

SUMMARY
The main purpose of our study is to understand how mycobacteria exert control over the biosynthesis of their membrane lipids and find out the key components of the regulatory network that control fatty acid biosynthesis at the transcriptional level. In this paper we describe the identification and purification of FasR, a transcriptional regulator from Mycobacterium sp. that controls the expression of the fatty acid synthase (fas) and the 4-phosphopantetheinyl transferase (acpS) encoding genes, whose products are involved in the fatty acid and mycolic acid biosynthesis pathways. In vitro studies demonstrated that fas and acpS genes are part of the same transcriptional unit and that FasR specifically binds to three conserved operator sequences present in the fas-acpS promoter region (Pfas). The construction and further characterization of a fasR conditional mutant confirmed that FasR is a transcriptional activator of the fas-acpS operon and that this protein is essential for mycobacteria viability. Furthermore, the combined used of Pfas-lacZ fusions in different fasR backgrounds and electrophoretic mobility shift assays experiments, strongly suggested that long-chain acyl-CoAs are the effector molecules that modulate the affinity of FasR for its DNA binding sequences and therefore the expression of the essential fas-acpS operon.
INTRODUCTION
Mycobacterium tuberculosis has a complex lifestyle and can modulate its metabolism in response to different environmental changes (Bacon and Marsh, 2007). The success of this pathogen largely stems from its remarkable capacity to survive within the infected host, where it can persist for several decades. The presence of its unusual cell wall is a key factor in this survival (Daffe and Draper, 1998). Despite extensive literature on the biosynthesis, structure, and biological function(s) of the major cell wall components of M. tuberculosis (Takayama et al., 2005, Kaur et al., 2009), very little is known regarding the mechanisms allowing the bacterium to modulate and adapt expression of its cell wall components in response to environmental changes. Therefore, uncovering cell wall regulatory processes represents a crucial step toward understanding the physiology and physiopathology of M. tuberculosis, as well as the interactions between mycobacteria and their environment in general.
Mycolic acids are essential components of the lipid-rich cell envelope of M. tuberculosis and related mycobacteria (Takayama et al., 2005); they are very long-chain α-alkyl β-hydroxylated fatty acids that play an important role in the reduced cell wall permeability (Daffe, 2008, Brennan and Nikaido, 1995), virulence (Bhatt et al., 2007b, Dubnau et al., 2000, Glickman et al., 2000, Rao et al., 2006), and acid fastness characteristic of M. tuberculosis (Bhatt et al., 2007a). The biosynthesis of mycolic acids depends on two distinct systems: the eukaryotic-like type I fatty acid synthase (FAS-I) and the prokaryotic-like type II fatty acid synthase (FAS-II)(Takayama et al., 2005). Although the structural organization of FAS-I and FAS-II is different, the chemical reactions and the catalytic mechanisms for fatty acid biosynthesis are essentially the same (Schweizer and Hofmann, 2004). In mycobacteria FAS-I performs de novo biosynthesis of acyl-CoAs (C16 and C24–C26) (Zimhony et al., 2004, Bloch, 1975). Long chain-length acyl-CoAs are used as primers by the FAS-II system and iteratively condensed with malonyl-acyl carrier protein (ACP) leading to very long-chain meromycolyl-ACPs (up to C56), which together with the C26 fatty acids synthesized by FAS-I, become the precursors of mycolic acids. An additional level of complexity comes from the ability of mycobacteria to utilize the acyl-CoAs generated by FAS-I not only for the biosynthesis of phospholipids or as precursors of mycolic acid, but also for the synthesis of the storage compound triacylglycerol (Daniel et al., 2004, Deb et al., 2009). Therefore, M. tuberculosis needs to coordinately integrate all these pathways in order to maintain membrane homeostasis tightly regulated.
The functional and biochemical relationship between FAS-I and FAS-II enzyme complexes has been largely studied and well documented in the literature due to the relevance of these metabolic pathways in the survival and pathogenicity of M. tuberculosis as well as for being important targets for tuberculosis (TB) treatment (Zhang et al., 2006, Zhang, 2005, Brennan and Crick, 2007). However, the molecular mechanisms used by the two FAS systems to communicate with each other in order to control the balance of fatty acids biosynthesis with that of mycolic acids and also with other complex lipids biosynthesis pathways, like phthiocerol dimycocerosate (PDIM) or sulpholipids, is still an open and challenging question. Our studies on the regulation of the fasII operon have provided strong evidence of the existence of a coordinate regulation of the two FAS systems of mycobacteria at the transcriptional level (Salzman et al., 2010). A bioinformatic search of the genome of M. tuberculosis led us to identify the transcriptional regulator MabR, located immediately upstream of the fasII operon. Biochemical and functional characterization of MabR confirmed that MabR is the transcriptional regulator of the fasII operon of Mycobacterium. This regulatory protein binds to a 21 bp palindrome (5′-TTTTGT-N9-ACAAAA-3′) located in fasII promoter region controlling the expression of the fasII operon, and therefore the synthesis of mycolic acids (Salzman et al., 2010). Interestingly, altering MabR levels led to changes not only in mycolic acid biosynthesis but also in the expression of the fas gene (and consequently in the biosynthesis of fatty acids), suggesting that common regulatory factors are involved in the coordination of the two FAS systems.
The complexity of lipid biosynthesis within the genus Mycobacterium led us to believe that a more sophisticated regulatory signalling cascade should be involved in the regulation of the FAS-I and FAS-II systems, in order to keep lipid homeostasis tightly regulated. In this work we present in vivo and in vitro studies on FasR, a new transcriptional regulator from M. tuberculosis that binds specifically to the fas promoter to regulate its expression. In addition, we also present the identification of the metabolic intermediates (ligand) that are most probably sensed by FasR in order to modulate its regulatory function in the expression of fas.
The identification and characterization of this regulatory protein and its ligand is another step towards a more complete understanding of the complex network of regulation that allow mycobacteria to finely control de novo fatty acid biosynthesis and its interaction with other more complex lipid biosynthesis pathways.
RESULTS
Identification of the fas gene promoter
In an attempt to identify a putative transcription factor(s) involved in the regulation of fatty acid biosynthesis in Mycobacterium, we first characterized the promoter sequence of the fas gene from M. smegmatis (MSMEG_4757, fasMS) and M. tuberculosis (Rv2524c, fasMT) by mapping their transcription start sites (TSS). Total RNA was isolated from exponential phase cultures of wild-type M. smegmatis mc2155 and M. tuberculosis H37Rv and used as templates for the TSS determination using RACE. A single PCR product was obtained for both genes and the analysis of the sequencing results revealed that the TSS is situated 43 bp and 5 bp upstream of the translation start codon of fasMS and fasMT, respectively (Figure 1A and 1C).
Figure 1.

A, C. Determination of fas TSS by 5′ RACE. Pfas probes were constructed from the defined Pfas promoter regions.
B, D. fas and acpS are part of a unique transcriptional unit. Primers were designed to amplify the 275 bp and 263 bp intergenic regions between fasMS or fasMT and acpS genes respectively, from wild type M. smegmatis mc2155 or M. tuberculosis H37Rv cDNA by RT-PCR, the amplification products spanning the intergenic regions of fasMS or fasMT and acpS are shown. RNA not treated with RT was used as control. Negative and positive controls were also performed with water and genomic DNA respectively.
The fas gene encodes the unique and essential type I FAS (FAS-I) required for the initiation of fatty acid biosynthesis in mycobacteria. Interestingly the fas gene is genetically linked to acpS, which codes for a 4-phosphopantetheinyl transferase (PPTase), which transfers the phosphopantetheine group from CoA to the ACP domain of FAS and to the soluble ACP of mycobacteria (AcpM). Thus, AcpS is the enzyme responsible of the posttranslational modification necessary for the activity of ACP, a central protein covalently bound to all fatty acyl intermediates during fatty acid and mycolic acid biosynthesis. The common metabolic pathway in which FAS and AcpS are involved and the genetic linkage of their encoding genes in the genome of mycobacteria, prompted us to investigate if fas and acpS were part or not of a unique transcriptional unit. For this, total RNA was isolated from exponential phase cultures of wild-type M. smegmatis mc2155 and M. tuberculosis H37Rv and subjected to reverse transcription followed by PCR using primers designed to amplify the intergenic region between fas and acpS from their corresponding cDNAs. As shown in Figure 1B and 1D, a predicted fragment spanning the fas-acpS intergenic region was detected for both microorganisms suggesting that the two genes form part of a single transcriptional unit. An extensive bioinformatic analysis showed a full conservation of the operon organization in Mycobacterium highlighting the biological significance of this operon across this genus.
Purification of a TetR transcription factor that binds to the promoter region of the fas gene in Mycobacterium
Microarray studies have indicated that fas expression responds to the presence of exogenously added fatty acids (http://www.tbdb.org/cgi-bin/data/prd.pl?e=22793). Furthermore, we also demonstrated that the transcription of fas, and consequently the levels of de novo FA biosynthesis, is affected by changes in the physiological concentrations of the MabR regulator, although this effect appeared not to be a consequence of a direct binding of this regulator to the fas promoter (Salzman et al., 2010). Based on this information and having identified the fas TSS we sought for a putative regulatory protein with a binding site in the promoter region of the fas-acpS operon (from now on Pfas). For this, we performed band shift assays with crude protein extracts obtained from M. smegmatis mc2155. After confirming that the protein extract was able to specifically shift the mobility of a 448 bp DNA fragment carrying the PfasMS region (Figure S1A and B), we followed the purification of the putative DNA-binding protein using the same assay (Figure 2A). The protocol used to purify the protein associated to Pfas was a modification of the procedure described by Jourlin-Castelli et al. (Jourlin-Castelli et al., 2000). The active fractions proved to contain four major proteins that were excised and analyzed by MALDI-TOF MS/MS (Figure 2B). The proteins corresponding to the three major bands were the nucleoid-associated proteins Hup (MSMEG_2389) and Lsr2 (MSMEG_1060 and MSMEG_6092). The fourth one was the product of the MSMEG_1935 gene, a putative TetR transcriptional regulator. Consistent with most TetR family of regulators whose molecular masses range from 21 to 25 kDa, MSMEG_1935 has a calculated molecular mass of 24,410 Da and consists of 219 amino acids. A helix-turn-helix DNA-binding motif, which is also a characteristic feature of the TetR family of regulators (Ramos et al., 2005) is present at the N-terminal region of this DNA-binding protein, corresponding to amino acids 51–72. The prototype of the TetR family of transcriptional regulators is TetR from the Tn10 transposon of E. coli; which regulates the expression of the tetracycline efflux pump in Gram-negative bacteria (Orth et al., 2000). These proteins often serve as repressors and are widely distributed among bacteria regulating a number of diverse processes including fatty acid biosynthesis (Ramos et al., 2005). A bioinformatic search of the orthologous protein in M. tuberculosis identified Rv3208. This M. tuberculosis protein has 84 % identity with MSMEG_1935 and they are both located in the same loci of the corresponding bacterial chromosome. Therefore, we identify orthologous proteins from M. tuberculosis and M. smegmatis whose most probable role is to regulate the expression of the fas gene by binding specifically to the corresponding Pfas regions.
