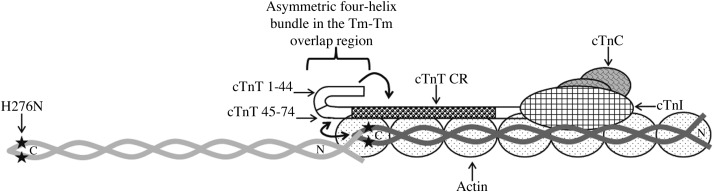
Schematic representation of the asymmetric 4-helix bundle in the head-to-tail overlapping region of 2 contiguous Tms. The 4-helix bundle (2 helices from the C termini of Tm, one from the N terminus of Tm, and one from the central region of cTnT) results from local disruption of coiled-coil regions in the N terminus of Tm (11). The precise configuration of this molecular swivel structure is considered to be important for the flexibility of Tm on actin filaments. Flexibility not only enables adjacent Tm to wind around actin properly, but it also permits the filamentous Tm to occupy discrete positions on actin during thin-filament activation and deactivation. The H276N mutation in the C terminus of Tm is denoted by black stars. The central helical region of cTnT (residues 78–193) is represented by CR. Amino acid residues 1–77 of cTnT are not known to interact with Tm, actin, TnC, or TnI; however, this region is known to synergistically modulate the ability of CR to affect Tm function (shown by single-headed arrow; Gollapudi et al., ref, 37). The intermolecular interaction between the N terminus of cTnT and the overlapping ends of Tm is denoted by the double-headed arrow. (Adapted from Gollapudi et al., ref. 37).
TmWT and TmH276N have divergent effects on how McTnT1–44Δ and McTnT45–74Δ attenuate Ca2+-activated maximal tension
TmH276N did not affect Ca2+-activated maximal tension, but slowed XB detachment rate (Table 1). McTnT1–44Δ attenuated Ca2+-activated maximal tension against both TmWT and TmH276N backgrounds (Fig. 2B), but sped XB detachment rate only against TmH276N background (Fig. 4B). McTnT45–74Δ attenuated Ca2+-activated maximal tension only against the TmH276N, but not against TmWT background (Fig. 2B). These observations demonstrate that TmH276N has divergent effects on how McTnT1–44Δ and McTnT45–74Δ tweak thin-filament activation, suggesting that the intermolecular interplay between the N terminus of cTnT and the head-to-tail overlapping region of two contiguous Tms influences different states of Tm on the actin filament. It is likely that the N-terminal end region of cTnT (residues 1–74 of McTnT) does not interact directly with other thin-filament proteins (35, 36). However, recent binding affinity studies from our laboratory (37) suggest that the N-terminal end of cTnT may still influence cardiac thin-filament activation by synergistically modulating the functional effect of the neighboring region of cTnT (represented by the central region residues 78–193 of cTnT in Fig. 8). Based on our recent studies (37), we believe that the effect of regions 1–44 and 45–74 of cTnT on tension and other contractile parameters is exerted via their ability to synergistically modulate the functional effect of the central region of cTnT, which is known to interact directly with Tm (9,38). Collectively, these observations suggest that the functional outcome depends on the interplay between the central region of cTnT and the overlapping ends of Tms. Just as 1–44 and 45–74 synergistically modulate the activity of CR-Tm interactions, a mutation in Tm (TmH276N in this study) may also modify contractile function by modulating the effects of cTnT mutants used in this study.
As for the effects of McTnT1–44Δ and McTnT45–74Δ, the mechanism underlying the depression of thin-filament activation was previously explained by the strengthening of charge-dependent interactions between TnT and Tm (32,36). Such strengthened TnT-Tm interactions may hinder the azimuthal movement of Tm on the actin filament, thereby attenuating the transition of the RU (Tm-Tn) to the fully activated state (20). The binding of the N terminus of cTnT to the head-to-tail linked contiguous Tm molecules is not only important for promoting the interaction of Tm with actin filaments (9, 39) but also essential for full cooperative activation of thin filaments (39,40). Thus, alterations in either the N terminus of cTnT or the overlapping ends of Tms are expected to affect thin-filament activation. If the cTnT-induced binding of Tm to actin is strengthened by McTnT1–44Δ and McTnT45–74Δ, as predicted by previous studies, it may limit the movement of Tm on actin or stiffen the actin filament such that the thin-filament activation is attenuated via one of the two following mechanisms: an indirect effect on XB cycling kinetics, whereby tension is attenuated by altered XB cycling kinetics caused by changes in the structure of actin; and a direct constraining effect on the movement of Tm across the myosin binding sites on actin, which not only decreases the fraction of myosin XBs bound to actin but also attenuates the activating effects of XBs via diminished XB-mediated cooperativity.
