

Abstract
Background
The antiinflammatory effects of hydrogen sulfide (H2S) and sodium sulfide (Na2S) treatment may prevent acute lung injury induced by high tidal volume (HVT) ventilation. However, lung protection may be limited by direct pulmonary toxicity associated with H2S inhalation. Therefore, the authors tested whether the inhalation of H2S or intravascular Na2S treatment can protect against ventilator-induced lung injury in mice.
Methods
Anesthetized mice continuously inhaled 0, 1, 5, or 60 ppm H2S or received a single bolus infusion of Na2S (0.55 mg/kg) or vehicle and were then subjected to HVT (40 ml/kg) ventilation lasting 4 h (n = 4–8 per group).
Results
HVT ventilation increased the concentrations of protein and interleukin-6 in bronchoalveolar lavage fluid, contributing to reduced respiratory compliance and impaired arterial oxygenation, and caused death from lung injury and pulmonary edema. Inhalation of 1 or 5 ppm H2S during HVT ventilation did not alter lung injury, but inhalation of 60 ppm H2S accelerated the development of ventilator-induced lung injury and enhanced the pulmonary expression of the chemoattractant CXCL-2 and the leukocyte adhesion molecules CD11b and L-selectin. In contrast, pretreatment with Na2S attenuated the expression of CXCL-2 and CD11b during HVT ventilation and reduced pulmonary edema. Moreover, Na2S enhanced the pulmonary expression of Nrf2-dependent antioxidant genes (NQO1, GPX2, and GST-A4) and prevented oxidative stress-induced depletion of glutathione in lung tissue.
Conclusions
The data suggest that systemic intravascular treatment with Na2S represents a novel therapeutic strategy to prevent both ventilator-induced lung injury and pulmonary glutathione depletion by activating Nrf2-dependent antioxidant gene transcription.
MECHANICAL ventilation can be life-saving in acute respiratory failure but may contribute to lung injury because of its side effects.1,2 Cyclic lung stretch during mechanical ventilation induces tissue disruption and activates proinflammatory pathways, promoting the formation of pulmonary edema and neutrophil infiltration.3 In addition, cyclic lung stretch is associated with generation of reactive oxygen species and production of redox imbalance in the lung.4–6 Together, these events may initiate and perpetuate oxidative stress and local and systemic inflammatory responses, enhance lung injury, and lead to multiorgan failure and mortality.7–9 Developing novel therapies targeted at oxidative stress and inflammation in ventilator-induced lung injury (VILI) could be useful clinically.
Hydrogen sulfide (H2S) is an endogenous gaseous transmitter.10 The antiinflammatory and antiapoptotic effects of H2S and other sulfides, such as sodium hydrogen sulfide or disodium sulfide (Na2S), have been studied in various animal models of inflammation (reviewed in Baumgart et al.11) and oxidative stress.12,13 In particular, Calvert et al. reported that Na2S protects against cardiac ischemia or reperfusion injury by up-regulating Nrf2-dependent antioxidant and detoxification proteins.14 Nrf2 (i.e., nuclear factor E2-related factor 2) is a key transcription factor that regulates antioxidant genes as an adaptive response to oxidative stress or pharmacologic stimuli. Downstream targets of Nrf2 include direct antioxidant proteins (such as glutathione peroxidase, NAD(P)H oxidoreductase, etc.), thiol-metabolism associated detoxifying enzymes (such as glutathione-S-transferase, glutamate–cysteine ligase, thioredoxin, etc.), stress-response genes (heme oxygenase, heat-shock proteins, ferritin, etc.), and others. In a murine model of VILI, it has been shown that Nrf2-dependent antioxidant genes play a key protective role in reducing oxidative stress.5 Whether or not H2S can protect against VILI by modulating the expression of Nrf2-dependent antioxidant genes has not been reported.
Inhalation of H2S gas is known to cause airway mucosa irritation and cytotoxicity,10,15 has been reported to exert proinflammatory effects in various models (reviewed in Szabó10 and Baumgart et al.11), and traditionally has been considered a health hazard. On the other hand, Faller et al. recently showed that inhaled H2S at 80 ppm prevents lung injury in mice ventilated with a tidal volume of 12 ml/kg (plateau pressure 10–13 cm H2O, i.e., moderate stretch).16 Nonetheless, molecular mechanisms responsible for the protective effects of H2S inhalation against VILI are not completely understood. In addition, it remains uncertain whether inhaled H2S is protective or deleterious during mechanical ventilation that produces increased lung stretch, such as in patients with acute respiratory distress syndrome.
To elucidate the role of H2S in VILI, we examined the effect of inhaled H2S in a model of lung injury induced by high tidal volume (HVT) ventilation. We found that H2S gas promotes VILI and enhances the pulmonary expression of leukocyte adhesion and chemoattractant molecules. Subsequently, we hypothesized that intravascular administration of Na2S, avoiding direct exposure of the lung to H2S gas, could provide better protection against VILI than could inhaled H2S. We report that intravascular Na2S both attenuates the pulmonary expression of chemoattractant and leukocyte adhesion molecules and enhances Nrf2-dependent expression of antioxidant genes, thereby attenuating VILI from HVT ventilation.
Materials and Methods
This study was approved by the Institutional Animal Care and Use Committee (Subcommittee on Research Animal Care, Massachusetts General Hospital, Boston, Massachusetts), and conforms to the revised Guide for the Care and Use of Laboratory Animals.
Mouse Model of VILI
Male C57BL/6 mice (23.3 ± 1.0 g, mean ± SD) were subjected to HVT ventilation as described previously6 with minor modifications of inspired oxygen fraction (FIO2) positive end-expiratory pressure, and alveolar recruitment. Briefly, after anesthesia was induced and tracheostomy performed, mice were ventilated in a volume-controlled mode (Inspira; Harvard Apparatus, Boston, MA) at a tidal volume (VT) of 10 ml/kg, respiratory rate of 90 breaths/min, positive endexpiratory pressure of 2 cm H2O, and FIO2 0.4 for 1 h (baseline ventilation). Immediately after the tracheostomy and after the initiation of ventilation, a carotid catheter was inserted for blood pressure monitoring, continuous infusion of anesthetics, and blood sampling. After 1 h of baseline ventilation, the ventilator settings were switched to an HVT of 40 ml/kg, positive end-expiratory pressure of 1 cm H2O, and respiratory rate of 60 breaths/min, and mice were ventilated for a maximum duration of 240 min of HVT ventilation. The FIO2 (0.4) was not changed. Because a plateau pressure exceeding 30 cm H2O is associated with increased mortality1 and commonly is used as a trigger for initiating rescue therapies in patients with acute respiratory distress syndrome, we aimed for plateau pressures of more than 30 cm H2O to induce VILI in mice. In this model using normal lungs, a tidal volume of 40 ml/kg was necessary to increase plateau pressure above 30 cm H2O, resulting in a peak pressure of ~35 cm H2O.
