

Abstract
Regulation of protein function by S-nitrosation of critical cysteines is known to be an important mechanism for nitric oxide signaling. Evidence for this comes from several different experimental approaches including the ascorbate-based biotin switch method. However technical problems with specificity and sensitivity of ascorbate reduction of S-nitrosothiols limit its usefulness and reliability. In the current study we report the use of triphenylphosphine ester derivatives to selectively reduce SNO bonds in proteins. After triphenylphosphine ester reduction thiols were tagged with biotin or fluorescently labeled maleimide reagents. Importantly we demonstrate that these compounds are specific reductants of SNO in complex biological samples and do not reduce protein disulfides or protein thiols modified by hydrogen peroxide. Reduction proceeds efficiently in cell extracts and in whole fixed cells. Application of this approach allowed us to demonstrate S-nitrosation of specific cellular proteins, label S-nitrosoproteins in whole fixed cells (especially the nuclear compartment) and demonstrate S-nitrosoprotein formation in cells expressing inducible nitric oxide synthase.
Keywords: S-nitrosoprotein, triphenylphosphine, nitric oxide
INTRODUCTION
S-Nitrosation represents an important cell signaling mechanism where addition of NO to cysteine thiols alters protein function [1]. As such, this form of posttranslational modification of proteins leads to modulation of structure, inhibition of active site cysteines, disruption of allosteric interactions with other molecules, etc. Numerous targets have been described including protein tyrosine phosphatases [2; 3], IκB kinase [4], NFκB [5], and ras [6; 7; 8; 9]. Specificity for formation of S-nitrosoproteins may be directed by peptide sequences surrounding sensitive cysteine residues, by exogenous versus endogenous NO sources or by compartmentalization of nitric oxide synthases (see [10; 11; 12; 13; 14] for review). In addition protein-protein interactions are also important since binding of iNOS to cyclooxygenase 2 is necessary for nitrosation of this enzyme and activation of prostaglandin synthesis [15]. Subsequent to S-nitrosation, proteins may undergo intermolecular and intramolecular reactions leading to disulfide formation or may be returned to the basal state via a process termed denitrosation. Denitrosation is also highly regulated and likely a determinant of the duration of cellular responses to nitrosothiol formation [16; 17; 18].
Critical to our understanding of the role of protein S-nitrosation in cell signaling are techniques which allow identification of modified proteins both in cell lysates and in cells. In this regard the biotin switch assay which relies on selective reduction of S-nitrosothiol (SNO) by ascorbate has been frequently used [19]. Since its original development, modification of ascorbate concentrations, reaction buffer composition, and catalytic metal ions [20; 21] have been made to improve selectivity and sensitivity. However, various groups have reported that ascorbate-based methods may be difficult to interpret due to inefficient SNO reduction [20], to artifacts resulting from sunlight exposure [22] and to potential reduction of disulfides [23]. For these reasons other sensitive, specific approaches would be important.
Recently Xian and others have developed a series of phosphine-mediated reactions which can selectively convert unstable S-nitrosothiols to stable or detectable conjugates [24; 25; 26; 27; 28]. One of these reactions, i.e. reductive ligation, produces sulfenamide as a stable intermediate [29]. However, with some phosphine substrates, the sulfenamide intermediate can be over-reduced in the presence of excess phosphine reagent leading to formation of the free thiol from the starting S-nitrosothiol. Thus we reasoned that this approach could be used to selectively reduce protein nitrosothiols in biological systems and the resulting free thiols labeled with biotinylated maleimide to produce tagged proteins which could easily be identified by standard labeling approaches.
In this study, we have used this approach to selectively reduce S-nitrosoproteins and have applied maleimide-based labeling strategies to demonstrate individual protein nitrosation and to follow compartmentalization of S-nitrosoprotein formation in cells exposed to nitric oxide (NO) derivatives. We show that the methods used detect S-nitrosoprotein without measurable interference from protein disulfides or sulfenic acid and are sufficiently sensitive to demonstrate protein S-nitrosation is cells producing NO endogenously.
METHODS
Reagents
N-(3-maleimidylpropionyl)biocytin (MPB), Alexa Fluor 488 C5 maleimide were purchased from Invitrogen (Carlsbad, CA). N-ethyl maleimide (NEM), lipopolysaccharide (LPS), tetrahydrofuran (THF) and avidin were purchased from Sigma (St. Louis, MO). CSNO was synthesized as described previously [2]. Recombinant mouse interferon gamma (INFγ) was purchased from R&D Systems (Minneapolis, MN).
