

Abstract
Inflammatory bowel disease (IBD) is a consequence of the complex, dysregulated interplay between genetic predisposition, environmental factors, and microbial composition in the intestine. Despite a great advancement in identifying host-susceptibility genes using genome-wide association studies (GWAS), the majority of IBD cases are still underrepresented. The immediate challenge in post-GWAS era is to identify other causative genetic factors of IBD. DNA methylation has received increasing attention for its mechanistical role in IBD pathogenesis. This stable, yet dynamic DNA modification, can directly affect gene expression that have important implications in IBD development. The alterations in DNA methylation associated with IBD are likely to outset as early as embryogenesis all the way until old-age. In this review, we will discuss the recent advancement in understanding how DNA methylation alterations can contribute to the development of IBD.
Keywords: Intestinal inflammation, Crohn’s disease, Colitis, DNA methyltransferase, Epi-therapy
Core tip: This review discuss the recent research advancement in the area of DNA methylation during the pathogenesis of inflammatory bowel disease (IBD) and IBD-associated cancer, with a focus on highlighting major players mediating DNA methylation alterations during IBD development. Temporal and spatial differential DNA methylation status that contributes to the disease, as well as epi-therapy treatment options for IBD patients, are also discussed. This emerging information will have important clinical significance, especially so in this post-genome-wide association studies era of IBD research.
INTRODUCTION
Inflammatory bowel disease (IBD) is a chronic intestinal inflammatory condition that affects the intestine of millions of individuals throughout their lifetime[1]. IBD is classified into two major forms, Crohn’s disease (CD) and ulcerative colitis (UC), which both exhibit etiologically and clinically distinct features. Patients with IBD have a 2-3 fold greater life time risk of developing IBD-associated colorectal cancer (IBD-CRC)[2]. Although numerous clinical and experimental reports have given large amount of insights on the pathogenesis of IBD, the complexity of the initiation of IBD renders an incomplete understanding. Recently, there has been significant progress in identifying risk loci that are associated with IBD patients through genome-wide association studies (GWAS). These robust analyses have identified 163 IBD susceptible gene loci[3-6]. Genome-wide meta-analysis has confirmed that 71 of these loci are associated with CD, but only accounts for 25% of disease heritability[4]. The immediate challenge of the post-GWAS era is to unravel other parameters that may be less obvious from a genetic point of view. One of such emerging fields is epigenetics, in particularly DNA methylation. In this review, we will discuss the recent progress in DNA methylation analysis in IBD and how it can be used as a potential therapeutic target.
DNA METHYLATION ENSEMBLE IN IBD
By definition, epigenetics refers to a heritable change in gene expression phenotype that does not involve alterations in DNA sequence. DNA methylation, histone modifications and non-coding RNA are the three major components involved in epigenetic mechanism. In DNA methylation, the addition of a methyl group at the 5th position of cytosine (5mC) is common on CpG dinucleotides in eukaryotic genomes[7]. Methylation of CpG rich regions (CpG islands) are relative lower and are usually associated with transcription silencing when the methylated CpG islands occur at gene promoters[8]. DNA methylation is catalyzed by enzymes known as DNA methyltransferases and the reaction is reversible. Methyl groups can be edited and removed via actions of DNA demethylases during specific time points such as gametogenesis and disease onset including IBD. In this section, we discuss the roles of players mediating DNA methylation in the context of IBD development.
DNA methylation authors
Highly heritable and bona fide DNA methylation is attributed towards the actions of DNA methyltransferase. In the pathogenesis of IBD and IBD-CRC, three major DNA methyltransferases (DNMT) have been proposed to be involved, including DNMT1, DNMT3a and DNMT3b (Figure 1).
Figure 1.
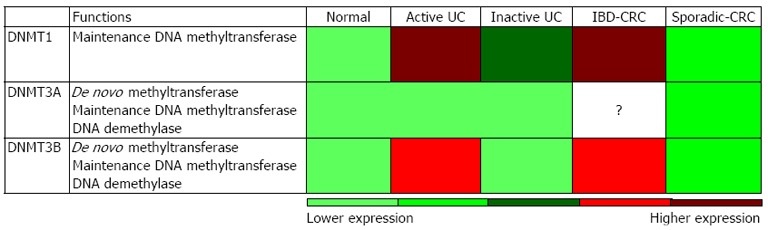
Potential relative expression levels of DNA methyltransferase in active-ulcerative colitis, inactive-ulcerative colitis, inflammatory bowel disease-associated colorectal cancer and sporadic-colorectal cancer patient specimens consolidate from several studies. DNA methyltransferase (DMNTs) is primarily responsible for DNA methylation maintenance, whereas DNMT3A/B have additional roles in de novo DNA methylation and demethylation functions. The relative DNMTs expressions were built on consolidated reports that were normalized to healthy controls to display potential relative expression in different inflammatory bowel disease associated diseases. UC: Ulcerative colitis; IBD-CRC: Inflammatory bowel disease-associated colorectal cancer.
DNMT1 is a key maintenance methyltransferase that primarily methylates hemimethylated DNA in the genome during DNA replication. During IBD and IBD-CRC development, DNMT1 activity is significantly up-regulated[9-11]. DNMT1 is highly expressed in actively inflamed colonic mucosa in UC patients as compared to normal or quiescent UC colonic mucosa[11]. In IBD-CRC, Foran et al[9] compared the methylation profiles of 36 IBD-CRC vs 44 sporadic CRC tumour specimens and demonstrated increased nuclear localization of DNMT1 in IBD-CRC than in sporadic-CRC, evidence linking inflammation-mediated DNMT1 activity. In addition, overexpression of DNMT1 is proposed to correlate with an abundance of CD68 positive macrophages, suggesting direct involvement of DNA methylation in a pro-inflammatory response[9]. Stimulation of HCT116 human colon cancer cells with interleukin (IL)-6 increases and stabilizes DNMT1 expression, leading to increase levels of global methylcytosine, especially at gene promoter regions[9]. This effect by IL-6 is mediated through AKT (Protein Kinase B), but not signal transducer and activator of transcription 3 (STAT3) or c-Jun N-terminal kinase (Jnk), pathway in Hela human cervical cancer cells[9]. Alternatively, another group showed that STAT3 binds directly onto the DNMT1 promoter in malignant T cell lymphoma that is responsible for inducing DNMT1 expression[12]. All these suggest that specific cell type, temporal, or even inflammatory vs non-inflammatory mechanisms, affect DNMT1 expression and activity.
DNMT1 binds to non-intronic upstream enhancer of Foxp3 (forkhead box P3), a locus required to induce the development of regulatory T cells (Treg) capable of suppressing broad ranges of inflammatory responses such as colitis[13]. Stimulation with IL-6 has been proposed to increase methylation in upstream enhancer regions of Foxp3 in Treg cells, resulting in down-regulation of both mRNA and protein expression. This effect was not observed in STAT3-deficient Treg, providing additional evidence on the involvement of the STAT3-signalling pathway in the methylation process[13]. In a separate study, Li et al[14] reported that IL-6-associated STAT3 signalling is highly dependent on DNMT1 enzymatic activity. They showed that IL-6-induced DNMT1 expression results in hypermethylation on the promoter of suppressor of cytokine-signaling-3 (SOCS3), a negative regulator of IL-6 signalling. The decreased SOCS3 expression may then promote full pro-oncogenic effects of STAT3.