Alveolar recruitment maneuvers were performed every 30 min during the baseline period and every 60 min during HVT ventilation. Body temperature was maintained at 37°C with a heating pad. See Materials and Methods in Supplemental Digital Content 1, http://links.lww.com/ALN/A775, which provides the detailed methodology of the mouse model of VILI.
Experimental Groups
After 30 min of baseline ventilation (i.e., 30 min before the onset of injurious HVT ventilation), mice were treated with either inhaled H2S or intravascular Na2S as follows: H2S gas (hydrogen sulfide 100 ppm, balance nitrogen, MedTech-Gases, Medford, MA) was diluted and added continuously to the inhaled gas mixture (FIO2 = 0.4) at a concentration of 0, 1, 5, or 60 ppm (n = 6 each) until the end of the study. Alternatively, mice received a single intraarterial bolus injection of Na2S (sodium sulfide nonahydrate, Sigma–Aldrich, St. Louis, MO; 0.55 mg/kg in 5.5 ml vehicle/kg body weight, n = 8) or vehicle alone (Dulbecco's phosphate buffered saline, Sigma-Aldrich; 5.5 ml/kg body weight, n = 8).
The animals were killed after 240 min of HVT ventilation or when their mean arterial pressure decreased to less than 60 mmHg for more than 5 min. Immediately after the animals were killed, respiratory mechanics were assessed and arterial blood was collected. Bronchoalveolar lavage (BAL) and the collection of lung tissues were performed as described in Supplemental Digital Content 1, http://links.lww.com/ALN/A775.
Control mice not subjected to HVT ventilation (n = 4) were killed after 5 min of baseline ventilation.
Evaluation of Respiratory Mechanics and Blood Gas Analysis
We measured VT, peak inspiratory airway pressure (PIP), inspiratory capacity, compliance of the respiratory system, pressure-volume curves, and arterial blood gas tensions, as described in Supplemental Digital Content 1, http://links.lww.com/ALN/A775.
Evaluation of Pulmonary Edema, Inflammation, and Histologic Changes
Protein concentration in BAL fluid was measured (Bradford assay; Sigma–Aldrich) to assess pulmonary edema formation. Lung inflammation was determined by measuring interleukin-6 (IL-6 enzyme-linked immunosorbent assay; R&D Systems, Minneapolis, MN) and leukocyte concentrations in BAL fluid.
Paraffin-embedded 6-μm lung sections were stained with hematoxylin and eosin or reacted with an antimouse neutrophil antibody (Cedarlane Laboratories, Burlington, Ontario, Canada) to evaluate histopathologic changes and neutrophil infiltration (see Materials and Methods in Supplemental Digital Content 1, http://links.lww.com/ALN/A775, which provides the detailed methodology of staining tissue sections).
Evaluation of Oxidative Stress and Pulmonary Gene Expression
Total, reduced (GSH) and oxidized (GSSG), glutathione concentrations in lung tissue were measured using an enzymatic assay (Cayman Chemical, Ann Arbor, MI).
Quantitative real-time polymerase chain reaction was used to determine the pulmonary expression of the neutrophil chemoattractant cytokine CXCL-2 (i.e., macrophage inflammatory protein 2), the leukocyte adhesion molecules CD11b (i.e., integrin α M), and L-selectin, as well as the following Nrf2-dependent antioxidant genes: NAD(P)H: quinone oxidoreductase (NQO1), glutathione-S-transferase A4 (GST-A4), and glutathione peroxidase 2 (GPX2) (see Materials and Methods in Supplemental Digital Content 1, http://links.lww.com/ALN/A775, which provides the detailed methodology of polymerase chain reaction).
Statistical Analysis
Animals subjected to HVT ventilation without H2S (0 ppm) were compared with H2S-treated animals (60 ppm) subjected to HVT ventilation and with animals not subjected to HVT ventilation (controls) using ANOVA and Bonferroni multiple comparison tests (two comparisons: controls vs. 0 ppm H2S; 0 vs. 60 ppm H2S). Na2S-treated animals subjected to HVT ventilation were compared with vehicle-injected animals using Student unpaired t test (between-group comparison). Survival curves were compared with the log-rank Mantel–Cox test. Compliance of the respiratory system and PIP values at the beginning (1 h of HVT) and end of HVT ventilation were compared with the Student paired t test (within-group comparison). All tests were two-tailed and were performed with GraphPad Prism® version 5.01 for Windows (GraphPad Software, San Diego, CA). Data are expressed as mean ± SEM, unless indicated otherwise.
Results
Duration of HVT Ventilation in Mice Treated with and without H2S or Na2S
Mice breathing 40% oxygen without added H2S died of acute lung injury or fulfilled the criteria for being killed after 224 min (median) of HVT ventilation (fig. 1). That is similar to the median survival time of mice ventilated with 1 ppm (236 min) or 5 ppm (215 min) of inhaled H2S. In contrast, the median survival time during HVT ventilation was shorter in mice breathing 60 ppm H2S (166 min; P = 0.0016 vs. 0 ppm H2S). The median survival time in vehicle-treated animals was 231 min. Remarkably, all Na2S-treated animals were alive at the end of the experiment (240 min of HVT ventilation) and were then killed.
Fig. 1.
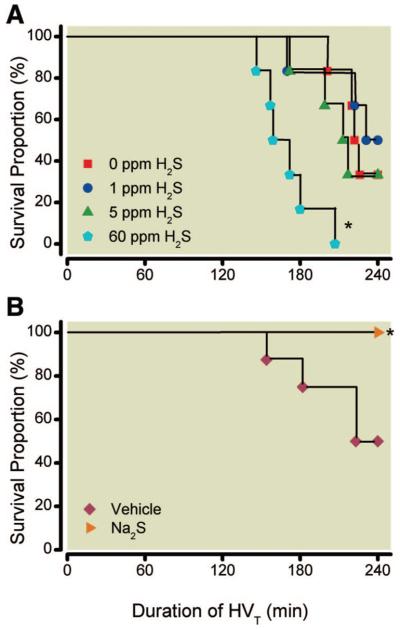
Survival of mice subjected to high tidal volume (HVT) ventilation (40 ml/kg) in the presence and absence of various concentrations of inhaled hydrogen sulfide (H2S) (n = 6 for each concentration) (A) or after intravascular administration of sodium sulfide (Na2S) (n = 8) (B). Mice were killed when their mean arterial pressure decreased to less than 60 mmHg (for more than 5 min) or after a maximum duration of 240 min of HVT ventilation. * P = 0.0016 for 0 versus 60 ppm H2S and P = 0.0256 for vehicle versus Na2S.