Attenuation of Ca2+-activated maximal tension in McTnT1–44Δ+ TmH276N fibers is caused by altered XB cycling kinetics
One of the major findings in our study is that McTnT1–44Δ decreased Ca2+-activated maximal tension (Fig. 2B) against both TmWT and TmH276N backgrounds, but it increased tension cost only against the TmH276N background (Fig. 4B). Thus, McTnT1–44Δ sped XB detachment rate against the TmH276N background, suggesting that the interplay between the N terminus of cTnT and the head-to-tail overlapping region of TmH276N affects the states of Tm on actin to alter XB detachment kinetics. This observation has striking similarity to our recent study, which showed that McTnT1–44Δ increased tension cost only in the presence of β-Tm (20). Interestingly, β-Tm contains 2 charged amino acid substitutions (S229E and H276N) when compared to α-Tm (41). We also observed a significant decrease in ktr in McTnT1–44Δ + TmH276N fibers (Fig. 5B). Based on a simple 2-state XB model (ktr=f+g), we predict that f decreases significantly in McTnT1–44Δ+ TmH276N fibers. Thus, our observations suggest that altered XB cycling kinetics causes a decrease in the fraction of strongly bound XBs (f/f+g) in McTnT1–44Δ+ TmH276N fibers.
Attenuation of Ca2+-activated maximal tension in McTnT45–74Δ + TmH276N fibers is caused by decreased thin-filament cooperativity
Another major finding was that McTnT45–74Δ decreased Ca2+-activated maximal tension only against the TmH276N background (Fig. 2B). This effect was surprising because neither McTnT45–74Δ (Fig. 2A) nor TmH276N (Table 1) had any effect on Ca2+-activated maximal tension (see TmWT group in Fig. 2A); other contractile parameters suggested no effect of McTnT45–74Δ on XB cycling kinetics (Figs. 4B and 5B). Therefore, the attenuation of Ca2+-activated maximal tension in McTnT45–74Δ + TmH276N fibers can only be ascribed to the interaction effect of McTnT45–74Δ and TmH276N on the thin-filament activation. To explain why the Ca2+-activated maximal tension is attenuated in McTnT45–74Δ + TmH276N fibers, but not in McTnT45–74Δ or TmH276N fibers, one must consider the individual effects of McTnT45–74Δ and TmH276N on the thin filament. McTnT45–74Δ is known to depress thin-filament cooperativity (20), and TmH276N decreases the rate of XB detachment (Table 1). Therefore, the combined actions of McTnT45–74Δ + TmH276N are expected to attenuate the XB-RU cooperative interactions.
A decrease in the strength of XB-RU interactions may impair Ca2+-activated maximal tension and myofilament Ca2+ sensitivity by decreasing the fraction of force-bearing XBs (34). As expected, both Ca2+-activated maximal tension (43 vs. 51 mN/mm2) and myofilament Ca2+ sensitivity (pCa50 of 5.93 vs. 6.00) of McTnT45–74Δ + TmH276N fibers were significantly lower than McTnT45–74Δ + TmWT fibers. Because nH of McTnT45–74Δ + TmH276N fibers was not altered when compared to McTnT45–74Δ + TmWT fibers, we speculate that the RU-mediated effect on thin-filament cooperativity (RU-RU) was not affected (34). Therefore, we conclude that the Ca2+-activated maximal tension in McTnT45–74Δ + TmH276N fibers is attenuated because of an abatement of mechanisms that underlie XB-RU-mediated thin-filament activation.