DNMT3 (includes three known members: DMNT3A, DMNT3B and DMNT3L) is another family of DNA methyltransferase. Although DNMT3 acts primarily for de novo methylation during gametogenesis and development, many reports have shown that DNMT3 can serve cooperatively with DNMT1 to regulate bona fide DNA methylation maintenance. Active UC colonic mucosa showed higher DNMT3B expression as compared to normal colonic samples or quiescent UC colon patient samples, but relatively lower than that of DNMT1[11]. Similarly, IBD-associated neoplasm lesions showed up-regulation of DNMT3B expression as compared to colonic epithelium without any neoplastic changes[15]. Conversely, human colorectal cancer cell lines (HCT15, DLD1, Col15, HT29, SW480 and RKO) are hypermethylated on the distal DNMT3B promoter as compared to healthy colon tissues, correlating it to the low expression level that results in hypomethylation of many of its target gene promoters[16]. These different observations may suggest that the etiology of IBD-CRC and sporadic-CRC are mechanistically distinct.
DNMT3A has been shown to play an important role in both innate and adaptive immune responses. For example, DNMT3A affects T cell polarization through IL-4 and interferon gamma (IFNγ) promoter methylation upon ligation of T cell receptors[17]. In UC patient’s peripheral T cells, levels of methylation within IFNγ promoter regions have been reported to correlate to the immune response against microbial antigens[18]. In addition, DNMT3A hypermethylates the CpG islands within the tumour necrosis factor alpha (TNFα) promoter region in the context of LPS stimulation[19]. However, another study has proposed no alteration of DNMT3A expression levels in colonic mucosa of UC patients[11]. It is possible that a modification of DNA methylation status during UC pathogenesis is mediated primarily via DNMT1 and DNMT3B. In contrast, meta-analysis of GWAS data has suggested DNMT3A as an important risk loci associated with CD[4]. Therefore, it is likely that methylation status in CD vs UC is controlled by different mechanisms.
In a clinical setting, the differential expression, involvement, and activities of DNMTs can provide additional options as a diagnosis marker tool to monitor IBD and IBD-CRC progression in patients.
DNA methylation editors
Editing and removal of methyl groups from 5mC can be actively or passively achieved through actions of DNA demethylases. Passive DNA demethylation blocks additional methylation during DNA replication by methylation dilution, or by inactivating DNMTs. Over the years, the search for active demethylases has been hindered by the fact that demethylation process is controlled by indirect multi-step mechanisms. DNA demethylation processes appear to be executed through DNA repair and base excision mechanisms, rather than direct removal of the methyl group from the 5mC moiety[20]. Recently, three proteins have been reported to potentially possess demethylase activity, including ten-eleven translocation (TET) methylcytosine dioxygenase, thymine DNA glycosylase (TDG), and activation-induced cytidine deaminase (AID).
TET converts 5mC to 5-hydroxymethylcytosine (5hmC) that is predicted to lift the repression of gene expression imposed by 5mC in both humans and mice[21]. Recently, Neves-Costa et al[22] demonstrated that TET1 negatively regulates the expression and secretion of a pro-inflammatory cytokine IL-1β in a THP-1 monocytic leukemia cell line. In addition, TET co-operates with TDG in the process of active DNA demethylation. TDG excises the mismatch bases at the deaminated 5mC or its derivatives caused by TET[23]. However to date, neither TET nor TDG has been implicated in the pathogenesis of IBD.
AID is another candidate involved in DNA demethylation[24,25]. AID belongs to the family of apolipoprotein B mRNA-editing catalytic polypeptide (APOBECs), which were extensively studied due to its master regulatory function in antibody diversification in B cells[26]. A process for this antibody diversification includes immunoglobulin class switch recombination (CSR), immunoglobulin somatic hypermutation (SHM), and gene conversion (GC)[27]. AID was originally demonstrated as an enzyme to convert cytosine (C) to uracil (U) for induction of SHM[28]. Subsequently, Morgan et al[24] unveiled an additional and unexpected ability of AID to convert 5mC to thymidine in vitro (5mC→T), suggesting the involvement of AID in DNA demethylation. This conversion of 5mC to T creates a T:G mismatch, which will be excised by T:G mismatch-specific glycosylases (i.e., TDG). The T position will then be replaced with unmethylated C through base excision repair process, thereby concluding a 5mC to unmethylated C transition[29]. Recently, AID has been implicated in the pathogenesis of IBD and IBD-CRC[30,31]. Endo and colleagues showed that colonic AID expression is up-regulated under Th2-mediated colonic inflammatory conditions seen in T cell receptor (TCR)-α knockout mice[30]. In addition, ectopic expression of AID in colonic epithelial cells (CECs) was elicited in UC (54%) and IBD-CRC (80%) patients[30]. In contrast, AID expression was seen in only 40% of sporadic colon cancer, indicating the differential pathogenesis between IBD-CRC and sporadic-CRC with respect to AID functions. AID expression may be induced via IKK (IκB kinase)-dependent NF-κB signalling and further enhanced by Th2 cytokines such as IL-4 and IL-13[30]. Functionally, overexpression of AID in CECs has been reported to tremendously increase mutations within some, but not all, oncogenes including p53. Importantly, such mutations were significantly reduced in AID deficient mice[30,31]. However, it still remains largely unknown whether AID plays any specific roles in IBD and/or IBD-CRC through its epigenetic (demethylation) modification ability rather than its classical functions (SHM, CSR, and/or GC). The co-relationship between aberrant AID expression and IBD/IBD-CRC progression suggests that further studies on the role of AID-mediated epithelial homeostasis can potentially be translated into a therapeutic strategy for IBD patients by targeting AID.
In 2008, Kangaspeska et al[32] and Métivier et al[33] reported that DNMT3A and DNMT3B are recruited to gene promoters during transcription and they directly mediate cyclical demethylation and also remethylation processes. The identification of deaminase activity in DNMT3A and DNMT3B has received tremendous attention in the epigenetic field. Since it is clear now that DNMT3A and DNMT3B have dual functions for demethylation and methylation, the idea of dynamic methylation patterns during transcription will be further discussed in the following section.
DYNAMICS OF DNA METHYLATION FROM AN IBD PERSPECTIVE
Covalent modification of DNA through the addition of methyl moieties on CpG dinucleotides is highly stable and conserved. These epigenetic marks, however, do undergo dynamic changes at specific time points, including embryonic development and during perturbed cellular homeostasis such as increased cellular stress and disease onset. Thus, these temporal changes will have important implications that are relevant to the development of IBD.
During germ cell specification and post-fertilization, 5mC undergo de novo erasure and subsequent reprogramming[34]. The consequences of such wholescale DNA methylation reprogramming include formation of parental specific gene expression, including X-linked effects and genomic imprinting, of which gene expression are predominately contributed by specific parental allele. Several lines of evidence have demonstrated the parent-of-origin effects in IBD. As one of the earliest reports, Akolkar et al[35] demonstrated a familial association of IBD. In this study, clinical data analysis of 135 families showed that offspring of IBD affected mothers had higher risk for CD than offspring of fathers with IBD (P = 0.00001). Indeed, sex of parent seemed to play a role in IBD susceptibility and genetic imprinting process, at least in part, by DNA methylation. Fransen et al[36] recently present limited evidence for genomic imprinting effects of IBD susceptibility genes. They analysed 28 IBD susceptibility gene locus and found that IL12B, PR domain containing 1 (PRDM1) and nucleotide-binding oligomerization domain containing 2 (NOD2; L1007fs variant) have genomic imprinting effect. Recently, Schaible et al[37] showed that the offspring from female mice fed with methyl donor supplements (folic acid, betaine and vitamin B12) had a striking susceptibility towards dextran sulfate sodium (DSS)-induced colitis as compared to control mice fed with regular diet. These effects were also reflected with colonic mucosal DNA methylation profile alterations and prolonged gene expression changes, as well as difference in bacteria microflora when compared to mice with control diet. Therefore, better characterization of the effects and mechanisms of imprinting and parent-of origin can be utilized as a clinical risk predictor of IBD for offsprings of IBD susceptible parents in the future.