Figure 2. Purification of PfasMS binding protein from M. smegmatis strain mc2155 crude extracts.
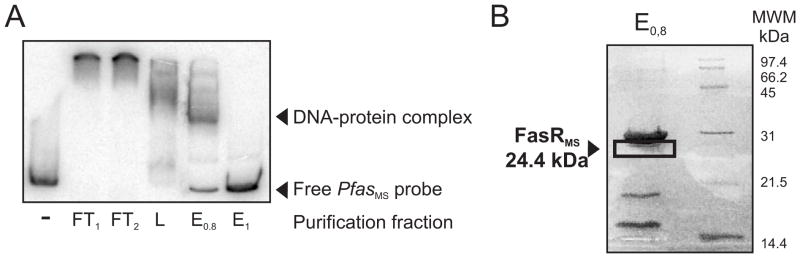
A. Gel shift assay was performed using a 32P-labeled 448 bp PfasMS probe and partially purified affinity column-eluted fractions in the presence of poly-dIdC. FT1 and FT2, first and second flow through column fractions respectively; L, column wash fraction; E0.8, protein fraction eluted at 0.8 N saline concentration; E1, remaining protein fraction eluted at 1 N saline concentration.
B. SDS-PAGE from the active fraction eluted at 0.8 N saline concentration proved to contain four major proteins that were identified using MS-MS mass spectrometry. A protein of 24.4 kDa is the product of the MSMEG_1935 gene.
M. tuberculosis FasR binds specifically to the fas gene promoter region
To find out if the transcription factor identified in M. tuberculosis was a DNA binding protein with the ability to bind specifically the Pfas promoter region and in that way control the expression of the fas-acpS operon genes, we constructed a His-tag version of Rv3208 in order to use it in electrophoretic mobility shift assays (EMSA). Recombinant His6-Rv3208 was expressed in E. coli and purified to homogeneity (Figure S2A). Analysis by size exclusion chromatography indicated that the Rv3208 solution structure was predominantly a dimer with an apparent molecular weight of ~52 kDa (Figure S2A and B).
Binding of Rv3208 to PfasMT was assayed by EMSA using recombinant His6-Rv3208 and a PCR-amplified fragment derived from the fas promoter region of M. tuberculosis (PfasMT). The 398 bp PCR probe extends 366 and 31 bp upstream and downstream of the TSS, respectively (Fig. 1C). As shown in Figure 3A, His6-Rv3208 binds to the PfasMT promoter region in a concentration-dependent manner forming a single protein-DNA complex. The specificity of Rv3208 binding to the PfasMT region was further investigated by competition experiments (Figure 3B) in which binding of Rv3208 to the 398 bp radiolabeled probe was challenged with a 60-fold excess of the unlabeled probe of PfasMT or PfasMS, with a 60-fold excess of a 260 bp non-related DNA fragment or with a 272 bp fragment containing the PfasIIMT promoter region. Figure 3B shows that Rv3208 binding to PfasMT was almost completely inhibited in the presence of the unlabeled probes corresponding to the Pfas promoters from M. tuberculosis or M. smegmatis but was unaffected by non-related DNA, confirming the specificity of the binding. These data demonstrate that His6-Rv3208 binds to the PfasMT and PfasMS promoters and strongly suggest that it could be a transcriptional regulator of the fas-acpS operon. Thus, based on the ability of Rv3208 to bind to PfasMT, we have named this gene fasR for fatty acid synthesis Regulator.
Figure 3. Purified FasRMT binds to the PfasMT promoter region.
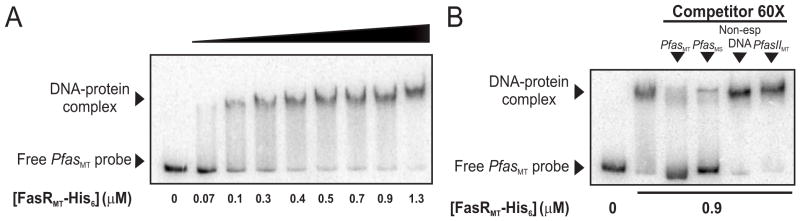
A. Gel shift assay was performed by incubating the 32P-labelled 398 bp PfasMT probe with increasing concentrations (from 0 to 1.3 μM) of His6-FasRMT in the presence of poly-dIdC.
B. The specificity of the binding was confirmed by competing labeled PfasMT probe with a 60-fold excess of unlabelled PfasMT or PfasMS probe, and with a 60-fold excess of non-related DNA or a PfasIIMT probe.
Identification of the FasR binding sites
In order to identify the FasR binding site, we carried out DNase I footprinting analysis on both strands of the 398 bp PfasMT promoter region, in the presence or in the absence of purified M. tuberculosis His6-FasRMT (Figure 4). As shown in Figure 4A and B, binding of FasR resulted in the protection of DNA sequences extending from positions − 324 to − 304 and −243 to −209 in the coding strand and from positions −324 to −304 and − 238 to −204 in the non-coding strand, relative to the TSS of fasMT. In addition to the protected regions, hypersensitive bands, presumably caused by bending of the 16DNA helix, were also detected.
Figure 4. Identification of FasRMT binding sites in the PfasMT promoter region.

A. Both strands containing the PfasMT promoter sequences were labelled with [γ-32P]-ATP and protected from DNase I nuclease activity with two different concentrations of FasRMT (0.5 and 0.9 μM for coding strand; 0.8 and 1.3 μM for non-coding strand). The protected regions in each strand are indicated with black bars. Lanes A–T: DNA sequence of the probe.
B. PfasMT sequence. The protected regions in each strand are underlined. Conserved inverted repeats are highlighted in grey.
C. Sequence analysis of the putative FasR binding regions of several species of mycobacteria with the motif-based sequence analysis tool MEME, led to the identification of a motif highly conserved in Mycobacterium.
The nucleotide sequence alignment of several Pfas promoters highlighted a remarkable conservation in the sequences that are specifically recognized by FasR (Figure S3). Analysis of the refined binding regions with the motif-based sequence analysis tool MEME (Bailey and Elkan, 1995) led to the identification of a 12 bp palindromic sequence highly conserved (Figure 4C) and shared by all fas promoters of Mycobacterium, strongly suggesting that this motif is part of the operator region recognized by the putative FasR orthologues. The inverted repeat sequence (IR) is present in two copies in the longest protected region and in one copy in the shortest one. They are located about 200–300 bp upstream of the TSS, a remarkable difference with most TetR-operator sites which are located around the TSS therefore preventing expression of the target genes by blocking RNA polymerase binding (Ramos et al., 2005). In order to analyze whether the 12 bp palindromic sequence identified is present within the upstream regions of additional genes, the generated motif profile (Figure 4C) was used to search a database of intergenic regions from M. tuberculosis and M. smegmatis using the program MAST (Bailey and Gribskov, 1998). The results obtained showed that the motif recognized by FasR is only present in the promoter region of the fas gene.
To further understand the role of the IRs in the binding of FasRMT, we constructed three DNA variants of the 398 bp PfasMT promoter region (named Mut1, Mut2 and Mut3) and assayed the DNA-binding properties of them by EMSA. In Mut1, both IRs present in the protected region 1 were replaced by the random sequence 5′-CGAATTATGAGCTCGTAACATGAGC-3′; in Mut2, the IR present in the protected region 2 was replaced by the random sequence 5′-CGAATTATGAGC-3′ and in Mut3, the three IRs identified were simultaneously replaced by the same random sequences used in Mut1 and Mut2. Incubation of the wild type and the mutated PfasMT promoter regions with 1.2 μM His6-FasRMT showed that neither of the probes containing mutated versions of the IR 1 (Mut1 and Mut3) were bound while the wild-type probe clearly shifted (Figure 5). However, FasRMT is still able to bind to the probe Mut2, although the DNA-protein complex formed has different mobility in the EMSA assay (Figure 5).
Figure 5.
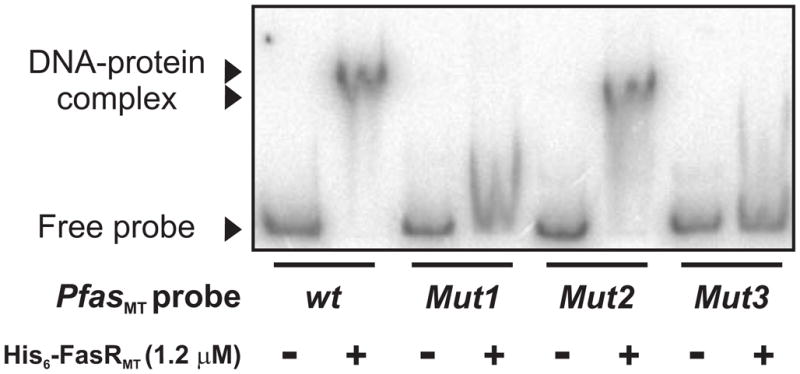
Gel shift assay was performed by incubating wild type and three DNA variants of the 398 bp PfasMT promoter region (Mut1, Mut2 and Mut3) with His6-FasRMT in the presence of poly-dIdC. In Mut1, both IRs present in the protected region 1 were replaced by the random sequence 5′-CGAATTATGAGCTCGTAACATGAGC-3′; in Mut2, the IR present in the protected region 2 was replaced by the random sequence 5′-CGAATTATGAGC-3′ and in Mut3, the three IRs identified were simultaneously replaced by the same random sequences used in Mut1 and Mut2.
