

Abstract
Clearance of apoptotic cells is critical for control of tissue homeostasis however the full range of receptor(s) on phagocytes responsible for recognition of apoptotic cells remains to be identified. Here we show that dendritic cells (DCs), macrophages and endothelial cells use scavenger receptor type F family member 1 (SCARF1) to recognize and engulf apoptotic cells via C1q. Loss of SCARF1 impairs uptake of apoptotic cells. Consequently, in SCARF1-deficient mice, dying cells accumulate in tissues leading to a lupus-like disease with the spontaneous generation of autoantibodies to DNA-containing antigens, immune cell activation, dermatitis and nephritis. The discovery of SCARF1 interactions with C1q and apoptotic cells provides insights into molecular mechanisms involved in maintenance of tolerance and prevention of autoimmune disease.
Clearance of apoptotic cells is one of the most important processes of the immune system and is necessary for the homeostatic maintenance of healthy tissues and removal of infected or damaged cells1–3. Several types of cells are capable of apoptotic cell uptake, including both professional scavengers (macrophages, DCs) and non-professional phagocytes (fibroblasts, endothelial, and epithelial cells). In vivo, phagocytes are responsible for the rapid removal of dying cells before necrosis, a post-apoptotic stage accompanied by loss of membrane integrity and leakage of noxious intracellular molecules into the surrounding tissues4,5. Phagocyte engulfment of apoptotic cells occurs via an immunologically silent process by activating immunosuppressive pathways and the production of anti-inflammatory cytokines to prevent an immune response against self-antigens6. Consequently, defects in recognition and/or engulfment of apoptotic cells can lead to chronic inflammatory diseases, such as systemic lupus erythematosus (SLE), rheumatoid arthritis, glomerulonephritis and atherosclerosis7. Several studies have shown that patients with the autoimmune disease SLE have increased numbers of circulating apoptotic cells, indicating a failure in the clearance of dying cells8. In addition, lupus patients have circulating autoantibodies with specificity to intracellular autoantigens, further emphasizing the importance of apoptotic cell clearance in maintaining tolerance and preventing autoimmunity.
During apoptosis, phosphatidylserine (PS) is externalized from the inner to the outer leaflet of the cell membrane where it serves as a primary recognition and ‘eat-me’ signal for phagocytes9. Several cell surface receptors (TIM-1, TIM-3, TIM-4, and BAI) and soluble bridging proteins such as milk fat globule epidermal growth factor 8 (MFG-E8), calreticulin (Calr), and the complement component C1q can specifically bind to PS exposed on the surface of dying cells and enhance the uptake of apoptotic cells by phagocytes10–15. The mechanism of these bridging molecules awaits further characterization; however, recent studies demonstrate that Calr acts as an eat-me signal on the surface of apoptotic cells, whereas this signal is overridden on live cells by ‘don’t eat-me’ signals involving CD4716. Furthermore, an important role for C1q and MFG-E8 in apoptotic cell clearance is supported by data demonstrating the accumulation of apoptotic cells and the development of lupus autoimmune disease in mice deficient in C1q or MFG-E817–19. Other studies have demonstrated that C1q, Calr and PS have a strong interaction with each other and suggest a combinatorial role for these three molecules in the recognition of apoptotic cells14. To date the identification of the full range of receptor(s) used by phagocytes to recognize and engulf C1q-, Calr- and/or MFG-E8-opsonized apoptotic cells in vivo has remained elusive.
Scavenger receptors are a large family of structurally diverse molecules that have been implicated in the recognition of endogenous host derived-ligands and microbial pathogens20. SCARF1, previously known as SREC-1 (scavenger receptor expressed by endothelial cell-1) after it was originally cloned from an endothelial cell cDNA library, is an 86 kDa single-pass type 1 transmembrane protein composed of 830 amino acids21. The extracellular domain is made up of 406 amino acids and contains 5 epidermal growth factor (EGF)-like cysteine-rich repeats, followed by a long C-terminal cytoplasmic tail (391 amino acids) composed of serine and proline-rich regions. EGF-like domains mediate homophilic and heterophilic protein-protein interactions, and these domains in SCARF1 have been postulated to contribute to oligomerization of the protein or serve as the ligand-binding domain. Although SCARF1 was first shown to bind acetylated low density lipoprotein (acLDL), SCARF1 is also an endocytic receptor for HSP70, HSP90, Calr, gp96, and GP222–26. In addition to recognizing these endogenous host proteins, SCARF1 also binds to and is involved in internalizing pathogenic fungi and Neisseria gonorrhoeae bacterium via its interaction with gp9627,28.
Scavenger receptors are also found in lower organisms including the nematode Caenorhabditis elegans, which expresses CED-1, a transmembrane protein homologous to mammalian SCARF1, particularly in the extracellular EGF-like cysteine-rich repeat domains and has an overall similarity of 24%29,30. Previous work described the CED-1 gene to be required for the capture of necrotic cells in C. elegans29. These observations prompted us to generate mice deficient in SCARF1 to determine if this receptor is the mammalian orthologue to CED-1 and shares an evolutionarily-conserved function in apoptotic cell capture. Here we show that SCARF1 binds and engulfs apoptotic cells by recognizing C1q bound to PS exposed on the surface of dying, but not live cells. Our findings demonstrate that SCARF1 has a critical evolutionarily-conserved role in removing apoptotic cells and that its failure can lead to autoimmune disease.
RESULTS
SCARF1 mediates recognition of dying cells
The C. elegans receptor CED-1 and its mammalian orthologue SCARF1 function in innate sensing of the fungal pathogen Cryptococcus neoformans27,30. To identify other ligands for SCARF1, we constructed a chimera comprised of the extracellular domain of SCARF1 fused to the intracellular domain of TNFRSF1A (also called TNF-R1) and expressed it in HEK293T cells (Supplementary Fig. 1a). Ligand binding to the chimeric receptor triggered signaling via a RIP-TRADD-TRAF2 pathway leading to activation of NF-κB and Il8 gene expression in these cells. For example, the addition of heat-killed C. neoformans to these reporter cells induced signaling by CED-1–TNF-R1, mouse SCARF1–TNF-R1, and by the fungal β-glucan receptor Dectin-1–TNF-R1 (Supplementary Fig. 1b). To determine whether SCARF1, like CED-1, is involved in the innate recognition of apoptotic cells, we added ultraviolet (UV)-irradiated mouse embryonic fibroblasts (MEFs) to the reporter cells. We observed activation of the reporter cell lines expressing mouse or human SCARF1–TNF-R1 and CED-1–TNF-R1, but not with Dectin-1–TNF-R1 (Fig. 1a). Furthermore, activation of cells expressing SCARF1–TNF-R1, but not Dectin-1–TNF-R1 correlated with the number of apoptotic cells placed in co-culture and this was blocked by addition of recombinant soluble extracellular human SCARF1 (Fig. 1b and Supplementary Fig. 1c). As an additional control we generated reporter cells expressing human SCARF2, a related scavenger receptor family member with 35% amino acid homology to SCARF131. In contrast to SCARF1, apoptotic cells failed to trigger signaling of SCARF2–TNF-R1 reporter cells, indicating specificity of SCARF1 in apoptotic cell sensing (Fig. 1b). Using a conventional flow cytometry assay, SCARF1–TNF-R1, but not Dectin-1–TNF-R1 expressing cells were shown to capture dye-labeled UV-MEFs, indicating a direct interaction between SCARF1 and apoptotic cells (Fig. 1c). Microscopic analysis of cells expressing SCARF1–TNF-R1 chimera revealed binding to apoptotic cells, but only cells expressing full-length SCARF1 phagocytosed apoptotic cells, indicating that the C-terminal cytoplasmic tail of SCARF1 was required to signal the actin cytoskeleton for internalization (data not shown). Since live cells fail to trigger SCARF1 signaling (Fig. 1a), we sought to determine at what apoptotic stage ligands for SCARF1 are exposed on dying cells. We found that both early apoptotic cells (1–3 h post-UV treatment) and late apoptotic cells (8–24 h post-UV treatment) that have undergone secondary necrosis, as assessed by permeability to the DNA stain propidium iodide (PI), could trigger SCARF1–TNF-R1 signaling, indicating that ligands for SCARF1 are exposed rapidly after cell death (Fig. 1d). We also found that SCARF1 signaling was independent of the method used to induce cell death. Addition of dying MEFs induced by osmotic shock or exposure to Cesium (gamma) irradiation to the reporter cells induced SCARF1–TNF-R1 signaling similar to UV-MEFs (Fig. 1e). Moreover, all apoptotic cell types tested (i.e. B cells, DCs, splenocytes, MEFs), regardless of the species of origin, induced SCARF1–TNF-R1 signaling, indicating that the ligand is neither cell-type nor species restricted (Fig. 1e and data not shown). MFG-E8 was demonstrated to bind to apoptotic cells and facilitate their clearance through interaction with phagocytes18. We found that treatment of the reporter cells with recombinant MFG-E8 alone or in combination with apoptotic cells did not trigger or enhance SCARF1–TNF-R1 signaling, indicating that MFG-E8 is not a ligand for SCARF1 (Supplementary Fig. 1c). Together these data demonstrate that the CED-1-like scavenger receptor SCARF1 shares an evolutionarily-conserved function in apoptotic cell recognition.
Figure 1. SCARF1 mediates recognition of apoptotic cells.