Altered interplay between the overlapping ends of Tm and the N terminus of cTnT produces different states of Tm on the actin filament
At first glance, altered XB cycling kinetics in McTnT1–44Δ + TmH276N fibers (Fig. 4B) seems to be at odds with the observation made by Mamidi et al. (20), who demonstrated that XB cycling kinetics were unaffected in McTnT1–44Δ + TmWT fibers. However, it must be noted that the effect of McTnT1–44Δ in the study by Mamidi et al. was tested against the background of TmWT, in which the structural integrity of the overlapping ends of 2 contiguous Tms was not altered. Because Ca2+-activated maximal tension decreases in both cases, our data suggests that different mechanisms bring about the attenuation of thin-filament activation in McTnT1–44Δ + TmH276N fibers and McTnT1–44Δ + TmWT fibers. The attenuation of tension in McTnT1–44Δ + TmWT appears be due to a constraining effect on the movement Tm brought about by increased TnT-Tm interactions, and the attenuation of tension in McTnT1–44Δ + TmH276N fibers is likely due to an effect on actin structure brought about by the altered overlapping ends of Tms. Indeed, recent studies demonstrate that mutations near the head-to-tail overlapping ends of Tms alter the structure of actin. For example, the magnitude of actin subdomain 1 movement, associated with the transition of XBs from weakly bound to strongly bound states, is attenuated by E40K and E54K mutations in Tm (42,43). Just as interesting as the effects of McTnT1–44Δ + TmH276N, the combined actions of McTnT45–74Δ and TmH276N produce a distinct effect on the thin-filament activation, which we believe is mediated by the abatement of cooperative mechanisms operating within the thin filament.
Inferences drawn from our observations suggest that, depending on whether the cTnT moiety is WT, McTnT1–44Δ, or McTnT45–74Δ, the interplay between the overlapping ends of 2 contiguous Tms and the N terminus of cTnT effectuates different states of Tm on the actin filament. These observations are indicative of significant conformational plasticity in the region where the N terminus of cTnT and the overlapping ends of Tm come together to form a 4-helix bundle (11). Conformational flexibility is what one would intuit from weak protein-protein interactions that play a nuanced role in contractile regulation. Thus, it is not surprising that only the end-to-end polymerized Tm, mediated by the N terminus of cTnT, has any measurable affinity for the actin filament. Such localized electrostatic interactions between Tm and actin are now considered to be relatively weak (14, 44, 45). Weak interactions not only afford flexibility to Tm on the actin filament, but also enable subtle variations in actin and Tm isoforms to produce disparate physiological functions (46, 47).
CONCLUSIONS
This is the first study to demonstrate that the interplay between the N terminus of cTnT and the overlapping ends of Tms leads to the emergence of different states of Tm on the actin filament. Specifically, McTnT1–44Δ and McTnT45–74Δ attenuated Ca2+-activated maximal tension in cardiac muscle fibers from TmH276N TG mouse hearts via independent mechanisms. Altered XB cycling kinetics decreased the fraction of strongly bound XB in McTnT1–44Δ + TmH276N fibers, whereas the diminished cooperative mechanisms attenuated Ca2+-activated maximal tension in McTnT45–74Δ + TmH276N fibers. Our observations are suggestive of a significant link between the structure of the 4-helix bundle near the overlapping ends of Tm and contractile function. Therefore, the functional effect of different cTnT/Tm isoforms, disease-related mutations in the N terminus of cTnT, and phosphorylation of Tm can now be considered in the framework of manifestation of their effects on different states of Tm on the actin filament.
Acknowledgments
The authors thank Sri Lakshmi Mallampalli for excellent technical assistance.
This work was funded by U.S. National Heart, Lung, and Blood Institute grants R01-HL-075643 (M.C.) and R01-HL-80526 (M.M.) and by a Poncin fellowship (R.M.).
The authors declare no conflicts of interest.