Effect of Inhaled H2S on Respiratory Mechanics during HVT Ventilation
Ventilation with a tidal volume of 40 ml/kg resulted in a PIP of 33–35 cm H2O in all groups during the first hour of HVT ventilation. In mice breathing 40% oxygen without added H2S, HVT ventilation induced a gradual increase of PIP to 43 cm H2O by the end of the experiment. A comparable increase in PIP was observed in mice ventilated with 1, 5, or 60 ppm H2S (table 1). This increased PIP was associated with a marked reduction of respiratory system compliance (table 1). Reduced respiratory system compliance was also reflected by a significant downward shift of the pressure-volume curves (fig. 2) measured in mice after HVT ventilation with and without H2S compared with those curves measured in mice not subjected to HVT ventilation.
Table 1.
Respiratory Mechanics
| Peak Inspiratory Pressure (cm H2O) |
Respiratory Compliance (μl/cm H2O) |
|||
|---|---|---|---|---|
| 1 h HVT | End of HVT | 1 h HVT | End of HVT | |
| Inhalation | ||||
| 0 ppm H2S | 35 ± 0.3 | 43 ± 0.1* | 38 ± 1.1 | 22 ± 1.6 |
| 1 ppm H2S | 34 ± 0.4 | 42 ± 0.7* | 40 ± 1.3 | 20 ± 2.2* |
| 5 ppm H2S | 35 ± 0.6 | 42 ± 0.1* | 40 ± 0.6 | 18 ± 1.3* |
| 60 ppm H2S | 33 ± 0.5 | 42 ± 0.2* | 39 ± 0.9 | 18 ± 0.7* |
| Intravascular infusion | ||||
| Vehicle | 34 ± 0.2 | 43 ± 0.3* | 40 ± 0.4 | 24 ± 1.4* |
| Na2S, 0.55 mg/kg | 35 ± 0.2 | 39 ± 0.6† | 39 ± 0.6 | 34 ± 2.3† |
Respiratory mechanics were measured in mice subjected to high tidal volume (HVT) ventilation. Values represent mean ± SEM after 60 min (1 h HVT) and at the end of HVT ventilation (maximum 240 min).
P < 0.05 vs. 1 h HVT.
P < 0.05 vs. vehicle.
H2S = hydrogen sulfide; Na2S = sodium sulfide.
Fig. 2.
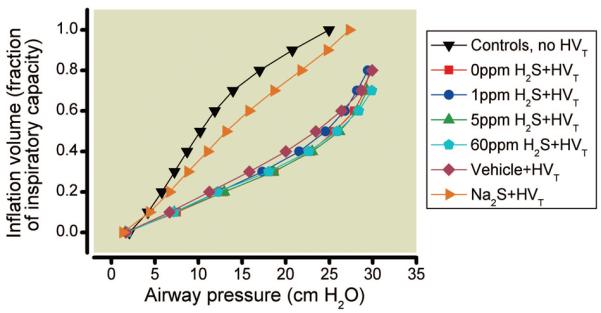
Compliance of the respiratory system was assessed in mice subjected to high tidal volume (HVT) ventilation in the presence and absence of various concentrations of inhaled hydrogen sulfide (H2S) (n = 6 for each concentration) or after HVT ventilation in mice pretreated with intravascular sodium sulfide (Na2S) (n = 8) or vehicle (n = 8). Pressure-volume curves were plotted as the inflation volume (expressed as a fraction of inspiratory capacity) as a function of airway pressure and were compared with control mice not subjected to HVT ventilation (n = 4). Deterioration of respiratory mechanics is indicated by a downward shift of the pressure-volume curve compared with controls. Na2S treatment prevents the deterioration of respiratory system compliance induced by HVT ventilation (40 ml/kg body weight). Pressure-volume curves are displayed as the mean of all curves in each experimental group. For clarity, only the mean values are displayed.
Effect of Inhaled H2S on Arterial Blood Gas Tensions during HVT Ventilation
In control mice, killed after 5 min of baseline ventilation (FIO2 0.4), the PaCO2 was 40 ± 4 mmHg, and the PaO2 was 239 ± 16 mmHg (fig. 3). After HVT ventilation (0 ppm H2S), PaCO2 did not change, but PaO2 decreased to 59 ± 17 mmHg at the end of the experiment. Similarly, in mice breathing 1 or 5 ppm H2S, PaCO2 did not change but PaO2 decreased to 60 ± 6 and 56 ± 8 mmHg, respectively. In contrast, in mice breathing 60 ppm H2S, PaCO2 increased to 88 ± 13 mmHg (P = 0.0001 vs. 0 ppm) and PaO2 decreased to 77 ± 10 mmHg (P = not significant vs. 0 ppm) in response to HVT ventilation.
Fig. 3.
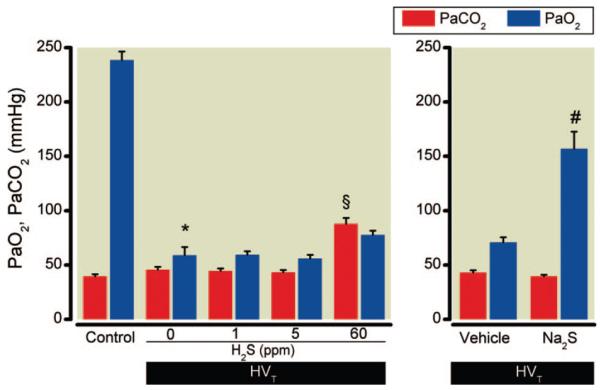
Arterial oxygen (Pao2) and carbon dioxide (Paco2) tensions of mice subjected to high tidal volume (HVT) ventilation (Fio2 0.4) in the presence and absence of various concentrations of inhaled hydrogen sulfide (H2S) (n = 6 for each concentration) or after intravascular administration of sodium sulfide (Na2S) or vehicle (n = 8 in each group). Control mice were not subjected to HVT ventilation but were briefly ventilated at tidal volume 10 ml/kg and Fio2 0.4 (n = 4). Pao2 and Paco2 were measured in arterial blood obtained from the carotid artery. Arterial oxygenation was impaired by HVT ventilation, but the decrease in Pao2 was attenuated by Na2S pretreatment. * P < 0.0001 versus control, § P = 0.0001 versus 0 ppm H2S, # P = 0.0001 versus vehicle. FIO2 = inspired oxygen fraction.