All these results confirm that FasR is a DNA-binding protein that specifically binds the Pfas region by recognizing a DNA motif highly conserved within Mycobacterium and related bacteria. Thus, FasR and the corresponding orthologues appear as suitable transcriptional regulators of the fas gene in mycobacteria and probably in most FAS-I containing actinomycetes.
FasR is a transcriptional activator of fatty acid biosynthesis in mycobacteria
To study the role of the DNA-binding protein FasR in the transcription of the fas gene, we investigated the effect of FasRMT on the Pfas dependent production of β-galactosidase. For this, an integrative plasmid carrying a PfasMT::lacZ transcriptional fusion (pFR47) was transformed into M. smegmatis that had been previously transformed with plasmid pFR9, which expresses FasRMT from the Pami promoter, generating the strain MSpFR47 pFR9. β-galactosidase activity was measured four hours after induction of FasRMT expression with acetamide 0.2 %. Overexpression of FasRMT increased up to 80 % the activity of β-galactosidase as compared to non-induced cells (Figure 6). To confirm the role of FasR in the increased levels of activity observed in the overexpressing strain, we constructed mutated versions of the PfasMT::lacZ transcriptional fusion (pFR48, pFR49 and pFR50). In pFR48, both IRs present in the protected region 1 were replaced by the random sequence 5′-CGAATTATGAGCTCGTAACATGAGC-3′; in pFR49, the IR present in the protected region 2 was replaced by the random sequence 5′-CGAATTATGAGC-3′ and in pFR50, the three IRs identified were simultaneously replaced by the same random sequences used for pFR48 and pFR49. These plasmids were transformed into M. smegmatis that had been previously transformed with plasmid pFR9, generating the strains MSpFR48 pFR9, MSpFR49 pFR9, MSpFR50 pFR9. β-galactosidase activity was measured four hours after induction of fasRMT expression with acetamide 0.2 %. Figure 6 shows that β-galactosidase activity was barely detectable in the strains bearing the mutated versions of PfasMT, confirming that FasR binding sites are required for regulation and suggesting that the basal activity of the fas promoter is very low. These results strongly suggest that FasR is a transcriptional activator of the fas-acpS operon in M. tuberculosis.
Figure 6. FasR is a positive regulator of the fas-acpS operon.

Intracellular β-galactosidase activity of strains MSpFR47 pFR9 (pFR47), MSpFR48 pFR9 (pFR48), MS pFR49 pFR9 (pFR49) and MSpFR50 pFR9 (pFR50) grown in 7H9 medium with or without acetamide 0.2%. Following 4 h of induction, samples of each culture were removed to assay β-galactosidase specific activity. Levels of activity are shown as nmol ONPG per min per mg of protein and are the means of the results of three independent experiments ± standard deviations (n=3). *, P <0.05.
It has been demonstrated that the FAS-I system of Mycobacterium has a bimodal behavior, releasing long chain length fatty-acyl CoAs for phospholipid and mycolic acid biosynthesis and also very long chain (C24) acyl-CoAs for the production of the α–branch of mycolic acids. Therefore, we assayed the ability of fatty acids to modulate the activity of Pfas promoter in vivo by supplementing the growth media with palmitic acidand by following β-galactosidase activity in the wild type strain M. smegmatis mc2 155 harboring the plasmid pFR47 (MSpFR47). Cells were grown in 7H9 medium containing or not palmitic acid 0.01 % and β-galactosidase activities were monitored four hours after the addition of the fatty acid. The presence of palmitic acid in the growth media led to decreased levels of β-galactosidase activity, suggesting that the expression of the fas-acpS operon is affected by the levels of the FAS-I products (Figure 7A). We also assayed the ability of fatty acids to modulate the activity of Pfas in the strain MSpFR47 pFR9. Cells were grown in 7H9 medium supplemented with acetamide 0.2 % and containing or not different chain length fatty acids at a concentration of 0.01 %; and β-galactosidase activities were monitored four hours post-induction (Figure 7B). The presence long chain length fatty acids led to two to three fold decreased levels of β-galactosidase activities, strongly suggesting that the expression of the fas-acpS operon is regulated as a response of the cell metabolism to the availability of long chain length fatty acids.
Figure 7.

A. Intracellular β-galactosidase activity of strain MSpFR47 grown in 7H9 medium with or without C16:0 0.01 %. Levels of activity are shown as nmol ONPG per min per mg of protein and are the means of the results of three independent experiments ± standard deviations (n=3). **, P = 0.015.
B. Intracellular β-galactosidase activity of strain MSpFR47 pFR9 grown in 7H9 medium without acetamide (control), MSpFR47 pFR9 grown in 7H9 medium with acetamide 0.2% (OE FasRMT) and MSpFR47 pFR9 grown in 7H9 medium with acetamide 0.2% and in the presence of different fatty acids at a final concentration of 0.01 % (OE FasRMT + C16:0 to OE FasRMT + C24:0). Samples of each culture were removed 4 h post-induction to assay β-galactosidase specific activity. Levels of activity are shown as nmol ONPG per min per mg of protein and are the means of the results of three independent experiments ± standard deviations (n=3). *, P <0.0001.
Long chain acyl-CoAs regulate FasR binding to Pfas
The fact that FasR is a transcriptional activator of the fas gene and that supplementation of cells with long-chain fatty acids proved to reduce the fas promoter activity suggests that either fatty acids or their CoA-activated derivatives could interact with FasR to prevent binding of the activator to its DNA target. Therefore, in order to identify the molecule that could serve as the metabolic signal for the regulation of fas expression we used gel shift assays to survey a set of compounds which are intermediates in fatty acid and lipid metabolism in mycobacteria for their ability to modulate binding of FasRMT to PfasMT. These included palmitic acid, palmitoleic acid, arachidic acid, acetyl-coA, malonyl-CoA, lauryl-CoA, myristoyl-CoA, palmitoyl-CoA, arachidoyl-CoA and behenoyl-CoA. In these experiments the reaction mixture contained, in addition to the test compound, 32P-labeled PfasMT DNA and 0.67 μM His6-FasRMT. As shown in Figure 8, where each compound was tested at a final concentration of 1 μM, long fatty-acyl CoA compounds (C14- to C22-CoA) clearly inhibited FasRMT-DNA binding while fatty acids and short chain acyl-CoAs had no effect at a final concentration of 1 μM. This result is in accordance with the effect observed in the experiment shown in Figure 7B. However, the release of FasR from DNA could be considered the result of denaturation of the protein by detergent action of the long chain acyl-CoAs. Therefore, we used purified acyl-CoA thioesterase to cleave the long chain acyl-CoAs in order to evaluate the reversibility of the acyl-CoA-mediated release of FasR from DNA (Figure S4). We found that dissociation of FasR from PfasMT mediated by long-chain acyl-CoAs was readily reversed upon addition of acyl-CoA thioesterase, indicating that long chain acyl-CoAs act as regulatory ligands instead of detergents.
Figure 8. Binding of FasR to fas promoter region is inhibited by long-chain fatty acyl-CoAs.
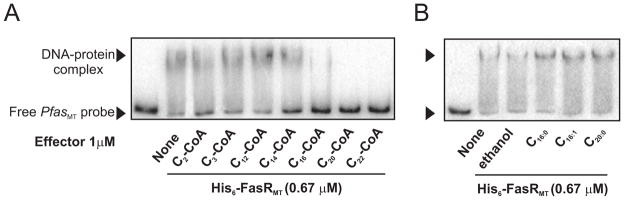
Gel shift assays were performed by incubating the 32P-labeled 398 bp probe with 0.67 μM of His6-FasRMT in the presence of A) acyl-CoAs of different chain length at a final concentration of 1 μM (C2-CoA to C22-CoA) or B) fatty acids (C16:0, C16:1, C20:0) at a final concentration of 1 μM. Because fatty acids are solubilized in ethanol, a lane containing the solvent was run as a control.
Considering the tight coordination that should exist between the two FAS systems present in mycobacteria, we also assayed the ability of FAS-II intermediates to modulate FasRMT binding to PfasMT. For this, we used a long chain acyl-AcpM and also AcpM as putative ligands but none of these had effect on FasR binding, suggesting that FasR activity is not affected by mycolic acid biosynthesis intermediates (data not shown).
FasR is essential for mycobacteria viability
The fasR gene has been proposed to be essential in M. tuberculosis based on a combination of high density mutagenesis and deep sequencing analysis (Griffin et al., 2011). Interestingly, most of the fatty acid biosynthesis regulators described so far in bacteria are not essential for survival (Simons et al., 1980, Lu and Rock, 2006, Schujman et al., 2003). The only exception is MabR, a novel regulatory protein involved in mycolic acid biosynthesis regulation in mycobacteria (Salzman et al., 2010). Therefore, to genetically test the essentiality of fasR gene in the viability of Mycobacterium, we undertook a genetic approach using M. smegmatis as a model system.
The essentiality of fasRMS was first suggested by our inability to generate a knock-out mutant strain through a two-step homologous recombination strategy (Pelicic et al., 1996). Knock out mutants were only obtained in the presence of an extra copy of fasR, using merodiploid strains of M. smegmatis expressing fasRMS or fasRMT under the control of Pami. Although the conditional mutants obtained were not useful for further analysis due to the high levels of fasR expression under non-inducing conditions, these experiments allowed us to confirm that fasR is an essential gene in M. smegmatis and strongly suggested that FasRMT and FasRMS have similar functions in M. tuberculosis and in M. smegmatis, respectively.
In order to obtain a conditional mutant in which we could tightly control the expression of fasR, we used an adaptation of the system developed by Boldrin et al (Boldrin et al., 2010). The system is based in two different repressors (TetR and Pip) encoded at the chromosomal level and the addition of anhydrotetracycline (ATc) allows tight repression of the gene under investigation.