Footnotes
- BDM
- 2, 3-butanedione monoxime
- BES
- N,N-bis-(2-hydroxyethyl)-2-aminoethanesulfonic acid
- cTnC
- cardiac troponin C
- cTnI
- cardiac troponin I
- cTnT
- cardiac troponin T
- HR
- high relaxing
- McTnC
- mouse cardiac troponin C
- McTnI
- mouse cardiac troponin I
- McTnT
- mouse cardiac troponin T
- McTnT1–44Δ
- mouse cardiac troponin T 1–44 deletion
- McTnT45–74Δ
- mouse cardiac troponin T 45–74 deletion
- NTG
- nontransgenic
- PEP
- phosphoenolpyruvate
- RU
- regulatory unit
- SL
- sarcomere length
- TG
- transgenic
- Tm
- tropomyosin
- TmWT
- wild-type α-tropomyosin
- Tn
- troponin
- WT
- wild-type
- XB
- crossbridge
REFERENCES
- 1. Gordon A. M., Homsher E., Regnier M. (2000) Regulation of contraction in striated muscle. Physiol. Rev. 80, 853–924 [DOI] [PubMed] [Google Scholar]
- 2. Lehman W., Hatch V., Korman V., Rosol M., Thomas L., Maytum R., Geeves M. A., Van Eyk J. E., Tobacman L. S., Craig R. (2000) Tropomyosin and actin isoforms modulate the localization of tropomyosin strands on actin filaments. J. Mol. Biol. 302, 593–606 [DOI] [PubMed] [Google Scholar]
- 3. Moss R. L., Razumova M., Fitzsimons D. P. (2004) Myosin crossbridge activation of cardiac thin filaments: implications for myocardial function in health and disease. Circ. Res. 94, 1290–1300 [DOI] [PubMed] [Google Scholar]
- 4. Smith D. A., Geeves M. A. (2003) Cooperative regulation of myosin-actin interactions by a continuous flexible chain II: actin-tropomyosin-troponin and regulation by calcium. Biophys. J. 84, 3168–3180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McLachlan A. D., Stewart M. (1976) The 14-fold periodicity in alpha-tropomyosin and the interaction with actin. J. Mol. Biol. 103, 271–298 [DOI] [PubMed] [Google Scholar]
- 6. Phillips G. N., Jr., Fillers J. P., Cohen C. (1986) Tropomyosin crystal structure and muscle regulation. J. Mol. Biol. 192, 111–131 [DOI] [PubMed] [Google Scholar]
- 7. McLachlan A. D., Stewart M. (1975) Tropomyosin coiled-coil interactions: evidence for an unstaggered structure. J. Mol. Biol. 98, 293–304 [DOI] [PubMed] [Google Scholar]
- 8. Ueno H., Tawada Y., Ooi T. (1976) Properties of non-polymerizable tropomyosin obtained by carboxypeptidase A digestion. J. Biochem. 80, 283–290 [DOI] [PubMed] [Google Scholar]
- 9. Heeley D. H., Golosinska K., Smillie L. B. (1987) The effects of troponin T fragments T1 and T2 on the binding of nonpolymerizable tropomyosin to F-actin in the presence and absence of troponin I and troponin C. J. Biol. Chem. 262, 9971–9978 [PubMed] [Google Scholar]
- 10. Pato M. D., Mak A. S., Smillie L. B. (1981) Fragments of rabbit striated muscle alpha-tropomyosin. II. Binding to troponin-T. J. Biol. Chem. 256, 602–607 [PubMed] [Google Scholar]
- 11. Murakami K., Stewart M., Nozawa K., Tomii K., Kudou N., Igarashi N., Shirakihara Y., Wakatsuki S., Yasunaga T., Wakabayashi T. (2008) Structural basis for tropomyosin overlap in thin (actin) filaments and the generation of a molecular swivel by troponin-T. Proc. Natl. Acad. Sci. U. S. A. 105, 7200–7205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Butters C. A., Willadsen K. A., Tobacman L. S. (1993) Cooperative interactions between adjacent troponin-tropomyosin complexes may be transmitted through the actin filament. J. Biol. Chem. 268, 15565–15570 [PubMed] [Google Scholar]
- 13. Hammell R. L., Hitchcock-DeGregori S. E. (1996) Mapping the functional domains within the carboxyl terminus of alpha-tropomyosin encoded by the alternatively spliced ninth exon. J. Biol. Chem. 271, 4236–4242 [DOI] [PubMed] [Google Scholar]
- 14. Li X. E., Tobacman L. S., Mun J. Y., Craig R., Fischer S., Lehman W. (2011) Tropomyosin position on F-actin revealed by EM reconstruction and computational chemistry. Biophys. J. 100, 1005–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Perry S. V. (1998) Troponin T: genetics, properties and function. J. Muscle Res. Cell Motil. 19, 575–602 [DOI] [PubMed] [Google Scholar]