Another incidence where DNA methylation dynamics is activated is when colonic cellular homeostasis is perturbed such as during oxidative stress, which results in global loss or gain in DNA methylation. Oxidative stress and damage are common phenomenon in IBD and IBD-CRC that are mainly contributed by the reactive oxygen species produced by inflammatory cells[38,39]. Oxidative damage in cells induces recruitment of DNMT1 to the affected chromatin and forms a complex consisting of DNMT3B and members of the polycomb repressive complex 4, including Sirtuin-1 (SIRT1), Enhancer of zeste homolog 2 (EZH2) and embryonic ectoderm development (EED), to re-establish DNA methylation pattern after the DNA is repaired[40]. These key components relocalize from non-GC-rich regions to GC-rich regions[40]. The observation was validated in an in vivo model of colitis where infection with human commensal enterotoxigenic Bacteroides fragilis (ETBF) into a mouse model of adenomatous polyposis coli in Multiple intestinal neoplasia (Min) mice induced inflammation and tumorigenesis[40]. This model may provide a good putative explanation on the mechanism of how certain specific genes are hypermethylated, whereby other loci are hypomethylated, within the same cell, during disease onset.
Despite well-established consensus that DNA methylation is a highly stable modification on the DNA under steady state, two recent reports have changed this conventional perspective. In the first report, Kangaspeska et al[32] showed that estrogen receptor α (ERα) induces waves of transcription of its target promoter that involves series of active and cyclical demethylation and remethylation during the course of transcriptional activation. DNA methylation status were quantified using glutathione S-transferase tagged methyl binding domain (GST-MBD) pull-down assay, which showed a periodicity of 100 min at the ERα target pS2 gene promoter. The second report by Métivier et al[33] showed similar cyclical demethylation-methylation effects at the pS2 promoter, and further provided evidence of DNMTs are present at the promoter during transcription activation and is involved in both demethylation and remethylation processes. Specifically, methylated CpG were deaminated by DNMT3A and DNMT3B, resulting in a base-pair mismatch that is subsequently repaired by base-excision machinery. These two reports pioneered a previously unreported cyclical methylation-demethylation association with transcription and that this process is mediated by DNMTs, proteins previously tightly linked to only methylation but not demethylation. The authors have validated this observation in other promoters including ERα, trefoil factor 3 (TFF3), and potassium inwardly-rectifying channel, subfamily J, member 8 (KCNJ8). Interestingly, these selected validated genes have previously been implicated in different studies of IBD and IBD-CRC in patients or animal models[41-44]. TFF3 is secreted by intestinal goblet cells that forms part of the enteric mucus layer and has a role in epithelial repair and restitution[45]. TFF3-/- mice are less reactive towards mounting repair response during colonic injury induced by chemical, hypoxia and radiation stress[46-48]. Another of the validated candidates is KCNJ8 (also known as KIR6.1), which forms the pore-forming sub-unit of the ATP-sensitive potassium channel (KATP). hydrogen sulphide (H2S), produced by colonic smooth muscles, neurons and other enteric cell types, activates and opens KATP channels in a 2,4,6-trinitrobenzene sulfonic acid (TNBS)-induced murine colitis model[44]. Similarly in TNBS-induced colitis in rats, the production and effects of H2S is associated with the resolution of colitis[49]. Nevertheless, whether cyclical demethylation-remethylation process plays any roles in the pathogenesis of IBD remains elusive, and further extensive studies will be required.
IMPACT OF ENTERIC MICROBES IN IBD HOST DNA METHYLATION
It has become increasingly apparent that dysregulated host microbial interactions contribute to the induction, exacerbation and perpetuation of IBD. Importantly, commensal microbes have an ability to alter DNA methylation status. Mice that were housed in germ free (GF) conditions exhibited hypermethylation of the chemokine ligand CXCL16 [chemokine (C-X-C motif) ligand 16] in the colon, as compared to mice kept under specific pathogen-free (SPF) environment[50]. CXCL16 expressed on the surface of antigen-presenting cells, including subsets of CD19+ B cells and CD14+ monocytes/macrophages, mediates the adhesion and phagocytosis of gram-negative and positive bacteria[51,52]. Soluble CXCL16 can also act as a strong chemo-attractant for CXCR6+ [chemokine (C-X-C motif) receptor 6] T cells[53,54]. Up-regulation of CXCL16 mRNA and protein has been reported in CD patients[55]. Hypermethylation of CXCL16 gene in GF mice leads to the gene activation and accumulation of invariant natural killer T (iNKT) cells, in the colonic lamina propria. iNKT cells are highly conserved subset of T cells expressing a semi-invariant T cell receptor, which is restricted to CD1d and specific for the glycosphingolipid antigen α-galactosylceramide. Furthermore, the activated CXCL16 pathway made GF mice more susceptible against oxazolone-induced Th2-type of acute colitis as compared to SPF mice[50]. Importantly, colonization of neonatal GF mice with a conventional microbiota reduced hypermethylation of CXCL16 to SPF level[50]. However, this phenomenon was not observed when adult GF mice were colonized with the same conventional microbiota, indicating that early-life microbial exposure has a significant impact on host epigenetic status[50]. In addition, recent studies showed that oral inoculation of lipoteichoic acid (LTA)-deficient Lactobacillus acidophilus bacteria (NCK2025), protect mice from colitis-associated cancer presumably by restoring aberrant DNA methylation pattern of cancer-specific genes[56,57]. LTA is a major immunostimulatory component of cell wall of Gram-positive bacteria, which can specifically bind to CD14 and toll-like receptors (TLRs) such as TLR2 on host cells. It is well known that host-microbial recognition is attributed to TLRs. Of note, TLR2 deficient (Tlr2-/-) mice were characterized by low abundance of intestinal Firmicutes and high proportion of Proteobacteria, Bacteroidetes and Actinonbacteria, as compared to wild-type mice[58]. This specific change in microbial composition was associated with epigenomic alterations. For instance, 1.4% of the interrogated genome in Tlr2-/- mice was differentially methylated[58]. Female wild-type C57BL/6J mice that were given methyl-donor supplemented diet produce offspring that exhibit different microbiome profile at postnatal day 30, as compared to control diet offspring[59]. All these data cumulatively suggest that the commensal microbiota can directly influence the status of host DNA methylation and therefore may have important implications in IBD development.
In addition to how bacteria affect the host DNA methylome, the status of DNA methylation on exogenous sources of DNA, in this case bacterial DNA and host self-DNA, also plays a role in the pathogenesis of autoimmune diseases such as IBD. Bacterial DNA has high CpG frequencies but is predominately unmethylated and has immunostimulatory effect[60]. It was originally shown that the introduction of bacterial CpG motifs oligodeoxynucleotides exacerbates existing intestinal inflammation in DSS-treated mice[61]. Recent studies showed that the unmethylation status of bacterial DNA is the predominate factor to induce human plasmacytoid dendritic cells to produce high levels of interferon-alpha (IFN-α), since methylation of the bacterial DNA abolished this induction[62]. These unmethylated CpG DNAs are recognised by the host toll-like receptor 9 (TLR9)[63]. Specific CpG motifs (purine-purine-CpG-pyrimidine-pyrimidine) common in microbial DNA, but which are rare in mammalian DNA, have the strongest activation potential of TLR9[64]. In contrast to bacterial DNA, mammalian DNA has lower CpG frequencies and is predominately methylated, with an exception of CpG islands. There are now increasing evidence that these mammalian self-DNA, presumably released from necrotic cells, can also be an effective TLR9 ligand[64]. Under normal circumstances, the host immune system is protected against self-DNA because of the intracellular location of TLR9. However, during IBD progression, natural antimicrobial peptide LL37 is expressed on the mucosa surfaces and form an immuno-complex with self-DNA, which may lead to the activation of TLR9[65]. Yasuda et al[64] showed that CpG-rich DNA from mammalian DNA, commonly found on CpG islands, are optimal sequence to activate TLR9 and suggested a possible contribution towards autoimmune diseases pathogenesis. However, this TLR9 activation by methylated self-DNA is still comparatively lower than those of unmethylated bacterial DNA[62]. As such, it was proposed that the initiation of autoimmune disease, such as IBD, is initiated by unmethylated microbial DNA whereas subsequent autoimmunity is mediated by methylated (or unmethylated) self-DNA[62]. Therefore, appropriately targeting self-DNA mediated immune responses may be another attractive option to reduce the perpetuation of inflammation in IBD.