For the construction of the fasR conditional mutant we cloned the 5′end of fasR under the control of the pristinamycin (Pip) dependent promoter Ptr in the ts plasmid pPR27 (Pelicic et al., 1997), obtaining the plasmid pFR20. This plasmid was used to transform M. smegmatis cells harboring the plasmid pFRA42B carrying the TetR/Pip OFF system (Boldrin et al., 2010), and one of the Str/Apra resistant transformants was grown at 30 °C and plated at 42 °C to promote plasmid recombination. The recombination event that left fasR under the control of ATc was confirmed by PCR, and the resultant strain named MSPtr:fasRMS. When plated on solid media containing 200 ng ml−1 of ATc, MSPtr:fasRMS was unable to grow, confirming that fasR is essential for the viability of M. smegmatis (data not shown). The mutant was grown in liquid media containing different ATc concentrations ranging from 50 to 200 ng ml−1. As shown in Figure S5, under these conditions the fasR mutant stopped growing after 20 h of incubation. However, growth was restored at different times, depending on ATc concentration in the media (Figure S5) probably due to ATc instability as suggested previously (Boldrin et al., 2010).
The MSPtr:fasRMS conditional mutant strain was grown for 40 h in 7H9 medium. The culture was then divided into two equal fractions and one of them was supplemented with ATc 200 ng ml−1. The mutant strain exhibited a typical exponential growth curve when grown in the absence of ATc but growth was inhibited after 3 h of ATc addition (Figure 9A). In order to correlate cell growth and expression of the fasR gene, the relative amount of fasR mRNA was measured by quantitative RT-PCR (qRT-PCR) after 9 h (T3) of exposure to ATc. As shown in Figure 9B, expression of fasR was severely repressed (~90 %) compared with the non-treated culture. The repression of the fasR gene expression was also confirmed by Western blot. Figure 9C shows a clear decrease in FasR protein levels after 6 (T2) and 9 (T3) h of ATc exposure.
Figure 9.

A. Growth curve of strain MSPtr:fasRMS in 7H9 medium. After 40 h, the culture was divided in two equal fractions and one of them was supplemented with ATc 200 ng ml−1 (indicated with an arrow). Samples were taken at different time points (T1, T2 and T3) for further analysis.
B. Changes in the relative amounts of fasR mRNA measured by quantitative RT-PCR. Values represent the mean difference between MSPtr:fasRMS strain grown with and without ATc 200 ng ml−1 and are normalized using sigA as an invariant transcript. Samples for RNA extraction were collected 9 h after addition of ATc (T3).
C. Western blot analysis of total crude lysates from MSPtr:fasRMS strain grown with (+) and without (−) ATc 200 ng ml−1. Detection was performed using anti-FasRMT antibodies elicited in rabbit (upper panel) and horseradish peroxidase conjugated streptavidin to detect the biotinylated protein AccA3 as loading control (lower panel).
Characterization of the M. smegmatis fasR conditional mutant MSPtr:fasRMS
To further characterize the role of FasR in fatty acid biosynthesis, we analyzed the expression of the fas-acpS operon genes in the M. smegmatis conditional mutant strain MSPtr:fasRMS. The relative amounts of fas and acpS mRNAs were measured by quantitative RT-PCR after 9 h (T3) of exposure to ATc and compared with non treated cells. As shown in Figure 10A, the transcription of both fas and acpS is reduced 40 % when cells are deprived of FasR. These results confirm that fas and acpS are part of the same transcriptional unit and that FasR is a transcriptional activator of the operon.
Figure 10.

A. The relative expression level of fas and acpS mRNA was measured by quantitative RT-PCR. Values represent the mean difference between MSPtr:fasRMS strain grown with and without ATc 200 ng ml−1 and are normalized using sigA as an invariant transcript. Samples for RNA extraction were collected 9 h after addition of ATc (T3).
B. Lipid composition of fasR conditional mutant MSPtr:fasRMS. Thin-layer chromatography (TLC) of 14C-labelled methyl esters of mycolic acids (MAMEs) and FA (FAMEs) extracted from MSPtr:fasRMS strain grown with (+) and without (-) ATc 200 ng ml−1. Aliquots were labeled with [14C]-acetate at T1 and T3 for 1 h at 42°C. The sample volumes loaded were normalized according to the optical densities of the cultures. Solvent system: hexane:ethyl acetate (9:1 v/v).
To study the physiological consequences of FasR depletion, we carried out [14C] acetate labeling experiments with the fasR conditional mutant strain MSPtr:fasRMS. The cultures were grown with and without ATc 200 ng ml−1 and their lipid content analyzed by TLC. As shown in Figure 10B, de novo synthesis of fatty acids and mycolic acids was progresively reduced after 3 (T1) and 9 (T3) h exposure to ATc. To confirm that the reduction observed in fatty acid and mycolic acid biosynthesis was specifically related to the deprivation of FasR, we performed [3H]-leucine and [3H]-uracil labeling experiments in order to determine the levels of metabolic activity in the cultures. As shown in Figure S6, 3 h after the conditional mutant stopped growing (after 9 h exposure to ATc 200 ng ml−1), the metabolic activity of the cells was still comparable to the control strain (without ATc). Altogether, these results demonstrate that at least in M. smegmatis, FasR is an essential transcriptional regulator of fatty acid biosynthesis.
DISCUSSION
Lipid metabolism plays a key role in the pathogenesis of M. tuberculosis and metabolic adaption to the host niche. Although mycobacteria can utilize a variety of carbon sources for in vitro growth, host fatty acids are thought to provide the major source of carbon during intracellular infection (McKinney et al., 2000, Munoz-Elias and McKinney, 2006, Schnappinger et al., 2003). This implies that the two fatty acid synthases present in mycobacteria, FAS-I and FAS-II, need to be regulated in a coordinate manner, both in vitro and in vivo, in order to preserve cell membrane homeostasis, optimize the high demand of energy required for fatty acid biosynthesis, and to modulate the host immune response during infection. Any perturbation of this tight regulation will result in a severe metabolic damage of the organism that probably leads to cell death. Therefore, understanding the molecular bases of such regulation will not only help us to comprehend better the physiology of this pathogen but might also give us some new tools to tackle this deadly bacterium.
Our studies of transcriptional regulation of the mycolic acid biosynthesis pathway identified MabR, a transcriptional regulator in charge of modulating the expression of the fasII operon. Our studies suggested that the coordinate regulation of the two FAS systems not only occurs at the post-translational levels, as it was previously reported (Molle et al., 2006, Veyron-Churlet et al., 2009, Slama et al., 2011), but also at the hierarchical upper level of transcription. Although overexpression of MabR also produced a negative effect on the expression levels of fas, we failed to demonstrate a direct role of MabR on the transcriptional regulation of fas, suggesting that a much complex network of regulation of fatty acid/mycolic acid biosynthesis would exist in mycobacteria. In this work, by using a classical approach for the purification of DNA binding proteins, we identified FasR, a TetR-like transcriptional regulator that specifically binds to the fas promoter region to activate its expression. As demonstrated by RT-PCR, the fas promoter (Pfas) not only transcribes fas but also acpS, two genes that are part of the same transcriptional unit both in M. smegmatis and in M. tuberculosis (Fig. 1B–C). acpS codes for a 4-phosphopantetheinyl transferase (PPTase), which transfers the phosphopantetheine group from CoA to the acyl carrier protein (ACP), a key protein involved in lipid biosynthesis (Lambalot et al., 1996, Walsh et al., 1997). Chalut et al demonstrated that PptT activates the Acp domains of the numerous type-I polyketide synthase (PKS) and non-ribosomal peptide synthetase (NRPS) present in M. tuberculosis, whereas AcpS is dedicated to the post-translational modification of FAS I and of the AcpM subunit of FAS II (Chalut et al., 2006). Thus, FasR, the regulatory protein of the fas-acpS operon could be considered as a key factor involved in the coordination of the activity of the two FAS systems that coexist in mycobacteria.
BLAST analysis, using the amino acid sequence of FasRMT, revealed a significant homology (above 45 % of identity along the whole sequence) with other hypothetical proteins present exclusively in actinomycetes, including members of the genera Frankia, Streptomyces, Rhodococcus and Nocardia. Notably, the highest percentages of identity (above 62 %) were observed within the FasRMT homologues of the mycolic-acid containing actinomycetes. Since the corresponding genes show a conserved genetic organization compared to that of fasRMT, we propose that they all might share the same physiological role and therefore could be considered orthologues. It is interesting noticing that although Corynebacterium sp. only employs a FAS-I system for the biosynthesis of both fatty acid and corynomycolic acids, our bioinformatic analysis could not find a FasRMT homologue on their genomes, indicating that corynebacteria followed a completely different pathway of evolution for the regulation of this essential metabolism. Recently, a transcriptional regulator of the two fas genes present in Corynebacterium was described (Nickel et al., 2010). Although this regulatory protein also belongs to the TetR-family of transcriptional regulators, it has no significant homology with fasRMT (< 20%), it is not essential for bacterial survival and it only affects the expression of fasA and fasB genes when acetate is the sole carbon source in the growth medium (Nickel et al., 2010).
TetR-like proteins normally bind to short palindromic DNA sequences (Grkovic et al., 1998, Orth et al., 2000, Ramos et al., 2005). Because protein binding constrains the evolution of these nucleotides, regulatory motifs may be identifiable through their conservation relative to neighboring DNA sequences. The alignment of the binding sites of FasRMT with the orthologous regions from other species (Figure S3) followed by bioinformatic analysis using MEME led us to identify a highly conserved consensus sequence: TAC[TG][CG][GAC][CTG][GC][A/C]GTA. Although in silico search of the FasR binding motif suggested that it is only present in the fas promoter region, we could not rule out that this protein could bind to a different motif in a different genetic context, or associated to other regulatory elements as it happens with other transcriptional regulators such as LexA of E. coli or AdpA of Streptomyces griseus (Butala et al., 2009, Higo et al., 2011). ChIP-on-chip experiments are being conducted in order to find out in vivo the complete regulon of FasRMT.
