

Abstract
BACKGROUND
The current understanding of the effects of hypoxia on early embryogenesis is limited. Potential mediators of hypoxic effects include adenosine, which increases dramatically during hypoxic conditions and activates A1 adenosine receptors (A1ARs).
METHODS
To examine the influences of hypoxia and adenosine signaling on cardiac development, chicken embryos were studied. Real time RT-PCR assay was used to examine the A1AR gene expression during embryogenesis and after siRNA- mediated knock down. Cell proliferation was determined by counting cell nuclei and PhosphoHistone H3 positive cells. Apoptosis was determined by TUNEL assay.
RESULTS
A1ARs were found to be expressed in chicken embryos during early embryogenesis. Treatment of Hamburger and Hamilton stage 4 embryos with the A1AR agonist N6-cyclopentyladenosine caused cardiac bifida and looping defects in 55% of embryos. Hamburger and Hamilton stage 4 embryos exposed to 10% oxygen for 6, 12, 18, and 24 h followed by recovery in room air until stage 11, exhibited cardia bifida and looping defects in 34, 45, 60, and 86% of embryos respectively. Hypoxia-induced abnormalities were reduced when A1AR signaling was inhibited by the A1AR antagonist 1,3 dipropyl-8-cyclopentylxanthine or by siRNA-targeting A1ARs. Hypoxia treatment did not increase apoptosis, but decreased embryonic cell proliferation.
CONCLUSIONS
These data indicate that hypoxia adversely influences cardiac malformations during development, in part by A1AR signaling.
Keywords: hypoxia, adenosine, adenosine receptor, chicken embryo, heart
INTRODUCTION
Hypoxia is a common fetal stressor (Zhang, 2005). Pregnancy at high altitude, smoking during pregnancy, anemia, and placental insufficiency can induce hypoxemic states (Zhang, 2005). These conditions are associated with an increased incidence of congenital heart disease (Uebing et al., 2006). Yet, the link between hypoxia and congenital heart disease is not clear.
Cardiovascular development begins early in gestation, with the heart being one of the first organs to form (Kirby, 2002; Moorman et al., 2004). The symmetrically located cardiac primordia in the anterior lateral regions of the embryo migrate ventrally and fuse in the midline to form a primitive heart tube, which asymmetrically bends to the right (Kirby, 2002; Moorman et al., 2004). With successive looping and turning, a four chambered heart develops (Kirby, 2002; Moorman et al., 2004).
Hypoxia exposure during critical periods of embryogenesis can lead to impaired heart development including cardiac bifida, as well as neural tube and somite defects (Altimiras and Phu, 2000; Chan and Burggren, 2005; Jaffee, 1974; Villamor et al., 2004). Our recent studies show that a period of transient hypoxia during early murine embryogenesis impairs cardiac development and leads to intrauterine growth retardation (Wendler et al., 2007). Prolonged hypoxia results in reduced heart function with ventricular hypertrophy (Chan and Burggren, 2005; Sharma et al., 2006; Villamor et al., 2004). Studies performed using cultured rat embryos and in ovo chicken embryos also suggest that hypoxia directs remodeling of the cardiac outflow tract (Chen et al., 1999; Lampl, 2005; Sugishita et al., 2004) as well as coronary vessel organization (Wikenheiser et al., 2006).
Potential mediators of hypoxia include the purine nucleoside adenosine. Adenosine is a small molecule produced by all cells (Livingston et al., 2004; Rivkees et al., 2001; Shryock and Belardinelli, 1997). The specificity of adenosine action is determined at the receptor level (Rivkees et al., 2001). Four types of adenosine receptors are known: A1 and A3 adenosine receptors inhibit adenylyl cyclase activity, and A2a and A2b receptors stimulate adenylyl cylase activity (Jacobson and Gao, 2006; Olah and Stiles, 1995). Of the different adenosine receptor subtypes, A1ARs have the highest affinity for adenosine and are activated by modest increases in adenosine levels (Poulsen and Quinn, 1998). A1ARs are expressed very early in mammalian heart development (Rivkees, 1995). A1ARs regulate heart function (Porter and Rivkees, 2001) and activation of A1ARs reduces heart wall thickness and myocyte proliferation in murine embryos (Zhao and Rivkees, 2004). Our recent studies using A1AR−/− mice indicate that A1ARs are involved in protecting embryos against the effects of hypoxia (Wendler et al., 2007).
To examine the influences of hypoxia and adenosine signaling on cardiac development, chicken embryos were studied. Avian embryos provide an excellent model system to study the effects of hypoxia due to easy access to desired embryonic stages and the advantage of culturing embryos away from potential maternal influences (Chen et al., 1999; Lampl, 2005).
Based on the above observations, we hypothesize that if the downstream effects of hypoxia on the adenosinergic pathway are inhibited, some of the hypoxia-induced abnormalities can be rescued. We demonstrate that acute hypoxia during gastrulation induces cardiac tube malformation and looping defects (LDs). Pharmacological activation of A1ARs induces cardiac defects similar to hypoxia, while partial inhibition of A1ARs reverses some of the hypoxia-associated abnormalities.
MATERIALS AND METHODS
In Ovo Culture
Chicken eggs were obtained from SPAFAS (North Franklin, CT). Fresh, fertilized chicken eggs were incubated at 37°C to obtain the desired stage chicken embryos as described by Hamburger and Hamilton (1951). Eggs were exposed to hypoxia for 2 to 24 h in a hypoxia chamber (Biospherix, Redfield, NY); normoxia controls were incubated in the same incubator under room air. After hypoxia treatment eggs were returned to room air in the same incubator. To deliver drugs to the developing embryo, a hole was created above the air space in the egg using a thumbtack. A defined volume of drug was injected into the air cavity using a 30 gauge needle. Unless otherwise noted, all the embryos were examined at stage 11.