Phosphine ester preparation and characterization
Compound A was prepared as described {Wang, 2008 #1735}. Compound B: Compound B was prepared as follows. 2-(Diphenylphosphino)benzoic acid (Aldrich) (1.22 g, 4 mmol) was dissolved into 30 mL DMF containing N,N′-dicyclohyxylcarbodiimide (DCC) (0.9 g, 4.4 mmol) and dimethylaminopyridine (DMAP) (488 mg, 4 mmol). After stirring at room temperature for 20 minutes, sodium 4-hydroxybenzenesulfonate dehydrate (Aldrich) (1.4 g, 6 mmol) was added. The mixture was stirred at room temperature for another 15 hours. The white solid was filtered off and the solvent was removed under reduced pressure. The resulted residue was dissolved in 20 mL H2O and extract with dichloromethane/methanol (8 times) (methanol was added to remove the emulsion generated during the extraction). The organic layers were collected, dried over MgSO4, and concentrated. The crude product was purified by flash chromatography and obtained as the salt with DMAP: 1H NMR (300 MHz, DMSO-d6) δ 8.18 (d, J = 6.6 Hz, 3H), 7.59 (m, 4H), 7.38 (m, 6H), 7.21 (m, 4H), 6.90 (t, J = 7.8 Hz, 5H), 3.13 (s, 6H); 13C NMR (75 MHz, DMSO-d6) δ 165.5, 157.3, 150.8, 146.7, 140.7, 137.8, 137.7, 134.4, 134.1, 133.6, 133.5, 131.8, 129.7, 129.5, 129.4, 127.5, 121.6, 107.6, 40.2; ESI-MS: m/z = 463.2 [as sulfonic acid RSO3H + H+], m/z = 461.3 [as sulfonate ion RSO3−].
Cell culture and treatment
COS7 and J744A.1 cells were obtained from ATCC (Manassas, VA). Cells were grown in Dulbecco’s modified Eagle’s medium with high glucose (DMEM) supplemented with 10% fetal bovine serum (FBS) (Invitrogen). To begin an experiment, confluent cells were rinsed twice with Hanks’ balanced salt solution (Invitrogen) buffered with 25 mM Hepes (HBSH, pH 7.4) and incubated at 37 °C with various agents in HBSH for the indicated times.
Determination of PTP1B thiol modification
After treatment, cells were washed three times with ice-cold HBSH and then scraped into RIPA buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), and 1 mM phenylmethylsulfonyl fluoride) containing 40 mM NEM. After brief sonication and incubation on ice for 30 min, samples were centrifuged at 14,000 rpm for 10 minutes at 4 °C and the supernatant was applied to a PD-10 desalting column (Amersham Biosciences, Piscataway, NJ) to remove excess NEM. The eluate that contained total cellular protein was incubated with 2 mM triphenylphosphine ester as described below (1:50 dilution of 100 mM stock, freshly prepared in THF) and 2 mM MPB for 2 hours at room temperature. Some samples were treated with 2 mM DTT for 30 minutes and, after removing excess DTT by a PD-10 column, treated for an additional 90 minutes with 2 mM MPB. After labeling, excess MPB was removed by elution from a PD-10 column. Avidin was added to some samples prior to electrophoresis to tightly bind biotinylated proteins, increase their apparent size and shift them to a much higher molecular weight region (or not enter the gel). For this avidin shift assay, 5 μg of the eluates were incubated without or with 50 μg avidin at room temperature for 5 minutes, then mixed with 4 × NuPAGE SDS Sample Buffer (Invitrogen) containing 200 mM DTT and heated at 65 °C for 10 minutes. The proteins were then separated by SDS-PAGE and immunoblotted with PTP1B monoclonal antibody (EMD Chemicals, Gibbstown, NJ).
For the immunoprecipitation studies, aliquots containing equivalent amounts of protein were immunoprecipitated using PTP1B monoclonal antibody. Immunoprecipitates were separated by SDS-PAGE. Biotin-labeled PTP1B was visualized by incubation with 1:4000 dilution of NeutrAvidin-linked horseradish peroxidase (Invitrogen) followed by incubation with the ECL Plus detection system (Amersham Biosciences). Blots were stripped using 100 mM mercaptoethanol, 2% SDS, 62.5 mM Tris-HCl, pH 6.7, at 50 °C for 30 min, reprobed using rabbit polyclonal PTP1B antibody (Santa Cruz Biotechnology), and visualized using anti-rabbit IgG coupled to horseradish peroxidase followed by visualization with ECL Plus.
Fluorescence Microscopy
After treatment or induction, cells on glass coverslips were washed with ice cold HBSH three times and fixed with −20 °C methanol for 15 minutes, permeablized with 0.2% (vol/vol) Triton X-100 in PBS (PBST) containing 1 mM EDTA and 40 mM NEM for 30 minutes followed by an additional 30 minute room temperature incubation with 40 mM NEM in DMSO. After washing for 5 times with PBST to remove access NEM, the cells were treated with 2 mM triphenylphosphine ester (in DMSO) for 30 minutes at room temperature. The excess phosphine ester was removed by washing twice with PBST and the reduced thiols were then labeled by incubation with 0.1 μg/ml Alexa Fluor 488 C5 maleimide for 30 minutes. Coverslips were washed twice and then mounted using ProLong Gold Antifade reagent containing DAPI (Invitrogen). Images were acquired using a Zeiss Axio Imager microscope (Carl Zeiss, Thornwood, NY).
Induction of iNOS expression
J744A.1 cells were treated with 1 μg/ml LPS plus 0.1 μg/ml INFγ in DMEM with 10% FBS at 37°C, 5% CO2 for 18 hours. The specific iNOS inhibitor, W1400, was added to medium at a final concentration of 100 μM as indicated.