The activator nature of FasR, demonstrated by the Pfas-lacZ transcriptional studies, highlights a sharp difference with most transcriptional regulators of fatty acid biosynthesis which are repressor proteins (Zhang and Rock, 2009). The only exceptions are FadR of E. coli that acts as a repressor of the β-oxidation genes and also as activator of the fatty acid biosynthesis genes fabA and fabB (specifically involved in the synthesis of unsaturated fatty acids) (Henry and Cronan, 1991), and FasR of Streptomyces coelicolor, that was the first activator of the core set of fab genes described in bacteria (Arabolaza et al., 2010). Furthermore, FasR differs from most TetR regulators described which mainly work as repressors of their target genes (Ramos et al., 2005). The activator nature of FasR is in agreement with the localization of the binding site of this regulator (200–300 bp) relative to the TSS of the fas gene. The main difference of FasR with all the other FA biosynthesis regulators described in bacteria is that FasR is essential for bacterial growth while all the others are not. It is important to state that the only exception of this is MabR, the transcriptional regulator of the fasII operon genes of mycobacteria (Salzman et al., 2010).
In several bacterial systems, the final product of the fatty acid biosynthesis pathways are the common effectors of the corresponding transcriptional regulators (DiRusso et al., 1992, Campbell and Cronan, 2001, Zhu et al., 2009, Feng and Cronan, 2011, Lu and Rock, 2006, Jerga and Rock, 2009, Schujman et al., 2006). On the other hand, FapR is a transcription factor regulated by a feed-forward mechanism by malonyl-CoA and malonyl-ACP, which are metabolites used at the beginning of the fatty acid biosynthesis pathway (Schujman et al., 2006). EMSA assays carried out in the presence of different chain-length acyl-CoAs gave as the clue that fatty acids esters > C16 were the ligands that are sensed by FasRMT in order to release its binding from the fas promoter and then stop its activation function. The strongest effect was seen with the C20 acyl-CoA, however, it is difficult to define with this technique if longer acyl-CoAs could have a stronger effect on the affinity of FasR for the DNA. The in vivo analysis of the PfasMT::lacZ transcriptional fusions carried out in growth media, supplemented or not with fatty acids of different chain-length, supported the in vitro studies suggesting that long-chain acyl-CoAs are the signal molecules sensed in vivo by FasR to modulate its activity. In this sense the negative effect on fas transcription, observed when MabR was overexpressed (Salzman et al., 2010), could reflect the effect of the long-chain acyl-CoAs accumulated due to the shut off of the fasII operon on FasR activity. The hypothesis would be that FasR senses the pool of long-chain acyl-CoAs and then relieve the activation of fas. We do not know what metabolites are sensed by MabR, however it is tempting to speculate that the product of FAS-I could also be sensed by MabR and then the levels of these compounds would be a key player on this complex network of regulation.
This study and our previous work on MabR have contributed to identify the minimal set of cis and trans elements that define the regulatory network that control fatty acid and mycolic acid biosynthesis in mycobacteria. Furthermore, our results suggest that a tight mechanism of transcriptional regulation of the genes involved in fatty acid and mycolic acid biosynthesis is needed in order to keep membrane homeostasis in mycobacteria. The identification and characterization of all the components of this regulatory network will not only help understanding the complex interaction of lipid metabolism in mycobacteria but it will also bring new opportunities for drug developments using FasR and MabR as potential targets.
EXPERIMENTAL PROCEDURES
Bacterial strains, culture, and transformation conditions
The E. coli strain DH5α (Hanahan, 1983) was used for routine subcloning and was transformed according to Sambrook et al (Sambrook et al., 1989). The transformants were selected on media supplemented with the appropriate antibiotics. Strain BL21 λ (DE3) is an E. coli B strain lysogenized with λ DE3, a prophage that expresses the T7 RNA polymerase from the isopropyl-β-D-thiogalactopyranoside (IPTG)-inducible lacUV5 promoter (Studier and Moffatt, 1986). Mycobacterium smegmatis mc2155 is an electroporation-efficient mutant of mc26 (Snapper et al., 1990). Liquid cultures of M. smegmatis were grown at 37 °C in 7H9 medium supplemented with 0.2% glycerol and 0.03% Tyloxapol. M. tuberculosis H37Rv was grown in 7H9 supplemented with 0.2% glycerol, 0.05% Tween 80 and 10% Middlebrook ADC (albumin-dextrose-catalase). Recombinant plasmids and strains genotypes are listed in Tables SI and SII.
DNA manipulation and plasmid construction
Isolation of plasmid DNA, restriction enzyme digestion, and agarose gel electrophoresis were carried out by conventional methods (Sambrook et al., 1989). Genomic DNA of M. smegmatis was obtained as described (Connell, 1994).
pFR3
fasRMT (rv3208) was PCR-amplified from genomic DNA of M. tuberculosis H37Rv using the oligonucleotides F-Rv3208 (5′-CATATGAGCGATCTCGCCAAGACA -3′) to introduce an NdeI site at the translational start codon of fasRgene, and R-Rv3208 (5′-GAATTCCTACGAGCGGGTAAGCGG -3′) to introduce an EcoRI site at the end of the ORF. To generate a fasRMT His tag fusion gene, the PCR product was digested with NdeI and EcoRI and cloned into NdeI/EcoRI cleaved pET24b, yielding pFR3.
pFR9
In order to express fasRMT in M. smegmatis in a multicopy plasmid, plasmid pFR3 was digested with NdeI and EcoRI and the fragment was cloned into the pMV306 derivative pMR24 (Salzman et al., 2010) previously digested with the same enzymes, yielding pFR5. Plasmid pFR5 was digested with KpnI and XbaI enzymes and the fragment, containing fasRMT as a His6-tag fusion under acetamidase promoter control, was cloned into KpnI/XbaI cleaved pJAM2 yielding plasmid pFR9.
pFR20
For the construction of the M. smegmatis mutant allele Ptr:fasRMS, the 5′ region of fasRMSwas amplified with oligos FasRMS-FNsiI (5′-TGCATGCATATGAGCGATCTCGCCAACAC-3′) and FasRMS-RXbaI (5′-CCTCTAGACGATGAAATCGAAGAACGCCT-3′). The 372 bp PCR product was cloned in pCR BluntII TOPO (Invitrogen) and digested with NsiI/XbaI. The fragment was inserted in a pMP349 derivative plasmid, that contains Ptr from pFRA50 (Bazet Lyonnet, unpublished results) cloned into EcoRI/PvuII sites, obtaining pFR40 plasmid. This plasmid has been sequenced in order to see that no errors were introduced during amplification. Finally, pFR40 was digested with SpeI/XbaI and cloned in the same sites of the ts plasmid pPR27 (Pelicic et al., 1997), generating pFR20 plasmid. This plasmid was used to obtain M. smegmatis MSPtr:fasRMS mutant strain.
pFR47
The fragment containing the promoter region PfasMT used for transcriptional fusion to lacZ, was generated by PCR amplification from M. tuberculosis H37Rv genomic DNA with primers N2Fas1Mt-prom (5′-CATAACGATTTGATAACAAAACTGC-3′) and CFas1Mt-prom (5′-CACCCGGTCGTGCTCGTGGATCGTC-3′). PCR-amplified fragment was digested with EcoRI, filled-in with Klenow and ligated into the ScaI site of the integrative promoter-probe vector pSM128 (Dussurget et al., 1999) generating the mycobacterial reporter plasmid pFR47. The pSM128 vector carries a cII-lacZ fusion, the mycobacteriophage L5 integrase gene, attachment sites, and a streptomycin/spectinomycin resistance cassette. DNA sequence of the cloned fragment was determined by automated sequencing to both, confirm the identity of the amplified product and check the in-frame transcriptional fusion of the insert to the promoterless lacZ gene.
pFR48-50
Synthetic versions of the promoter region PfasMT with ScaI flanking sites and a replacement of one or both of the FasRMT binding sites (IR 1 and IR 2) by a nonrelated sequence, were obtained from Epoch Biolabs and cloned into pBluescript-SK to yield pBSK-Mut1 (replacement of wt IR 1 5′-TACCCGTACGTAGAACTCGCCAGTA-3′ with the random sequence 5′-CGAATTATGAGCTCGTAACATGAGC-3′), pBSK-Mut2 (replacement of wt IR 2 5′-TACTCCACCGTA-3′ with 5′-CGAATTATGAGC-3′) and pBSK-Mut3 (replacement of both wt IRs with the sequences shown above). Fragments used for transcriptional fusion to lacZ, were generated digesting pBSK-Mut1, pBSK-Mut2 and pBSK-Mut3 with ScaI. Each fragment was ligated into the ScaI site of the integrative promoter-probe vector pSM128 (Dussurget et al., 1999) generating the mycobacterial reporter plasmids pFR48, pFR49 and pFR50 respectively. DNA sequence of the cloned fragments were determined by automated sequencing to confirm the identity of the synthesized products and check the in-frame transcriptional fusion of the inserts to the promoterless lacZ gene.
Isolation and Identification of FasR
To purify Pfas binding proteins we used a modification of the procedure described by Jourlin-Castelli et al. (Jourlin-Castelli et al., 2000). M. smegmatis mc2155 was grown at 37 °C in 400 ml of 7H9 medium. When the culture reached an OD600 of 0.9, the cells were harvested by centrifugation at 10000 × g (10min, 4 °C), and washed with buffer A (20 mM Tris-HCl (pH 8), 0.5 mM EDTA, 1 mM DTT, 5% (v/v) glycerol) supplemented with 50 mM NaCl and 1mM phenylmethylsulfonyl fluoride (PMSF). The pellet was resuspended in the same buffer and the cells were disrupted by sonication (10 pulses, 20 s ON and 30 s OFF, 20% amplitude). After centrifugation at 23000 × g (30 min, 4 °C) and ultracentrifugation at 160000 × g (1 h; 4 °C), the crude extract was recovered and subjected to two steps of precipitation in ammonium sulfate (25% and 75% saturation). The pellet recovered was resuspended and dialyzed against buffer A to reduce ionic strength. A streptavidin column, to which DNA fragments containing the PfasMS or PfasMT promoter region were bound, was prepared as follows. A biotinylated version of the promoter DNA fragment of M. smegmatis (PfasMS) was generated by PCR amplification from M. smegmatis genomic DNA with primers N2fasI MS (5′-GATAACGATTAGATAACAATGCTGC-3′) and CfasI MT prom BIOT (5′-Biotin-CACCCGGTCGTGCTCGTGGATCGTC -3′). Once purified, the biotinylated promoter was fixed to a streptavidin column (Streptavidin MagneSphere® Paramagnetic Particles, Promega) and the column was equilibrated with binding buffer (25 mM Tris/HCl pH 8, 1 mM PMSF, 5% (v/v) glycerol, 5 mM MgCl2, 150 mM NaCl and 1 μg poly-dIdC) at room temperature. The dialyzed fraction was then loaded onto the Pfas-streptavidin column. After one wash with binding buffer and two washes with binding buffer without poly-dIdC, the DNA- bound proteins were eluted with 0.8 M NaCl. The eluted proteins were precipitated with 80% acetone and submitted to electrophoresis in a 12% SDS-PAGE. After Coomassie blue staining, bands were excised from the gel and analyzed by mass spectrometry. Mass spectrometric data were obtained using a MALDI-TOF-TOF spectrometer, Ultraflex II (Bruker), in the mass spectrometry facility CEQUIBIEM, Argentina.