a, Transfected HEK293T cells expressing mouse or human SCARF1–TNF-R1, Dectin-1–TNF-R1, or CED-1–TNF-R1 were untreated (UT) or treated with live or UV irradiated apoptotic MEFs at 1:1 ratio for 3 hours. RNA was extracted and human Il8 mRNA measured by qPCR. b, Human SCARF1–TNF-R1, human SCARF2–TNF-R1, and Dectin-1–TNF-R1 reporter cells were treated with UV-irradiated apoptotic MEFs at apoptotic cell to reporter cell ratios of 0:1, 1:5, 1:1, and 5:1 for 3 hours. Reporter cell activity was measured as in a. c, HEK293T reporter cells expressing mSCARF1–TNF-R1 or Dectin-1–TNF-R1 were treated with a 1:1 ratio of dye-labeled apoptotic UV-MEFs for 3 hours. Capture of dye-labeled apoptotic MEFs by the reporter cells was quantified by flow cytometry and analyzed using FlowJo software. d, MEFs were UV-irradiated and allowed to rest in culture for 1, 3, 8, and 24 hours. Early apoptotic cells (1 and 3 hour post-UV exposure) were Annexin V positive and propidium iodide negative, while cells undergoing secondary necrosis (8 and 24 hour post-UV exposure) stained positive for both as assessed by flow cytometry (not shown). Mouse SCARF1–TNF-R1 reporter cells were treated with a 1:1 ratio of early apoptotic cells or necrotic cells for 3 hours and Il8 expression measured as above. e, Apoptotic cells were generated by exposure to UV, Cesium-137 γ-irradiation (30 Gy), or hypotonic buffers. mSCARF1–TNF-R1 and Dectin-1–TNF-R1 reporter cells were treated with a 1:1 ratio of apoptotic Cesium-irradiated (Cs-Rad) MEFs, hypotonic MEFs, hypotonic primary B cells, and UV-irradiated DCs for 3 hours, measured as in a. All data shown are from one representative experiment of at least three performed and error bars denote the means and s.d. of at least triplicate measurements. *P<0.001; Student’s t-test.
SCARF1 binds to C1q
Exposure of PS on the outer leaflet of the plasma membrane of apoptotic cells is considered the primary eat-me signal recognized by phagocytes9. Since SCARF1 signaling and binding to apoptotic cells coincided with their rapid exposure of PS, we hypothesized that PS is a ligand for SCARF1. To examine direct interactions between SCARF1 and PS, we used a protein-lipid overlay dot blot assay in which hydrophobic membranes pre-spotted with 15 different lipids found in cell membranes were probed with SCARF1 protein. In contrast to MFG-E8, we found that SCARF1 failed to bind to PS or any other lipids, except for 3-sulfogalactosylceramide, a known scavenger receptor ligand (Fig. 2a and Supplementary Fig. 2a)32. To determine whether SCARF1 recognition of apoptotic cells was mediated by 3-sulfogalactosylceramide, we added UV-MEFs in the presence of 3-sulfogalactosylceramide to the SCARF1–TNF-R1 reporter cells. We observed that 3-sulfogalactosylceramide failed to enhance or inhibit SCARF1 activation by apoptotic cells, indicating that 3-sulfogalactosylceramide (sulfatide) is not an important contributor in mediating SCARF1-apoptotic cell interactions (Supplementary Fig. 2b). In addition to PS, several soluble bridging proteins have been shown to be involved in the recognition of apoptotic cells, including C1q, Calr, and MFG-E812,16,19. Using a protein-protein overlay dot blot assay, SCARF1 specifically bound to C1q in a concentration-dependent manner, but not to Calr or MFG-E8 (Fig. 2b). We further examined direct binding of SCARF1 with C1q using antigen-capture enzyme linked immunosorbent assays (ELISA) with C-coated microtiter plate wells. These experiments showed strong binding of SCARF1 to C1q and to the classic scavenger receptor ligand acLDL, whereas negligible binding was observed to bovine serum albumin (BSA), PS, phosphatidylcholine (PC) or Calr (Fig. 2c). Moreover, a similar interaction pattern was observed when the ELISA was performed in the reverse configuration with C1q binding to plate-bound SCARF1 (data not shown). To confirm these findings and determine the affinity of the SCARF1-C1q interaction, we measured SCARF1 binding to a C1q-immobilized sensor chip by Biacore SPR analysis using a BIAcore 3000 instrument. No binding (NB) of SCARF1 to PS or Calr was observed; however, affinity dissociation constants revealed a Kd of 1.24 × 10−7 M for the interaction of SCARF1 and C1q (Table 1). These results demonstrate that C1q is a ligand for SCARF1.
Figure 2. SCARF1 binds C1q.

a, Dot blot representing the binding capacity of SCARF1 to triglycerides (TG), diacylglycerol (DAG), phosphatidic acid (PA), PS, phosphatidylethanolamine (PE), phosphatidylcholine (PC), phosphatidylglycerol (PG), cardiolipin, phosphatidylin (PtdIns), ptdlns(4)P, ptdlns(4,5)P2, ptdlns(3,4,5)P3, cholesterol, sphingomyelin or 3-sulfogalactosylceramide. Representative dot blot of three independent experiments performed. b, Dot blot representing binding of SCARF1 to immobilized Calr, MFG-E8, C1q, PS, PC or acLDL. c, Binding of SCARF1 to BSA, C1q, PS, PC, Calr and acLDL as measured by ELISA. d, HEK293T cells expressing mouse SCARF1–TNF-R1 were incubated with live cells from C57BL/6 mice, apoptotic B cells from Mfge8+/+ or Mfge8−/− mice, apoptotic Calr+/+ or Calr−/− MEFs, and apoptotic wild-type MEFs transduced with shRNAs targeting GFP (shControl) or C1q (shC1q #1) for 3 hours. Knockdown efficiency was confirmed by measuring abundance of C1q mRNA in transduced MEFs by qPCR (Supplementary Fig. 2c). RNA was extracted and Il8 mRNA expression was measured by qPCR. e–h, HEK293T expressing mouse SCARF1–TNF-R1 were incubated with recombinant C1q, Calr, Annexin V or liposomes containing PC or PS at indicated concentrations. Treated cells were then incubated with UV-apoptotic MEFs for 3 hours, RNA was extracted and the levels of Il8 mRNA were measured by qPCR. Data presented represent one experiment of at least three performed and error bars represent the means and s.d. of triplicate samples. *P<0.001, **P <0.0001; Student’s t-test.
Table 1.
SPR analysis
| Analyte | KD, M |
|---|---|
| C1q | 1.24 × 10−7 |
| PS | NB |
| Calr | NB |
SCARF1-C1q-apoptotic cell interaction is PS-dependent
To further investigate the interaction of SCARF1 with apoptotic cell ligands, we co-cultured SCARF1-TNF-R1 reporter cells with apoptotic cells deficient in either C1q, MFG-E8, or Calr. Apoptotic cells deficient in C1q, but not MFG-E8 or Calr were significantly impaired in their ability to induce SCARF1 signaling (Fig. 2d and Supplementary Fig. 2c). In addition, C1q alone or in the presence of apoptotic cells triggered SCARF1 signaling (IL-8 expression) in a concentration-dependent manner (Fig. 2e). In contrast, Calr treatment was not sufficient to enhance or inhibit SCARF1-mediated recognition of apoptotic cells (Fig. 2e). Together these data demonstrate that SCARF1 recognition of apoptotic cells requires C1q. Since C1q has been shown to interact with PS on apoptotic cells, we hypothesized that SCARF1 recognition of apoptotic cells occurs via a C1q-PS-apoptotic cell interaction13. To test this hypothesis, reporter cells were co-cultured with apoptotic MEFs pretreated with Annexin V, a phospholipid binding protein that has high affinity for PS, which binds to exposed PS on apoptotic cells. Pretreatment of apoptotic MEFs with Annexin V, where PS is blocked, inhibited signaling of SCARF1–TNF-R1 reporter cells in a dose-dependent manner (Fig. 2f). Moreover, to further determine whether SCARF1 recognition of apoptotic cells requires PS, we performed a liposome competition experiment. PS liposomes, but not phosphatidylcholine (PC) liposomes, inhibited apoptotic MEF-induced SCARF1–TNF-R1 signaling in a dose-dependent manner, while PS treatment alone failed to activate SCARF1 signaling (Fig. 2g). Finally, to demonstrate whether the SCARF1-C1q-apoptotic cell interaction is dependent on PS, SCARF1–TNF-R1 reporter cells were co-cultured with Annexin V-treated apoptotic MEFs in the presence of C1q. We found that Annexin V-treated apoptotic MEFs failed to activate SCARF1-dependent signaling even in the presence of C1q (Fig. 2h). Together these results demonstrate that SCARF1 mediated recognition of apoptotic cells occurs by SCARF1 binding to C1q via PS-exposed apoptotic cells.
SCARF1 mediates apoptotic cell recognition in vitro
To determine where SCARF1 might play a functional role we performed quantitative real time PCR (qPCR) analyses to examine its expression in various organs of mice (Supplementary Fig. 3a). SCARF1 was expressed in a variety of organs, most predominantly in the spleen and lung. Expression of SCARF1 in circulating immune cell types (neutrophils, T cells and B cells) was very low, consistent with previous reports demonstrating its restricted expression on endothelial cells, DCs, and macrophages (Fig. 3a)21,22,26,30. SCARF1 expression was also observed in peritoneal B1a and B1b cells at levels equivalent to CD8α+ DCs. Addition of apoptotic cells to the cell culture media robustly increased mRNA and surface protein expression of SCARF1 in CD8α+ DCs, but not in other cell types (Fig. 3b,c). The CD8α+ DC subset is specialized to capture dying cells33 so to further examine the role of SCARF1 in mediating recognition and responses to apoptotic cells, we generated Scarf1−/− mice in which SCARF1 expression is ablated by replacing exons 1–8, which code for the entire extracellular and transmembrane domains, with a neomycin cassette (Supplementary Fig. 4a–c). To test whether SCARF1 expression on CD8α+ DCs is necessary for the capture and engulfment of apoptotic cells, we used a flow cytometry assay to determine the percentage of dye-labeled apoptotic cells captured by CD8α+ DCs isolated from Scarf1+/+ and Scarf1−/− mice. CD8α+ DCs from Scarf1−/− mice had significant impairment in their ability to acquire dye-labeled apoptotic cells as compared to Scarf1+/+ CD8α+ DCs (Fig. 3d). These data demonstrate that SCARF1 is required for recognition of apoptotic cells by CD8α+ DCs. In addition to CD8α+ DCs, Scarf1−/− macrophages, endothelial cells, and B1a cells, but not neutrophils, B cells, T cells, or B1b cells had a significant impairment in the uptake of apoptotic cells, consistent with the expression of SCARF1 on these cell types (Fig. 3d). These data demonstrate that SCARF1 expression contributes approximately 70%, 32%, 74%, and 52% in the clearance of apoptotic cells by CD8α+ DCs, macrophages, endothelial cells, and B1a cells, respectively. Since SCARF1 expression only had a moderate contribution to macrophage uptake of apoptotic cells, it suggests that other apoptotic cell clearance pathways play a more dominant role in this cell type. Indeed, expression of MFG-E8, TIM-4, CD91, c-mer, and ITAGV were significantly higher in macrophages as compared to CD8α+ DCs and endothelial cells, and suggests that these pathways may partially compensate for the loss of SCARF1 (Supplementary Fig. 3b).