Determination of S-nitrosothiols by chemiluminescence
Intracellular S-nitrosothiols was determined by ozone-based chemiluminescence using acidic triiodide to release NO as previously described [30]. Briefly activated J744A.1 cells were washed three times with ice cold HBSH and scraped into cold HBSH. Cells were collected by centrifugation at 1,000×G for 10 minutes, resuspended in PBS containing 1 mM EDTA and 40 mM NEM and sonicated for 10 seconds. After incubation for 30 minutes on ice and centrifugation at 14,000 rpm for 10 minutes, the supernatant was treated with 1/10 volume of 5% acidified sulfanilamide for 5 minutes to remove nitrite. Samples were then injected into a mixture of KI and I2 in glacial acetic acid and the NO released was analyzed by chemiluminescence using the Nitric Oxide Analyzer (Sievers, Boulder, CO).
RESULTS AND DISCUSSION
In these studies, we used triphenylphosphine esters modified to contain either polyethylene glycol (A, PE PEG) or sulfonate (B, PE SO3). Both compounds were found to reduce SNO proteins (see below) although the efficiency appeared to be higher with PE SO3 due to better water solubility. In the present experiments we wished to establish the general applicability in complex biological matrices and investigate whether the approach could be used to detect protein as shown in Scheme 1. We choose protein tyrosine phosphatase 1B (PTP1b) as a model since we had previously shown that PTP1b is S-nitrosated in cells exposed to S-nitrosocysteine (CSNO) [2]. In these experiments, COS7 cells were incubated for 15 minutes with 200 uM CSNO, washed and lysates prepared in the presence of 40 mM NEM to block all free thiols (conditions previously by us to block efficiently block free thiols in cell lysates [2]). PE PEG was then added and samples incubated for 30 minutes. Resulting thiols represent those originally occurring as protein SNO in the lysate and were tagged by incubation of PE PEG-treated samples with biotinylated maleimide. The biotinylated proteins were separated by SDS-PAGE. Avidin was added to bind tightly to biotinylated proteins forming large complexes which do not enter the gel. Blots were then analyzed using PTP1b antibodies. Loss of PTP1b immunoreactivity at the appropriate molecular size due to the avidin shift represents the fraction of PTP1b-SNO which was present in the original sample now trapped in a avidin-biotiynlated protein complex. The ratio of immunoreactivity between the avidin-treated and non-treated lanes represents the fraction of SNO in the total PTP1b isolated from cells. As shown in Figure 1A, PE PEG efficiently reduced PTP1b-SNO (compare lanes 3 & 4) and was nearly as effective in this assay as dithiothreitol (DTT, a stronger reductant which reduces protein SNO, protein disulfides and protein sulfenic acid).
Scheme 1.
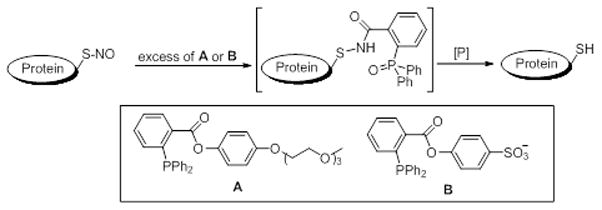
Figure 1.
Detection of PTP1B nitrosylation. COS 7 cells were treated with oxidants (200 uM CSNO, 1 mM H2O2 or 1 mM diamide) as indicated for 15 min at 37 °C. Cell extracts were prepared in RIPA buffer containing NEM, reduced with either PE PEG or DTT, and then labeled with MPB. A and B: MPB-labeled (oxidized) PTP1B was shifted by avidin as described in the methods. The total (without avidin added) and reduced PTP1B (with avidin added, which shifted oxidized PTP1B away) were visualized by immunoblotting with PTP1B antibody. C: PTP1B was immunoprecipitated from treated extracts using a specific monoclonal antibody. MPB-labeled (oxidized) PTP1B was visualized by avidin-HRP (upper blot).
In the same experiments we also investigated whether PE PEG would reduce other forms of thiol modification. Exposure of cells to H2O2 should lead to formation of protein-SOH or protein-S-S-R (disulfides) while incubation with diamide should lead to protein disulfides. In experiments where cells were exposed to 200 uM CSNO, 1 mM H2O2 or 1 mM diamide, modification of protein thiols was seen as revealed by reduction with DTT and shift with avidin (Figure 1B) (compare with and without avidin lanes). As in Figure 1A, reduction using PE PEG and shift with avidin appears to be nearly quantitative (compare lanes 1 and 2). Importantly PE PEG does not appear to reduce either sulfenic acid (H2O2 treatment, lane 4) or disulfides (diamide treatment, lane 6). The inability of PE PEG to reduce disulfides is in agreement with previously published data showing that these triphenylphosphine ester derivatives do not reduce oxidized glutathione [29]. Failure of DTT reduction/avidin shift to completely shift PTP1b in cells treated with H2O2 may reflect formation of higher oxidation thiols states (sulfonic acid for example) which cannot be reduced by DTT (lane 10). While reduction with PE PEG occurs efficiently, it should be mentioned that the small residual PTP1b that was not shifted by avidin following PE PEG reduction (lane 4, Fig. 1A and lane 2, Fig. 1B) may represent small levels of protein mixed disulfide resulting from reactions between nitrosothiols and nearby/cellular thiols (glutathione mixed disulfide or intramolecular proteins disulfides for example).