In Vitro Culture
Stage 4 embryos were cultured using the New (1955) culture method. Briefly, stage 4 embryos were opened in a dish containing Dulbecco’s phosphate buffered saline (PBS) solution, dissected from the yolk by an equatorial cut, and placed in a watch glass. A glass ring was placed over the embryo and the vitelline membrane was stretched over the ring. The entire assembly was placed in a Petri dish and incubated at 37°C. Unless otherwise noted, all the embryos were examined at stage 11.
Reverse Transcriptase Polymerase Chain Reaction (RT-PCR)
Hamburger and Hamilton (HH) stage 3 to 14 embryos were collected for RNA isolation. Using glass needles, heart forming regions (HFRs) and Hensen’s nodes were isolated from stage 4 embryos under a dissecting microscope. RNA was isolated from embryos and tissues with Trizol reagent (Invitrogen) and treated with DNaseI. cDNAs were prepared from 1 μg total RNA with the Superscript III RT kit (Invitrogen) in a 20 μL reaction. PCR primers were designed from the chicken A1AR sequence (accession number NM204316), forward primer: ctgcgggatgccactttct and reverse primer: ccagcgcgagggacttg, to amplify 598 bp bands. A1AR primer assay (Qiagen; Cat log Number QT00596484) amplifying 69 base pair product was also used. GAPDH primers used were as follows: GAPDH forward: tatgatgatatcaagagggtagt, and reverse: tgtatccaaactcattgtcatac to amplify 214 bp bands. PCR was performed with 1 μL of the 20 μL RT reaction, primers, and 2X Mastermix (Promega) in a Gene Amp machine (Applied Biosystems) at 55°C annealing for 35 cycles. Amplification products were separated by electrophoresis on 1% agarose gels. PCR products were sequenced at the Keck Biotechnology Center at Yale University.
Real Time RT-PCR
Real time RT-PCR was performed using cDNA and primers, as described above. One microgram total RNA from different stage embryos was used to prepare cDNA. The PCR protocol included 95°C denaturation (10 min), followed by denaturation at 95°C (20 s), annealing at 55°C (20 s), extension at 72°C (30 s), for 35 cycles with a final extension of 10 min at 70°C. Real time PCR was performed using the DyNAmoTM HS SYBR Green qPCR kit (Finnzymes) in the DNA engine Opticon 2 cycler (MJ Research). A1AR product (598 bp) was cloned in PCRII TOPO vector (Invitrogen) and used in real-time PCR to generate a standard curve from which copy number calculations were performed. Data were processed using Opticon software. qRT-PCR was repeated three times with three different sets of embryos.
Antisense RNA Treatment
New cultured stage 4 chicken embryos were treated with 10 μM (final concentration) of stealth antisense A1AR siRNA plus lipofectamine (Invitrogen); GGAUCAAGAAGUUCCGGACAGCCUU. siRNA sequences were designed using Invitrogen’s patented algorithm. Control embryos were treated with control siRNA; GGAAG AAUUGAGGCCGACACCUCUU. Treated embryos were incubated at room temperature for 1 h and further incubated at 37°C in 10% hypoxia or room air until HH stage 11.
Drug Treatment
A stock solution of 30 mM N6-cyclopentyladenosine (CPA; Sigma) was prepared in DMSO and diluted in PBS for injection. Assuming 50 mL as an egg volume, 50 μL of 1 mM CPA was injected in the air cavity to achieve final concentration of 1 μM. 1,3 dipropyl-8-cyclopentyl-xanthine (DPCPX; Sigma) was dissolved in DMSO and diluted in PBS. Fifty microliters of 1 μM DPCPX was used for in ovo injection to achieve a final concentration of 1 nM. CPA and DPCPX doses are based on the studies of Zhou and Rivkees (2001). Control embryos were treated with appropriately diluted DMSO as vehicle.
Bead Treatment
Heparin-acrylic beads (100–160 μM diameter; Sigma) were washed several times in PBS and soaked in 1 μM CPA for 1 h at room temperature. Control beads were soaked in PBS. Using glass needles, beads were implanted in the Hensen’s node and in the heart forming regions, in cultured stage 4 embryos. After implantation, embryos were incubated at room temperature for 1 h before being returned to 37°C. All the embryos were allowed to develop until stage 11. Unless otherwise noted, all the embryos were examined at stage 11.
Myosin Heavy Chain (MHC) Immunostaining
Embryos were fixed at room temperature for 1 h with freshly prepared 4% paraformaldehyde in PBS. Embryos were permeabilized in PBS containing 0.5% Triton X-100 for 2 h at room temperature. Permeabilized embryos were incubated in blocking solution (PBS containing 2% bovine serum albumin; Sigma) for 2 h at room temperature. Sarcomeric MHC expression was assessed in intact embryos using the monoclonal MF20 antibody (1:50, overnight at 4°C; Developmental Studies Hybridoma Bank, Iowa City, IA). Embryos were rinsed in PBS/Triton X-100 followed by incubation with Cy3-conjugated rabbit antimouse IgG (Molecular Probes; 1:200 in PBS containing 0.5% Triton X-100 and 2% BSA) at room temperature. Stained embryos were washed for 1 h in PBS then PBS-T (4 × 20 min each) and observed using an epifluorescence Olympus microscope equipped with a digital camera.
PhosphoHistone H3 Immunostaining
Paraffin sections (7 μm thick) were prepared from embryos. After deparafinization and rehydration sections were permeabilized in PBS containing 5% BSA, 0.5% Triton X-100 for 2 h at room temperature. Sections were incubated overnight at 4°C in anti-PhosphoHistone H3 (ser 10) antibody (1:500 dilution; Upstate, Lake Placid, NY). After washing (3X, PBS) sections were incubated in goat antirabbit Alexa 488 secondary antibody (1:200; Molecular Probes) for 2 h at room temperature. After washing (3X, PBS) sections were mounted in 4′,6-diamidino-2-phenylindole (DAPI) containing mounting medium and viewed under fluorescence. PhosphoHistone H3 positive cells were counted per 100 DAPI positive cells in five separate fields and presented as percent proliferative cells.