In summary, bacterial genetics have a direct impact on host epigenetics. Similarly, bacterial and host (self-DNA) epigenetics can also directly affect host genetics to trigger inflammatory responses (Figure 2).
Figure 2.
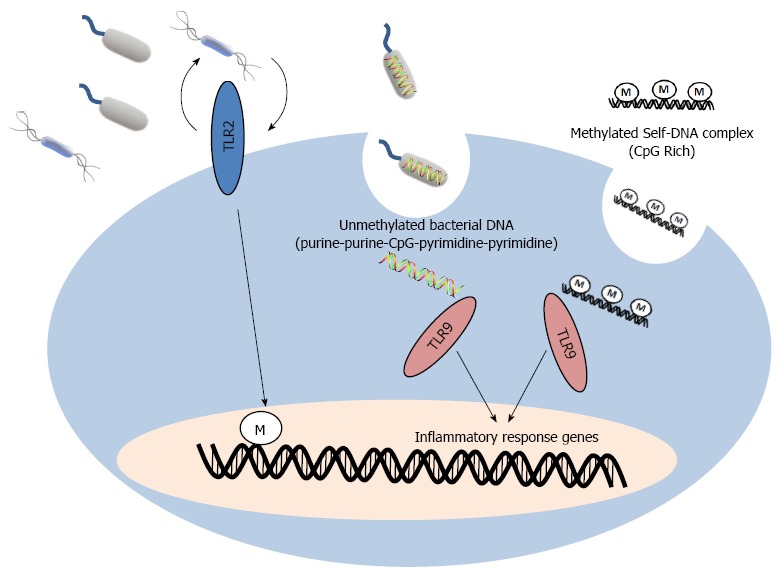
Host genetics and epigenetics alterations by commensal bacterial and self-DNA. Alterations in intestinal microflora or host pathogen recognition functions, such as toll-like receptor (TLR)2, directly affect host DNA methylation. Endocytosis of bacterial and release of unmethylated bacterial DNA into host cell triggers inflammatory response via TLR9. Strong activation requires a purine-purine-CpG-pyrimidine-pyrimidine bacterial DNA motif. Endocytosis of CpG rich methylated self-DNA also activates TLR9 to induce similar inflammatory response via TLR9, but with on a less magnitude compared to stimulation via bacterial DNA.
GENOME-WIDE DNA METHYLOME PROFILES IN IBD
Recent advances on genomic/epigenetic technologies targeting the “omics” level have contributed to a plethora of reports on genome-wide DNA methylome analysis to study the pathogenesis of IBD. The information derived from the analyses in IBD will provide significant rationale to open up a new avenue to develop novel diagnostic and therapeutic strategies. Indeed, genome-wide altered methylation patterns have been shown to be enriched around GWAS identified loci[66,67]. In addition, methylome profiling may also resolve the differences in etiology and pathogenesis of UC vs CD.
Nimmo et al[67] recently profiled the methylome of whole blood genomic DNA from 21 ileal CD patients and 19 healthy controls. They identified 1117 CpG sites that are differentially methylated. Within the list, 35 genes overlapped with previous GWAS identified CD loci, including NOD2, TNFα and caspase recruitment domain family, member 9 (CARD9). Comparative analysis of these gene hits showed that differentially methylated CpG sites are located within 25-100 kb of the 71 previously identified GWAS CD loci. Importantly, sex, environmental and individual lifestyle (e.g., non-smoking and immunomodulatory therapy status) factors were taken into consideration for the selection of cohort in this study because these factors are influential in determining IBD, as well as epigenetic changes. This is especially apparent as seen from the high discordance rate of CD (68%) and UC (85%) in monozygotic twins, who had identical genomes[68]. The immediate question is how these identical genomes in monozygotic twins divert into different phenotypes outcome. A recent report studied 20 monozygotic twins discordant for UC and investigated the genomic profile based on three-layers of genome-wide scans, including transcriptome profiling, genome-wide methylation variable positions (MVPs) and genome-wide differentially methylation regions (DMRs)[69]. In this study, they identified 61 disease loci defined by differential gene expression profile and at least one MVP or DMR position within 50 kb from the transcription start site. Promoter regions of these hits showed prominent hypomethylation, whereas gene-intronic regions were more frequently hypermethylated. However, none of these 61 loci overlapped with the previously reported 47 UC GWAS risk loci[5]. Nevertheless, environmental factors and lifestyle surely contribute to the pathogenesis of IBD and provide the most direct clues to understand how identical genomes from monozygotic twins can have distinct susceptibility to IBD. IBD usually occurs during young adulthood and the peak age of onset is around 15-30 years old[70,71]. Thus, identification of the changes in methylome during crucial developmental time point can provide great insights on IBD risk. Studies showed that postnatal day 90 mice had increase susceptibility to DSS-induced colitis as compared to postnatal day 30 mice[59]. Methylation specific amplification microarray (MSAM) revealed 271 differential methylation genomic intervals between the above two mice groups[59]. These results suggest that age-dependent methylation dynamics is another important aspect to consider in the risk of IBD.
In addition to prying into the individual genomic status in UC or CD as compared to normal individuals, epigenome-wide profiling can also dissect the differences in disease-associated loci between UC and CD. Cooke et al[66] recently characterized the genome-wide methylation changes in the rectal samples obtained from patients with inflamed UC/CD and non-inflamed UC/CD. Consistent with other reports, many identified loci in this study overlapped with GWAS-identified risk loci, including CARD9, intercellular adhesion molecule 3 (ICAM3) and cadherin 1 (CDH1). Inflamed UC and CD, as well as non-inflamed UC formed individual methylome signatures when compared to normal control individuals. Interestingly, there was no difference in the methylation profile between inflamed UC and inflamed CD. In contrast, 13 differentially methylated loci were identified between non-inflamed UC and non-inflamed CD. These multiple comparison suggests that the different sub-types of IBD, as well as disease severity, may be distinguished by their methylome status. In addition, Lin et al[72,73] also reported the methylome profiles of UC and CD patients derived B cells and intestinal tissues. Therefore, the methylome may be one of the useful clinical diagnostic biomarkers in IBD (Table 1). However, much more careful attention would be necessary in this regard because different cell types exhibit different methylomes in IBD.
Table 1.
High throughput DNA methylome profiling in inflammatory bowel disease
| Ref. | Disease | Tissue/cell | Array platform | Significant differential methylation | GWAS overlap |
| Human patients | |||||
| Lin et al[73] | UC and CD | Intestinal | Illumina goldengate | 7 CpG sites | Not reported |
| Cooke et al[66] | UC and CD | Rectal | Illumina infinium human methylation 27 | 3604 (UC) and 472 (CD) loci | Yes |
| Lin et al[72] | UC and CD | B cell | Illumina goldengate | 24 (UC) and 14 (CD) CpG sites | Not reported |
| Häsler et al[69] | UC | Intestinal | Illumina human methylation 27 and nimblegen custom 385K | 61 loci | No |
| Nimmo et al[67] | CD | Whole blood | Illumina human methylation 27 | 1117 CpG sites | Yes |
| Mouse | |||||
| Kellermayer et al[59] | DSS colitis (postnatal day 30 vs day 90) | Colon | Custom array (Agilent) | 271 intervals | Not reported |
| Kellermayer et al[58] | Tlr2-/- | Colon | Custom array (Agilent) | 387 intervals | Not reported |
UC: Ulcerative colitis; CD: Crohn’s disease; GWAS: Genome-wide association studies; DSS: Dextran sulfate sodium.