In Figure 1C we used a different approach to demonstrate selectivity. Cells were incubated with either CSNO or H2O2, lysates prepared in buffer containing NEM, modified/oxidized thiols reduced with PE PEG and labeled with biotin. After removing reagents with a size exclusion column, PTP1b was immunoprecipitated with a PTP1b antibody, separated by SDS-PAGE and visualized using horseradish peroxidase tagged neutravidin. As seen in lane 2 only those cells treated with CSNO contained PTP1b that could be biotinylated after triphenylphosphine ester reduction indicating CSNO-mediated nitrosation of this protein. It should be pointed out that NEM appears to completely block free thiols in cell lysates since there is no band in the control lane of Figure 1C. Together, the data in Figure 1 show that PE PEG efficiently and selectively reduced protein nitrosothiols and that this reductant can be applied to detection of nitrosated proteins in complex biological systems such as cell lysates.
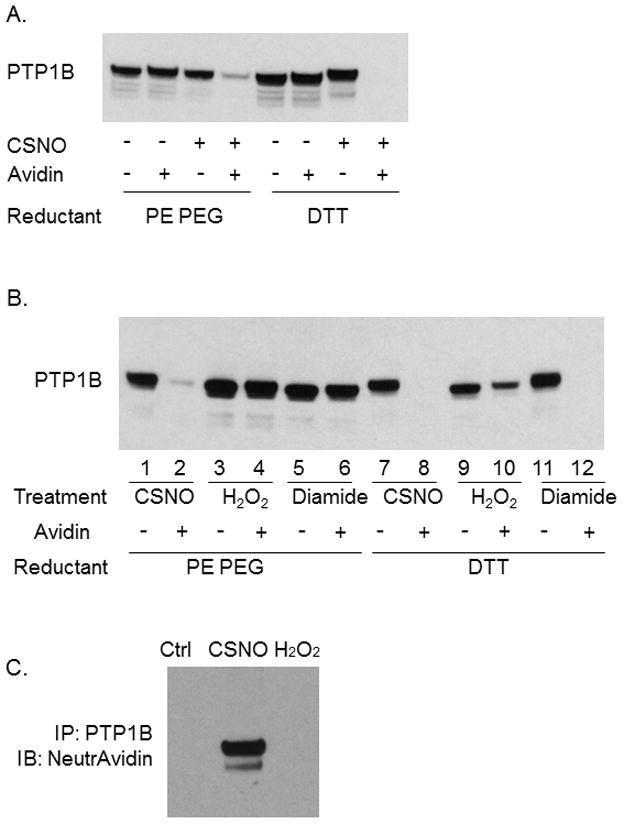
Knowing that triphenylphosphine ester reduction could be used to identify protein nitrosothiols, we used this technique to investigate nitrosation of proteins in intact cells. For these experiments, nitrosation of cellular proteins was induced by incubation with CSNO. Cells were rinsed, fixed with cold methanol and treated with NEM. After 5 additional rinses with buffer, fixed/NEM-blocked cells were treated with PE PEG to reduce any protein nitrosothiol present. Thiols revealed by triphenylphosphine ester reduction were labeled by treating with maleimide linked to the fluorescent molecule, A488. Cells were then treated with DAPI to fluorescently label nuclei and visualized by fluorescence microscopy. As shown in Figure 2, incubation of intact cells with NEM appears to completely block free thiols since treatment reduced A488 labeling to a level indistinguishable from background.
Figure 2.
Thiol modification by NEM in intact cells. COS 7 cells were fixed with methanol, permeabilized with triton X-100. For NEM treated cells, thiols were alkylated by a 30 minute incubation with 40 mM NEM in PBS followed by a 30 minute incubation with 40 mM NEM in DMSO. Free thiols were then labeled with Alexa Fluor 488 C5 maleimide as described in methods. DAPI was used for staining nuclei and shown in the lower panels.
Using these approaches we examined labeling in cells treated for 15 minutes with different concentrations of CSNO. As shown in Figure 3, CSNO-treated cells exhibited A488 fluorescence after PE PEG reduction indicating formation of protein nitrosothiols. The increase in fluorescence was easily seen above background and was clearly evident at 30 uM CSNO. Interestingly, while S-nitrosoproteins appeared throughout the cell, higher amounts we clearly evident in the nuclear compartment (compare DAPI nuclear stain to corresponding S-nitrosoproteins labeling for each CSNO concentration). We did not explore reasons for background fluorescence. Reduction in this background might allow visualization of labeling at concentrations of CSNO below 30 uM since background fluorescence could limit sensitivity.
Figure 3.