Embryonic Cell Counting
Three to five embryos were suspended in TENM2-sucrose buffer (10 mM NaCl, 1 mM MgCl2, 1 mM MnCl2, 15 mM triethanolamine, pH 7.6, and 0.3 M sucrose) and homogenized in a Dounce homogenizer with a loose piston as described (Ghatpande et al., 1990). For counting cell nuclei, ethidium bromide was added to a final concentration of 2 μg/mL to 100 μL of homogenate. The homogenate was appropriately diluted and examined in a Neubauer’s cell counting chamber under an epifluorescence Olympus microscope. Fluorescent nuclei were counted in the appropriate fields and total number of nuclei/embryo was estimated.
TUNEL Assay
Paraffin sections of embryos were prepared and used for TUNEL assay as per manufacturer’s instructions (Roche) using a fluorescein based in situ cell death kit. Sections were mounted in a medium containing DAPI as a counter stain and examined under epifluorescence using an Olympus microscope. Digital photographs were taken.
Statistical Analysis
The data are presented as percent and mean ± SEM. Analyses were performed by using Microsoft Excel and GraphPad Prism. Statistical comparisons between groups were performed with chi-square test ANOVA (for PCR data analysis), and nonparametric t test.
RESULTS
Effects of Hypoxia on Chicken Embryo Development
We first tested if hypoxia exposure during embryogenesis induces cardiac defects. The array of cardiac abnormalities induced by 10% O2 in ovo was characterized at different stages of cardiac development and evaluated at stage HH11 (Table 1). Exposure of stage 4 embryos to 2 h of hypoxia induced defects in 30% (p < .05) of the embryos, and 6 h of hypoxia exposure caused defects in 34% (p < .05) of embryos (Table 1). These defects included cardia bifida and abnormal looping. At stage 7, 27% (p < .05) of embryos exposed to hypoxia for 4 h displayed abnormal looping (Table 1). At stage 10, 20% (p < .05) of embryos exposed to hypoxic conditions for 6 h developed conotruncal defects (Table 1), which included swollen or thickened conotruncal region. Noncardiac defects included open neural tubes, wavy neural tubes, reduced optic vesicle size, and retarded blastoderm expansion. Somatic defects included loss of segmentation and winged or smaller somites.
Table 1.
Stage Specific Effects of Hypoxia In Ovo
| Treatment | Stage (HH) | Duration hours | Number of embryos | Abnormal (%) | Cardiac abnormalities
|
||
|---|---|---|---|---|---|---|---|
| FD | LD | Other | |||||
| Room air | 4 | 2–6 | 10 | 0% | 0 | 0 | 0 |
| 10% oxygen | 4 | 2 | 10 | 30%** | 1 (33%) | 2 (66%) | 0 |
| 10% oxygen | 4 | 6 | 15 | 34%** | 2 (40%) | 3 (60%) | 0 |
| Room air | 7 | 4 | 5 | 0% | 0 | 0 | 0 |
| 10% oxygen | 7 | 4 | 15 | 27%** | 0 | 4 (100%) | 0 |
| Room air | 10 | 6 | 10 | 10% | 0 | 0 | 1 (100%) |
| 10% oxygen | 10 | 6 | 20 | 20%* | 0 | 4 (100%) | |
All the embryos were evaluated at stage HH11 unless otherwise noted. FD, fusion defect; LD, looping defect; Other, other defects.
p < .05 (Chi-square) compared to room air controls.
An array of cardiac abnormalities was observed with hypoxia exposure at stage 4 (Table 2). These abnormalities are broadly categorized as fusion defects (FD), LDs, and other defects. FDs included cardiac primordia failing to fuse partially or completely in the embryonic midline. Hypoxia exposure starting at HH stage 4 for 6, 12, 18, and 24 h induced 34, 45, 60, and 86% (p < .05) cardiac malformations at HH stage 11, respectively (Table 2). The typical findings include butterfly-shaped hearts, which results when the bilateral heart primordia migrate to the midline and partially fuse (Fig. 1C,D). This butterfly-shaped heart was associated with a shortened conus region, shortened embryonic axis, and smaller somites, compared to normoxia embryos (Fig. 1). Some embryos displayed cardia bifida in which bilateral cardiac primordia migrated to the midline but failed to fuse altogether (Fig. 1E,F). The bifid hearts remained in the midline and were beating at the time of observation (Fig. 1E,F).
Table 2.
Cardiac Defects Induced by Hypoxia and CPA In Ovo
| Treatment | Stage (HH) | Duration hours | Number of embryos | Abnormal (%) | Cardiac abnormalities
|
||
|---|---|---|---|---|---|---|---|
| FD | LD | Others | |||||
| Room air | 4 | 6 to 24 | 50 | 6% | 0 (0%) | 1 (33%) | 2 (67%) |
| 10% oxygen | 4 | 6 | 50 | 34%* | 8 (47%) | 7 (41%) | 2 (12%) |
| 10% oxygen | 4 | 12 | 20 | 45%* | 4 (44%) | 5 (66%) | 0 (0%) |
| 10% oxygen | 4 | 18 | 20 | 60%* | 6 (50%) | 5 (42%) | 1 (8%) |
| 10% oxygen | 4 | 24 | 30 | 86.6%* | 12 (46%) | 9 (35%) | 5 (19%) |
| 1 nM DPCPX | 4 | 24 | 20 | 10% | 0 (0%) | 0 (0%) | 2 (100%) |
| 1 nM DPCPX + 10% oxygen | 4 | 6 | 25 | 12% | 1 (33%) | 2 (67%) | 0 (0%) |
| 1μM CPA | 4 | 24 | 20 | 55%* | 4 (36%) | 6 (55%) | 1 (9%) |
All the embryos were evaluated at stage HH11 unless otherwise noted. FD, fusion defect; LD, looping defect; Other, other defects.
p < .05 (Chi-square) compared to room air controls.
Figure 1.