Figure 3. Impaired apoptotic cell engulfment by SCARF1-deficient CD8α+ DCs cells.

a, b, qPCR analyses. RNA was obtained from CD8α+ DCs, macrophages, endothelial cells, B cells, T cells (spleen), neutrophils (bone marrow), B1a and B1b cells (peritoneum) isolated from Scarf1+/+ mice in the presence or absence of Scarf1−/− apoptotic B cells and analyzed for expression of SCARF1 by qPCR. Copies of Scarf1 mRNA were normalized to copies of B2m. Bars denote means and s.d. of three independent experiments performed with triplicate measurements. c, Splenic DCs from Scarf1+/+ and Scarf1−/− mice were co-cultured in vitro with apoptotic Scarf1−/− B cells for 4 hours and either stained with fluorescent-conjugated antibodies to SCARF1 or isotype control and examined by flow cytometry to measure surface expression. d, Cells isolated from Scarf1+/+ and Scarf1−/− mice were incubated with dye-labeled apoptotic MEFs for 2 hour at 37°C. Cells were harvested and samples were analyzed by flow cytometry (n=8 mice per group). e, CD11c+CD8α+ DCs (red) were isolated from spleens of Scarf1+/+ and Scarf1−/− mice by magnetic selection and incubated with dye-labeled apoptotic B cells (green) for 2 hours before visualization by confocal microscopy; % of DCs that phagocytose apoptotic cells was determined by counting 5 random high powered views per slide (n=8 slides). f, Apoptotic or live MEFs were labeled with the pH-sensitive dye pHrodo and incubated with CD8α+ DCs for 2 hours at 4°C or 37°C. Cells were then harvested and stained for CD11c and CD8α and mean fluorescence intensity (MFI) for each sample was determined by flow cytometry. g, Splenic CD8α+ DCs isolated from Scarf1+/+ and Scarf1−/− mice were co-cultured in C1q-depleted serum with dye-labeled apoptotic UV-MEFs in the presence or absence of exogenous C1q for 2 hours at 37°C. Uptake was measured by flow cytometry. Data presented represent one experiment of at least three performed and error bars represent the means and s.d. of triplicate samples. *P<0.01, **P <0.001; Student’s t-test.
As a control and to determine whether SCARF1 is a ligand for itself, we co-cultured Scarf1+/+ CD8α+ DCs with apoptotic cells isolated from Scarf1+/+ and Scarf1−/− mice. Scarf1+/+ CD8α+ DCs engulfed an equivalent percentage of Scarf1+/+ and Scarf1−/− apoptotic cells, indicating that one of the shared ligands for SCARF1 on apoptotic cells is not SCARF1 itself (data not shown). To confirm uptake of apoptotic cells, we performed confocal microscopy on CD8α+ DCs incubated with dye-labeled apoptotic B cells. Scarf1+/+, but not Scarf1−/− CD8α+ DCs engulfed apoptotic cells and cleared them from the culture medium (Fig. 3e). To further confirm the impairment in internalization of apoptotic cells by Scarf1−/− CD8α+ DCs, we incubated CD8α+ DCs with apoptotic cells labeled with pHrodo, a pH-sensitive fluorescent dye that makes engulfed cells highly fluorescent. Scarf1−/− CD8α+ DCs showed a dramatic inability to engulf pHrodo-labeled apoptotic cells (Fig. 3f). In contrast, Scarf1+/+ and Scarf1−/− CD8α+ DCs engulfed an equivalent, but small percentage of pHrodo-labeled live cells. To further test our hypothesis that SCARF1 mediates recognition of apoptotic cells via C1q, we incubated Scarf1+/+ and Scarf1−/− CD8α+ DCs with dye-labeled apoptotic cells in the presence or absence of C1q. C1q significantly enhanced apoptotic cell uptake by Scarf1+/+, but not Scarf1−/− CD8α+ DCs (Fig. 3g). Together these experiments indicate that SCARF1 is required for optimal capture of apoptotic cells and can specifically ‘sense’ exposed ligands on apoptotic versus live cells.
SCARF1 is required for apoptotic cell recognition
To confirm that the apoptotic cell clearance deficiency observed in SCARF1-deficient cells is not due to a secondary defect unrelated to SCARF1 ablation, we reconstituted CD8α+ DCs, macrophages and endothelial cells from Scarf1−/− mice with a cDNA for mouse Scarf1 by nucleofection. Ectopic expression of Scarf1, which we confirmed by immunoblot and microscopy, restored the ability of Scarf1−/− CD8α+ DCs, macrophages and endothelial cells to efficiently capture apoptotic cells (Fig. 4a–c and data not shown). Moreover, Scarf1+/+ CD8α+ DCs transfected with mouse Scarf1 had a >25% increase in uptake of apoptotic cells as compared to mock transfected CD8α+ DCs, indicating that upregulation and/or overexpression of Scarf1 may be a useful therapeutic strategy for the clearance of dead cells that occurs in some diseases1.
Figure 4. SCARF1 is necessary for apoptotic cell uptake.
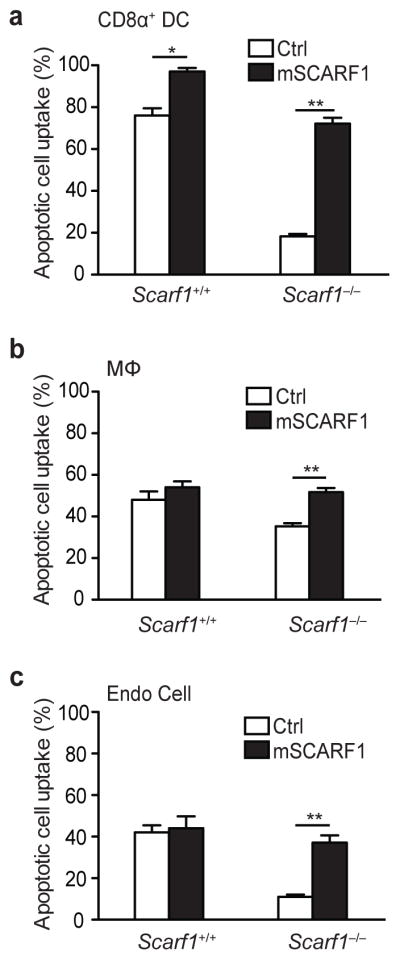
a, b, c, Splenic CD8α+ DCs, macrophages, or endothelial cells isolated from Scarf1+/+ and Scarf1−/− mice were nucleofected with a vector encoding for mSCARF1-GFP (C-terminal GFP fusion) or GFP alone. SCARF1 and control transfected cells were treated with dye-labeled apoptotic MEFs for 2 hours at 37°C. Cells were harvested, and uptake of apoptotic cells by GFP-positive cells was analyzed by flow cytometry. SCARF1 expression was confirmed by GFP expression using flow cytometry, immunoblot, and microscopy (not shown). All data shown are from one representative experiment of at least three performed and error bars denote the mean and s.d. of triplicate measurements. *P<0.01, **P <0.001; Student’s t-test.