Formation of protein S-nitrosothios in whole fixed cells incubated with increasing concentrations of CSNO. COS 7 cells were treated with concentrations of CSNO as indicated. Cells were fixed with methanol, blocked with NEM, reduced by 2 mM PE PEG and then labeled with Alexa Fluor 488 C5 maleimide as described in methods. DAPI was used for staining nuclei and shown in the left panels.
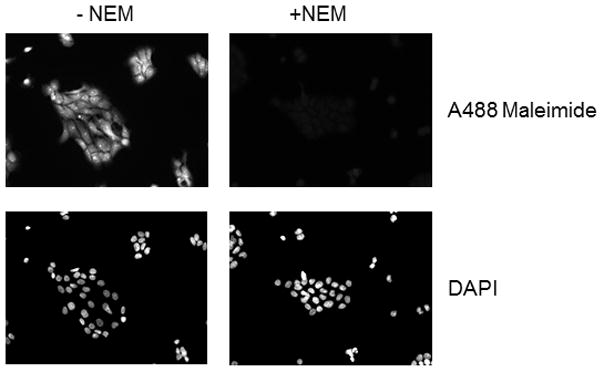
In other experiments we used PE SO3 due to its better aqueous solubility. As was the case with PE PEG, PE SO3 reduced SNO bonds allowing us to label S-nitrosated proteins in cells exposed to CSNO (Figure 4). PE SO3 concentrations around 2 mM appeared to efficiently reduce any nitrosothiols present. Fluorescence labeling was both dependent on PE SO3 reduction (top panels) and on pretreatment with CSNO (bottom panels) suggesting that reduction of CSNO-mediated modification is required for labeling. We also studied time dependence of reduction using 2 mM PE SO3. Reduction proceeded rapidly with peak fluorescence occurring after 10–15 minutes of incubation with the triphenylphosphine ester (Figure 5A). Longer reduction times did not increase labeling. As in Figure 4, labeling was shown to be dependent on CSNO pretreatment (Fig. 5B) and on PE SO3 reduction (Fig. 5C).
Figure 4.
Effect of PE SO3 concentration on S-nitrosothiol reduction in whole fixed cells. Cellular S-nitrosylation was induced by incubation of COS 7 cells with 200 μM CSNO for 15 minutes. Cells were fixed with methanol and blocked with NEM. S-nitrosothiols were reduced using different concentrations of PE SO3 by incubation for 30 minutes. Reduced thiols were then labeled with Alexa Fluor 488 C5 maleimide.
Figure 5.
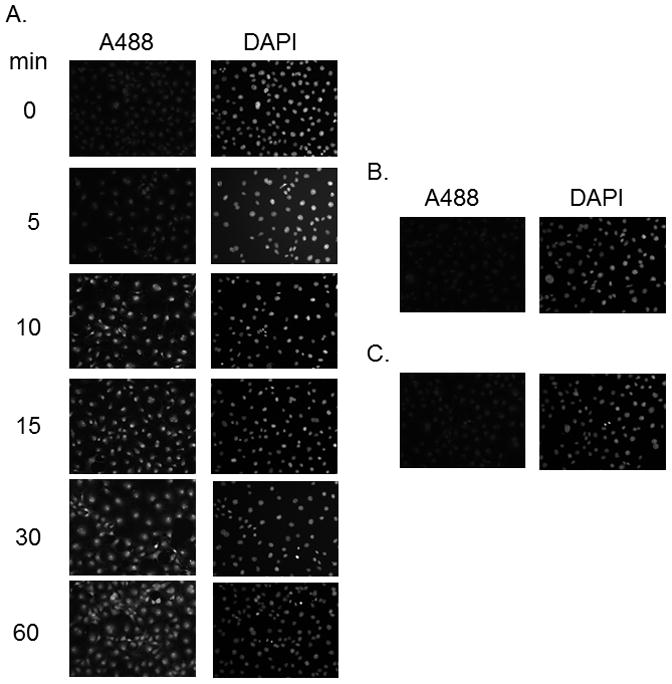
Effect of incubation time on S-nitrosothiol reduction in whole fixed cells. Cellular S-nitrosylation was induced by incubation of COS 7 cells with 200 μM CSNO for 15 minutes. Cells were fixed with methanol and blocked with NEM. 2 mM PE SO3 was added for the time indicated to reduce S-nitrosothiols and reduced thiols labeled with Alexa Fluor 488 C5 maleimide.
We next examined the selectivity of PE SO3 reduction in cells exposed to 200 uM CSNO, 1 mM H2O2 or 1mM diamide as was done for PTP1b in Figure 1. After exposure, cells were labeled with A488 maleimide and with DAPI and examined by fluorescence microscopy. As shown in Figure 6, only cells exposed to CSNO led to an A488 fluorescence signal. Fluorescence in diamide or H2O2-treated cells was indistinguishable from background. These data are consistent with those in Figure 1 and demonstrate selective reduction of nitrosothiols by this technique. We also demonstrated selectivity for nitrosothiols using the known sensitivity of S-NO bonds to UV photolysis [22]. To accomplish this, cells were treated with CSNO rinsed and then exposed to UV light for 5 minutes. After exposure, cells were rinsed and treated with PE SO3 to reduce nitrosothiols and labeled with A488. As seen in Figure 7, exposure of cells to UV light prior to PE SO3 reduction rapidly decomposed any SNO bonds present in cells and prevented labeling following PE SO3 reduction (fluorescence in CSNO-treated cells subsequently treated with UV light was not different from background). Together, data in Figures 5–7 strongly support selectivity of this technique for S-nitrosothiols detection.