Effect of Inhaled H2S on the Formation of Pulmonary Edema during HVT Ventilation
To investigate whether alveolar-capillary barrier disruption with pulmonary edema formation contributes to the observed impairment of arterial oxygenation, the protein concentration in BAL fluid was measured (fig. 4). BAL fluid protein concentrations were consistently higher in all mice subjected to HVT ventilation with and without inhaled H2S than in controls.
Fig. 4.
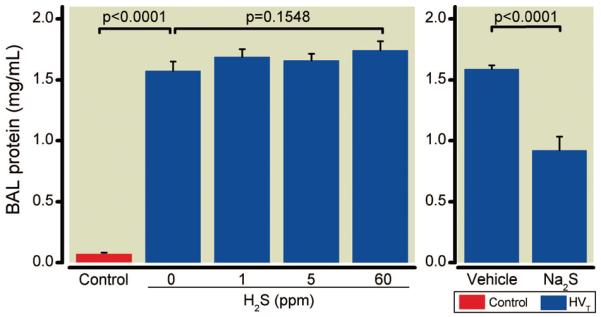
Protein concentrations in bronchoalveolar lavage (BAL) fluid obtained from mice after high tidal volume (HVT) ventilation. BAL fluid protein concentrations were measured subsequent to HVT ventilation in the presence and absence of various concentrations of inhaled hydrogen sulfide (H2S) (n = 6 for each concentration) or after intravascular administration of sodium sulfide (Na2S) or vehicle (n = 8 in each group), and in control mice not subjected to HVT ventilation (n = 4).
Effects of Inhaled H2S on BAL Fluid IL-6 and Leukocyte Concentrations during HVT Ventilation
To evaluate pulmonary inflammation, we measured the concentrations of the proinflammatory cytokine IL-6 and the concentration of leukocytes in BAL fluid (fig. 5). BAL fluid IL-6 concentrations were greater in mice subjected to HVT ventilation than in controls. In contrast, BAL fluid IL-6 concentrations were decreased in mice ventilated with 60 ppm H2S than without H2S (fig. 5A). BAL fluid leukocyte concentrations (fig. 5B) invariably were reduced by HVT ventilation in mice compared with controls, independent of whether the mice were ventilated with or without H2S.
Fig. 5.
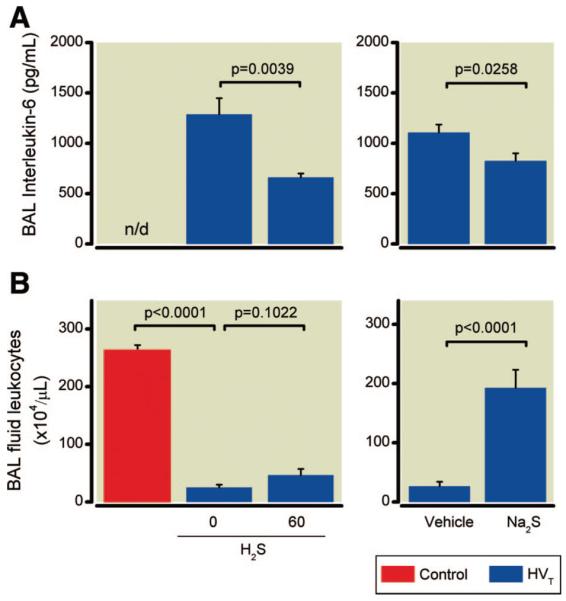
Concentrations of interleukin-6 (IL-6) (A) and leukocytes in bronchoalveolar lavage (BAL) fluid (B) obtained from mice after high tidal volume (HVT) ventilation. IL-6 and leukocyte concentrations were measured subsequent to HVT ventilation in the presence and absence of inhaled hydrogen sulfide (H2S) (n = 6 for each concentration) or after intravascular administration of sodium sulfide (Na2S) or vehicle (n = 8 in each group), and in control mice not subjected to HVT ventilation (n = 4). n/d = not detectable.
Effects of Inhaled H2S on Pulmonary Expression of Leukocyte Chemoattractant and Adhesion Molecules during HVT Ventilation
The pulmonary messenger RNA (mRNA) concentrations of CXCL-2, CD11b, and L-selectin in mice subjected to HVT ventilation were approximately 7-, 3-, and 1.5-fold greater than in controls (fig. 6). Inhalation of 60 ppm H2S during HVT ventilation augmented the increase in CXCL-2, CD11b, and L-selectin mRNA concentrations (15-, 6-, and 3-increase vs. controls).
Fig. 6.

Pulmonary messenger RNA (mRNA) concentrations of CXCL-2 (A), CD11b (B), and L-selectin (C) in mice subjected to high tidal volume (HVT) ventilation in the presence and absence of hydrogen sulfide (H2S) (n = 6 for each concentration) or after intravascular administration of sodium sulfide (Na2S) or vehicle (n = 8 in each group). mRNA concentrations are expressed as fold change relative to the average expression values in control mice not subjected to HVT ventilation (n = 4).
Effects of Inhaled H2S on Oxidative Stress and Pulmonary Expression of Antioxidant Genes
The concentration of total glutathione and the ratio of reduced to oxidized glutathione (GSH/GSSG) were significantly lower in lungs subjected to HVT ventilation than in controls (fig. 7). This decrease was independent of whether mice were ventilated with or without H2S.
Fig. 7.
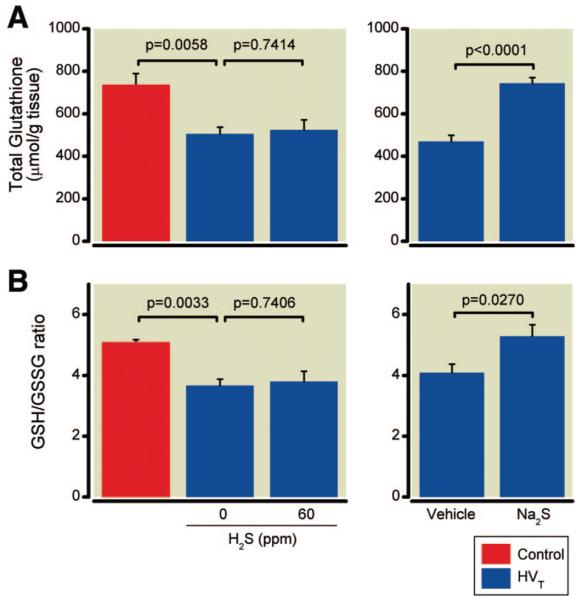
Total glutathione concentrations (A) and the ratio of reduced to oxidized glutathione (GSH/GSSG) (B) in lung tissues of mice after high tidal volume (HVT) ventilation in the presence and absence of inhaled hydrogen sulfide (H2S) (n = 6 for each concentration) or after intravascular administration of sodium sulfide (Na2S) or vehicle (n = 8 in each group) and in control mice not subjected to HVT ventilation (n = 4).