EPI-THERAPY TARGETING DNA METHYLATION IN IBD
Several compounds targeting DNA methylation status has been demonstrated to have potential therapeutic effects on animal models of IBD and/or human IBD patients. One of these compounds is folate, a methyl donor that exerts an effect to increase global methylation. Chronic UC patients that were given dietary folinic acid, a vitamer of folic acid, supplementation (15 mg/d) had a lower risk of colon cancer[74]. Kominsky et al[75] recently also showed that intraperitoneal injection of folate (50 mg/kg) into DSS-treated mice results in less severe colitis. In addition, dietary folate deficiency led to aggravation of DSS-induced colitis in rats[76]. These results are consistent with clinical reports showing that folate deficiencies are common in IBD patients[77-79]. However, oral dietary supplementation of folate did not seem to have an effect on the suppression of IBD-CRC in an azoxymethane/DSS-associated cancer model[80]. In this model, diet supplementation with folic acid (8 mg/kg) did not show any alterations in intestinal microflora or difference in tumor initiation, growth and progression as compared to the control mice without receiving folic acid supplement. One possible reason for this failure is that the chronic inflammation that has transited into tumorigenic stage would have acquired more stable genetic changes including chromosomal instability and translocation, as well as genetic mutations, as compared to acute intestinal inflammation. These alterations in DNA sequence may occur at critical DNA methylated CpG sites and hence global methylation effects of folate can no longer re-establish methylation at these mutated CpG target sites.
Development of small compounds that can directly or indirectly affect DNA methylated mediated gene expression may also be useful targets of IBD treatment. Meng et al[81] demonstrate that using a combination of a novel tylophorine analog W-8, together with TGF-β (transforming growth factor), demethylates Foxp3 promoter. Tylophorine analogs, including W-8, are phenanthroindolizidine alkaloids that have anti-cancer and anti-inflammatory effects. The effect of W-8 is mediated through ERK (extracellular signal-regulated kinase) pathway inhibition that results in the down-regulation of DNMT1 expression. Therefore, W-8 appears to up-regulate Foxp3 expression by demethylating the promoter in the presence of TGF-β and promotes differentiation of naïve CD4+ T cells into Foxp3+ Treg cells with immunosuppression capabilities.
The challenge in developing innovative therapies for IBD has been on-going. Currently, oral and topical aminosalicylates are usually the first-line medication to treat IBD. Other immunosuppressive agents including azathioprine, methotrexate and cyclosporine are also in used. However, the beneficial effects of these drugs are accompanied with detrimental side effects, such as allergy. In addition, not all patients respond to these treatments. Recently, the use of anti-TNFα antibody has also been deployed to control IBD in patients. However, on top of the adverse side effects of anti-TNFα antibody, the administration of the treatment requires invasive intravenous infusion or subcutaneous injection and the high cost of this form of medication, which range from US$3000 to US$8000 per infusion, is a major disadvantage. Therefore epi-therapy drug design is an attractive alternative method to develop an effective, low-cost and non-invasive therapy for IBD patients.
CONCLUSION
DNA methylation has great heuristic potential in improving our understanding of the IBD pathogenesis in the post-GWAS era. Individuals who inherited a normal set of DNA may still be susceptible to IBD depending on epigenetic changes during their course of life. As described in this review, epigenetics changes that may account for IBD risk begin right from the fertilized egg to entire life period (Figure 3). Further advancements in this promising field would allow the discovery of new mediators to control DNA methylation/demethylation, aiming to improve the lives of patients with IBD and IBD-CRC.
Figure 3.
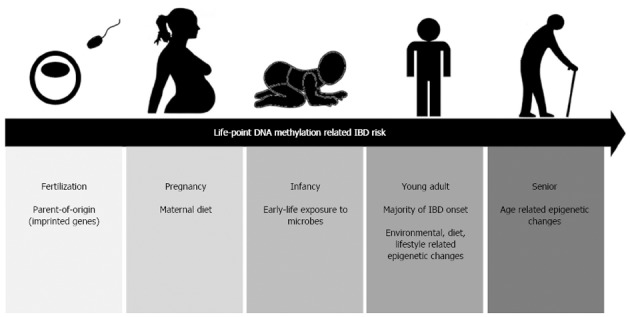
Life-stages with an impact on epigenetic changes that increase inflammatory bowel disease risk. Alterations in DNA methylome in inflammatory bowel disease (IBD) pathogenesis begin right from the fertilized egg. The risk alleles are inherited, and its expression is dependent on the parent-of-origin (imprinting). Maternal diet during pregnancy may also potentially alter the fetal IBD-associated-methylome. Exposures to certain microbes during infancy can also have lasting effects on DNA methylation alteration towards IBD susceptibility. Environment-, lifestyle- and diet- associated DNA methylation changes are important aspects during young adulthood where the majority of IBD onset occurs.
ACKNOWLEDGMENTS
The authors are grateful to Dr. Michael Choi for his helpful discussion and advice. We would like to thank Mr. Alan Kamba’s assistance for preparing this manuscript.