Hypoxia affects cardiac morphogenesis. Embryos were exposed to hypoxia (10% O2) or room air (21% O2) for 6 h followed by recovery in room air until stage 11. Embryos were then stained for myosin heavy chain protein with MF 20 antibody (A,C,E,G,I). (A,B) Control embryos (room air oxygen) developed normally with rightward looping heart tubes. Examples of hypoxia-induced malformations include: (C,D) a butterfly shaped beating heart with smaller somites, (E,F) cardia bifida, (G,H) incompletely looped heart, (I,J) swollen heart tube with severely thickened somites. All embryos are shown as ventral views with anterior embryonic end at the top. Arrow points to developing heart tube.
We also observed a high percentage of LDs. These embryos had fused cardiac primordia, but no dextral looping (Table 2, Fig. 1G,H).
Other cardiac defects were identified as well; these include situs inversus (Fig. 1I,J), incomplete migration of primordia, and other atypical defects. On average, 10% of the hypoxic embryos had cardiac defects other than FDs and LDs (Table 2).
Exposure of stage 4 chicken embryos to 6 to 24 h of hypoxia resulted in an array of cardiac abnormalities. To test for MHC gene expression, embryos were stained at the heart looping stages with the MF20 antibody. The expression of MHC was observed in all hypoxic embryos although the expression appeared to be lower than in the control embryos (Fig 1).
Cardiac abnormalities were further examined by preparing histological sections stained with hematoxylin and eosin (Fig 2). Normal embryos have a single endocardial tube forming a C-shaped heart loop by HH stage 11 (Fig. 2A,E). Examination of embryos with defective looping showed a single heart tube that failed to form a C-shaped loop and the heart tube remained close to the midline (Fig. 2B,F). In fusion defective embryos, butterfly-shaped hearts were seen and the cross section passing through the heart region showed two endocardial tubes side by side (Fig. 2C). In a severely abnormal embryo, bent and short embryonic axis with a failure of cardiac primordia migration and fusion was seen (Fig. 2D). Hypoxia did not affect foregut formation even in severely abnormal embryos (Fig. 2).
Figure 2.

Hypoxia induces cardiac defects. Embryos were exposed to room air (A,E) or hypoxia (10% O2, B–H) for 24 h. Embryos were then embedded in paraffin, sectioned, and stained. (A,E) Control embryo with C-looped heart showing a single heart tube (arrow). (B,F) Hypoxia-exposed embryo with a retarded heart showing incomplete looping. (C,G) Hypoxia-exposed embryo showing fusion defect (cardia bifida, arrows). (D,H) A severely abnormal embryo with incomplete migration and fusion (arrows) of cardiac primordia and an open neural tube (arrowhead). Hypoxia did not affect foregut morphogenesis (dotted arrow in E–H). Dotted line in panels (A–D) depicts the level of cross section in panels (E–H). S, somites.
Next, New-cultured stage 4 embryos were exposed to 10% hypoxia for 6 h followed by recovery until stage 11 in room air (Table 3). Culturing embryos in vitro allows precise determination of developmental stage as well as direct exposure to a teratogenic agent. Hypoxia exposure of embryos in vitro resulted in cardiac abnormalities in 50% (p < .05) of the embryos (n = 20) compared to 10% heart defects in normoxia embryos (n = 21). Hypoxia-induced cardiac malformations included cardia bifida and heart tube LDs. Noncardiac defects included neural tube abnormalities and somite defects. Although hypoxia in New cultures produced slightly more cardiac malformation than in ovo (50 vs. 34%), the types of abnormalities observed were similar.
Table 3.
Cardiac Defects Induced by Hypoxia and CPA In Vitro
| Treatment | Stage (HH) | Duration hours | Number of embryos | Abnormal (%) | Cardiac abnormalities
|
||
|---|---|---|---|---|---|---|---|
| FD | FD | Others | |||||
| Room air | 4 | 6 to 24 | 21 | 10% | 0 (0%) | 1 (50%) | 1 (50%) |
| 10%-oxygen | 4 | 6 | 20 | 50%* | 5 (50%) | 4 (40%) | 1 (10%) |
| Vehicle | 4 | 18–24 | 10 | 00% | 0 (0%) | 0 (0%) | 0 (0%) |
| 1 μM CPA (systemic) | 4 | 18–24 | 15 | 60%* | 5 (56%) | 4 (44%) | 0 (0%) |
| PBS beads @ node | 4 | 18–24 | 10 | 10% | 0 (0%) | 1 (100%) | 0 (0%) |
| 1 μM CPA beads @ node | 4 | 18–24 | 10 | 60%* | 2 (33%) | 3 (50%) | 1 (17%) |
| PBS beads @ HFR | 4 | 18–24 | 10 | 00% | 0 (0%) | 0 (0%) | 0 (0%) |
| 1 μM CPA beads @ HFR | 4 | 18–24 | 10 | 30%* | 0 (0%) | 2 (100%) | 0 (0%) |
| Control siRNA + room air | 4 | 18–24 | 5 | 00% | 0 (0%) | 0 (0%) | 0 (0%) |
| A1AR siRNA + room air | 4 | 6 | 8 | 12.5% | 0 (0%) | 0 (0%) | 1 (100%) |
| Control siRNA + 10% oxygen | 4 | 6 | 15 | 60%* | 4 (44%) | 4 (44%) | 1 (11%) |
| A1AR siRNA + 10% oxygen | 4 | 6 | 15 | 33%* | 3 (60%) | 2 (40%) | 0 (0%) |
All the embryos were evaluated at stage HH11 unless otherwise noted. FD, fusion defect; LD, looping defect; Other, other defects.
p < .05 (Chi-square) compared to corresponding controls.
Effects of A1AR Activation on Embryos
A1AR gene expression in the chicken embryo was examined by real time RT-PCR. A1AR expression was detected in whole embryos from HH stage 3 to 14 (Fig. 3). To examine A1AR expression in the different regions of gastrulation stage embryos, heart forming regions and Hensen’s nodes were dissected from stage 4 embryos (Fig. 3B). A1AR expression was detected in the Hensen’s node and the heart forming regions (Fig. 3C). Nonradioactive in situ hybridization assays to detect the distribution and expression of A1AR were unsuccessful due to low levels of A1AR transcripts.
Figure 3.