The pulmonary mRNA concentrations of NQO1 and GPX2 in mice subjected to HVT ventilation were approximately 1.6- (not significant) and 2-fold higher than in controls (fig. 8). Inhalation of 60 ppm H2S during HVT ventilation attenuated the increase in NQO1 and GPX2 mRNA concentrations (1.1- and 1.3-fold increase vs. controls). HVT ventilation with 0 or 60 ppm H2S reduced the pulmonary mRNA concentrations of GST-A4 to 66% and 54%, respectively, of the concentrations measured in control mice.
Fig. 8.

Pulmonary messenger RNA (mRNA) concentrations of the Nrf2-dependent antioxidant genes NAD(P)H:quinone oxidoreductase, NQO1 (A), glutathione peroxidase 2, GPX2 (B), and glutathione-S-transferase A4, GST-A4 (C) in mice subjected to high tidal volume (HVT) ventilation in the presence and absence of hydrogen sulfide (H2S) (n = 6 for each concentration) or after intravascular administration of sodium sulfide (Na2S) or vehicle (n = 8 in each group). mRNA concentrations are expressed as fold change relative to the average expression values in control mice not subjected to HVT ventilation (n = 4).
Effect of Na2S on Respiratory Mechanics during HVT Ventilation
In vehicle-treated animals, PIP and respiratory system compliance deteriorated to the same extent as in mice receiving HVT ventilation alone (table 1). In contrast, treatment with Na2S attenuated the PIP increase, preserved respiratory system compliance, and reduced the downward shift of the pressure-volume curve (fig. 2).
Effect of Na2S on Arterial Blood Gas Tensions during HVT Ventilation
In vehicle-treated animals, PaCO2 was stable and PaO2 decreased after HVT ventilation (fig. 3). Compared with vehicle-treatment, Na2S treatment attenuated the decrease in PaO2 in mice subjected to HVT ventilation. At the end of the experiment, PaO2 was higher in Na2S-treated animals than in vehicle-treated animals (157 ± 45 vs. 71 ± 14 mmHg, P = 0.0001).
Effect of Na2S on the Formation of Pulmonary Edema during HVT Ventilation
BAL fluid protein concentrations were greater in vehicle-treated mice subjected to HVT ventilation than in controls (fig. 4). In contrast, BAL fluid protein concentrations were lower in Na2S-treated mice than in vehicle-treated mice.
Effects of Na2S on BAL Fluid IL-6 and Leukocyte Concentrations during HVT Ventilation
BAL fluid IL-6 concentrations were greater in vehicle-treated animals subjected to HVT ventilation than in controls. Na2S treatment reduced BAL fluid IL-6 concentrations in mice subjected to HVT ventilation (fig. 5A).
High tidal volume ventilation reduced the leukocyte concentration in BAL fluid obtained from vehicle-treated mice compared with that from controls. In contrast, Na2S treatment restored the concentration of leukocytes in BAL fluid of mice subjected to HVT ventilation (fig. 5B).
Effects of Na2S on Pulmonary Expression of Leukocyte Chemoattractant and Adhesion Molecules during HVT Ventilation
The CXCL-2 and CD11b mRNA concentrations measured in the lungs of vehicle-treated animals subjected to HVT ventilation (7- and 3-fold increase vs. controls) were significantly attenuated by Na2S treatment (3- and 0.8-fold increase vs. controls, fig. 6, A and B). In contrast, the pulmonary mRNA concentrations of L-selectin measured in vehicle-treated animals (1.5-fold increase vs. controls) did not differ from those measured in Na2S-treated animals (1.3-fold increase vs. controls, fig. 6C).
Effects of Na2S on Oxidative Stress and Pulmonary Expression of Antioxidant Genes
Total glutathione concentrations and the GSH/GSSG in lung tissues obtained from mice subjected to HVT ventilation were significantly higher in Na2S-treated animals than in vehicle-treated animals (fig. 7). The increase of antioxidant NQO1 and GPX2 mRNA concentrations measured in the lungs of vehicle-treated animals subjected to HVT ventilation (1.7- and 1.9-fold increase vs. controls) was significantly enhanced by Na2S treatment (2.5- and 2.9-fold increase vs. controls, fig. 8). Moreover, Na2S treatment prevented the reduction of GST-A4 mRNA concentrations measured in vehicle-treated animals subjected to HVT ventilation.
Pulmonary Histopathologic Changes from HVT Ventilation
Histologic analysis of lung sections from mice treated with either 60 ppm inhaled H2S or with injection of Na2S revealed that HVT ventilation induced signs of edema formation and airway epithelial disruption (fig. 9). These pathologic signs were similar in mice ventilated with or without 60 ppm H2S. Fewer signs of edema and epithelial disruption were observed after Na2S pretreatment. The number of neutrophils in lung tissue sections taken from three mice subjected to HVT ventilation was found to be greater than the number of lung tissue neutrophils determined in two control animals not subjected to HVT ventilation (fig. 10). In addition, the numbers of lung tissue neutrophils found after HVT ventilation in three mice with and three mice without inhalation of 60 ppm H2S were similar. In contrast, the number of neutrophils found in lung tissue sections taken from three mice subjected to HVT ventilation after Na2S pretreatment was smaller. Despite the small sample size of tissue sections used for neutrophil staining, the variance of lung tissue neutrophil concentrations is small. The interpretation of these data must be made with great caution.
Fig. 9.

Representative lung sections of mice subjected to high tidal volume (HVT) ventilation in the presence and absence of 60 ppm hydrogen sulfide (H2S) or after intravascular administration of sodium sulfide (Na2S). These sections were stained with hematoxylin and eosin (H/E) or reacted with antibodies (AB) against neutrophils (inset: higher magnification) and were compared with sections from nonventilated control mice. In control mice, the airway epithelium (open arrows) is intact, no edema surrounding the airway is seen, and few neutrophils are visible (thin black arrows). HVT ventilation induces airway disruption, edema formation between airways and adjacent vessels (thick black arrows), and neutrophil infiltration. Similar pathologic changes were present in lungs subjected to HVT ventilation in the presence of 60 ppm H2S. Less epithelial disruption and edema and fewer neutrophils are seen in lung tissue sections after Na2S pretreatment.
Fig. 10.