Figure 6.
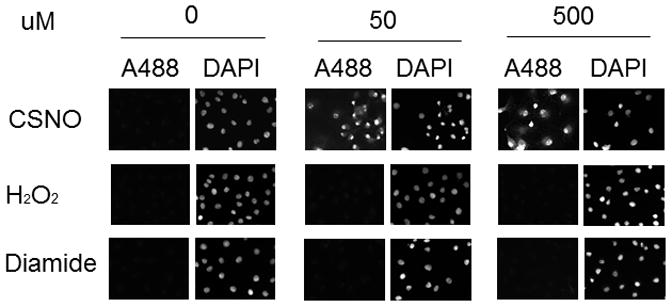
Specificity of PE SO3 for reduction of S-nitrosoproteins. COS 7 cells were treated with 1 mM of different oxidants as indicated for 15 min. Cells were rinsed, fixed and blocked with NEM as described in methods. 2 mM PE SO3 was used for reduction (30 minutes) and reduced thiols were labeled with Alexa Fluor 488 C5 maleimide.
Figure 7.
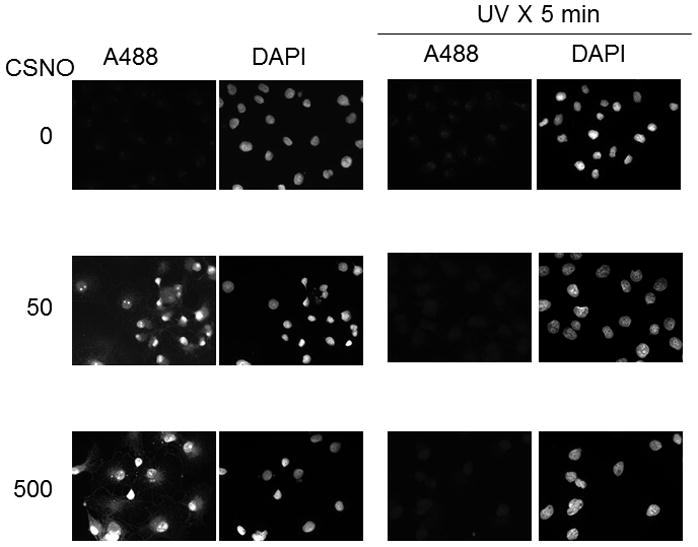
UV photolysis of protein nitrosothiols in whole fixed cells. Cellular protein S-nitrosylation was induced by incubation of COS 7 cells with CSNO as indicated. After treatment, cells were rinsed and fixed and then either kept in the dark or exposed to UV light to photolyze SNO bonds. After 5 minutes, cells were blocked with NEM, reduced by 2 mM PE SO3 for 30 minutes and then labeled with Alexa Fluor 488 C5 maleimide.
Using these approaches, we have also studied S-nitrosothiol formation in cells synthesizing NO endogenously. The macrophage cell line, J744, was treated with a combination of LPS and INFγ for 18 hours to induce iNOS. For an experiment, cells were rinsed and S-nitrosoproteins were determined in whole cell protein extracts using the triiodide method modified as previously described [30]. As show in Figure 8A, treated cells contained significantly higher levels of cellular S-nitrosoprotein than non-treated cells. The formation of S-nitrosoproteins was inhibitable by 100 μM W1400, a selective inhibitor of iNOS, suggesting that S-nitrosoprotein formation was iNOS-dependent. When similarly treated cells were fixed as above and PE-SO3 used to selectively reduce nitrosothiols, A488 fluorescence indicated that cells expressing active iNOS contained levels of S-nitrosoproteins which were clearly distinguishable from non-induced cells (Figure 8B). Again, incubation with W1400 reduced levels of fluorescence confirming data in Figure 8A which suggest iNOS-dependent S-nitrosoprotein formation. Together, these data show that PE-SO3 reduction of S-nitrosothiols allows for detection of S-proteins in cells treated to express iNOS. Further, they clearly show that endogenously generated NO leads to protein nitrosation in cells, suggesting that all components necessary for nitrosation is present in these cells.
Figure 8.