A1AR transcripts are present in early chicken embryogenesis. (A) Total RNA from stage 3–14 whole embryos was isolated for real time RT-PCR. A1AR specific primers were used to amplify a 598 bp band. Copy number per embryo was calculated using a standard curve obtained from using an A1AR plasmid (n = 3). (B) Using glass needles, Hensen’s node (arrowhead) and heart forming regions (HFR) were dissected from stage 4 embryos (n = 5). (C) Total RNA from these dissected tissues was isolated for RT-PCR analysis. A1AR gene expression is seen in HFR and Hensen’s node of stage 4 chicken embryo. PS, primitive streak.
We next assessed if A1AR mediates the effects of hypoxia on cardiac development. Stage 4 chick embryos were treated with 1 μM (final concentration) of the A1AR agonist CPA in ovo. Eggs were incubated at 37°C in room air until embryos reached stage 11. CPA treatment (single dose) resulted in cardiac defects in 55% (p < .05) of the embryos (Table 2), which included cardiac bifida, unlooped heat tubes, and incorrectly looped hearts (Fig. 4). These cardiac defects were similar to those seen with hypoxia exposure.
Figure 4.

Activation of A1AR in chicken embryos results in cardiac defects. Gastrula stage chicken embryos were cultured by the method of New (1955). (A,E) Control embryos were treated with vehicle and grown until stage 11. (B,F) Sixty percent of 1 μM CPA treated embryos developed abnormally, including incompletely looped heart as shown. (C,G) In ovo control embryos treated with vehicle grew normally. (D,H) In ovo CPA (1 μM) treatment resulted in heart looping defects (55%). All embryos are shown as ventral views with anterior embryonic end at the top. Arrow points to developing heart tube. S, somites. Panels (E–H) are line tracings of hearts shown in panels (AD) respectively.
Effects of CPA treatment were then examined with New cultures. Embryos were treated with 1 μM CPA at stage 4 for 18 to 24 h until stage 11 (Table 3). CPA treatment induced cardiac defects in 60% (n = 15, p < .05) of the embryos, including cardia bifida and LDs (Fig. 4). Vehicle-treated control embryos displayed no embryonic abnormality (n = 10). These cardiac malformations were similar to those induced by in ovo CPA treatment and hypoxia exposure (Fig. 4).
To identify the sites of A1AR action during early chick development, we selectively activated A1ARs in the Hensen’s node and the HFRs. Stage 4 embryos were grafted with beads soaked in 1 μM CPA, which were placed in the Hensen’s node pit (n = 10), HFRs (n = 10), and cultured until stage 11 in vitro (Fig. 5). Control embryos (n = 20) were treated with beads soaked in PBS and placed in node (n = 10) and HFRs (n = 10). Treatment with CPA in Hensen’s node resulted in cardiac defects including bifida and looping abnormalities in 60% (p < .05) of the embryos (Fig. 5). Only 10% of the embryos developed abnormally with PBS beads grafted in the node (Table 3). We also placed CPA-loaded beads in the HFRs and found that 30% (p < .05) of the embryos had cardiac LDs (n = 10). PBS- loaded beads placed in the HFRs induced no obvious cardiac FDs or LDs (n = 10). CPA treatment at the node induced more LDs than CPA treatment at HFRs (p < .05).
Figure 5.
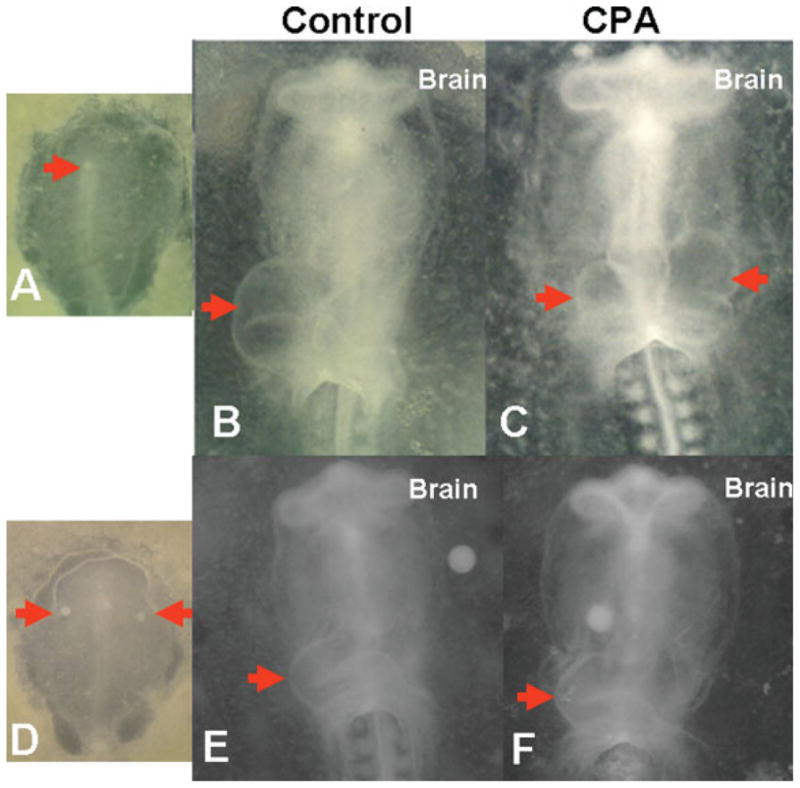
CPA treatment at Hensen’s node results in cardiac looping defects. (A) New cultured stage 4 chicken embryos were grafted with Heparin acrylic beads in the node region of the embryo (red arrow). Embryos were then cultured until stage 11 and digital images were captured. (B,D) Control embryos were implanted with beads soaked in PBS. (C,E) Beads soaked in 1 μM CPA produced cardia bifida (arrows). All embryos are shown as ventral views with anterior embryonic end at the top. Arrows point to developing heart tube. S, somites.