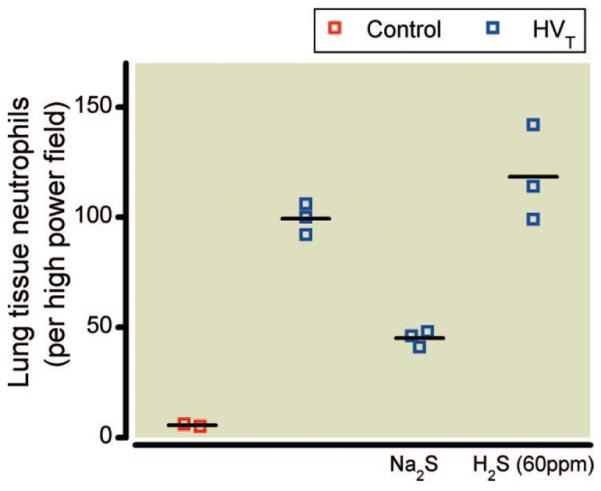
Pulmonary infiltration with neutrophils in mice subjected to high tidal volume (HVT) ventilation in the presence and absence of inhaled hydrogen sulfide (H2S) (60 ppm) or after intravascular administration of sodium sulfide (Na2S) (n = 3 in each group). Lung tissue sections were reacted with antibodies against neutrophils and were compared with lung sections obtained from control mice not subjected to HVT ventilation (n = 2). Representative examples of lung sections are displayed in figure 9. Neutrophil concentrations are reported as the number of neutrophils per high power field (magnification ×40).
Additional figures describing the effect of various concentrations of inhaled H2S on BAL fluid leukocyte and IL-6 concentrations, pulmonary expression of leukocyte chemoattractant and adhesion molecules, and oxidative stress parameters and antioxidant gene expression are provided in Supplemental Digital Content 2, http://links.lww.com/ALN/A776, figures 1–4.
Discussion
This study was performed to evaluate whether inhalation of H2S gas protects against lung injury from HVT ventilation in mice. We found that inhalation of 1 or 5 ppm H2S produces neither beneficial nor deleterious effects, whereas inhalation of 60 ppm H2S accelerates the development of lung injury and death from HVT ventilation. In contrast, treatment with a bolus infusion of Na2S attenuates pulmonary edema formation, thereby limiting the deterioration of respiratory system compliance and arterial oxygenation. Our results suggest that these protective effects of Na2S in VILI are linked to attenuated expression of chemoattractant and leukocyte adhesion molecules, enhanced expression of Nrf2-dependent antioxidant genes, and improved availability of reduced glutathione in the lung.
Our findings underscore the potential toxicity of H2S gas inhalation in a rodent model of VILI. Lopez et al. reported cytotoxic effects and edema formation in respiratory tract tissues of rats breathing 10–400 ppm H2S.17 The primary biochemical mechanism responsible for the toxicity of H2S gas is believed to involve the inhibition of cytochrome c oxidase.18 Inhibition of cytochrome c oxidase blocks oxidative phosphorylation, a major source of cellular adenosine triphosphate synthesis. In turn, the abolishment of oxidative phosphorylation implies functional hypoxia (inability to use available oxygen for oxidative metabolism).18 Both hypoxia and a lack of adenosine triphosphate are associated with pulmonary vasoconstriction, impaired alveolar fluid clearance, and pulmonary edema (reviewed in Bärtsch et al.19). Based on these considerations, the inhalation of 60 ppm H2S is likely to have contributed to the formation of pulmonary edema in our murine model.
Faller et al. reported that inhalation of 80 ppm H2S limited cytokine release in a model of lung injury in C57BL/6 mice caused by ventilation with a moderate tidal volume (12 ml/kg). Similarly, we found that BAL fluid IL-6 concentrations were lower when mice developed lung injury in the presence of 60 ppm H2S (fig. 5). However, the current study demonstrates that under conditions of higher mechanical lung stretch (peak airway pressure 35 cm H2O, as opposed to ~12 cm H2O in the Faller et al. study), the potential antiinflammatory effects of inhaled H2S are outweighed by the deleterious effects of inhaling 60 ppm H2S, including enhanced pulmonary expression of chemoattractant and leukocyte adhesion molecules and earlier deaths (figs. 1 and 6). In addition, H2S inhalation prevented the up-regulation of antioxidant gene expression in mice subjected to HVT ventilation (fig. 8) and caused a decrease in both the GSH/GSSG and total glutathione concentrations in the lung (fig. 7).
Mechanical stretching of the lung may initiate intravascular pulmonary leukocyte sequestration in mice.20 Various chemoattractant and adhesion molecules that are derived from epithelial and endothelial cells, as well as alveolar macrophages and fibroblasts, are responsible for inflammatory recruitment and migration of leukocytes from the circulation into injured lung tissue.21 In particular, the neutrophil chemoattractant CXCL-2 and the adhesion molecules CD11b and L-selectin are reported to play a critical role in the pathogenesis of VILI and for control of leukocyte trafficking into the lung.22,23 Ultimately, pulmonary neutrophil activation and accumulation promotes microvascular permeability and edema formation.3,22 Our results demonstrate that HVT ventilation increases the pulmonary expression of CXCL-2, CD11b, and L-selectin. Moreover, the HVT-induced increase in CXCL-2, CD11b, and L-selectin was enhanced markedly by concomitant inhalation of 60 ppm H2S (fig. 7). These findings suggest that the pathophysiologic changes leading to an earlier death in mice ventilated with 60 ppm H2S appear to be linked to enhanced expression of chemoattractant and leukocyte adhesion molecules in the lung.
As an alternative to the inhalation of potentially toxic H2S gas, we examined whether intravascular administration of Na2S can attenuate the development of VILI during HVT ventilation. We found that systemic treatment with Na2S protects against VILI and reduces GSH/GSSG imbalances (fig. 7) in mice subjected to HVT ventilation. Our results also suggest that Na2S attenuates the pulmonary expression of CXCL-2 and CD11b during VILI (fig. 6). This finding is supported by other reports demonstrating that both Na2S and sodium hydrogen sulfide can impair leukocyte adherence to endothelium and subsequent diapedesis during inflammation in vivo by reducing the expression of endothelial and leukocyte adhesion molecules.24,25 Increased pulmonary leukocyte adhesion in mice subjected to HVT ventilation may also account for the low concentrations of BAL leukocytes reported here (fig. 5B) and elsewhere.26 The protective effect of Na2S in this model of lung injury produced by HVT ventilation is consistent with the beneficial effect of sodium hydrogen sulfide in other rodent models of lung injury induced by oleic acid27 or a skin burn and smoke inhalation.28
The current study demonstrates that Na2S pretreatment, but not H2S inhalation during HVT ventilation, enhances the pulmonary expression of Nrf2-dependent antioxidant genes and stabilizes the concentration of reduced glutathione in the lung (fig. 6). A transcription factor for several antioxidant genes, Nrf2 is a master regulator of antioxidant response mechanisms (reviewed in Maher and Yamamoto29). Activation of Nrf2 results in the induction of many cytoprotective proteins. In the lung, Nrf2-dependent genes regulate the cellular redox status and have been reported to modulate pulmonary cellular responses and oxidative stress induced by mechanical ventilation.5 The reasons inhaled H2S and intravenous Na2S have distinct effects in VILI in the current study are likely to be multifactorial. It is possible that pretreatment with Na2S before injurious ventilation is required to induce the Nrf2-dependent antioxidant defense mechanisms that contribute to protecting against VILI. It is also conceivable that intravascular administration of Na2S may preferentially target activation of Nrf2-dependent signaling in different cell types compared with inhaled H2S (e.g., activating vascular endothelial vs. airway epithelial cells). Analyzing the candidate cellular and molecular targets of H2S and Na2S in future studies may provide additional insights into their differential biologic effects.