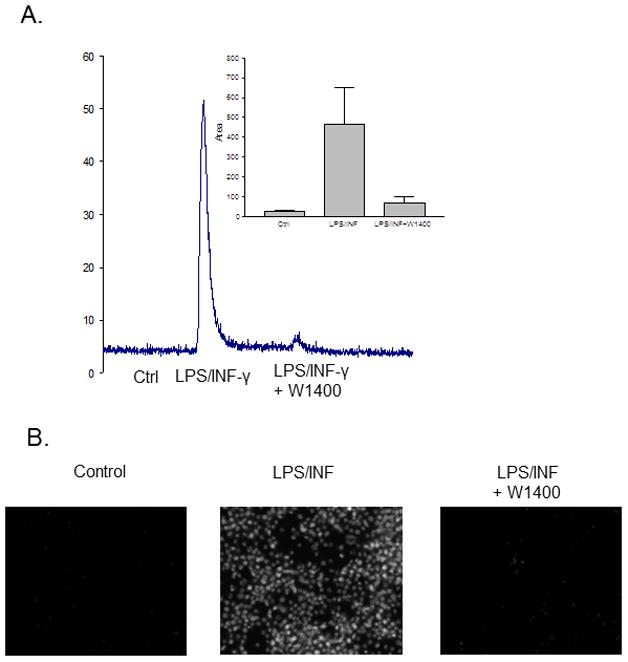
Detection of endogenous S-nitrosothiol formation in activated macrophages. J744A.1 cells were treated without (control) or with LPS plus INF-γ (LPS/INF-γ) overnight to induce iNOS expression. W1400 was added to inhibit iNOS activity as indicated. A. Cells were rinsed three times in ice cold HBSH, scraped and sonicated in 40 mM of NEM. Nitrosothiol context of the extracts was analyzed by the ozone-based chemiluminescence as described in methods. B. After activation, cells were washed, fixed, blocked with NEM, reduced by 2 mM PE SO3 and labeled with Alexa Fluor 488 C5 maleimide.
In summary we have demonstrated that triphenylphosphine ester derivatives can be used to selectively reduce SNO bonds in proteins. In the case of PTP1b the yield is nearly quantitative and can be accomplished in biological matrices. S-nitrosoproteins were readily formed after treatment of cells with exogenous CSNO or following endogenous production of nitric oxide by iNOS. In both cases, these approaches indicated widespread formation of S-nitrosoproteins in most cellular compartments with higher amounts in the nucleus.
Highlights.
We use newly described triphenylphosphine esters to demonstrate SNO protiens.
Reductive ligation using phosphine esters is selective for SNO adducts.
We demonstrate SNO proteins in cells expressing iNOS using phosphine ester reduction.
Acknowledgments
This work was supported by NIH grants R01HL088559 (ARW) and R01GM088226 (MX).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Stamler JS, Lamas S, Fang FC. Nitrosylation. The prototypic redox-based signaling mechanism. Cell. 2001;106(6):675–683. doi: 10.1016/s0092-8674(01)00495-0. [DOI] [PubMed] [Google Scholar]
- 2.Li S, Whorton AR. Regulation of protein tyrosine phosphatase 1B in intact cells by S-nitrosothiols. Arch Biochem Biophys. 2003;410:269–279. doi: 10.1016/s0003-9861(02)00696-3. [DOI] [PubMed] [Google Scholar]
- 3.Yu CX, Li S, Whorton AR. Redox regulation of PTEN by S-nitrosothiols. Mol Pharmacol. 2005;68:847–54. doi: 10.1124/mol.104.010504. [DOI] [PubMed] [Google Scholar]
- 4.Reynaert NL, Ckless K, Korn SH, Vos N, Guala AS, Wouters EF, van der Vliet A, Janssen-Heininger YM. Nitric oxide represses inhibitory kappaB kinase through S-nitrosylation. Proc Natl Acad Sci U S A. 2004;101:8945–50. doi: 10.1073/pnas.0400588101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marshall HE, Stamler JS. Inhibition of NF-kappa B by S-nitrosylation. Biochemistry. 2001;40:1688–93. doi: 10.1021/bi002239y. [DOI] [PubMed] [Google Scholar]
- 6.Deora AA, Hajjar DP, Lander HM. Recruitment and activation of Raf-1 kinase by nitric oxide-activated Ras. Biochemistry. 2000;39:9901–9908. doi: 10.1021/bi992954b. [DOI] [PubMed] [Google Scholar]
- 7.Heo J, Prutzman KC, Mocanu V, Campbell SL. Mechanism of free radical nitric oxide-mediated Ras guanine nucleotide dissociation. J Mol Biol. 2005;346:1423–40. doi: 10.1016/j.jmb.2004.12.050. [DOI] [PubMed] [Google Scholar]
- 8.Heo J, Campbell SL. Mechanism of p21Ras S-nitrosylation and kinetics of nitric oxide-mediated guanine nucleotide exchange. Biochemistry. 2004;43:2314–22. doi: 10.1021/bi035275g. [DOI] [PubMed] [Google Scholar]
- 9.Williams JG, Pappu K, Campbell SL. Structural and biochemical studies of p21Ras S-nitrosylation and nitric oxide-mediated guanine nucleotide exchange. Proc Natl Acad Sci U S A. 2003;100:6376–81. doi: 10.1073/pnas.1037299100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gaston BM, Carver J, Doctor A, Palmer LA. S-nitrosylation signaling in cell biology. Mol Interv. 2003;3:253–263. doi: 10.1124/mi.3.5.253. [DOI] [PubMed] [Google Scholar]
- 11.Gow AJ, Farkouh CR, Munson DA, Posencheg MA, Ischiropoulos H. Biological significance of nitric oxide-mediated protein modifications. Am J Physiol Lung Cell Mol Physiol. 2004;287:L262–L268. doi: 10.1152/ajplung.00295.2003. [DOI] [PubMed] [Google Scholar]
- 12.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–66. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 13.Hess DT, Matsumoto A, Nudelman R, Stamler JS. S-nitrosylation: spectrum and specificity. Nat Cell Biol. 2001;3:E46–E49. doi: 10.1038/35055152. [DOI] [PubMed] [Google Scholar]