Expression and purification of FasRMT
Expression of fasRMT was carried out following IPTG induction of BL21 λ (DE3) E. coli transformed with pFR3. His6-FasRMT was then affinity-purified from BL21 λ (DE3) lysates using Ni-NTA agarose (Qiagen, Inc) according to manufacturer’s instructions.
Protein Methods
Purified proteins were analysed by SDS-PAGE (Laemmli, 1970). Coomassie brilliant blue was used to stain protein bands. Protein contents were determined using Quant-iT™ Protein Assay Kits and Qubit® fluorometer (Invitrogen).
Proteins were electroblotted onto nitrocellulose membranes (Bio-Rad) for Western blot analysis. To recognize FasRMS, the membrane was probed with 1:100 dilution of polyclonal anti- FasRMT serum and antigenic polypeptides were visualized using a horseradish peroxidase conjugated secondary antibody. To identify the biotinylated protein AccA3MS, the membrane was probed with 1:10000 dilution of a horseradish peroxidase conjugated streptavidin. Antiserum against His6-FasRMT was elicited in rabbits.
Electrophoretic mobility shift assays (EMSAs)
EMSA with cell crude extracts
M. smegmatis mc2155 cells from 15 ml mid-log phase cultures were washed and disrupted by sonication (10 pulses, 20 s ON and 30 s OFF, 20% amplitude) in 0.3ml of lysis buffer (50mM Tris/HCl pH8, 100mM NaCl, 20% (v/v) glycerol, 1mM DTT and 1mM PMSF). The cell wall fraction was pelleted by centrifugation at 23000 × g (30 min, 4°C) and the resulting supernatant was used to assess protein binding to PfasMS (448 bp) or to PfasMT (398 bp) promoter fragments. The promoter DNA fragment for these assays was generated by PCR amplification from M. smegmatis genomic DNA with primers N2fasI MS (5′-GATAACGATTAGATAACAATGCTGC-3′) and CfasI MS (5′-GTCGTGTTCGTAGATCGTCACTGGG -3′) or from M. tuberculosis genomic DNA with primers N2Fas1Mt-prom (5′-CATAACGATTTGATAACAAAACTGC-3′) and CFas1Mt-prom (5′-CACCCGGTCGTGCTCGTGGATCGTC -3′). N2fasI MS or N2Fas1Mt-prom primers were end-labelled with [γ-32P] ATP (3,000 Ci mmol−1) using T4 polynucleotide kinase and the PCR product obtained was purified from agarose gels. Increasing concentrations of cell crude extracts were incubated with the appropriate 32P-labelled probe (6000 c.p.m.) in a total volume of 25 μl binding buffer (25 mM Tris/HCl pH 8, 1 mM PMSF, 5% (v/v) glycerol, 5 mM MgCl2, 150 mM NaCl and 1 μg poly-dIdC) at room temperature for 15 min. DNA–protein complexes were resolved by electrophoresis on a 6% (w/v) non-denaturing polyacrylamide gel in 1X TBE (89 mM Tris Base; 89 mM Boric Acid; 2 mM EDTA), 5% (v/v) glycerol at 150 V at 4°C and were visualized and digitalized with a Storm167 840 scanner (Amersham).
Unlabeled specific and nonspecific competitor DNA (85 fold molar excess) were incubated with 52 μg cell crude extract for 10 min at room temperature, followed by the addition of the labelled probe and incubation for 15 min at room temperature. The resulting DNA-protein complexes were then subjected to electrophoresis. Results were developed and digitalized with a Storm167 840 scanner (Amersham).
EMSA with purified His6-FasRMT protein
Purified recombinant His6-FasRMT was used to assess protein binding to PfasMT promoter fragment (398 bp). The promoter DNA fragment (PfasMT) for these assays was generated as described above. Once purified from agarose gel, the 32P-labelled probe (3000 c.p.m.) was incubated with different concentrations of His6-FasRMT in a total volume of 25 μl binding buffer at room temperature for 15 min. When indicated, an acyl-CoA, acyl-ACP or fatty acid was added to the binding buffer and incubated with the protein for 5 min at 4 °C before the addition of the labelled probe. DNA–protein complexes were resolved by electrophoresis on a 6% (w/v) non-denaturing polyacrylamide gel in 1X TBE, 5% (v/v) glycerol at 150 V at 4 °C and were visualized and digitalized with a Storm167 840 scanner (Amersham).
Unlabeled specific and nonspecific competitor DNA (60 fold molar excess) were incubated with His6-FasRMT for 10 min at room temperature, followed by the addition of the labelled probe and incubation for 15 min at room temperature. The resulting DNA-protein complexes were then subjected to electrophoresis. Results were developed and digitalized with a Storm167 840 scanner (Amersham).
To assess protein binding to the mutated PfasMT promoters Mut1, Mut2 and Mut3, DNA fragments were generated by PCR amplification from plasmids pBSK-Mut1–3 (see construction of plasmids pFR48–50), with primers N2Fas1Mt-prom (5′-CATAACGATTTGATAACAAAACTGC-3′) end-labelled with [γ-32P] ATP, and CFas1Mt-promMUT (5′-GGTCTATGTCTCCCTATGTGCATC-3′). In the Mut1 probe, the two IR from the protected region 1 were replaced by the random sequence 5′-CGAATTATGAGCTCGTAACATGAGC-3′. In the Mut2 probe, the IR from the protected region 2 was replaced by the random sequence 5′-CGAATTATGAGC-3′, and in the Mut3 probe, the three IR were replaced by the same random sequences described. Once purified from the gel, the wild type and mutated 32P-labelled probes (1000 c.p.m.) were incubated with 1.2 μM His6-FasRMT in a total volume of 25 μl binding buffer at room temperature for 15 min. DNA–protein complexes were resolved and visualized as described before.
DNase I footprinting
Radiolabeled PfasMT promoter fragment (50000 c.p.m.) was incubated at room temperature for 15 min with different amounts of purified His6-FasRMT in 25 μl binding buffer. DNA was partially digested with DNaseI (Promega) for 3 min at room temperature and digestion was stopped by adding 204 μl of stop solution (20 mM EDTA pH 8.0, 200 mM NaCl, 100 μg μl−1 Yeast RNA) and 235 μl phenol/chloroform (1:1). After ethanol precipitation, the pellet was washed with 70% (v/v) ethanol, dissolved in 6 μl formamide-dye mixture, heat denatured (94 °C for 2 min) and immediately placed on ice. Digestion products were resolved on a 6% (w/v) denaturing polyacrylamide gel by electrophoresis. Appropriate sequencing reactions were loaded onto the gels along with the footprinting samples and used as a size ladder for identification of the sequences of protected sites. Results were developed and digitalized with a Storm167 840 scanner (Amersham).
β-Galactosidase assays
Saturated cultures of M. smegmatis grown in 7H9 with the corresponding antibiotic were diluted 100 fold into the same medium and incubated at 37 °C. At the early-log phase, they were supplemented (when indicated) with 0.2% acetamide and/or fatty acids at a final concentration of 0.01%. Following 4 h of incubation, 10 ml of each culture were pelleted, washed and finally resuspended in Z buffer (Miller, 1972). The collected cells were disrupted by sonication (5 pulses of 30 s duration, with intervals of 30 s, maximum potential) and the cell wall fractions were pelleted by centrifugation at 23,000 × g (30 min, 4°C). The resulting supernatants were quantified with Quant-iT™ Protein Assay kit according to manufacturer’s instructions (Invitrogen) and assayed for β-galactosidase activity (Miller, 1972). The data were recorded in triplicates for at least three independent experiments (n= 3). Levels of activity are expressed as nmol ONPG per min per mg of protein, and values are the means of the results of three independent experiments ± standard deviations. The average β-galactosidase level produced by the parental plasmid pSM128 was subtracted from each data set to account for background activity. For this, the promoterless lacZ fusion (pSM128) was integrated in M. smegmatis mc2155 and MSpFR9, generating the control strains MSpSM128 and MSpSM128 pFR9 respectively.
RNA techniques
RNA extraction
RNA was extracted from mid-log phase cultures of M. smegmatis or M. tuberculosis H37Rv using the SV total RNA isolation system (Promega) and treated when needed with RQ1 RNase-Free DNase (Promega).
RT-PCR
To assess the operon nature of fas and acpS, RNA was extracted from mid-log phase cultures of M. smegmatis mc2155 and M. tuberculosis H37Rv. Reverse transcription reaction was carried out using SuperScript III Reverse Transcriptase (Invitrogen) and random primers. PCR amplification of the intergenic regions on cDNA were performed using specific oligos on fas and acpS (For fasMS-acpSMS = oligos F-MS acpS-fas 5′-CTCGGCGAGAACGATCAGTA-3′ and R-MS acpS-fas 5′-GCCTTGATCACGGCTTCCTT-3′; For fasMT-acpSMT = oligos F-TB acpS-fas 5′-CGAGGCGTATATCGGCTGAC-3′ and R-TB acpS-fas 5′-CGGCGAAATCGGGAATGGAG-3′).