SCARF1 mediates apoptotic cell clearance in vivo
To determine whether SCARF1 is necessary for engulfment of apoptotic cells in vivo, we intravenously injected Scarf1+/+ and Scarf1−/− mice with either dye-labeled apoptotic B cells or 1 μM FITC-latex spheres, as previously described33. B cells were subjected to UV irradiation to induce apoptosis, as assessed by Annexin V-positive and PI-negative staining. One hour after injecting apoptotic cells or FITC-latex beads, we harvested spleen and enriched for DCs using anti-CD11c magnetic microbeads. Flow cytometry with antibodies to CD11c and CD8α was then used to identify DCs that had captured apoptotic cells or FITC-latex beads. Both CD8α+ and CD8α− DCs from Scarf1+/+ and Scarf1−/− mice captured equivalent amounts of FITC-latex beads (Fig. 5a). In contrast, apoptotic cells were engulfed selectively by the CD8α+ DC subset (Fig. 5b). Moreover, CD8α+ DCs from SCARF1-deficient mice captured significantly fewer apoptotic cells than those from Scarf1+/+ mice (Fig. 5b). These data demonstrate that SCARF1 is necessary on CD8α+ DCs to capture dying cells and may be involved in homeostatic clearance of apoptotic cells in vivo. To test this hypothesis, we isolated peripheral blood mononuclear cells from the whole blood of 20-week old Scarf1+/+ and Scarf1−/− mice. Flow cytometry with Annexin V and propidium iodide (PI) staining was then used to identify the number of circulating apoptotic and necrotic cells. We found a significant increase in the number of early apoptotic cells (Annexin V+ and PI−) and secondary necrotic cells (Annexin V+ and PI+), >4- and 10-fold respectively, in the circulation of Scarf1−/− mice as compared to Scarf1+/+ mice, indicating that SCARF1 plays an important role in maintaining apoptotic cell clearance in vivo (Fig. 5c). In wild-type mice the removal of apoptotic cells is usually a very efficient process and few apoptotic cells are found even in tissues with high rates of cell death. In contrast, dying cells accumulated in the tissues of Scarf1−/− mice. Notably, spleens taken from Scarf1−/− mice had significantly higher numbers of apoptotic cells (green), as assessed by TUNEL staining, compared to spleens from Scarf1+/+ mice and while splenic CD11c+ DCs (red) in Scarf1−/− mice were localized near apoptotic cells (green), they had reduced uptake (Fig. 5d). To ensure that the accumulation of apoptotic cells in Scarf1−/− mice was due solely to the impairment in apoptotic cell clearance and not to increased cell turnover, we monitored the rate of apoptosis in Scarf1+/+ and Scarf1−/− cells. The rate and percentage of Scarf1−/− splenocytes, B cells, and bone marrow cells undergoing apoptosis was similar to Scarf1+/+ cells (Supplementary Fig. 5a–c). Furthermore, expression of other known apoptotic cell recognition receptors C1q, MFG-E8, TIM1, TIM3, TIM4, CD91, MARCO, CD14, LOX-1, SR-A1, GAS6, ITGAV and c-mer were not affected in Scarf1−/− DCs (Supplementary Fig. 5d). From these results we conclude that SCARF1 is necessary for the ability of CD8α+ DCs to engulf and clear apoptotic cells in vivo.
Figure 5. SCARF1 is necessary for the homeostatic clearance of apoptotic cells in vivo.

a, b, Groups of six mice were injected intravenously with PBS, FITC-latex beads (0.2 mL of a 0.27% solution), or apoptotic B cells (20 × 106 CFSE-labeled). One hour later spleens were harvested and splenocytes stained for CD11c and CD8α, and samples analyzed by flow cytometry. Panels of dot plots show DCs pre-gated for CD11c expression and analyzed for CD8α and CFSE fluorescent stain. b, Graphical representation of apoptotic B cell uptake by CD11c+CD8α+ splenic DCs (n=5 mice per group). c, Whole blood from >16 week-old Scarf1+/+ and Scarf1−/− mice was collected and PBMCs were isolated by Ficoll gradient. PBMCs were stained with Annexin V and propidium iodide and analyzed by flow cytometry. Graphs depict the total number of circulating apoptotic or secondary necrotic cells per mL of blood and represent the average of 3 independent experiments (n=8 mice per group). d, Apoptotic cells in spleen sections from >16 week-old Scarf1+/+ and Scarf1−/− mice were stained with TUNEL (green) and CD11c (red) and analyzed by fluorescent microscopy. TUNEL quantification was determined by analyzing images with a DAPI-TUNEL staining automated analysis macro for ImageJ software. Pictures and graph are representative of 3 independent experiments (4 mice per group). *P<0.001, **P <0.0001; Student’s t-test.
SCARF1 deficiency results in spontaneous autoimmunity
Defective clearance of apoptotic cells can increase susceptibility to the autoimmune disease SLE and autoantigens derived from these apoptotic cells can lead to the production of autoantibodies, a hallmark feature of lupus34–36. We hypothesized that the inability of Scarf1−/− mice to clear apoptotic cells efficiently would result in spontaneous lupus-associated autoimmune disease. To test this, we used the fluorescent HEp-2 cell anti-nuclear antibody (ANA) assay as a sensitive detection method for antibodies to both RNA- and DNA-containing autoantigens (Fig. 6a). Beginning at ~20 weeks of age a majority (~62%) of sera from Scarf1−/− mice produced a homogenous (~63%) or speckled (~37%) nuclear ANA staining pattern (Fig. 6b). There was a significantly higher prevalence of ANA-positive sera from female Scarf1−/− mice (17 of 22, ~77%) than male Scarf1−/− mice (11 of 25, ~48%) (Fig. 6c). Indeed, a strong gender bias exists in human SLE and for multiple animal models of lupus in which females are more susceptible to disease37,38. Sex hormones and other genetic effects conferred by the female background may be involved in the generation of higher amounts of systemic autoantibodies than those found in male SCARF1-deficient mice11. In contrast, none of the sera from Scarf1+/+ littermate control mice produced any type of staining, indicating the absence of nuclear autoantibodies (Fig. 6a). The homogenous staining pattern found in the majority of ANA-positive sera from Scarf1−/− mice reflects autoantibodies to nucleoproteins and DNA, which have been reported to correlate with clinical diagnosis of SLE. The presence of autoantibodies responsible for speckled staining patterns also occurs in SLE and other disorders, notably Sjogren’s syndrome. However, the specificity of autoantibodies resulting in the speckled pattern is less well defined, but is often indicative of the presence of autoantibodies to the nuclear proteins Sm, RNP, and/or La. To further define the autoantibody profile, we used ELISA to quantitate the increase in autoantibodies to DNA-containing antigens in Scarf1−/− sera. An anti-nucleosome autoantibody ELISA, in which the detection antigen was the combination of histones and double-stranded DNA (dsDNA), demonstrated a significant increase in specific DNA-histone autoantibodies in approximately ~72% of Scarf1−/− mice (Fig. 6d). Consistent with the ANA data, female Scarf1−/− mice generated significantly higher amounts of anti-nucleosome autoantibodies than their male counterparts. To determine if there are autoantibodies specifically to dsDNA, we performed the highly specific Crithida luciliae immunofluorescence assay. Male (13%) and female (22%) Scarf1−/− mice had low, but readily detectable amounts of anti-dsDNA autoantibodies in their serum compared to Scarf1+/+ mice (Fig. 6e). Together these data indicate that autoantibodies in Scarf1−/− mice preferentially bind to DNA-histone epitopes rather than non-chromatin DNA, suggesting that chromatin is the physiologically relevant autoantigen. We also determined amounts of autoantibodies to the RNA-containing antigen Smith (SmRNP) and La. Unlike DNA-containing antigens, we did not detect the presence of serum antibodies to SmRNP or La, thus the autoantigen responsible for the speckled ANA pattern found in ~23% of Scarf1−/− mice remains to be identified (Fig. 6f and data not shown).
Figure 6. Autoantibody generation in Scarf1−/− mice.

a, ANA immunofluorescence was performed using HEp-2 cells, with sera from 20-week old Scarf1+/+ and Scarf1−/− mice (25 male and 25 female mice per group). b, Serum ANAs were classified as either nuclear homogenous, nuclear speckled, or cytoplasmic staining patterns. None detected (ND). c, Percentage of male and female Scarf1−/− mice staining positive (>1:320 dilution) for ANA. Staining was scored by an observer blinded to the genotype of the mice. **P <0.0001; Student’s t-test. d, Anti-nucleosome (dsDNA-histone) autoantibodies in the serum of Scarf1+/+ and Scarf1−/− male and female mice were determined by ELISA (n=14 mice per group). Bars represent median values with s.d. **P<0.0001; Mann-Whitney test. e, Crithidia luciliae immunofluorescence was performed with serum from Scarf1−/− male and female mice at 1:80 dilution (n=14 mice per group). *P<0.001, **P <0.0001; Student’s t-test. f, Anti-Smith-RNP autoantibodies in the serum of Scarf1+/+ and Scarf1−/− male and female mice were determined by ELISA (n=14 mice per group).
Spontaneous inflammation in Scarf1−/− mice
Since we observed a significant effect of Scarf1 deficiency on autoantibody production, we next determined whether the absence of this receptor had a significant impact on other clinical manifestations of lupus-like autoimmune disease. Scarf1−/− mice also had a significant increase in the incidence and severity of skin disease, which does not occur in wild-type mice (Fig. 7a). Beginning at ~20 weeks of age a majority of Scarf1−/− mice developed skin inflammation on their back, head, and face, often accompanied by hair and whisker loss. Female Scarf1−/− mice were more susceptible to skin disease than male mice. Cryosections of skin lesions from 20 week-old Scarf1−/− female mice showed cellular infiltration and the presence of apoptotic cells (Fig. 7b). In addition, we observed increased splenomegaly in >20 week old Scarf1−/− mice compared to wild-type age-matched littermates (Fig. 7c). Cryosection of spleens from Scarf1−/− mice demonstrated expanded germinal centers as compared to Scarf1+/+ mice (Fig. 7d). Scarf1−/− mice had significantly increased numbers of CD4+ T cells and B cells in the spleen (Fig. 7e). Increased spleen weight in Scarf1−/− mice was accompanied by accumulation of lymphocytes expressing an activated phenotype. Scarf1−/− mice had a significant increase in activated CD44+CD4+ T cells and CXCR5+Blimp−Bcl6+CD4+ follicular helper T cells (Fig. 7f–g). As a measure of global B cell activation, we determined the total amounts of serum IgG. Scarf1−/− mice had significantly higher concentrations of total serum IgG than Scarf1+/+ mice independent of gender (Fig. 7h). Together these data demonstrate that genetic absence of Scarf1 leads to spontaneous autoimmune disease activity and global immune cell activation.
Figure 7. Increased immune activation in Scarf1−/− mice.