Footnotes
Supported by National Institute of Health (DK80070, DK74454, and DK64289); the Eli and Edythe L. Broad Medical Foundation and American Gastroenterological Association Foundation to Mizoguchi E; and the Singapore A*STAR Graduate Academy (BM/AIF/13/001) to Low D
P- Reviewers Capasso R, Koyama N, Sipos F S- Editor Zhai HH L- Editor A E- Editor Zhang DN
References
- 1.Podolsky DK. Inflammatory bowel disease (1) N Engl J Med. 1991;325:928–937. doi: 10.1056/NEJM199109263251306. [DOI] [PubMed] [Google Scholar]
- 2.Bernstein CN, Blanchard JF, Kliewer E, Wajda A. Cancer risk in patients with inflammatory bowel disease: a population-based study. Cancer. 2001;91:854–862. doi: 10.1002/1097-0142(20010215)91:4<854::aid-cncr1073>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 3.Franke A, Balschun T, Karlsen TH, Hedderich J, May S, Lu T, Schuldt D, Nikolaus S, Rosenstiel P, Krawczak M, et al. Replication of signals from recent studies of Crohn’s disease identifies previously unknown disease loci for ulcerative colitis. Nat Genet. 2008;40:713–715. doi: 10.1038/ng.148. [DOI] [PubMed] [Google Scholar]
- 4.Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, Lees CW, Balschun T, Lee J, Roberts R, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 2010;42:1118–1125. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anderson CA, Boucher G, Lees CW, Franke A, D’Amato M, Taylor KD, Lee JC, Goyette P, Imielinski M, Latiano A, et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat Genet. 2011;43:246–252. doi: 10.1038/ng.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ehrlich M, Gama-Sosa MA, Huang LH, Midgett RM, Kuo KC, McCune RA, Gehrke C. Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res. 1982;10:2709–2721. doi: 10.1093/nar/10.8.2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 9.Foran E, Garrity-Park MM, Mureau C, Newell J, Smyrk TC, Limburg PJ, Egan LJ. Upregulation of DNA methyltransferase-mediated gene silencing, anchorage-independent growth, and migration of colon cancer cells by interleukin-6. Mol Cancer Res. 2010;8:471–481. doi: 10.1158/1541-7786.MCR-09-0496. [DOI] [PubMed] [Google Scholar]
- 10.Fujii S, Katake Y, Tanaka H. Increased expression of DNA methyltransferase-1 in non-neoplastic epithelium helps predict colorectal neoplasia risk in ulcerative colitis. Digestion. 2010;82:179–186. doi: 10.1159/000311064. [DOI] [PubMed] [Google Scholar]
- 11.Saito S, Kato J, Hiraoka S, Horii J, Suzuki H, Higashi R, Kaji E, Kondo Y, Yamamoto K. DNA methylation of colon mucosa in ulcerative colitis patients: correlation with inflammatory status. Inflamm Bowel Dis. 2011;17:1955–1965. doi: 10.1002/ibd.21573. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Q, Wang HY, Woetmann A, Raghunath PN, Odum N, Wasik MA. STAT3 induces transcription of the DNA methyltransferase 1 gene (DNMT1) in malignant T lymphocytes. Blood. 2006;108:1058–1064. doi: 10.1182/blood-2005-08-007377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lal G, Zhang N, van der Touw W, Ding Y, Ju W, Bottinger EP, Reid SP, Levy DE, Bromberg JS. Epigenetic regulation of Foxp3 expression in regulatory T cells by DNA methylation. J Immunol. 2009;182:259–273. doi: 10.4049/jimmunol.182.1.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Y, Deuring J, Peppelenbosch MP, Kuipers EJ, de Haar C, van der Woude CJ. IL-6-induced DNMT1 activity mediates SOCS3 promoter hypermethylation in ulcerative colitis-related colorectal cancer. Carcinogenesis. 2012;33:1889–1896. doi: 10.1093/carcin/bgs214. [DOI] [PubMed] [Google Scholar]
- 15.Ueda H, Tanaka H, Ichikawa K, Itabashi M, Kameoka S, Fujii S, Saito N, Kimura R, Shida Y, Fujimori Y, et al. Immunohistochemical analysis of the DNA methyltransferase 3b expression is associated with significant improvements in the discrimination of ulcerative colitis-associated neoplastic lesions. Surg Today. 2013:Epub ahead of print. doi: 10.1007/s00595-012-0456-6. [DOI] [PubMed] [Google Scholar]
- 16.Huidobro C, Urdinguio RG, Rodríguez RM, Mangas C, Calvanese V, Martínez-Camblor P, Ferrero C, Parra-Blanco A, Rodrigo L, Obaya AJ, et al. A DNA methylation signature associated with aberrant promoter DNA hypermethylation of DNMT3B in human colorectal cancer. Eur J Cancer. 2012;48:2270–2281. doi: 10.1016/j.ejca.2011.12.019. [DOI] [PubMed] [Google Scholar]
- 17.Jones B, Chen J. Inhibition of IFN-gamma transcription by site-specific methylation during T helper cell development. EMBO J. 2006;25:2443–2452. doi: 10.1038/sj.emboj.7601148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gonsky R, Deem RL, Landers CJ, Derkowski CA, Berel D, McGovern DP, Targan SR. Distinct IFNG methylation in a subset of ulcerative colitis patients based on reactivity to microbial antigens. Inflamm Bowel Dis. 2011;17:171–178. doi: 10.1002/ibd.21352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.El Gazzar M, Yoza BK, Chen X, Hu J, Hawkins GA, McCall CE. G9a and HP1 couple histone and DNA methylation to TNFalpha transcription silencing during endotoxin tolerance. J Biol Chem. 2008;283:32198–32208. doi: 10.1074/jbc.M803446200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bhutani N, Burns DM, Blau HM. DNA demethylation dynamics. Cell. 2011;146:866–872. doi: 10.1016/j.cell.2011.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neves-Costa A, Moita LF. TET1 is a negative transcriptional regulator of IL-1β in the THP-1 cell line. Mol Immunol. 2013;54:264–270. doi: 10.1016/j.molimm.2012.12.014. [DOI] [PubMed] [Google Scholar]
- 23.Cortellino S, Xu J, Sannai M, Moore R, Caretti E, Cigliano A, Le Coz M, Devarajan K, Wessels A, Soprano D, et al. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell. 2011;146:67–79. doi: 10.1016/j.cell.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morgan HD, Dean W, Coker HA, Reik W, Petersen-Mahrt SK. Activation-induced cytidine deaminase deaminates 5-methylcytosine in DNA and is expressed in pluripotent tissues: implications for epigenetic reprogramming. J Biol Chem. 2004;279:52353–52360. doi: 10.1074/jbc.M407695200. [DOI] [PubMed] [Google Scholar]
- 25.Popp C, Dean W, Feng S, Cokus SJ, Andrews S, Pellegrini M, Jacobsen SE, Reik W. Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature. 2010;463:1101–1105. doi: 10.1038/nature08829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Conticello SG, Langlois MA, Yang Z, Neuberger MS. DNA deamination in immunity: AID in the context of its APOBEC relatives. Adv Immunol. 2007;94:37–73. doi: 10.1016/S0065-2776(06)94002-4. [DOI] [PubMed] [Google Scholar]
- 27.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–563. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 28.Petersen-Mahrt SK, Harris RS, Neuberger MS. AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature. 2002;418:99–103. doi: 10.1038/nature00862. [DOI] [PubMed] [Google Scholar]
- 29.Hardeland U, Bentele M, Jiricny J, Schär P. The versatile thymine DNA-glycosylase: a comparative characterization of the human, Drosophila and fission yeast orthologs. Nucleic Acids Res. 2003;31:2261–2271. doi: 10.1093/nar/gkg344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Endo Y, Marusawa H, Kou T, Nakase H, Fujii S, Fujimori T, Kinoshita K, Honjo T, Chiba T. Activation-induced cytidine deaminase links between inflammation and the development of colitis-associated colorectal cancers. Gastroenterology. 2008;135:889–898, 898.e1-3. doi: 10.1053/j.gastro.2008.06.091. [DOI] [PubMed] [Google Scholar]