RACE
The method that we used for mapping transcription initiation sites in Mycobacterium is critically dependent on examining the differences between amplification products produced with TAP-treated vs untreated RNA samples as templates for cDNA synthesis and PCR. To map the transcription initiation site of Mycobacterium fas gene we used the GeneRacer™ kit without treating the RNA samples with CIP, but starting the protocol with the removal of the pyrophosphate with TAP. Duplicate samples of RNA were untreated or treated with TAP to convert 5′ triphosphates to monophosphates. An RNA oligonucleotide of known sequence was then ligated to the 5′ monophosphate ends and cDNA was made using random primers and SuperScript III Reverse Transcriptase (Invitrogen). Reverse transcription was followed by two rounds of PCR reactions in which an abridged anchor primer and two sets of specific primers were employed: the first PCR was performed using RACE-GSP fasI 5′-ACTCGATACCGGCCGAGGACACCAG-3′ for 5′fasMS and RACE fas MT 5′-CACCGAATGCGACAGCGTAGGG-3′ for 5′fasMT; and the second semi- nested PCR amplification was performed using a nested gene-specific primer (RACE-GSP fasI nested 5′-GGTCAGATCCGGACTCGTCGTT-3′ for 5′fasMS; RACE fas MT nested 5′-TGAGGCGATCGACCAGAGCGTG-3′ for 5′fasMT). The PCR products were cloned into pCR BluntII TOPO (Invitrogen) and the DNA sequence of several clones was determined. Any product appearing in TAP-treated samples that were not present in the untreated samples would have had 5′ triphosphates, indicating that the RNA oligonucleotide tagged a true initiation point. The first nucleotide following the 5′-RACE adaptor was taken as the transcription start site.
qRT-PCR
The expression of fasRMS, fasMS, and acpSMS was quantitated after normalization of RNA levels to the expression of the sigAMS gene. A saturated culture of MSPtr:fasRMS grown in 7H9 medium supplemented with streptomycin and apramycin was diluted 200 fold into the same medium and incubated for approximately 40 h at 42 °C. The culture was then divided in two equal fractions and one of them was supplemented with ATc 200 ng ml−1. Both cultures were incubated at 42°C for 9 h and RNA was extracted from each culture. Second strand cDNA generated with SuperScript III Reverse Transcriptase (Invitrogen) and random primers, was used in qRT-PCR with green fluorochrome as the indicator dye (qPCR master mix, Biodynamics). qRT-PCR cycling conditions were as follows: 95 °C for 10 min followed by 40 cycles of 95 °C for 30 s, 58 °C for 30 s, and 68 for 30 s. qRT-PCR data are presented as fold difference of expression in MSPtr:fasR without ATc over that in the same strain with ATc 200 ng ml−1, using the Pfafll method (Pfaffl, 2001). sigAMS was used as normalizing gene.
Lipid Analysis
Fatty acid and mycolic acid biosynthesis was analyzed by incorporation of [14C] acetate. A saturated culture of MSPtr:fasRMS grown in 7H9 medium supplemented with streptomycin and apramycin was diluted 200 fold into the same medium and incubated for approximately 40 h at 42 °C. The culture was then divided in two equal fractions and one of them was supplemented with ATc 200 ng ml−1. After 3 h and 9 h of incubation, 5 ml of each culture was labeled for 1 h with [1-14C] acetate (59 mCi/mmol) (New England Nuclear) at a concentration of 1 μCi ml−1. Fatty acid methyl esters (FAMEs) and mycolic acid methyl esters (MAMEs) were extracted as reported previously (Kremer et al., 2000). The resulting solution of FAMEs and MAMEs was assayed for radioactivity in a Beckman liquid scintillation counter and then subjected to TLC using silica gel plates (5735 silica gel 60 F254; Merck) normalizing by optic densities. Plates were developed in hexane:ethyl acetate (9:1 v/v). Autoradiograms were produced by overnight exposure to Kodak X-Omat AR films.
Statistical analysis
Data are reported as arithmetic means of the results obtained from three independent experiments ± standard deviations. Statistical significance was calculated using ANOVA (Figure 6 and 7B) and Mann Whitney test (Figure 7A). Statistical significance was accepted at P <0.05.
Supplementary Material
Acknowledgments
We thank Dr. C. Sala for critically reading of the manuscript and Hebe Bottai and Liliana Racca for the generous help in the statistical analysis. This work has received funding from ANPCyT (grants 2008-1245 and 2011-0245) and NIH (grant 1R01AI095183-01).
References
- Arabolaza A, D’Angelo M, Comba S, Gramajo H. FasR, a novel class of transcriptional regulator, governs the activation of fatty acid biosynthesis genes in Streptomyces coelicolor. Mol Microbiol. 2010;78:47–63. doi: 10.1111/j.1365-2958.2010.07274.x. [DOI] [PubMed] [Google Scholar]
- Bacon J, Marsh PD. Transcriptional responses of Mycobacterium tuberculosis exposed to adverse conditions in vitro. Curr Mol Med. 2007;7:277–286. doi: 10.2174/156652407780598566. [DOI] [PubMed] [Google Scholar]
- Bailey TL, Elkan C. The value of prior knowledge in discovering motifs with MEME. Proc Int Conf Intell Syst Mol Biol. 1995;3:21–29. [PubMed] [Google Scholar]
- Bailey TL, Gribskov M. Methods and statistics for combining motif match scores. J Comput Biol. 1998;5:211–221. doi: 10.1089/cmb.1998.5.211. [DOI] [PubMed] [Google Scholar]
- Bhatt A, Fujiwara N, Bhatt K, Gurcha SS, Kremer L, Chen B, et al. Deletion of kasB in Mycobacterium tuberculosis causes loss of acid-fastness and subclinical latent tuberculosis in immunocompetent mice. Proc Natl Acad Sci USA. 2007a;104:5157–5162. doi: 10.1073/pnas.0608654104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt A, Molle V, Besra GS, Jacobs WR, Jr, Kremer L. The Mycobacterium tuberculosis FAS-II condensing enzymes: their role in mycolic acid biosynthesis, acid-fastness, pathogenesis and in future drug development. Mol Microbiol. 2007b;64:1442–1454. doi: 10.1111/j.1365-2958.2007.05761.x. [DOI] [PubMed] [Google Scholar]
- Bloch K. Fatty acid synthases from Mycobacterium phlei. Methods Enzymol. 1975;35:84–90. doi: 10.1016/0076-6879(75)35141-0. [DOI] [PubMed] [Google Scholar]
- Boldrin F, Casonato S, Dainese E, Sala C, Dhar N, Palu G, et al. Development of a repressible mycobacterial promoter system based on two transcriptional repressors. Nucleic Acids Res. 2010;38:e134. doi: 10.1093/nar/gkq235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan PJ, Crick DC. The cell-wall core of Mycobacterium tuberculosis in the context of drug discovery. Curr Top Med Chem. 2007;7:475–488. doi: 10.2174/156802607780059763. [DOI] [PubMed] [Google Scholar]
- Brennan PJ, Nikaido H. The envelope of mycobacteria. Annu Rev Biochem. 1995;64:29–63. doi: 10.1146/annurev.bi.64.070195.000333. [DOI] [PubMed] [Google Scholar]
- Butala M, Zgur-Bertok D, Busby SJ. The bacterial LexA transcriptional repressor. Cell Mol Life Sci. 2009;66:82–93. doi: 10.1007/s00018-008-8378-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JW, Cronan JE., Jr Escherichia coli FadR positively regulates transcription of the fabB fatty acid biosynthetic gene. J Bacteriol. 2001;183:5982–5990. doi: 10.1128/JB.183.20.5982-5990.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connell ND. Mycobacterium: isolation, maintenance, transformation, and mutant selection. Methods Cell Biol. 1994;45:107–125. doi: 10.1016/s0091-679x(08)61848-8. [DOI] [PubMed] [Google Scholar]
- Chalut C, Botella L, de Sousa-D’Auria C, Houssin C, Guilhot C. The nonredundant roles of two 4′-phosphopantetheinyl transferases in vital processes of Mycobacteria. Proc Natl Acad Sci USA. 2006;103:8511–8516. doi: 10.1073/pnas.0511129103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daffe M. The global architecture of the mycobacteria cell envelope. In: Daffe M, Reyrat JM, editors. The Mycobacterial Cell Envelope. ASM Press; Washington, DC: 2008. pp. 3–12. [Google Scholar]
- Daffe M, Draper P. The envelope layers of mycobacteria with reference to their pathogenicity. Adv Microb Physiol. 1998;39:131–203. doi: 10.1016/s0065-2911(08)60016-8. [DOI] [PubMed] [Google Scholar]
- Daniel J, Deb C, Dubey V, Sirakova T, Abomoelak B, Morbidoni H, Kolattukudy P. Induction of a novel class of diacylglycerol acyltransferases and triacylglycerol accumulation in Mycobacterium tuberculosis as it goes into a dormancy-like state in culture. J Bacteriol. 2004;186:5017–5030. doi: 10.1128/JB.186.15.5017-5030.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deb C, Lee CM, Dubey VS, Daniel J, Abomoelak B, Sirakova TD, et al. A novel in vitro multiple-stress dormancy model for Mycobacterium tuberculosis generates a lipid-loaded, drug-tolerant, dormant pathogen. PLoS One. 2009;4 (6):e6077. doi: 10.1371/journal.pone.0006077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiRusso CC, Heimert TL, Metzger AK. Characterization of FadR, a global transcriptional regulator of fatty acid metabolism in Escherichia coli. Interaction with the fadB promoter is prevented by long chain fatty acyl coenzyme A. J Biol Chem. 1992;267:8685–8691. [PubMed] [Google Scholar]
- Dubnau E, Chan J, Raynaud C, Mohan VP, Laneelle MA, Yu K, et al. Oxygenated mycolic acids are necessary for virulence of Mycobacterium tuberculosis in mice. Mol Microbiol. 2000;36:630–637. doi: 10.1046/j.1365-2958.2000.01882.x. [DOI] [PubMed] [Google Scholar]