a, Incidence and severity of skin disease in Scarf1−/− mice (analyzed mice: 47 total, 22 female and 25 male). b, Cryosections of skin from 20 week-old Scarf1+/+ and Scarf1−/− female mice were stained for H&E, for TUNEL (green) to detect apoptotic cells and nuclear stain DAPI (blue), and analyzed by fluorescent microscopy. TUNEL quantification was determined by analyzing images with a DAPI-TUNEL staining automated analysis macro for ImageJ software (8 mice per group). c, Average weights of harvested spleens from Scarf1+/+ and Scarf1−/− mice at 8 and 20 weeks of age. Bars denote the means and s.d. of measurements (n=8 mice per group) P<0.01 by Student’s t-test. d, H&E stained cryosections of spleens showing enlarged germinal centers in Scarf1−/− mice (6 mice per group). e, Absolute numbers of immune cells in the spleen of Scarf1+/+ and Scarf1−/− mice determined by surface marker staining and flow cytometry analysis (6 mice per group). f, g, Absolute number of CD44+CD4+ activated T cells and follicular helper CXCR5hiBlimp−Bcl6+CD4+ T cells in the spleens of Scarf1+/+ and Scarf1−/− mice (6 mice per group). h, Concentration of total serum IgG in 20-week old female and male Scarf1+/+ and Scarf1−/− mice. Data represent the average of three independent experiments performed in triplicate (12 mice per group). *P<0.05, **P<0.01, ***P<0.001, Student’s t-test.
Nephritis in Scarf1−/− mice
One of the important end-organ pathologies in lupus is the presence of lupus nephritis. In agreement with other evidence of increased disease activity, Scarf1−/− mice develop kidney disease. We observed increased glomerular size, cellularity, and PAS-positive protein deposition in Scarf1−/− kidney sections (Fig. 8a). One of the major clinical pathogenic outcomes of circulating autoantibodies in lupus is their deposition as immune complexes within the kidney, leading to glomerulonephritis39. Examination of kidney cryosections revealed high amounts of IgG immune complex deposition within the glomeruli of Scarf1−/− mice (Fig. 8b). Meanwhile, female Scarf1−/− mice had significantly higher incidence and severity of renal disease as compared to male mice (Fig. 8c). Consistent with kidney disease, we found by 24 hour quantitative urine collection that >90% of Scarf1−/− mice have at least moderate proteinuria (Fig. 8d). Moreover, a majority of female Scarf1−/− mice also had pyuria and hematuria (Fig. 8e–f). As another measure of kidney function, we also found that Scarf1−/− mice had significantly elevated blood urea nitrogen (BUN) as compared to controls, an indication of ongoing kidney disease in these mice (Fig. 8g). There was no statistical difference in the survival rate between male or female Scarf1−/− mice or between Scarf1+/+ mice out to one year, suggesting that other pathways can partially compensate for Scarf1 deficiency thereby allowing these mice to tolerate this chronic autoimmune disease.
Figure 8. Lupus nephritis in Scarf1−/− mice.

a, Cryosections of kidneys from 24 week-old Scarf1+/+ and Scarf1−/− female mice were fixed with 4% paraformaldehyde and stained with periodic acid-Schiff (PAS, 12 mice per group). b, Deposition of immune complexes in glomeruli. Cryosections of kidneys from 24 week-old Scarf1+/+ and Scarf1−/− female mice were stained with AlexaFluor 488-conjugated antibody to mouse IgG (green) and nuclear stain DAPI (blue), and analyzed by fluorescent microscopy. c, Incidence and severity of renal disease in Scarf1−/− mice (24 mice per group). d, e, f, Scarf1+/+ and Scarf1−/− mice were placed in a metabolic chamber for 24 hours and the urine collected was analyzed for proteinuria, percentage of blood, and leukocytes in the urine of mice were determined by Roche Chemstrip 5OB for urinalysis (12 mice per group). g, Blood urea nitrogen levels in Scarf1+/+ and Scarf1−/− mice. Data represent average of three independent experiments (n=10 mice), error bars denote the means and s.d. of at least triplicate measurements. None detected (ND). *P<0.01, ** P<0.001; Student’s t test.
DISCUSSION
The data presented in this manuscript fill several major gaps in our understanding of apoptotic cell clearance and regulation of autoimmunity. Here we show the first experimental evidence that SCARF1 is a receptor that is critical for recognition and capture of apoptotic cells via binding to C1q that is adhered to newly exposed PS on the apoptotic cell surface, a process that is necessary to prevent lupus-like disease. This conclusion is based on the following observations: 1) SCARF1 recognizes and engulfs apoptotic cells at early stages of apoptosis before the loss of membrane integrity, a process that is critical for maintaining immune tolerance. 2) SCARF1 binding and uptake of apoptotic cells requires C1q and PS exposure on dying cells. 3) Dot blots, ELISAs, and SPR analyses show that SCARF1 recognizes C1q specifically and avidly (KD = 1.24 × 10−7 M). 4) SCARF1-deficient mice accumulate apoptotic and necrotic cells in their blood and tissues. 5) SCARF1-deficient mice spontaneously develop clinical manifestations of lupus disease, including generation of autoantibodies against chromatin, immune cell activation, skin inflammation, and kidney disease exemplified by glomerular immune complex deposition with proteinuria. Together these data demonstrate that SCARF1 plays a significant role in apoptotic cell removal in vitro and that its failure in vivo results in spontaneous development of autoimmune disease.
The pathways that mediate engulfment of dying cells are conserved across evolution. In the worm two partially redundant pathways exist for corpse removal, with ced-1, ced-6, and ced-7 genes functioning in one pathway and ced-2, ced-5, ced-10, and ced-12 genes functioning in the other40. In Drosophila, dead cells are engulfed by macrophages expressing Draper, a receptor that is structurally and functionally similar to CED-141,42. In mammals five proteins, MEGF10, MEGF11, Jedi-1, SCARF1, and CD91 have been identified that share homology to CED-1 and DRAPER29,43. Recently, expression of MEGF10 or Jedi-1 by glia cells has been shown to confer the ability to bind and engulf apoptotic neurons in vitro44,45. However, a MEGF10 transgene failed to rescue the engulfment defect in CED-1 deficient worms, the ligand(s) on apoptotic cells recognized by MEGF10 and Jedi-1 are not known, and knockout mice for these receptors to confirm these findings have yet to be described. The interaction of CD91 (also called LRP1) with apoptotic cells has been shown to occur via binding to Calr that is adhered to the apoptotic cell surface by C1q and/or PS46. Loss of CD91 impairs the uptake of apoptotic cells in vitro, but its role in vivo has not been examined since CD91-knockout mice die early during gestation. We previously demonstrated that CED-1 and SCARF1 share an evolutionarily-conserved function in the recognition and host defense to pathogenic fungi27. Here we show that SCARF1 also shares with CED-1, an evolutionarily-conserved function in the capture of apoptotic cells. Furthermore, SCARF1 is the first C1q receptor identified that has been shown to be necessary for removal of apoptotic cells in vivo. Moreover, the susceptibility and severity of spontaneous autoimmune disease that develops in SCARF1-deficient mice is remarkably similar to what occurs in C1q-deficient mice47.
Many receptors and soluble bridging molecules have been implicated in the clearance of apoptotic cells, and mice with a deficiency in C1q, MFG-E8, TIM4, or c-mer develop spontaneous autoimmune disease depending on their genetic background10,18,47–49. CD91 and Calr have also been shown to be involved in apoptotic cell uptake in vitro, however their role in vivo remains unclear as mice deficient in Calr or CD91 are embryonic lethal, suggesting these pathways may play an important role during development. Our in vitro and in vivo data demonstrate that the SCARF1 pathway for apoptotic cell clearance is not completely non-redundant, but contributes ~40–70% to the capture of apoptotic cells depending on the phagocytic cell type. These data indicate that other apoptotic cell clearance pathways, such as PS-MFG-E8-αvβ3/β5, PS-Calr-CD91, and/or PS-TIM-3/4 likely contribute to the other 30–60%. Indeed, previously published studies indicate that macrophages deficient in MFG-E8, Calr, or C1q have a ~75%, ~60%, or ~50% reduction in the capture of apoptotic cells, respectively10,16,18,46. This growing body of evidence, including our findings described herein, suggests that all of these receptors partially contribute to the clearance of apoptotic cells, which may represent a novel collective paradigm to explain apoptotic cell clearance in vivo that has not been clearly recognized prior to our current work. This hypothesis has been recently tested by showing that double-deficiency of TIM4 and MFG-E8 has a more severe phenotype than single deficiencies50. Similarly, we would hypothesize that double-deficiency of SCARF1 and MFG-E8 or SCARF1 and TIM4 would have a more severe phenotype than the single deficiencies alone. To fully understand the relative contribution of each molecule to apoptotic cell clearance it will be necessary in future studies to generate such double-knockout mice.
Together, these data indicate that the immune system has developed a failsafe mechanism involving several receptors and bridging proteins expressed by different cell types and in specific organs for the removal of dying cells to maintain tolerance and prevent autoimmunity. Identifying the primary molecules and receptors, and how they function to internalize and process apoptotic debris could lead to therapeutic exploitation, not only to remove unwanted cells, but as an immunosuppressive or anti-inflammatory strategy. As in human patients, the lupus phenotype observed in SCARF1-deficient mice is more prevalent and severe in females, indicating that SCARF1-deficient mice may be useful as a new spontaneous model system for the study of human lupus that can be utilized to provide a better understanding of apoptotic cell clearance mechanisms in healthy and diseased states in order to reveal targets for new therapeutic strategies.
METHODS
Reagents
Reagents were from Sigma-Aldrich unless otherwise stated. Complete media consisted of RPMI-1640 (Invitrogen) or DMEM (Invitrogen) supplemented with 100U/ml penicillin, 100U/ml streptomycin, 2mM L-glutamine, and 10% fetal calf serum (FCS). HEK293T cell line (Cat#: HCL4517) was obtained from Fisher Scientific and mouse embryonic fibroblasts (MEF, Cat#: SCRC-1008) were obtained from ATTC. All cell lines were determined to be free of mycoplasma as determined by a Mycoplasma detection PCR Kit (Sigma-Aldrich). Human and mouse anti-SCARF1 antibodies were purchased from R&D Systems (Monoclonal Mouse IgG2B Clone# 373606) and Proteintech (Rabbit polyclonal, Cat#: 13702-1-AP), respectively. All other antibodies were from eBioscience, unless otherwise specified.