- 14.Hogg N. The biochemistry and physiology of S-nitrosothiols. Annu Rev Pharmacol Toxicol. 2002;42:585–600. doi: 10.1146/annurev.pharmtox.42.092501.104328. [DOI] [PubMed] [Google Scholar]
- 15.Kim SF, Huri DA, Snyder SH. Inducible nitric oxide synthase binds, S-nitrosylates, and activates cyclooxygenase-2. Science. 2005;310:1966–70. doi: 10.1126/science.1119407. [DOI] [PubMed] [Google Scholar]
- 16.Benhar M, Forrester MT, Hess DT, Stamler JS. Regulated protein denitrosylation by cytosolic and mitochondrial thioredoxins. Science. 2008;320:1050–4. doi: 10.1126/science.1158265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu L, Yan Y, Zeng M, Zhang J, Hanes MA, Ahearn G, McMahon TJ, Dickfeld T, Marshall HE, Que LG, Stamler JS. Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell. 2004;116:617–28. doi: 10.1016/s0092-8674(04)00131-x. [DOI] [PubMed] [Google Scholar]
- 18.Forrester MT, Seth D, Hausladen A, Eyler CE, Foster MW, Matsumoto A, Benhar M, Marshall HE, Stamler JS. Thioredoxin-interacting protein (Txnip) is a feedback regulator of S-nitrosylation. J Biol Chem. 2009;284:36160–6. doi: 10.1074/jbc.M109.057729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jaffrey SR, Snyder SH. The biotin switch method for the detection of S-nitrosated proteins. Sci STKE. 2001;2001:PL1. doi: 10.1126/stke.2001.86.pl1. [DOI] [PubMed] [Google Scholar]
- 20.Wang X, Kettenhofen NJ, Shiva S, Hogg N, Gladwin MT. Copper dependence of the biotin switch assay: modified assay for measuring cellular and blood nitrosated proteins. Free Radic Biol Med. 2008;44:1362–72. doi: 10.1016/j.freeradbiomed.2007.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Y, Keszler A, Broniowska KA, Hogg N. Characterization and application of the biotin-switch assay for the identification of S-nitrosated proteins. Free Radic Biol Med. 2005;38:874–81. doi: 10.1016/j.freeradbiomed.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 22.Forrester MT, Foster MW, Stamler JS. Assessment and application of the biotin switch technique for examining protein S-nitrosylation under conditions of pharmacologically induced oxidative stress. J Biol Chem. 2007;282:13977–83. doi: 10.1074/jbc.M609684200. [DOI] [PubMed] [Google Scholar]
- 23.Landino LM, Crews BC, Timmons MD, Morrow JD, Marnett LJ. Peroxynitrite, the coupling product of nitric oxide and superoxide, activates prostaglandin biosynthesis. Proc Natl Acad Sci U S A. 1996;93:15069–15074. doi: 10.1073/pnas.93.26.15069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang J, Wang H, Xian M. An unexpected Bis-ligation of S-nitrosothiols. J Am Chem Soc. 2009;131:3854–5. doi: 10.1021/ja900370y. [DOI] [PubMed] [Google Scholar]
- 25.Zhang J, Li S, Zhang D, Wang H, Whorton AR, Xian M. Reductive ligation mediated one-step disulfide formation of S-nitrosothiols. Org Lett. 2010;12:4208–11. doi: 10.1021/ol101863s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang D, Devarie-Baez NO, Pan J, Wang H, Xian M. One-pot thioether formation from S-nitrosothiols. Org Lett. 2010;12:5674–6. doi: 10.1021/ol102491n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bechtold E, Reisz JA, Klomsiri C, Tsang AW, Wright MW, Poole LB, Furdui CM, King SB. Water-soluble triarylphosphines as biomarkers for protein S-nitrosation. ACS Chem Biol. 2010;5:405–14. doi: 10.1021/cb900302u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang H, Zhang J, Xian M. Facile formation of dehydroalanine from S-nitrosocysteines. J Am Chem Soc. 2009;131:13238–9. doi: 10.1021/ja905558w. [DOI] [PubMed] [Google Scholar]
- 29.Wang H, Xian M. Fast reductive ligation of S-nitrosothiols. Angew Chem Int Ed Engl. 2008;47:6598–601. doi: 10.1002/anie.200801654. [DOI] [PubMed] [Google Scholar]
- 30.Li S, Whorton AR. Functional characterization of two S-nitroso-L-cysteine transporters, which mediate movement of NO equivalents into vascular cells. Am J Physiol Cell Physiol. 2007;292:C1263–71. doi: 10.1152/ajpcell.00382.2006. [DOI] [PubMed] [Google Scholar]