- 16. McKillop D. F., Geeves M. A. (1993) Regulation of the interaction between actin and myosin subfragment 1: evidence for three states of the thin filament. Biophys. J. 65, 693–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gollapudi S. K., Mamidi R., Mallampalli S. L., Chandra M. (2012) The N-terminal extension of cardiac troponin T stabilizes the blocked state of cardiac thin filament. Biophys. J. 103, 940–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tobacman L. S., Nihli M., Butters C., Heller M., Hatch V., Craig R., Lehman W., Homsher E. (2002) The troponin tail domain promotes a conformational state of the thin filament that suppresses myosin activity. J. Biol. Chem. 277, 27636–27642 [DOI] [PubMed] [Google Scholar]
- 19. Maytum R., Geeves M. A., Lehrer S. S. (2002) A modulatory role for the troponin T tail domain in thin filament regulation. J. Biol. Chem. 277, 29774–29780 [DOI] [PubMed] [Google Scholar]
- 20. Mamidi R., Mallampalli S. L., Wieczorek D. F., Chandra M. (2013) Identification of two new regions in the N-terminus of cardiac troponin T that have divergent effects on cardiac contractile function. J. Physiol. 591, 1217–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gaffin R. D., Tong C. W., Zawieja D. C., Hewett T. E., Klevitsky R., Robbins J., Muthuchamy M. (2004) Charged residue alterations in the inner-core domain and carboxy-terminus of alpha-tropomyosin differentially affect mouse cardiac muscle contractility. J. Physiol. 561, 777–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chandra M., Tschirgi M. L., Ford S. J., Slinker B. K., Campbell K. B. (2007) Interaction between myosin heavy chain and troponin isoforms modulate cardiac myofiber contractile dynamics. Am. J. Physiol. 293, R1595–1607 [DOI] [PubMed] [Google Scholar]
- 23. Chandra M., Tschirgi M. L., Rajapakse I., Campbell K. B. (2006) Troponin T modulates sarcomere length-dependent recruitment of cross-bridges in cardiac muscle. Biophys. J. 90, 2867–2876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Montgomery D. E., Tardiff J. C., Chandra M. (2001) Cardiac troponin T mutations: correlation between the type of mutation and the nature of myofilament dysfunction in transgenic mice. J. Physiol. 536, 583–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tardiff J. C., Factor S. M., Tompkins B. D., Hewett T. E., Palmer B. M., Moore R. L., Schwartz S., Robbins J., Leinwand L. A. (1998) A truncated cardiac troponin T molecule in transgenic mice suggests multiple cellular mechanisms for familial hypertrophic cardiomyopathy. J. Clin. Invest. 101, 2800–2811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mamidi R., Gollapudi S. K., Mallampalli S. L., Chandra M. (2012) Alanine or aspartic acid substitutions at serine23/24 of cardiac troponin I decrease thin filament activation, with no effect on crossbridge detachment kinetics. Arch. Biochem. Biophys. 525, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chandra M., Mamidi R., Ford S., Hidalgo C., Witt C., Ottenheijm C., Labeit S., Granzier H. (2009) Nebulin alters cross-bridge cycling kinetics and increases thin filament activation: a novel mechanism for increasing tension and reducing tension cost. J. Biol. Chem. 284, 30889–30896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Michael J. J., Gollapudi S. K., Ford S. J., Kazmierczak K., Szczesna-Cordary D., Chandra M. (2013) Deletion of 1–43 amino acids in cardiac myosin essential light chain blunts length dependency of Ca(2+) sensitivity and cross-bridge detachment kinetics. Am. J. Physiol. Heart Circ. Physiol. 304, H253–H259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fabiato A., Fabiato F. (1979) Calculator programs for computing the composition of the solutions containing multiple metals and ligands used for experiments in skinned muscle cells. J. Physiol. (Paris) 75, 463–505 [PubMed] [Google Scholar]
- 30. De Tombe P. P., Stienen G. J. (1995) Protein kinase A does not alter economy of force maintenance in skinned rat cardiac trabeculae. Circ. Res. 76, 734–741 [DOI] [PubMed] [Google Scholar]