Mice
All mice were maintained under microisolation in specific pathogen-free conditions at the Massachusetts General Hospital animal facility under a protocol approved by the Institutional Animal Care and Use Committee. Wild-type C57Bl/6 mice were purchased from The Jackson Laboratories. To generate Scarf1−/− mice, we constructed a targeting vector that contained 4.42-kb of 5′ sequence upstream of the transcriptional start site, a neomycin resistance cassette replacing exons 1-8 (which encode the entire extracellular and transmembrane domains), and 1.85-kb of 3′ sequence spanning exons 9-10. The primers used to amplify the 5′ long arm and the 3′ short arm were: 5Arm Fw1 5′-CCCAGTCCTCCTGGAACTACAACTT-3′; 5Arm Rv1 5′-CCACAGGTCCCTGAAACACTAGAGA-3′; 3Arm Fw1 5′-AAGAAAAGTGAAATCAGAGCCGACA-3′; 3Arm Rv1 5′-CCAAAACACCATTACTACTGCCGAA-3′. These genomic regions were ligated into the targeting vector OSDupDel.Neo from Open Biosystems. The linearized vector was electroporated into C57BL/6 ES cells by inGenious Targeting Laboratory, and targeted clones that were selected in the presence of the antibiotic neomycin were identified by PCR and Southern blot analysis. Targeted ES clones were injected into C57BL/6 mice blastocysts, yielding several lines of chimeric mice that transmitted the disrupted allele through the germline DNA. The Scarf1−/− mice bred at normal Mendelian ratios. Mice were not randomized or placed in specific groups for these studies.
SCARF1 Genotype and Southern Blot Analysis
Homologous recombination in electroporated ES cell clones or genomic tail DNA was determined by PCR and Southern blot. PCR primer pairs: WT Fw1 5′-AGCCTGGAACTGCATGTCTT-3′; WT Rv1 5′-TGAATGCCTTACAGCACAGC-3′; KO Fw1 5′-CTTCTATCGCCTTCTTGACGAGT-3′; KO Rv1 5′-TAGATACCTCTCCTTCGCTCT-3′. Southern Blot probe (326 bp) primer pairs were: South Fw1 5′-CAAGATGTCACCACTCATGCCAAAA-3′; South Rv1 5′-ACAGGAGCATCTGAACCCTCTCGTA-3′. For Southern blot screening, gDNA was digested with BamHI before gel electrophoresis. The DNA was transferred to nitrocellulose and incubated with radio-labeled probe. The digest pattern was wild-type band at 5110 bp and gene-targeted band at 6122 bp.
Generation of apoptotic cells
Mouse primary B cells were isolated by negative selection from the spleen using EasySep Mouse B Cell enrichment kit (STEMCELL Technology) following the manufacturer’s instructions. Apoptotic cells from primary B cells, HEK293T, and MEFs were generated following the same method described below. For all assays, unless otherwise specified, apoptotic cells were labeled with CFSE or pHrodo (Invitrogen).
For generation of apoptotic cells by osmotic shock, cells were first centrifuged at 800xg for 5 minutes. Cells were then washed once with PBS and resuspended in hypertonic media (DMEM supplemented with 10% w/v polyethylene glycol 1000, 0.5M Sucrose, 10 mM Hepes) for 10 minutes. Immediately afterwards, 30 mL of hypotonic media (60% DMEM, 40% water) was added and incubated for 5 minutes. Cells were then washed, resuspended in DMEM-C, and cultured at 37C for 3 hours. Generation of apoptotic cells by UV radiation was performed by placing a flask of cells into a UV transilluminator set to 100 Joules/m2, then cultured for 3 hours as above. Generation of apoptotic cells by Cesium-137 irradiation was performed by exposing cells to 3000-rads. Apoptotic cells were generated from Mfge8−/− mice (kind gift from YuFeng Peng, PhD; University of Washington), K41 (Calr+/+) and K42 (Calr−/−) MEFs (kind gift from Marek Michalak, PhD; University of Alberta). Cells were used when a majority of cells was apoptotic as defined by Annexin V+/PI− staining by flow cytometry.
Generation of Liposomes
Small unilamellar vesicles (liposomes) were prepared as previously described9. Briefly, pure phospholipids were sonicated and specific phospholipids were prepared in sodium phosphate buffer by extrusion through a 0.1 μm polycarbonate filter using a mini extruder set (Avanti polar lipids) at 37C. Liposomes were composed of 100% mol PC or a mixture of 70% mol PC/30% mol PS.
Dot blot and Binding Assays
Membrane lipid strips (Echelon Bioscience, UT) were spotted with 100 pmol of ten membrane lipids. Membranes were blocked for 1 hour with 3% BSA in TBS-T (50 mM Tris, 0.5 M NaCl, 0.05% Tween-20, pH 7.4). Following blocking, membranes were incubated with soluble SCARF1 protein for 1 hour, washed and incubated with anti-SCARF1, followed by an HRP-labeled secondary antibody for 1 hour. Binding was detected by chemiluminescent detection. ELISA was used to validate the interaction of SCARF1 with other apoptotic cell markers. PVC microtiter plates were coated with acetylated LDL, C1q, BSA, PS, PC or Calr overnight at 4C. Plates were washed, blocked and incubated with SCARF1 protein, followed by primary and HRP-conjugated secondary antibodies. The optical density (OD) at 450 nm was measured using a Molecular Devices plate reader and Softmax Pro software. Biacore binding assays were performed following the manufacturer’s instructions. Briefly, C1q, PS and Calr were immobilized onto a CM5 sensor chip and binding affinity of SCARF1 was measured using Biacore 3000 instrument and software (GE Healthcare).
Nucleofection
Endothelial cells were transfected using Nucleofector primary mammalian endothelial cell kit, and DCs and macrophages using the Nucleofector kit V according to the manufacturer’s instructions (Lonza). Briefly, 2 × 106 primary CD8α+ DCs or macrophages or 5 × 105 endothelial cells isolated from Scarf1+/+ and Scarf1−/− mice were transfected using a Nucleofector II with nucleofector program D-32 or M-002 using 2 μg of mammalian expression vectors encoding GFP-C-terminal tagged mouse SCARF1 or GFP alone (pMAX-GFP, Amaxa). Experiments were performed 24 hours post-nucleofection at which time the transfection efficiency was determined to be over 50% and cell viability over 80% as determined by GFP and AnnexinV/PI staining, respectively. For flow cytometric analysis, GFP was pre-gated to analyze the % of GFP and SCARF1-GFP expressing cells that captured red dye-labeled apoptotic cells.
Real-Time quantitative PCR
Total RNA was extracted using the RNeasy kit and DNase treated according to the manufacturer’s protocol (Qiagen), and each sample was reverse transcribed using multiscribe reverse transcriptase (Applied Biosystems). The 25-μl qPCR reaction contained 2 μl of cDNA, 12.5 μl of 2× SYBR green master mix (Applied Biosystems), and 500 nM of sense and antisense primers. Oligonucleotide primer sequences designed on the PrimerBank website obtained from Integrated DNA Technologies were as follows: human GAPDH, 5′-GAAGGTGAAGGTCGGAGTC-3′ and 5′-GAAGATGGTGATGGGATTTC-3′; human IL8, 5′-CTGGCCGTGGCTCTCTTG-3′ and 5′-CCTTGGCAAAACTGCACCTT-3′; mouse B2M, 5′-TTCTGGTGCTTGTCTCACTGA -3′ and 5′-CAGTATGTTCGGCTTCCCATTC-3′; LOX-1, 5′-AAGCGAACCTTACTCAGCAGG-3′ and 5′-TGGATTTCTCATTCAGCTTCTGG; MARCO, 5′-CTGTGGCAATGGATCACTAGC-3′ and 5′-CTCCTGGCTGGTATGGACC-3′; SR-A1, 5′-TGAACGAGAGGATGCTGACTG-3′ and 5′-GGAGGGGCCATTTTTAGTGC-3′; CD14, 5′-GCACACTCACTCAACTTTTCCT-3′ and 5′-GCTGAGATCAGTCCTCTCTCG-3′; SCARF1, 5′-AGCCTGGAACTGCATGTCTT-3′ and 5′-TGAATGCCTTACAGCACAGC-3′; TIM3, 5′-TCAGGTCTTACCCTCAACTGTG-3′ and 5′-GGGCAGATAGGCATTTTTACCA-3′; TIM4, 5′-GTGTACTGCTGCCGTATAGAGG-3′ and 5′-TGGTGGTTGGGAGAACAGATG-3′; TIM1, 5′-ACATATCGTGGAATCACAACGAC-3′ and 5′-ACAAGCAGAAGATGGGCATTG-3′; ITGAV, 5′-CCGTGGACTTCTTCGAGCC-3′ and 5′-CTGTTGAATCAAACTCAATGGGC-3′; MFGE8, 5′-AGATGCGGGTATCAGGTGTGA-3′ and 5′-GGGGCTCAGAACATCCGTG-3′; CD91, 5′-ACTATGGATGCCCCTAAAACTTG-3′ and 5′-GCAATCTCTTTCACCGTCACA-3′; GAS6, 5′-TGCTGGCTTCCGAGTCTTC-3′ and 5′-CGGGGTCGTTCTCGAACAC-3′; c-mer, 5′-ACCCAGTTGCTAGAGAGCTG-3′ and 5′-TGGTGAGTCTGTCTCCGGTAA-3′; C1q, 5′-AAAGGCAATCCAGGCAATATCA-3′ and 5′-TGGTTCTGGTATGGACTCTCC-3′.