Role of A1ARs in Mediating the Effects of Hypoxia
We next examined if A1ARs mediate hypoxia-induced effects on cardiac development. Pretreatment of embryos with the A1AR antagonist DPCPX reduced the incidence of cardiac abnormalities from 34 to 12% (p < .05) in stage 4 embryos exposed to hypoxia for 6 h (Table 2). Stage 4 embryos treated with DPCPX (1 nM) alone in room air had 10% morphological defects.
As a complementary approach, cultured stage 4 embryos were pretreated with control siRNA or siRNA against chicken A1AR for 1 h at room temperature. Embryos were then exposed to hypoxia for 6 h followed by recovery in room air (until stage 11). Total RNA was isolated from siRNA-treated embryos and used in real time RT-PCR to assess expression of A1AR. siRNA against A1AR reduced expression of A1AR gene by fourfold (p < .05, one way ANOVA; Fig 6). siRNA treatment reduced the incidence of cardiac malformations from 60% seen using control siRNA (n = 15) to 33% (p < .05) with A1AR-specific siRNA (n = 15) under hypoxic conditions. Control siRNA (n = 5) and A1AR-specific siRNA (n = 8) treated embryos cultured in room air did not develop cardiac malformations (Table 3).
Figure 6.

siRNA decreases embryonic A1AR expression. New cultured stage 4 embryos were treated with siRNA that targets A1AR. At stage 11, total RNA was extracted and used in real time RT-PCR. Control siRNA treated embryos had normal levels of A1AR expression (set here as 100%). However, a fourfold decrease in the A1AR gene expression was observed with A1AR siRNA treatment (**p < .05, ANOVA).
Effect of Hypoxia on Embryonic Cell Apoptosis and Proliferation
We found that exposure of chicken embryos to hypoxia at stage 4 resulted in embryonic and cardiac malformations. We therefore examined the cellular proliferation and apoptosis in these embryos. Histological sections of control, hypoxic, and CPA-treated embryos were examined for cell proliferation. PhosphoHistone H3 antibody positive cells were counted at 6, 12, and 24 h during hypoxia and CPA treatment (Table 4). We found 5, 3, and 3.33% cells were positive for PhosphoHistone H3 staining at 6 h for control, hypoxia, and CPA treatment respectively (n = 3 embryos per group). Although the percentage of PhosphoHistone H3 positive cells in hypoxia and CPA-treated embryos were lower than corresponding control, there is statistical difference in values. At 12 h of hypoxia and CPA treatment we found 3.5 and 1% (p < .05) of cells positive for PhosphoHistone H3 staining, respectively, compared to 6.33% positive cells from control embryos (n = 3 embryos). Thus hypoxia and CPA treatment at 12 h lowered cell proliferation compared to controls. At 24 h PhosphoHistone H3 positive cells were examined only in the heart (Table 4). Approximately 1% of cells were positive in the hearts of stage 11 embryos (n = 3 embryos per treatment) irrespective of the treatment. Thus at 24 h there is no difference in cell proliferation rates as examined by PhosphoHistone H3 positive cells.
Table 4.
Cell Proliferation Analysis during Hypoxia
| Treatment | Percent phosphohistone H3 positive cells | Percent apoptotic cells |
|---|---|---|
| Control-6 h | 5% | 1% |
| Hypoxia-6 h | 3% | 1% |
| CPA-6 h | 3.33% | 1.66% |
| Control-12 h | 6.33% | 2.33% |
| Hypoxia-12 h | 3.5% | 1.25% |
| CPA-12 h | 1%* | 3.66% |
| DPCPX + hypoxia 12 h | 4.66% | 2% |
| Control-24 h | 1% (heart) | 1.66% (heart) |
| Hypoxia-24 h | 1.33% (heart) | 2.33% (heart) |
| CPA-24 h | 0.66% (heart) | 3.0% (heart) |
| DPCPX + hypoxia 24 h | 1.33% (heart) | 2.33% (heart) |
p < .05 (nonparametric t test) compared to controls.
Histological sections of control, hypoxic, and CPA-treated embryos were examined for apoptosis using TUNEL analysis (n = 3 embryos per treatment). We found 1, 1, and 1.66% TUNEL-positive cells at 6 h in control, hypoxia, and CPA-treated embryos. At 12 h we found a slight increase in TUNEL-positive cells (although not statistically significant) from embryos treated with CPA (3.66%) compared to 2.33% of cells from control embryos and 1.25% of cells from hypoxia embryos. At 24 h we found 1.66, 2.33, and 2.33% TUNEL-positive cells in control, hypoxia, and CPA-treated embryonic hearts, respectively. So, at all time points we demonstrate that hypoxia has no effect on cell death in these early chicken embryos.
Total cell proliferation in early chicken embryos can be determined by counting cells in the whole embryo (Ghatpande et al., 1990, 1991). We counted cells as described by Ghatpande et al. (1990). Stage 4 embryos had an average cell number of 4.1 ± 0.34 × 105 per embryo, which grew to 4.9 ± 0.63 × 106 in control embryos and hypoxic embryos attained 4.1 ± 0.511 × 106, while embryos treated with CPA attained 2.8 ± 0.83 × 106 average cell numbers, at the end of the experiment (stage 11). Cell counts indicated that hypoxia and CPA treatment slowed total cell proliferation.
DISCUSSION
Morphological abnormalities induced by hypoxia (5 to 15% oxygen) in early chicken embryos have been documented (Altimiras and Phu, 2000; Chan and Burggren, 2005). Studies examining the effects of hypoxia at the onset of cardiac tube morphogenesis are, however, lacking. We now report that hypoxia adversely affects cardiac structural development and that these effects are mediated in part by A1AR activation.
At first, we observed that hypoxia exposure during critical periods of embryogenesis lead to embryonic malformations including impaired heart development, cardia bifida, neural tube, and somite defects. Under hypoxic conditions the cardiac primordia were able to migrate to the midline of the embryo but failed to fuse properly, resulting in an array of defects including bifida and butterfly-shaped hearts. All the hearts, irrespective of the treatment, were beating at the time of observation. However, intensity of MF20 immunostaining for cardiac MHC proteins appeared lower in hypoxic or CPA-treated embryos. At this point, we cannot say whether this is due to lower expression or a reduction in the differentiated myocytes.