- 31. Brenner B., Eisenberg E. (1986) Rate of force generation in muscle: correlation with actomyosin ATPase activity in solution. Proc. Natl. Acad. Sci. U. S. A. 83, 3542–3546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chandra M., Montgomery D. E., Kim J. J., Solaro R. J. (1999) The N-terminal region of troponin T is essential for the maximal activation of rat cardiac myofilaments. J. Mol. Cell. Cardiol. 31, 867–880 [DOI] [PubMed] [Google Scholar]
- 33. Brenner B. (1988) Effect of Ca2+ on cross-bridge turnover kinetics in skinned single rabbit psoas fibers: implications for regulation of muscle contraction. Proc. Natl. Acad. Sci. U. S. A. 85, 3265–3269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Razumova M. V., Bukatina A. E., Campbell K. B. (2000) Different myofilament nearest-neighbor interactions have distinctive effects on contractile behavior. Biophys. J. 78, 3120–3137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Oliveira D. M., Nakaie C. R., Sousa A. D., Farah C. S., Reinach F. C. (2000) Mapping the domain of troponin T responsible for the activation of actomyosin ATPase activity. Identification of residues involved in binding to actin. J. Biol. Chem. 275, 27513–27519 [DOI] [PubMed] [Google Scholar]
- 36. Pan B. S., Gordon A. M., Potter J. D. (1991) Deletion of the first 45 NH2-terminal residues of rabbit skeletal troponin T strengthens binding of troponin to immobilized tropomyosin. J. Biol. Chem. 266, 12432–12438 [PubMed] [Google Scholar]
- 37. Gollapudi S. K., Gallon C. E., Chandra M. (2013) The tropomyosin binding region of cardiac troponin T modulates crossbridge recruitment dynamics in rat cardiac muscle fibers. J. Mol. Biol. 425, 1565–1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pearlstone J. R., Smillie L. B. (1977) The binding site of skeletal alpha-tropomyosin on troponin-T. Can. J. Biochem. 55, 1032–1038 [DOI] [PubMed] [Google Scholar]
- 39. Perry S. V. (2001) Vertebrate tropomyosin: distribution, properties and function. J. Muscle Res. Cell Motil. 22, 5–49 [DOI] [PubMed] [Google Scholar]
- 40. Schaertl S., Lehrer S. S., Geeves M. A. (1995) Separation and characterization of the two functional regions of troponin involved in muscle thin filament regulation. Biochemistry 34, 15890–15894 [DOI] [PubMed] [Google Scholar]
- 41. Gaffin R. D., Gokulan K., Sacchettini J. C., Hewett T., Klevitsky R., Robbins J., Muthuchamy M. (2004) Charged residue changes in the carboxy-terminus of alpha-tropomyosin alter mouse cardiac muscle contractility. J. Physiol. 556, 531–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Borovikov Y. S., Avrova S. V., Karpicheva O. E., Robinson P., Redwood C. S. (2011) The effect of the dilated cardiomyopathy-causing Glu40Lys TPM1 mutation on actin-myosin interactions during the ATPase cycle. Biochem. Biophys. Res. Commun. 411, 496–500 [DOI] [PubMed] [Google Scholar]
- 43. Borovikov Y. S., Karpicheva O. E., Avrova S. V., Robinson P., Redwood C. S. (2009) The effect of the dilated cardiomyopathy-causing mutation Glu54Lys of alpha-tropomyosin on actin-myosin interactions during the ATPase cycle. Arch. Biochem. Biophys. 489, 20–24 [DOI] [PubMed] [Google Scholar]
- 44. Brown J. H., Cohen C. (2005) Regulation of muscle contraction by tropomyosin and troponin: how structure illuminates function. Adv. Protein Chem. 71, 121–159 [DOI] [PubMed] [Google Scholar]
- 45. Holmes K. C., Lehman W. (2008) Gestalt-binding of tropomyosin to actin filaments. J. Muscle. Res. Cell Motil. 29, 213–219 [DOI] [PubMed] [Google Scholar]
- 46. Bach C. T., Schevzov G., Bryce N. S., Gunning P. W., O'Neill G. M. (2010) Tropomyosin isoform modulation of focal adhesion structure and cell migration. Cell. Adh. Migr. 4, 226–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gunning P. (2008) Emerging issues for tropomyosin structure, regulation, function and pathology. Adv. Exp. Med. Biol. 644, 293–298 [DOI] [PubMed] [Google Scholar]