Emitted fluorescence for each reaction was measured three times during the annealing-extension phase, and amplification plots were analyzed with MX4000 software, version 3.0 (Stratagene). The quantity of gene expression was generated by comparison of the fluorescence generated by each sample with standard curves of known quantities, and the calculated number of copies was divided by the number of copies of the housekeeping genes Gapdh or B2m (encoding β2-microglobulin).
Apoptotic cell uptake in vitro and in vivo
DCs, macrophages, and neutrophils were isolated from Scarf1+/+ and Scarf1−/− mice using EasySep magnetic isolation kits following the manufacturer’s instructions (STEMCELL Technologies). These cells were incubated with CFSE- or pHrodo-labeled apoptotic cells for 0.5, 1, or 2 hours at 37°C. Cells were stained with fluorescently-labeled antibodies to CD11c, Ly6G, F4/80, CD11b, and CD8a and analyzed by flow cytometry. For in vivo apoptotic cell uptake assays, dye-labeled apoptotic B cells or MEFs were injected intravenously into Scarf1+/+ and Scarf1−/− mice. After 1 hour, the spleen was harvested and the CD11c DC population isolated by negative selection using EasySep kit. The enriched DC population was stained with fluorescently labeled antibodies against CD11c and CD8α. Splenocytes that phagocytosed pHrodo-labeled MEFs exhibited a change in florescence from 488 nm (FITC) to 550 nm (PE). Samples were analyzed using Becton Dickinson FACSCalibur, data was collected using CellQuest Software and analyzed using FlowJo 9.5 version for Mac.
Autoantibody Profiles
ANA and Crithidia luciliae immunofluorescence assays (Bio-Rad) were performed according to the manufacturer’s instructions using mouse serum at 1:160–1:5120 dilutions and 1:50–1:200 dilutions for C. luciliae, respectively. Staining was scored by an observer blinded to the genotype of the mice. For nucleosome, smRNP, and La ELISAs, polystyrene plates were coated with either dsDNA and histones, smRNP complex antigens, or La, respectively (Immunovision). After blocking with 1% BSA in PBS, serial dilutions of serum from 1:160 to 1:5120 were added. Specific antibodies were detected with HRP-conjugated goat anti-mouse IgG and absorbance at 405/630 nm was compared with monoclonal antibodies to quantitate.
TUNEL Assay
Frozen sections were allowed to warm to room temperature. TUNEL Assay (Invitrogen) for detection of apoptotic cells was performed following manufacturer’s instructions. Paraformaldehyde-fixed tissue was permeabilized with PBS 0.25% Triton X-100 and washed twice with PBS. Slides were incubated with TdT reaction cocktail, washed twice, and incubated with the reaction cocktail containing AlexaFluor-488 florescent label for 45 minutes at RT and protected from light. Actin filaments of the cells were stained with 1:500 dilution of Phalloidan Alexa 546 antibody (Molecular Probes, Invitrogen) in PBS for 10 minutes at RT. Samples were washed twice with PBS. Finally, DNA was stained with 1:1000 dilution of Hoechst 33342 (Molecular Probes, Invitrogen) for 10 minutes at RT. Slides were mounted using Vectamount mounting media (Vector Laboratories) and visualized using a Nikon Eclipse ME600 fluorescent microscope equipped with a high resolution DXM1200C Nikon digital camera. Data were analyzed using NIS-Elements software (Nikon) and Adobe Photoshop.
Immune Complex Imaging Assay
Frozen sections were allowed to warm to room temperature and then fixed with 4% paraformaldehyde. Slides were washed twice with PBS at RT; each wash was performed for 5 minutes to allow tissue rehydration. After this step, slides were blocked with PBS supplemented with 10% Goat Serum (Invitrogen) for 45 minutes at room temperature. PBS washed slides were stained with 1μL per slide of Alexa Fluor 488 goat anti-mouse IgG (Invitrogen) for 20 minutes at RT and protected from light. Following staining, slides were washed twice, mounted with Vectashield containing DAPI and visualized by fluorescent microscopy.
Lentivirus production and infection
Plasmids encoding lentiviruses expressing shRNA molecules were obtained from The RNAi Consortium shRNA Library (Broad Institute). The shRNA target 21-mer sequences were: shControl, CCTAAGGTTAAGTCGCCCTCG; shC1q #1, CGGCTTCTATTACTTCAACTT; shC1q #2, CCTGAGTTTCTCTAACACCAA; shC1q #3, CGACAGCATCTTCAGCGGATT; shC1q #4, GCTTGGCAACGTGGTTATCTT. Plasmids were purified with a QIAprep Miniprep kit (Qiagen) and then transfected into HEK293T cells along with pCMV-dR8.2 dvpr and pCMV-VSVG for the production of lentivirus. MEFs were placed in 24-well tissue culture dishes (2 × 105 cells per well) and infected with 100 μl unconcentrated shRNA lentiviral supernatant and polybrene (7.5 μg/ml). Cells were spun for 30 min at 800g, media replaced, and incubated for 2 days. Infected cells were selected in complete RPMI medium containing 10% (vol/vol) FBS and puromycin (3 μg/ml) and were tested 1 week after infection. The shRNA knockdown efficiency was determined by qPCR.
Histological assessment and clinical disease score
Histopathologic examination of skin, spleen and kidney samples was done after routine fixation and paraffin embedding of the tissues. Tissue sections from the skin were cut and stained with hematoxylin and eosin. All slides were coded and evaluated in a blinded manner with regard to identity of the sample. Severity of skin inflammation was scored on a scale of 0–4, where 0 = normal, 1 = hyperplasia of the epidermis, and 2–4 = increasing numbers of infiltrating inflammatory cells in the skin. Mice were also scored for the extent of lesions on the face, ears, head, neck and back. Macroscopic surface area was scored on a scale from 0–4, where 0 = normal, 1 = mild lesion, and 2–4 = increasing severity of lesion and hair/whisker loss of the affected area up to 2 cm2. A composite score of skin disease was generated from the microscopic and macroscopic analysis. Pathologic changes in the kidney were graded according to the presence of glomerular, interstitial, perivascular inflammation and immune complex deposition. Scores ranging from 0 (normal) to 4 (most severely inflamed) were assigned for each of the 4 features. A minimum of 50 glomeruli were assessed to determine the glomerular index in each mouse.
Assessment of urine
Mice in each group were placed for 24 hours in sterilized Nalgene metabolic cages to collect urine. Urine was measured with Roche Chemstrip 5OB for urinalysis. Proteinuria was graded on a scale of 0–4, where 0 = none, 1 = 30–100 mg/dL, 2 = 100–300 mg/dL, 3 = 300–2,000 mg/dL, and 4 = >2,000 mg/dL.
Statistical Analysis
Statistical calculations were performed using a statistical software package (GraphPad Prism 5.0d). For comparisons of two groups, means ± SE were analyzed by the two-tailed unpaired Student’s t-test with the Bonferroni correction applied when making multiple comparisons. For comparisons of greater than two groups, significance was determining using the one- or two-way analysis of variance (ANOVA) with correction. The investigators were not blinded to the genotype of the mice unless otherwise indicated. Samples sizes were selected based on preliminary results to ensure a power of 80% with 95% confidence between populations.
Supplementary Material
Acknowledgments
This work was supported by grants from NIAID R01-AI084884, NIAMS K01-AR051367, Lupus Research Institute, and Alliance for Lupus Research. J.E.K. was supported by NIAID grant U24 AI082660. Z.G.R was supported by NIAID training grant T32-AI007061. W.F.P. III was supported by an American Society of Nephrology Research Fellowship Grant and NIDDK F32-DK097891. We thank Drs. Mark J. Shlomchik and Peter Mundel and members of their laboratories for technical assistance and stimulating discussions.
Footnotes
Note: Supplementary information is available on the Nature Immunology website.