Chicken embryos treated with CPA in vitro and in ovo developed cardia bifida and incorrectly looped hearts. These defects were similar to those observed with hypoxia exposure. Inhibiting A1AR signaling by DPCPX or by reducing expression of A1AR with siRNA treatment reduced the incidence of hypoxia-induced defects.
Hensen’s node is considered to be the organizer of anterior tissues including the heart (Kirby et al., 2003). Using CPA-soaked beads, we observed that activation of A1ARs in the node and possibly in the surrounding areas (because CPA may diffuse away from the site of application) could induce cardiac fusion and LDs. CPA-loaded beads in the HFRs did not induce equal or more cardiac defects as compared to node alone CPA treatment. This indicates that Hensen’s node is the primary site of A1AR action during early cardiac formation.
Cell proliferation analysis indicates that by 12 h, CPA treatment reduced cell proliferation significantly while hypoxic embryos had slightly lower cell proliferation rates. Pretreatment with DPCPX followed by hypoxia for 12 h had cell proliferating at a similar rate as controls. Cell proliferation rates by 24 h were similar among all embryos irrespective of the treatment (hypoxia, CPA). Thus, reduction in cell proliferation early on appears to be one causative factor in the genesis of abnormalities.
TUNEL analysis of the whole embryos indicated that hypoxia and CPA treatment did not alter the levels of apoptosis during early treatments. In addition, the percentage of TUNEL-positive cells in the heart was not significantly different at 24 h between all treatment groups. It appears hypoxia acting as a teratogen slows down the cell proliferation as reported for other known teratogens like Vitamin A or trypan blue, which induce abnormal growth (Ghatpande et al., 1991, 1993; Modak et al., 1993). Although cell apoptosis is not altered, a reduction in cell proliferation may be responsible for growth defects induced by hypoxia exposure and CPA treatment. Because the apoptosis assay was done after the treatment it is possible that any effect on apoptosis during the treatment might have been missed with the present study design. However, consistent with these findings are studies of murine embryos exposed to hypoxia (Wendler et al., 2007) or CPA treatment (Zhao and Rivkees, 2004) during the heart forming stages, which demonstrated reduced cell proliferation in the absence of apoptosis.
Dilated cardiomyopathy was observed in mice over-expressing cardiac specific A1ARs (Funakoshi et al., 2006), indicating that increased signaling can be detrimental to heart function, although some protection from ischemia was observed in isolated heart preparations (Gauthier et al., 1998). A complete absence of A1ARs followed by hypoxia is also deleterious (Wendler et al., 2007). Thus a threshold of A1ARs is required for protection in hypoxic episodes during embryogenesis and may be involved in stabilization of HIF protein (Wendler et al., 2007). It has been observed that loss of A1AR function during times of stress (hypoxia) adversely affects cardiac and embryonic development (Wendler et al., 2007). Others have shown that A1ARs can regulate cardiac function in the developing heart (Porter and Rivkees, 2001). Our results in this report support the idea that elevated A1AR signaling is detrimental to heart development, also indicated by Zhao and Rivkees (2001). However, A1AR−/− mouse embryos cannot respond well to stress due to total loss of A1AR signaling (Wendler et al., 2007). Thus a limited A1AR signaling is needed for normal development and either elevated signaling or loss of signaling during stress has adverse effects on cardiac development.
Overall, we find that hypoxia induces cardiac defects including cardia bifida and abnormal looping in chicken embryos and that these defects are mediated in part by A1AR signaling. Hypoxia and A1AR activation may cause these defects in part by altering cell proliferation rates. These data, collectively, identify adenosine as an important mediator of hypoxia during heart tube formation in the chicken embryo.
Acknowledgments
Grant sponsor: American Heart Association.
Grant sponsor: Charles Hood Foundation (S. K. G.).
Grant sponsor: National Institutes of Health; Grant number: HL58442 (S. A. R.)
Footnotes
This work has been presented, in part, at Weinstein Cardiovascular Development Conference at Tucson, AZ, May 19–21, 2005.
References
- Altimiras J, Phu L. Lack of physiological plasticity in the early chicken embryo exposed to acute hypoxia. J Exp Zool. 2000;286:450–456. [PubMed] [Google Scholar]
- Chan T, Burggren W. Hypoxic incubation creates differential morphological effects during specific developmental critical windows in the embryo of the chicken (Gallus gallus) Respir Physiol Neurobiol. 2005;145:251–263. doi: 10.1016/j.resp.2004.09.005. [DOI] [PubMed] [Google Scholar]
- Chen E, Fujinaga M, Giaccia A. Hypoxic microenvironment within an embryo induces apoptosis and is essential for proper morphological development. Teratology. 1999;60:215–225. doi: 10.1002/(SICI)1096-9926(199910)60:4<215::AID-TERA6>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Funakoshi H, Chan TO, Good JC, et al. Regulated overexpression of the A1-adenosine receptor in mice results in adverse but reversible changes in cardiac morphology and function. Circulation. 2006;114:2240–2250. doi: 10.1161/CIRCULATIONAHA.106.620211. [DOI] [PubMed] [Google Scholar]
- Gauthier N, Headrick J, Matherne G. Myocardial function in the working mouse heart overexpressing cardiac A1 adenosine receptors. J Mol Cell Cardiol. 1998;30:187–193. doi: 10.1006/jmcc.1997.0585. [DOI] [PubMed] [Google Scholar]