- 31.Takai A, Marusawa H, Minaki Y, Watanabe T, Nakase H, Kinoshita K, Tsujimoto G, Chiba T. Targeting activation-induced cytidine deaminase prevents colon cancer development despite persistent colonic inflammation. Oncogene. 2012;31:1733–1742. doi: 10.1038/onc.2011.352. [DOI] [PubMed] [Google Scholar]
- 32.Kangaspeska S, Stride B, Métivier R, Polycarpou-Schwarz M, Ibberson D, Carmouche RP, Benes V, Gannon F, Reid G. Transient cyclical methylation of promoter DNA. Nature. 2008;452:112–115. doi: 10.1038/nature06640. [DOI] [PubMed] [Google Scholar]
- 33.Métivier R, Gallais R, Tiffoche C, Le Péron C, Jurkowska RZ, Carmouche RP, Ibberson D, Barath P, Demay F, Reid G, et al. Cyclical DNA methylation of a transcriptionally active promoter. Nature. 2008;452:45–50. doi: 10.1038/nature06544. [DOI] [PubMed] [Google Scholar]
- 34.Smallwood SA, Kelsey G. De novo DNA methylation: a germ cell perspective. Trends Genet. 2012;28:33–42. doi: 10.1016/j.tig.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 35.Akolkar PN, Gulwani-Akolkar B, Heresbach D, Lin XY, Fisher S, Katz S, Silver J. Differences in risk of Crohn’s disease in offspring of mothers and fathers with inflammatory bowel disease. Am J Gastroenterol. 1997;92:2241–2244. [PubMed] [Google Scholar]
- 36.Fransen K, Mitrovic M, van Diemen CC, Thelma BK, Sood A, Franke A, Schreiber S, Midha V, Juyal G, Potocnik U, et al. Limited evidence for parent-of-origin effects in inflammatory bowel disease associated loci. PLoS One. 2012;7:e45287. doi: 10.1371/journal.pone.0045287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schaible TD, Harris RA, Dowd SE, Smith CW, Kellermayer R. Maternal methyl-donor supplementation induces prolonged murine offspring colitis susceptibility in association with mucosal epigenetic and microbiomic changes. Hum Mol Genet. 2011;20:1687–1696. doi: 10.1093/hmg/ddr044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dincer Y, Erzin Y, Himmetoglu S, Gunes KN, Bal K, Akcay T. Oxidative DNA damage and antioxidant activity in patients with inflammatory bowel disease. Dig Dis Sci. 2007;52:1636–1641. doi: 10.1007/s10620-006-9386-8. [DOI] [PubMed] [Google Scholar]
- 39.Roessner A, Kuester D, Malfertheiner P, Schneider-Stock R. Oxidative stress in ulcerative colitis-associated carcinogenesis. Pathol Res Pract. 2008;204:511–524. doi: 10.1016/j.prp.2008.04.011. [DOI] [PubMed] [Google Scholar]
- 40.O’Hagan HM, Wang W, Sen S, Destefano Shields C, Lee SS, Zhang YW, Clements EG, Cai Y, Van Neste L, Easwaran H, et al. Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG Islands. Cancer Cell. 2011;20:606–619. doi: 10.1016/j.ccr.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Konishi K, Shen L, Wang S, Meltzer SJ, Harpaz N, Issa JP. Rare CpG island methylator phenotype in ulcerative colitis-associated neoplasias. Gastroenterology. 2007;132:1254–1260. doi: 10.1053/j.gastro.2007.01.035. [DOI] [PubMed] [Google Scholar]
- 42.Podolsky DK, Gerken G, Eyking A, Cario E. Colitis-associated variant of TLR2 causes impaired mucosal repair because of TFF3 deficiency. Gastroenterology. 2009;137:209–220. doi: 10.1053/j.gastro.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mathias R, von der Weid PY. Involvement of the NO-cGMP-K(ATP) channel pathway in the mesenteric lymphatic pump dysfunction observed in the guinea pig model of TNBS-induced ileitis. Am J Physiol Gastrointest Liver Physiol. 2013;304:G623–G634. doi: 10.1152/ajpgi.00392.2012. [DOI] [PubMed] [Google Scholar]
- 44.Gade AR, Kang M, Akbarali HI. Hydrogen sulfide as an allosteric modulator of ATP-sensitive potassium channels in colonic inflammation. Mol Pharmacol. 2013;83:294–306. doi: 10.1124/mol.112.081596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Taupin D, Podolsky DK. Trefoil factors: initiators of mucosal healing. Nat Rev Mol Cell Biol. 2003;4:721–732. doi: 10.1038/nrm1203. [DOI] [PubMed] [Google Scholar]
- 46.Mashimo H, Wu DC, Podolsky DK, Fishman MC. Impaired defense of intestinal mucosa in mice lacking intestinal trefoil factor. Science. 1996;274:262–265. doi: 10.1126/science.274.5285.262. [DOI] [PubMed] [Google Scholar]
- 47.Furuta GT, Turner JR, Taylor CT, Hershberg RM, Comerford K, Narravula S, Podolsky DK, Colgan SP. Hypoxia-inducible factor 1-dependent induction of intestinal trefoil factor protects barrier function during hypoxia. J Exp Med. 2001;193:1027–1034. doi: 10.1084/jem.193.9.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Beck PL, Wong JF, Li Y, Swaminathan S, Xavier RJ, Devaney KL, Podolsky DK. Chemotherapy- and radiotherapy-induced intestinal damage is regulated by intestinal trefoil factor. Gastroenterology. 2004;126:796–808. doi: 10.1053/j.gastro.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 49.Wallace JL, Vong L, McKnight W, Dicay M, Martin GR. Endogenous and exogenous hydrogen sulfide promotes resolution of colitis in rats. Gastroenterology. 2009;137:569–78, 578.e1. doi: 10.1053/j.gastro.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 50.Olszak T, An D, Zeissig S, Vera MP, Richter J, Franke A, Glickman JN, Siebert R, Baron RM, Kasper DL, et al. Microbial exposure during early life has persistent effects on natural killer T cell function. Science. 2012;336:489–493. doi: 10.1126/science.1219328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wilbanks A, Zondlo SC, Murphy K, Mak S, Soler D, Langdon P, Andrew DP, Wu L, Briskin M. Expression cloning of the STRL33/BONZO/TYMSTRligand reveals elements of CC, CXC, and CX3C chemokines. J Immunol. 2001;166:5145–5154. doi: 10.4049/jimmunol.166.8.5145. [DOI] [PubMed] [Google Scholar]
- 52.Shimaoka T, Nakayama T, Kume N, Takahashi S, Yamaguchi J, Minami M, Hayashida K, Kita T, Ohsumi J, Yoshie O, et al. Cutting edge: SR-PSOX/CXC chemokine ligand 16 mediates bacterial phagocytosis by APCs through its chemokine domain. J Immunol. 2003;171:1647–1651. doi: 10.4049/jimmunol.171.4.1647. [DOI] [PubMed] [Google Scholar]
- 53.Nanki T, Shimaoka T, Hayashida K, Taniguchi K, Yonehara S, Miyasaka N. Pathogenic role of the CXCL16-CXCR6 pathway in rheumatoid arthritis. Arthritis Rheum. 2005;52:3004–3014. doi: 10.1002/art.21301. [DOI] [PubMed] [Google Scholar]