AUTHORS CONTRIBUTIONS
T.K.M., Z.G.R., and W.F.P. III planned the research, analyzed and interpreted data and wrote the manuscript. Z.G.R. performed most of the experiments. C.J.B helped with mouse breeding and genotyping. W.F.P. III, A.P., and T.I. performed and analyzed ELISAs, PCR, and mouse pathology. N.H. and A.D.L. analyzed and interpreted data. T.K.M., J.E.K., and M.H.B. contributed to the generation of SCARF1-deficient mice. All authors participated in editing the manuscript into its final form.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Elliott MR, Ravichandran KS. Clearance of apoptotic cells: implications in health and disease. J Cell Biol. 2010;189:1059–1070. doi: 10.1083/jcb.201004096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Devitt A, Marshall LJ. The innate immune system and the clearance of apoptotic cells. J Leukoc Biol. 2011;90:447–457. doi: 10.1189/jlb.0211095. [DOI] [PubMed] [Google Scholar]
- 3.Lauber K, Blumenthal SG, Waibel M, Wesselborg S. Clearance of apoptotic cells: getting rid of the corpses. Mol Cell. 2004;14:277–287. doi: 10.1016/s1097-2765(04)00237-0. [DOI] [PubMed] [Google Scholar]
- 4.Erwig LP, Henson PM. Clearance of apoptotic cells by phagocytes. Cell death and differentiation. 2008;15:243–250. doi: 10.1038/sj.cdd.4402184. [DOI] [PubMed] [Google Scholar]
- 5.Munoz LE, et al. Apoptosis in the pathogenesis of systemic lupus erythematosus. Lupus. 2008;17:371–375. doi: 10.1177/0961203308089990. [DOI] [PubMed] [Google Scholar]
- 6.Ravichandran KS, Lorenz U. Engulfment of apoptotic cells: signals for a good meal. Nature reviews. Immunology. 2007;7:964–974. doi: 10.1038/nri2214. [DOI] [PubMed] [Google Scholar]
- 7.Nagata S, Hanayama R, Kawane K. Autoimmunity and the clearance of dead cells. Cell. 2010;140:619–630. doi: 10.1016/j.cell.2010.02.014. [DOI] [PubMed] [Google Scholar]
- 8.Shao WH, Cohen PL. Disturbances of apoptotic cell clearance in systemic lupus erythematosus. Arthritis Res Ther. 2011;13:202. doi: 10.1186/ar3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fadok VA, et al. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol. 1992;148:2207–2216. [PubMed] [Google Scholar]
- 10.Kobayashi N, et al. TIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity. 2007;27:927–940. doi: 10.1016/j.immuni.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park D, et al. BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature. 2007;450:430–434. doi: 10.1038/nature06329. [DOI] [PubMed] [Google Scholar]
- 12.Hanayama R, et al. Identification of a factor that links apoptotic cells to phagocytes. Nature. 2002;417:182–187. doi: 10.1038/417182a. [DOI] [PubMed] [Google Scholar]
- 13.Paidassi H, et al. C1q binds phosphatidylserine and likely acts as a multiligand-bridging molecule in apoptotic cell recognition. Journal of immunology. 2008;180:2329–2338. doi: 10.4049/jimmunol.180.4.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paidassi H, et al. Investigations on the C1q-calreticulin-phosphatidylserine interactions yield new insights into apoptotic cell recognition. Journal of molecular biology. 2011;408:277–290. doi: 10.1016/j.jmb.2011.02.029. [DOI] [PubMed] [Google Scholar]
- 15.Galvan MD, Greenlee-Wacker MC, Bohlson SS. C1q and phagocytosis: the perfect complement to a good meal. J Leukoc Biol. 2012;92:489–497. doi: 10.1189/jlb.0212099. [DOI] [PubMed] [Google Scholar]
- 16.Gardai SJ, et al. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell. 2005;123:321–334. doi: 10.1016/j.cell.2005.08.032. [DOI] [PubMed] [Google Scholar]
- 17.Manderson AP, Botto M, Walport MJ. The role of complement in the development of systemic lupus erythematosus. Annual review of immunology. 2004;22:431–456. doi: 10.1146/annurev.immunol.22.012703.104549. [DOI] [PubMed] [Google Scholar]
- 18.Hanayama R, et al. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science. 2004;304:1147–1150. doi: 10.1126/science.1094359. [DOI] [PubMed] [Google Scholar]
- 19.Taylor PR, et al. A hierarchical role for classical pathway complement proteins in the clearance of apoptotic cells in vivo. The Journal of experimental medicine. 2000;192:359–366. doi: 10.1084/jem.192.3.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mukhopadhyay S, Pluddemann A, Gordon S. Macrophage pattern recognition receptors in immunity, homeostasis and self tolerance. Advances in experimental medicine and biology. 2009;653:1–14. doi: 10.1007/978-1-4419-0901-5_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adachi H, Tsujimoto M, Arai H, Inoue K. Expression cloning of a novel scavenger receptor from human endothelial cells. J Biol Chem. 1997;272:31217–31220. doi: 10.1074/jbc.272.50.31217. [DOI] [PubMed] [Google Scholar]
- 22.Tamura Y, et al. Scavenger receptor expressed by endothelial cells I (SREC-I) mediates the uptake of acetylated low density lipoproteins by macrophages stimulated with lipopolysaccharide. J Biol Chem. 2004;279:30938–30944. doi: 10.1074/jbc.M313088200. [DOI] [PubMed] [Google Scholar]
- 23.Berwin B, Delneste Y, Lovingood RV, Post SR, Pizzo SV. SREC-I, a type F scavenger receptor, is an endocytic receptor for calreticulin. The Journal of biological chemistry. 2004;279:51250–51257. doi: 10.1074/jbc.M406202200. [DOI] [PubMed] [Google Scholar]
- 24.Jeannin P, et al. Complexity and complementarity of outer membrane protein A recognition by cellular and humoral innate immunity receptors. Immunity. 2005;22:551–560. doi: 10.1016/j.immuni.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 25.Holzl MA, et al. The zymogen granule protein 2 (GP2) binds to scavenger receptor expressed on endothelial cells I (SREC-I) Cellular immunology. 2011;267:88–93. doi: 10.1016/j.cellimm.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murshid A, Gong J, Calderwood SK. Heat shock protein 90 mediates efficient antigen cross presentation through the scavenger receptor expressed by endothelial cells-I. J Immunol. 2010;185:2903–2917. doi: 10.4049/jimmunol.0903635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Means TK, et al. Evolutionarily conserved recognition and innate immunity to fungal pathogens by the scavenger receptors SCARF1 and CD36. The Journal of experimental medicine. 2009;206:637–653. doi: 10.1084/jem.20082109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rechner C, Kuhlewein C, Muller A, Schild H, Rudel T. Host glycoprotein Gp96 and scavenger receptor SREC interact with PorB of disseminating Neisseria gonorrhoeae in an epithelial invasion pathway. Cell host & microbe. 2007;2:393–403. doi: 10.1016/j.chom.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 29.Zhou Z, Hartwieg E, Horvitz HR. CED-1 is a transmembrane receptor that mediates cell corpse engulfment in C. elegans. Cell. 2001;104:43–56. doi: 10.1016/s0092-8674(01)00190-8. [DOI] [PubMed] [Google Scholar]
- 30.Means TK. Fungal pathogen recognition by scavenger receptors in nematodes and mammals. Virulence. 2010;1:37–41. doi: 10.4161/viru.1.1.10228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ishii J, et al. SREC-II, a new member of the scavenger receptor type F family, trans-interacts with SREC-I through its extracellular domain. J Biol Chem. 2002;277:39696–39702. doi: 10.1074/jbc.M206140200. [DOI] [PubMed] [Google Scholar]
- 32.Yoshiizumi K, Nakajima F, Dobashi R, Nishimura N, Ikeda S. Studies on scavenger receptor inhibitors. Part 1: synthesis and structure-activity relationships of novel derivatives of sulfatides. Bioorganic & medicinal chemistry. 2002;10:2445–2460. doi: 10.1016/s0968-0896(02)00120-7. [DOI] [PubMed] [Google Scholar]
- 33.Iyoda T, et al. The CD8+ dendritic cell subset selectively endocytoses dying cells in culture and in vivo. J Exp Med. 2002;195:1289–1302. doi: 10.1084/jem.20020161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tan EM, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25:1271–1277. doi: 10.1002/art.1780251101. [DOI] [PubMed] [Google Scholar]
- 35.Tsokos GC. Systemic lupus erythematosus. N Engl J Med. 2011;365:2110–2121. doi: 10.1056/NEJMra1100359. [DOI] [PubMed] [Google Scholar]
- 36.Casciola-Rosen LA, Anhalt G, Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med. 1994;179:1317–1330. doi: 10.1084/jem.179.4.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zandman-Goddard G, Peeva E, Shoenfeld Y. Gender and autoimmunity. Autoimmun Rev. 2007;6:366–372. doi: 10.1016/j.autrev.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 38.Xu Y, et al. Pleiotropic IFN-dependent and -independent effects of IRF5 on the pathogenesis of experimental lupus. Journal of immunology. 2012;188:4113–4121. doi: 10.4049/jimmunol.1103113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Winfield JB, Faiferman I, Koffler D. Avidity of anti-DNA antibodies in serum and IgG glomerular eluates from patients with systemic lupus erythematosus. Association of high avidity antinative DNA antibody with glomerulonephritis. J Clin Invest. 1977;59:90–96. doi: 10.1172/JCI108626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reddien PW, Cameron S, Horvitz HR. Phagocytosis promotes programmed cell death in C. elegans. Nature. 2001;412:198–202. doi: 10.1038/35084096. [DOI] [PubMed] [Google Scholar]
- 41.Awasaki T, et al. Essential role of the apoptotic cell engulfment genes draper and ced-6 in programmed axon pruning during Drosophila metamorphosis. Neuron. 2006;50:855–867. doi: 10.1016/j.neuron.2006.04.027. [DOI] [PubMed] [Google Scholar]
- 42.Hamon Y, et al. Cooperation between engulfment receptors: the case of ABCA1 and MEGF10. PloS one. 2006;1:e120. doi: 10.1371/journal.pone.0000120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Su HP, et al. Interaction of CED-6/GULP, an adapter protein involved in engulfment of apoptotic cells with CED-1 and CD91/low density lipoprotein receptor-related protein (LRP) The Journal of biological chemistry. 2002;277:11772–11779. doi: 10.1074/jbc.M109336200. [DOI] [PubMed] [Google Scholar]
- 44.Wu HH, et al. Glial precursors clear sensory neuron corpses during development via Jedi-1, an engulfment receptor. Nature neuroscience. 2009;12:1534–1541. doi: 10.1038/nn.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scheib JL, Sullivan CS, Carter BD. Jedi-1 and MEGF10 signal engulfment of apoptotic neurons through the tyrosine kinase Syk. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2012;32:13022–13031. doi: 10.1523/JNEUROSCI.6350-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ogden CA, et al. C1q and mannose binding lectin engagement of cell surface calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. The Journal of experimental medicine. 2001;194:781–795. doi: 10.1084/jem.194.6.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Botto M, et al. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat Genet. 1998;19:56–59. doi: 10.1038/ng0598-56. [DOI] [PubMed] [Google Scholar]
- 48.Wong K, et al. Phosphatidylserine receptor Tim-4 is essential for the maintenance of the homeostatic state of resident peritoneal macrophages. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:8712–8717. doi: 10.1073/pnas.0910929107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cohen PL, et al. Delayed apoptotic cell clearance and lupus-like autoimmunity in mice lacking the c-mer membrane tyrosine kinase. The Journal of experimental medicine. 2002;196:135–140. doi: 10.1084/jem.20012094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miyanishi M, Segawa K, Nagata S. Synergistic effect of Tim4 and MFG-E8 null mutations on the development of autoimmunity. International immunology. 2012;24:551–559. doi: 10.1093/intimm/dxs064. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.