Limitations
In patients, ventilator-associated lung injury usually occurs in lungs with reduced compliance caused by preexisting conditions (two-hit or multifactorial lung injury), including septic inflammation. The current study uses a one-hit model of VILI in healthy mouse lungs. Thus, the current study has limited clinical relevance. Although the current study demonstrates that Na2S pretreatment protects against VILI, this study does not elucidate the potential dose-dependency of this protective effect and does not investigate the potential toxicity of Na2S to central or peripheral organs. Lung tissue H2S concentrations were not measured in this study. Comparing the lung tissue concentrations of sulfide species in future studies may help explain the differential biologic effects of inhaled H2S and intravascular Na2S and their dose-dependency.
Summary
Our study demonstrates that continuous inhalation of H2S gas enhances the expression of leukocyte adhesion and chemoattractant molecules (CXCL-2, CD11b, and L-selectin), accelerates pulmonary edema formation, and promotes lung injury when it is inhaled at high concentrations (60 ppm) during mechanical ventilation with a high tidal volume. The deleterious effects of H2S gas inhalation reemphasize that H2S can cause pulmonary toxicity and do not suggest a substantial role for H2S in the prevention and treatment of VILI in patients. In contrast, our data suggest that systemic intravascular treatment with Na2S may represent a novel therapeutic strategy to prevent both VILI and GSH/GSSG imbalance by activating Nrf2-dependent antioxidant gene transcription.
Supplementary Material
What We Already Know about This Topic
Sulfides such as sodium sulfide and hydrogen sulfide have both protective and detrimental effects on cells
High tidal volume ventilation can produce acute lung injury
What This Article Tells Us That Is New
Inhalation of hydrogen sulfide has no beneficial effects on ventilator-induced lung injury in mice
Intravascular administration of sodium sulfide attenuates the development of ventilator-induced lung injury through antioxidative signaling pathways
Acknowledgments
Supported by fellowship grant FR 2555/3-1 (to Dr. Francis) from the German Research Foundation (Deutsche Forschungsgemeinschaft, Bonn, Germany); royalties from Ikaria Inc., Hampton, New Jersey, and Linde Corp, Murray Hill, New Jersey (to Dr. Zapol); and grants HL074352 (to Dr. Bloch) and HL101930 (to Dr. Ichinose) from the United States Public Health Service, Washington, D.C.
The authors thank their collaborators from the Department of Anesthesia, Critical Care and Pain Medicine, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts: Rosemary C. Jones, Ph.D. (Associate Professor of Pathology), and Diane E. Capen and Yuko Beppu (Research Assistants) for their skillful assistance and advice in the preparation and interpretation of histologic lung sections, and Andrea U. Steinbicker, M.D., M.P.H., Matthias Derwall, M.D., and Kentaro Tokuda, M.D. (Research Fellows), for advice and technical assistance in quantitative real-time PCR techniques. The authors also thank Yasuko Nagasaka, M.D., Ph.D. (Instructor), for multifaceted advice and assistance with the study.
Footnotes
This article may be accessed for personal use at no charge through the Journal Web site, www.anesthesiology.org.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are available in both the HTML and PDF versions of this article. Links to the digital files are provided in the HTML text of this article on the Journal's Web site (www.anesthesiology.org).
References
- 1.Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. The Acute Respiratory Distress Syndrome Network. N Engl J Med. 2000;342:1301–8. doi: 10.1056/NEJM200005043421801. [DOI] [PubMed] [Google Scholar]
- 2.Brower RG, Lanken PN, MacIntyre N, Matthay MA, Morris A, Ancukiewicz M, Schoenfeld D, Thompson BT, National Heart, Lung, and Blood Institute ARDS Clinical Trials Network Higher versus lower positive end-expiratory pressures in patients with the acute respiratory distress syndrome. N Engl J Med. 2004;351:327–36. doi: 10.1056/NEJMoa032193. [DOI] [PubMed] [Google Scholar]
- 3.Lionetti V, Recchia FA, Ranieri VM. Overview of ventilator-induced lung injury mechanisms. Curr Opin Crit Care. 2005;11:82–6. doi: 10.1097/00075198-200502000-00013. [DOI] [PubMed] [Google Scholar]
- 4.Birukov KG. Cyclic stretch, reactive oxygen species, and vascular remodeling. Antioxid Redox Signal. 2009;11:1651–67. doi: 10.1089/ars.2008.2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Papaiahgari S, Yerrapureddy A, Reddy SR, Reddy NM, Dodd-O JM, Crow MT, Grigoryev DN, Barnes K, Tuder RM, Yamamoto M, Kensler TW, Biswal S, Mitzner W, Hassoun PM, Reddy SP. Genetic and pharmacologic evidence links oxidative stress to ventilator-induced lung injury in mice. Am J Respir Crit Care Med. 2007;176:1222–35. doi: 10.1164/rccm.200701-060OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vaporidi K, Francis RC, Bloch KD, Zapol WM. Nitric oxide synthase 3 contributes to ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2010;299:L150–9. doi: 10.1152/ajplung.00341.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Belperio JA, Keane MP, Lynch JP, 3rd, Strieter RM. The role of cytokines during the pathogenesis of ventilator-associated and ventilator-induced lung injury. Semin Respir Crit Care Med. 2006;27:350–64. doi: 10.1055/s-2006-948289. [DOI] [PubMed] [Google Scholar]