- 54.Matsumura S, Wang B, Kawashima N, Braunstein S, Badura M, Cameron TO, Babb JS, Schneider RJ, Formenti SC, Dustin ML, et al. Radiation-induced CXCL16 release by breast cancer cells attracts effector T cells. J Immunol. 2008;181:3099–3107. doi: 10.4049/jimmunol.181.5.3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Diegelmann J, Seiderer J, Niess JH, Haller D, Göke B, Reinecker HC, Brand S. Expression and regulation of the chemokine CXCL16 in Crohn’s disease and models of intestinal inflammation. Inflamm Bowel Dis. 2010;16:1871–1881. doi: 10.1002/ibd.21306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Khazaie K, Zadeh M, Khan MW, Bere P, Gounari F, Dennis K, Blatner NR, Owen JL, Klaenhammer TR, Mohamadzadeh M. Abating colon cancer polyposis by Lactobacillus acidophilus deficient in lipoteichoic acid. Proc Natl Acad Sci USA. 2012;109:10462–10467. doi: 10.1073/pnas.1207230109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lightfoot YL, Yang T, Sahay B, Mohamadzadeh M. Targeting aberrant colon cancer-specific DNA methylation with lipoteichoic acid-deficient Lactobacillus acidophilus. Gut Microbes. 2013;4:84–88. doi: 10.4161/gmic.22822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kellermayer R, Dowd SE, Harris RA, Balasa A, Schaible TD, Wolcott RD, Tatevian N, Szigeti R, Li Z, Versalovic J, et al. Colonic mucosal DNA methylation, immune response, and microbiome patterns in Toll-like receptor 2-knockout mice. FASEB J. 2011;25:1449–1460. doi: 10.1096/fj.10-172205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kellermayer R, Balasa A, Zhang W, Lee S, Mirza S, Chakravarty A, Szigeti R, Laritsky E, Tatevian N, Smith CW, et al. Epigenetic maturation in colonic mucosa continues beyond infancy in mice. Hum Mol Genet. 2010;19:2168–2176. doi: 10.1093/hmg/ddq095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Krieg AM, Yi AK, Matson S, Waldschmidt TJ, Bishop GA, Teasdale R, Koretzky GA, Klinman DM. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature. 1995;374:546–549. doi: 10.1038/374546a0. [DOI] [PubMed] [Google Scholar]
- 61.Obermeier F, Dunger N, Deml L, Herfarth H, Schölmerich J, Falk W. CpG motifs of bacterial DNA exacerbate colitis of dextran sulfate sodium-treated mice. Eur J Immunol. 2002;32:2084–2092. doi: 10.1002/1521-4141(200207)32:7<2084::AID-IMMU2084>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 62.Coch C, Busch N, Wimmenauer V, Hartmann E, Janke M, Abdel-Mottaleb MM, Lamprecht A, Ludwig J, Barchet W, Schlee M, et al. Higher activation of TLR9 in plasmacytoid dendritic cells by microbial DNA compared with self-DNA based on CpG-specific recognition of phosphodiester DNA. J Leukoc Biol. 2009;86:663–670. doi: 10.1189/jlb.0509314. [DOI] [PubMed] [Google Scholar]
- 63.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, et al. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 64.Yasuda K, Richez C, Uccellini MB, Richards RJ, Bonegio RG, Akira S, Monestier M, Corley RB, Viglianti GA, Marshak-Rothstein A, et al. Requirement for DNA CpG content in TLR9-dependent dendritic cell activation induced by DNA-containing immune complexes. J Immunol. 2009;183:3109–3117. doi: 10.4049/jimmunol.0900399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schauber J, Rieger D, Weiler F, Wehkamp J, Eck M, Fellermann K, Scheppach W, Gallo RL, Stange EF. Heterogeneous expression of human cathelicidin hCAP18/LL-37 in inflammatory bowel diseases. Eur J Gastroenterol Hepatol. 2006;18:615–621. doi: 10.1097/00042737-200606000-00007. [DOI] [PubMed] [Google Scholar]
- 66.Cooke J, Zhang H, Greger L, Silva AL, Massey D, Dawson C, Metz A, Ibrahim A, Parkes M. Mucosal genome-wide methylation changes in inflammatory bowel disease. Inflamm Bowel Dis. 2012;18:2128–2137. doi: 10.1002/ibd.22942. [DOI] [PubMed] [Google Scholar]
- 67.Nimmo ER, Prendergast JG, Aldhous MC, Kennedy NA, Henderson P, Drummond HE, Ramsahoye BH, Wilson DC, Semple CA, Satsangi J. Genome-wide methylation profiling in Crohn’s disease identifies altered epigenetic regulation of key host defense mechanisms including the Th17 pathway. Inflamm Bowel Dis. 2012;18:889–899. doi: 10.1002/ibd.21912. [DOI] [PubMed] [Google Scholar]
- 68.Spehlmann ME, Begun AZ, Burghardt J, Lepage P, Raedler A, Schreiber S. Epidemiology of inflammatory bowel disease in a German twin cohort: results of a nationwide study. Inflamm Bowel Dis. 2008;14:968–976. doi: 10.1002/ibd.20380. [DOI] [PubMed] [Google Scholar]
- 69.Häsler R, Feng Z, Bäckdahl L, Spehlmann ME, Franke A, Teschendorff A, Rakyan VK, Down TA, Wilson GA, Feber A, et al. A functional methylome map of ulcerative colitis. Genome Res. 2012;22:2130–2137. doi: 10.1101/gr.138347.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Loftus EV. Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology. 2004;126:1504–1517. doi: 10.1053/j.gastro.2004.01.063. [DOI] [PubMed] [Google Scholar]
- 71.Herrinton LJ, Liu L, Lewis JD, Griffin PM, Allison J. Incidence and prevalence of inflammatory bowel disease in a Northern California managed care organization, 1996-2002. Am J Gastroenterol. 2008;103:1998–2006. doi: 10.1111/j.1572-0241.2008.01960.x. [DOI] [PubMed] [Google Scholar]
- 72.Lin Z, Hegarty JP, Yu W, Cappel JA, Chen X, Faber PW, Wang Y, Poritz LS, Fan JB, Koltun WA. Identification of disease-associated DNA methylation in B cells from Crohn’s disease and ulcerative colitis patients. Dig Dis Sci. 2012;57:3145–3153. doi: 10.1007/s10620-012-2288-z. [DOI] [PubMed] [Google Scholar]
- 73.Lin Z, Hegarty JP, Cappel JA, Yu W, Chen X, Faber P, Wang Y, Kelly AA, Poritz LS, Peterson BZ, et al. Identification of disease-associated DNA methylation in intestinal tissues from patients with inflammatory bowel disease. Clin Genet. 2011;80:59–67. doi: 10.1111/j.1399-0004.2010.01546.x. [DOI] [PubMed] [Google Scholar]
- 74.Biasco G, Zannoni U, Paganelli GM, Santucci R, Gionchetti P, Rivolta G, Miniero R, Pironi L, Calabrese C, Di Febo G, et al. Folic acid supplementation and cell kinetics of rectal mucosa in patients with ulcerative colitis. Cancer Epidemiol Biomarkers Prev. 1997;6:469–471. [PubMed] [Google Scholar]
- 75.Kominsky DJ, Keely S, MacManus CF, Glover LE, Scully M, Collins CB, Bowers BE, Campbell EL, Colgan SP. An endogenously anti-inflammatory role for methylation in mucosal inflammation identified through metabolite profiling. J Immunol. 2011;186:6505–6514. doi: 10.4049/jimmunol.1002805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen M, Peyrin-Biroulet L, George A, Coste F, Bressenot A, Bossenmeyer-Pourie C, Alberto JM, Xia B, Namour B, Guéant JL. Methyl deficient diet aggravates experimental colitis in rats. J Cell Mol Med. 2011;15:2486–2497. doi: 10.1111/j.1582-4934.2010.01252.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zezos P, Papaioannou G, Nikolaidis N, Vasiliadis T, Giouleme O, Evgenidis N. Hyperhomocysteinemia in ulcerative colitis is related to folate levels. World J Gastroenterol. 2005;11:6038–6042. doi: 10.3748/wjg.v11.i38.6038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Erzin Y, Uzun H, Celik AF, Aydin S, Dirican A, Uzunismail H. Hyperhomocysteinemia in inflammatory bowel disease patients without past intestinal resections: correlations with cobalamin, pyridoxine, folate concentrations, acute phase reactants, disease activity, and prior thromboembolic complications. J Clin Gastroenterol. 2008;42:481–486. doi: 10.1097/MCG.0b013e318046eab0. [DOI] [PubMed] [Google Scholar]
- 79.Yakut M, Ustün Y, Kabaçam G, Soykan I. Serum vitamin B12 and folate status in patients with inflammatory bowel diseases. Eur J Intern Med. 2010;21:320–323. doi: 10.1016/j.ejim.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 80.Macfarlane AJ, Behan NA, Matias FM, Green J, Caldwell D, Brooks SP. Dietary folate does not significantly affect the intestinal microbiome, inflammation or tumorigenesis in azoxymethane-dextran sodium sulphate-treated mice. Br J Nutr. 2012:1–9. doi: 10.1017/S0007114512001857. [DOI] [PubMed] [Google Scholar]
- 81.Meng X, Zhang Y, Jia Z, Huo X, He X, Tian G, Wu M, Wang Z, Zhou X, Xiong S, et al. A novel tylophorine analog W-8 up-regulates forkhead boxP3 expression and ameliorates murine colitis. J Leukoc Biol. 2013;93:83–93 ]. doi: 10.1189/jlb.0812402. [DOI] [PubMed] [Google Scholar]