- Ghatpande S, Mulherkar L, Modak S. Cell population growth and area expansion in early chick embryos during normal and abnormal morphogenesis in vitro. Develop Growth Differ. 1991;33:605–615. doi: 10.1111/j.1440-169X.1991.00605.x. [DOI] [PubMed] [Google Scholar]
- Ghatpande S, Vaidya P, Mulherkar L, et al. Lithium Chloride and Trypan Blue induce abnormal morphogenesis by suppressing cell population growth. Develop Growth Differ. 1993;35:409–419. doi: 10.1111/j.1440-169X.1993.00409.x. [DOI] [PubMed] [Google Scholar]
- Ghatpande SK, Guttikar GS, Paranjape SG, et al. Cell population growth in chick blastoderms cultured in vitro. Indian J Exp Biol. 1990;28:526–530. [PubMed] [Google Scholar]
- Hamburger V, Hamilton H. A series of normal stages in the development of the chick embryo. J Morphol. 1951;88:49–92. [PubMed] [Google Scholar]
- Jacobson K, Gao Z. Adenosine receptors as therapeutic targets. Nat Rev Drug Discov. 2006;5:247–264. doi: 10.1038/nrd1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffee O. The effects of moderate hypoxia and moderate hypoxia plus hypercapnea on cardiac development in chick embryos. Teratology. 1974;10:275–281. doi: 10.1002/tera.1420100310. [DOI] [PubMed] [Google Scholar]
- Kirby M. Molecular embryogenesis of the heart. Pediatr Dev Pathol. 2002;5:516–543. doi: 10.1007/s10024-002-0004-2. [DOI] [PubMed] [Google Scholar]
- Kirby M, Lawson A, Stadt HA, et al. Hensen’s node gives rise to the ventral midline of the foregut: implications for organizing head and heart development. Dev Biol. 2003;253:175–188. doi: 10.1016/s0012-1606(02)00024-6. [DOI] [PubMed] [Google Scholar]
- Lampl M. Cellular life histories and bow tie biology. Am J Hum Bio. 2005;17:166–180. doi: 10.1002/ajhb.20094. [DOI] [PubMed] [Google Scholar]
- Livingston M, Heaney LG, Ennis M. Adenosine, inflammation and asthma—a review. Inflamm Res. 2004;53:171–178. doi: 10.1007/s00011-004-1248-2. [DOI] [PubMed] [Google Scholar]
- Modak S, Ghatpande S, Rane R, et al. Caudalization by retinoic acid is correlated with inhibition of cell population growth and expansion of chick blastoderms cultured in vitro. Int J Dev Biol. 1993;37:601–607. [PubMed] [Google Scholar]
- Moorman A, Soufan A, Hagoort J, et al. Development of the building plan of the heart. Ann NY Acad Sci. 2004;1015:171–181. doi: 10.1196/annals.1302.014. [DOI] [PubMed] [Google Scholar]
- New DAT. A new technique for the cultivation of the chick embryo in vitro. J Embryol Exp Morphol. 1955;3:326–331. [Google Scholar]
- Olah M, Stiles G. Adenosine receptor subtypes: characterization and therapeutic regulation. Annu Rev Pharmacol Toxicol. 1995;35:581–606. doi: 10.1146/annurev.pa.35.040195.003053. [DOI] [PubMed] [Google Scholar]
- Porter GJ, Rivkees S. Ontogeny of humoral heart rate regulation in the embryonic mouse. Am J Physiol Regul Integr Comp Physiol. 2001;281:R401–407. doi: 10.1152/ajpregu.2001.281.2.R401. [DOI] [PubMed] [Google Scholar]
- Poulsen S, Quinn R. Adenosine receptors: new opportunities for future drugs. Bioorg Med Chem. 1998;6:619–641. doi: 10.1016/s0968-0896(98)00038-8. [DOI] [PubMed] [Google Scholar]
- Rivkees S. The ontogeny of cardiac and neural A1 adenosine receptor expression in rats. Brain Res Dev Brain Res. 1995;89:202–213. doi: 10.1016/0165-3806(95)00120-3. [DOI] [PubMed] [Google Scholar]
- Rivkees S, Zhao Z, Porter G, et al. Influences of adenosine on the fetus and newborn. Mol Genet Metab. 2001;74:160–171. doi: 10.1006/mgme.2001.3217. [DOI] [PubMed] [Google Scholar]
- Sharma S, Lucitti J, Nordman C, et al. Impact of hypoxia on early chick embryo growth and cardiovascular function. Pediatr Res. 2006;59:116–120. doi: 10.1203/01.pdr.0000191579.63339.90. [DOI] [PubMed] [Google Scholar]
- Shryock J, Belardinelli L. Adenosine and adenosine receptors in the cardiovascular system: biochemistry, physiology, and pharmacology. Am J Cardiol. 1997;79:2–10. doi: 10.1016/s0002-9149(97)00256-7. [DOI] [PubMed] [Google Scholar]
- Sugishita Y, Watanabe M, Fisher S. The development of the embryonic outflow tract provides novel insights into cardiac differentiation and remodeling. Trends Cardiovasc Med. 2004;14:235–241. doi: 10.1016/j.tcm.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Uebing L, Steer P, Yentis S, et al. Pregnancy and congenital heart disease. BMJ. 2006;332:401–406. doi: 10.1136/bmj.332.7538.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villamor E, Kessels C, Ruijtenbeek K, et al. Chronic in ovo hypoxia decreases pulmonary arterial contractile reactivity and induces biventricular cardiac enlargement in the chicken embryo. Am J Physiol Regul Integr Comp Physiol. 2004;287:R642–651. doi: 10.1152/ajpregu.00611.2003. [DOI] [PubMed] [Google Scholar]
- Wendler C, Amayta S, McClaskey C, et al. A1 Adenosine receptors play an essential role in protecting the embryo against hypoxia. Proc Natl Acad Sci USA. 2007;104:9697–9702. doi: 10.1073/pnas.0703557104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikenheiser J, Doughman Y, Fisher S, et al. Differential levels of tissue hypoxia in the developing chicken heart. Dev Dyn. 2006;235:115–123. doi: 10.1002/dvdy.20499. [DOI] [PubMed] [Google Scholar]
- Zhang L. Prenatal hypoxia and cardiac programming. J Soc Gynecol Investig. 2005;12:2–13. doi: 10.1016/j.jsgi.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Rivkees S. Inhibition of cell proliferation in the embryonic myocardium by A1 adenosine receptor activation. Dev Dyn. 2001;221:194–200. doi: 10.1002/dvdy.1130. [DOI] [PubMed] [Google Scholar]