- 8.Gurkan OU, O'Donnell C, Brower R, Ruckdeschel E, Becker PM. Differential effects of mechanical ventilatory strategy on lung injury and systemic organ inflammation in mice. Am J Physiol Lung Cell Mol Physiol. 2003;285:L710–8. doi: 10.1152/ajplung.00044.2003. [DOI] [PubMed] [Google Scholar]
- 9.Slutsky AS, Tremblay LN. Multiple system organ failure: Is mechanical ventilation a contributing factor? Am J Respir Crit Care Med. 1998;157:1721–5. doi: 10.1164/ajrccm.157.6.9709092. [DOI] [PubMed] [Google Scholar]
- 10.Szabó C. Hydrogen sulphide and its therapeutic potential. Nat Rev Drug Discov. 2007;6:917–35. doi: 10.1038/nrd2425. [DOI] [PubMed] [Google Scholar]
- 11.Baumgart K, Radermacher P, Wagner F. Applying gases for microcirculatory and cellular oxygenation in sepsis: Effects of nitric oxide, carbon monoxide, and hydrogen sulfide. Curr Opin Anaesthesiol. 2009;22:168–76. doi: 10.1097/ACO.0b013e328328d22f. [DOI] [PubMed] [Google Scholar]
- 12.Jha S, Calvert JW, Duranski MR, Ramachandran A, Lefer DJ. Hydrogen sulfide attenuates hepatic ischemia-reperfusion injury: Role of antioxidant and antiapoptotic signaling. Am J Physiol Heart Circ Physiol. 2008;295:H801–6. doi: 10.1152/ajpheart.00377.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kimura Y, Kimura H. Hydrogen sulfide protects neurons from oxidative stress. Faseb J. 2004;18:1165–7. doi: 10.1096/fj.04-1815fje. [DOI] [PubMed] [Google Scholar]
- 14.Calvert JW, Jha S, Gundewar S, Elrod JW, Ramachandran A, Pattillo CB, Kevil CG, Lefer DJ. Hydrogen sulfide mediates cardioprotection through Nrf2 signaling. Circ Res. 2009;105:365–74. doi: 10.1161/CIRCRESAHA.109.199919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Milby TH, Baselt RC. Hydrogen sulfide poisoning: Clarification of some controversial issues. Am J Ind Med. 1999;35:192–5. doi: 10.1002/(sici)1097-0274(199902)35:2<192::aid-ajim11>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 16.Faller S, Ryter SW, Choi AM, Loop T, Schmidt R, Hoetzel A. Inhaled hydrogen sulfide protects against ventilator-induced lung injury. Anesthesiology. 2010;113:104–15. doi: 10.1097/ALN.0b013e3181de7107. [DOI] [PubMed] [Google Scholar]
- 17.Lopez A, Prior M, Yong S, Albassam M, Lillie LE. Biochemical and cytologic alterations in the respiratory tract of rats exposed for 4 hours to hydrogen sulfide. Fundam Appl Toxicol. 1987;9:753–62. doi: 10.1016/0272-0590(87)90182-5. [DOI] [PubMed] [Google Scholar]
- 18.Khan AA, Schuler MM, Prior MG, Yong S, Coppock RW, Florence LZ, Lillie LE. Effects of hydrogen sulfide exposure on lung mitochondrial respiratory chain enzymes in rats. Toxicol Appl Pharmacol. 1990;103:482–90. doi: 10.1016/0041-008x(90)90321-k. [DOI] [PubMed] [Google Scholar]
- 19.Bärtsch P, Mairbäurl H, Maggiorini M, Swenson ER. Physiological aspects of high-altitude pulmonary edema. J Appl Physiol. 2005;98:1101–10. doi: 10.1152/japplphysiol.01167.2004. [DOI] [PubMed] [Google Scholar]
- 20.Choudhury S, Wilson MR, Goddard ME, O'Dea KP, Takata M. Mechanisms of early pulmonary neutrophil sequestration in ventilator-induced lung injury in mice. Am J Physiol Lung Cell Mol Physiol. 2004;287:L902–10. doi: 10.1152/ajplung.00187.2004. [DOI] [PubMed] [Google Scholar]
- 21.Jafari B, Ouyang B, Li LF, Hales CA, Quinn DA. Intracellular glutathione in stretch-induced cytokine release from alveolar type-2 like cells. Respirology. 2004;9:43–53. doi: 10.1111/j.1440-1843.2003.00527.x. [DOI] [PubMed] [Google Scholar]
- 22.Belperio JA, Keane MP, Burdick MD, Londhe V, Xue YY, Li K, Phillips RJ, Strieter RM. Critical role for CXCR2 and CXCR2 ligands during the pathogenesis of ventilator-induced lung injury. J Clin Invest. 2002;110:1703–16. doi: 10.1172/JCI15849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Doerschuk CM. Leukocyte trafficking in alveoli and airway passages. Respir Res. 2000;1:136–40. doi: 10.1186/rr24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fiorucci S, Antonelli E, Distrutti E, Rizzo G, Mencarelli A, Orlandi S, Zanardo R, Renga B, Di Sante M, Morelli A, Cirino G, Wallace JL. Inhibition of hydrogen sulfide generation contributes to gastric injury caused by anti-inflammatory nonsteroidal drugs. Gastroenterology. 2005;129:1210–24. doi: 10.1053/j.gastro.2005.07.060. [DOI] [PubMed] [Google Scholar]
- 25.Zanardo RC, Brancaleone V, Distrutti E, Fiorucci S, Cirino G, Wallace JL. Hydrogen sulfide is an endogenous modulator of leukocyte-mediated inflammation. Faseb J. 2006;20:2118–20. doi: 10.1096/fj.06-6270fje. [DOI] [PubMed] [Google Scholar]
- 26.Frank JA, Wray CM, McAuley DF, Schwendener R, Matthay MA. Alveolar macrophages contribute to alveolar barrier dysfunction in ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2006;291:L1191–8. doi: 10.1152/ajplung.00055.2006. [DOI] [PubMed] [Google Scholar]
- 27.Li T, Zhao B, Wang C, Wang H, Liu Z, Li W, Jin H, Tang C, Du J. Regulatory effects of hydrogen sulfide on IL-6, IL-8 and IL-10 levels in the plasma and pulmonary tissue of rats with acute lung injury. Exp Biol Med (Maywood) 2008;233:1081–7. doi: 10.3181/0712-RM-354. [DOI] [PubMed] [Google Scholar]
- 28.Esechie A, Kiss L, Olah G, Horváth EM, Hawkins H, Szabo C, Traber DL. Protective effect of hydrogen sulfide in a murine model of acute lung injury induced by combined burn and smoke inhalation. Clin Sci (Lond) 2008;115:91–7. doi: 10.1042/CS20080021. [DOI] [PubMed] [Google Scholar]
- 29.Maher J, Yamamoto M. The rise of antioxidant signaling-the evolution and hormetic actions of Nrf2. Toxicol Appl Pharmacol. 2010;244:4–15. doi: 10.1016/j.taap.2010.01.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.