- Dussurget O, Timm J, Gomez M, Gold B, Yu S, Sabol SZ, et al. Transcriptional control of the iron-responsive fxbA gene by the mycobacterial regulator IdeR. J Bacteriol. 1999;181:3402–3408. doi: 10.1128/jb.181.11.3402-3408.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Cronan JE. Complex binding of the FabR repressor of bacterial unsaturated fatty acid biosynthesis to its cognate promoters. Mol Microbiol. 2011;80:195–218. doi: 10.1111/j.1365-2958.2011.07564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickman MS, Cox JS, Jacobs WR., Jr A novel mycolic acid cyclopropane synthetase is required for cording, persistence, and virulence of Mycobacterium tuberculosis. Mol Cell. 2000;5:717–727. doi: 10.1016/s1097-2765(00)80250-6. [DOI] [PubMed] [Google Scholar]
- Griffin JE, Gawronski JD, Dejesus MA, Ioerger TR, Akerley BJ, Sassetti CM. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathogens. 2011;7:e1002251. doi: 10.1371/journal.ppat.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grkovic S, Brown MH, Roberts NJ, Paulsen IT, Skurray RA. QacR is a repressor protein that regulates expression of the Staphylococcus aureus multidrug efflux pump QacA. J Biol Chem. 1998;273:18665–18673. doi: 10.1074/jbc.273.29.18665. [DOI] [PubMed] [Google Scholar]
- Hanahan D. Studies on transformation of Escherichia coli with plasmids. J Mol Biol. 1983;166:557–580. doi: 10.1016/s0022-2836(83)80284-8. [DOI] [PubMed] [Google Scholar]
- Henry MF, Cronan JE., Jr Escherichia coli transcription factor that both activates fatty acid synthesis and represses fatty acid degradation. J Mol Biol. 1991;222:843–849. doi: 10.1016/0022-2836(91)90574-p. [DOI] [PubMed] [Google Scholar]
- Higo A, Horinouchi S, Ohnishi Y. Strict regulation of morphological differentiation and secondary metabolism by a positive feedback loop between two global regulators AdpA and BldA in Streptomyces griseus. Mol Microbiol. 2011;81:1607–1622. doi: 10.1111/j.1365-2958.2011.07795.x. [DOI] [PubMed] [Google Scholar]
- Jerga A, Rock CO. Acyl-Acyl carrier protein regulates transcription of fatty acid biosynthetic genes via the FabT repressor in Streptococcus pneumoniae. J Biol Chem. 2009;284:15364–15368. doi: 10.1074/jbc.C109.002410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jourlin-Castelli C, Mani N, Nakano MM, Sonenshein AL. CcpC, a novel regulator of the LysR family required for glucose repression of the citB gene in Bacillus subtilis. J Mol Biol. 2000;295:865–878. doi: 10.1006/jmbi.1999.3420. [DOI] [PubMed] [Google Scholar]
- Kaur D, Guerin ME, Skovierova H, Brennan PJ, Jackson M. Biogenesis of the cell wall and other glycoconjugates of Mycobacterium tuberculosis. Adv Appl Microbiol. 2009;69:23–78. doi: 10.1016/S0065-2164(09)69002-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lambalot RH, Gehring AM, Flugel RS, Zuber P, LaCelle M, Marahiel MA, et al. A new enzyme superfamily - the phosphopantetheinyl transferases. Chem Biol. 1996;3:923–936. doi: 10.1016/s1074-5521(96)90181-7. [DOI] [PubMed] [Google Scholar]
- Lu YJ, Rock CO. Transcriptional regulation of fatty acid biosynthesis in Streptococcus pneumoniae. Mol Microbiol. 2006;59:551–566. doi: 10.1111/j.1365-2958.2005.04951.x. [DOI] [PubMed] [Google Scholar]
- McKinney JD, Honer zu Bentrup K, Munoz-Elias EJ, Miczak A, Chen B, Chan WT, et al. Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature. 2000;406:735–738. doi: 10.1038/35021074. [DOI] [PubMed] [Google Scholar]
- Miller JH. Experiments in Molecular Genetics. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1972. [Google Scholar]
- Molle V, Brown AK, Besra GS, Cozzone AJ, Kremer L. The condensing activities of the Mycobacterium tuberculosis type II fatty acid synthase are differentially regulated by phosphorylation. J Biol Chem. 2006;281:30094–30103. doi: 10.1074/jbc.M601691200. [DOI] [PubMed] [Google Scholar]
- Munoz-Elias EJ, McKinney JD. Carbon metabolism of intracellular bacteria. Cell Microbiol. 2006;8:10–22. doi: 10.1111/j.1462-5822.2005.00648.x. [DOI] [PubMed] [Google Scholar]
- Nickel J, Irzik K, van Ooyen J, Eggeling L. The TetR-type transcriptional regulator FasR of Corynebacterium glutamicum controls genes of lipid synthesis during growth on acetate. Mol Microbiol. 2010;78:253–265. doi: 10.1111/j.1365-2958.2010.07337.x. [DOI] [PubMed] [Google Scholar]
- Orth P, Schnappinger D, Hillen W, Saenger W, Hinrichs W. Structural basis of gene regulation by the tetracycline inducible Tet repressor-operator system. Nat Struct Biol. 2000;7:215–219. doi: 10.1038/73324. [DOI] [PubMed] [Google Scholar]
- Pelicic V, Jackson M, Reyrat JM, Jacobs WR, Jr, Gicquel B, Guilhot C. Efficient allelic exchange and transposon mutagenesis in Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 1997;94:10955–10960. doi: 10.1073/pnas.94.20.10955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos JL, Martinez-Bueno M, Molina-Henares AJ, Teran W, Watanabe K, Zhang X, et al. The TetR family of transcriptional repressors. Microbiol Mol Biol Reviews. 2005;69:326–356. doi: 10.1128/MMBR.69.2.326-356.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao V, Gao F, Chen B, Jacobs WR, Jr, Glickman MS. Trans-cyclopropanation of mycolic acids on trehalose dimycolate suppresses Mycobacterium tuberculosis -induced inflammation and virulence. J Clin Invest. 2006;116:1660–1667. doi: 10.1172/JCI27335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzman V, Mondino S, Sala C, Cole ST, Gago G, Gramajo H. Transcriptional regulation of lipid homeostasis in mycobacteria. Mol Microbiol. 2010;78:64–77. doi: 10.1111/j.1365-2958.2010.07313.x. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. 2. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, N.Y: 1989. [Google Scholar]
- Schnappinger D, Ehrt S, Voskuil MI, Liu Y, Mangan JA, Monahan IM, et al. Transcriptional Adaptation of Mycobacterium tuberculosis within Macrophages: Insights into the Phagosomal Environment. J Exp Med. 2003;198:693–704. doi: 10.1084/jem.20030846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schujman GE, Guerin M, Buschiazzo A, Schaeffer F, Llarrull LI, Reh G, et al. Structural basis of lipid biosynthesis regulation in Gram-positive bacteria. EMBO J. 2006;25:4074–4083. doi: 10.1038/sj.emboj.7601284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schujman GE, Paoletti L, Grossman AD, de Mendoza D. FapR, a bacterial transcription factor involved in global regulation of membrane lipid biosynthesis. Dev Cell. 2003;4:663–672. doi: 10.1016/s1534-5807(03)00123-0. [DOI] [PubMed] [Google Scholar]
- Schweizer E, Hofmann J. Microbial type I fatty acid synthases (FAS): major players in a network of cellular FAS systems. Microbiol Mol Biol Rev. 2004;68:501–517. doi: 10.1128/MMBR.68.3.501-517.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons RW, Egan PA, Chute HT, Nunn WD. Regulation of fatty acid degradation in Escherichia coli: isolation and characterization of strains bearing insertion and temperature-sensitive mutations in gene fadR. J Bacteriol. 1980;142:621–632. doi: 10.1128/jb.142.2.621-632.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slama N, Leiba J, Eynard N, Daffe M, Kremer L, Quemard A, Molle V. Negative regulation by Ser/Thr phosphorylation of HadAB and HadBC dehydratases from Mycobacterium tuberculosis type II fatty acid synthase system. Biochem Biophys Res Com. 2011;412:401–406. doi: 10.1016/j.bbrc.2011.07.051. [DOI] [PubMed] [Google Scholar]
- Snapper SB, Melton RE, Mustafa S, Kieser T, Jacobs WR., Jr Isolation and characterization of efficient plasmid transformation mutants of Mycobacterium smegmatis. Mol Microbiol. 1990;4:1911–1919. doi: 10.1111/j.1365-2958.1990.tb02040.x. [DOI] [PubMed] [Google Scholar]
- Studier FW, Moffatt BA. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J Mol Biol. 1986;189:113–130. doi: 10.1016/0022-2836(86)90385-2. [DOI] [PubMed] [Google Scholar]
- Takayama K, Wang C, Besra GS. Pathway to synthesis and processing of mycolic acids in Mycobacterium tuberculosis. Clin Microbiol Rev. 2005;18:81–101. doi: 10.1128/CMR.18.1.81-101.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veyron-Churlet R, Molle V, Taylor RC, Brown AK, Besra GS, Zanella-Cleon I, et al. The Mycobacterium tuberculosis beta-ketoacyl-acyl carrier protein synthase III activity is inhibited by phosphorylation on a single threonine residue. J Biol Chem. 2009;284:6414–6424. doi: 10.1074/jbc.M806537200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh CT, Gehring AM, Weinreb PH, Quadri LE, Flugel RS. Post-translational modification of polyketide and nonribosomal peptide synthases. Curr Opin Chem Biol. 1997;1:309–315. doi: 10.1016/s1367-5931(97)80067-1. [DOI] [PubMed] [Google Scholar]
- Zhang Y. The magic bullets and tuberculosis drug targets. Annu Rev Pharmacol Toxicol. 2005;45:529–564. doi: 10.1146/annurev.pharmtox.45.120403.100120. [DOI] [PubMed] [Google Scholar]
- Zhang YM, Rock CO. Transcriptional regulation in bacterial membrane lipid synthesis. J Lipid Res. 2009;50:S115–119. doi: 10.1194/jlr.R800046-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YM, White SW, Rock CO. Inhibiting bacterial fatty acid synthesis. J Biol Chem. 2006;281:17541–17544. doi: 10.1074/jbc.R600004200. [DOI] [PubMed] [Google Scholar]
- Zhu K, Zhang YM, Rock CO. Transcriptional regulation of membrane lipid homeostasis in Escherichia coli. J Biol Chem. 2009;284:34880–34888. doi: 10.1074/jbc.M109.068239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimhony O, Vilcheze C, Jacobs WR., Jr Characterization of Mycobacterium smegmatis expressing the Mycobacterium tuberculosis fatty acid synthase I (fas1) gene. J Bacteriol. 2004;186:4051–4055. doi: 10.1128/JB.186.13.4051-4055.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.