

Abstract
We evaluated the sensitivity and specificity of a nested-polymerase chain reaction (PCR) method for detection of Mycobacterium leprae DNA from whole blood. Whole-blood specimens were subjected to nested-PCR amplification of M. leprae repeat DNA sequences in 49 multibacillary (MB) and 30 paucibacillary (PB) leprosy patients, 96 household contacts (HHCs), 18 tuberculosis (TB) patients, and 35 normal healthy individuals. M. leprae DNA was detected in 95.92% (47/49) of MB, 70% (21/30) of PB, and 6.25% (6/96) of HHC, but it was not detected in 18 TB or 35 normal controls. The sensitivities of the anti-bovine serum albumin (ND-O-BSA) immunoglobulin M (IgM) and antifusion protein of ML0405-ML2331 IgG for MB were 97.96% and 89.8%, and these values for PB were 70% and 53.33%. However, the ND-O-BSA enzyme-linked immunosorbent assay (ELISA) had lower specificity, with relatively high false-positive results for TB patients (16.67%) and normal healthy controls (10%). Based on these promising findings, we propose the use of nested PCR of whole-blood samples along with ELISA test for early detection of leprosy cases.
Introduction
Leprosy is an infectious disease caused by Mycobacterium leprae, which was identified by A. Hansen in 1873. It has a long incubation period and a wide spectrum of clinical manifestations, which make it difficult to diagnose in the early stages of the disease. The leprosy bacillus has not been successfully cultured in vitro to date. Recently, various serological and cell-mediated immune methods have been developed, including a serological enzyme-linked immunosorbent assay (ELISA) test that detects antibodies against an epitope on phenolic glycolipid-1 (PGL-1) antigen, a natural disaccharide with octyl linkage conjugated to bovine serum albumin (ND-O-BSA), and a fusion protein of ML0405-ML2331 (LID-1) of M. leprae.1 These two antigens are specific to this bacillus, and the test can potentially lead to early diagnosis and predict patient outcome. The antibodies are found in the tissues and circulating blood of multibacillary (MB) patients. The test has been used worldwide,1,2 and positivity is proportional to the bacillary load, especially for MB patients. However, the potential drawback of the ELISA test is its cross-reactivity to other mycobacterial cell wall components and its low sensitivity for detection of paucibacillary disease.
DNA amplification using polymerase chain reaction (PCR) seems to be more specific and sensitive for detecting bacilli in biopsies.3 Woods and Cole4 detected small numbers of M. leprae by amplifying a specific repetitive sequence, whereas Plikaytis and others5 detected as little as 3 fg M. leprae genomic DNA using nested PCR targeting limited sequences of the groEL gene. Although PCR has already been applied to the detection of M. leprae for some years, it has been used mainly on biopsies and slit skin smears (SSSs) from suspected cases. Biopsies and SSSs are invasive procedures for the patient, and typical lesions may not always be seen (for example, in indeterminate leprosy, where anesthesia is not clear and early detection and proper multidrug therapy [MDT] is often missed).
To improve the sensitivity and specificity of the current detection methods for diagnosing leprosy at an early stage, we applied a nested-PCR approach to whole-blood specimens from different groups to detect M. leprae by amplifying specific repetitive DNA sequences5,6 and compared this method with the ELISA assays based on detection of antibodies to certain M. leprae antigens for diagnosis of leprosy.
Methods
Study populations.
This study was conducted at an endemic site in the Honghe Prefecture (detection rate at 5-year average, 2007–2011, of 1.08 per 100,000 populations) of Yunnan Province in southwest China. The newly diagnosed patients were assessed by clinical signs of skin lesions, nerve involvement, and bacteriological (bacterial index by acid-fast staining) and histopathological methods, and they were classified according to the Ridley–Jopling scale7 carried out by qualified personnel. Specimens included 49 MB (9 lepromatous leprosy [LL], 38 borderline lepromatous [BL], and 2 borderline borderline [BB]) and 30 paucibacillary (PB; 1 indeterminate [I], 3 paucibacillary tuberculoid [TT], and 26 borderline tuberculoid [BT]; 26 BT included 24 acid-fast bacilli [AFB] -positive and 2 AFB-negative). Household contacts (HHCs) were defined as individuals who lived in the same dwelling (i.e., sharing the same kitchen or social/recreational area). The HHC contacts were living with the leprosy patients during the treatment; 96 HHC were from MB patient families, 10 normal individuals were from the food handlers from Honghe Prefecture, Yunnan Province, and 25 normal individuals were from the medical staff, Beijing Friendship Hospital, during their annual physical checkup. The control including 18 treated tuberculosis patients. Whole-blood and plasma samples were collected after obtaining written informed consent as per the standards of the Ethical Committee of Beijing Friendship Hospital Institutional Committee, which approved this study.
Bacterial strains and antigens.
M. leprae strain NHDP63, M. leprae DNA, and semisynthetic antigen ND-O-BSA were obtained from CSU, Fort Collins, CO, and LID-1 was obtained from IDRI, Seattle, WA. Seventeen other Mycobacterium species (M. tuberculosis, M. lufu, M. avium, M. marinum, M. bovis BCG-Pasteur, M. chelonei, M. bovis [Ravenel], M. flavescens, M. smegmatis, M. gordonae, M. ulcerans, M. intracellulare, M. simiae, M. bovis [AFZ/ZZ/97], M. lepraemurium, M. kansasii, and M. phlei) and four other bacterial species (Streptococcus pyogenes, Clostridium perfringens, Escherichia coli, and Staphylococcus epidermidis) from NHDP, Baton Rouge, LA, were also included as controls for this study.
DNA extraction from whole blood.
Blood (3–5 mL) was drawn from individuals into a vacutainer containing anticoagulant (BD Vacutainer #367884; BD, Sparks, MD) and kept for 4 hours at room temperature. The upper plasma layer was collected (0.5 mL), and the whole blood was thoroughly mixed. All serum and whole-blood specimens were aliquoted and stored at −40°C before the assay. DNA was extracted and purified using a DNeasy Blood & Tissue Kit (catalog number 69504; QIAGEN Inc., Valencia, CA) according to the manufacturer's instructions.
Nested PCR.
Amplification of a 372-bp fragment of M. leprae-specific repetitive DNA sequence was performed as described4 using the outer set of primers. The primers used were sense 5′-GCACGTAAGCCTGTCGGTGG-3′ (ML1) and antisense 5′-CGGCCGGATCCTCGATGCAC-3′ (ML2). An inner nested set of primers was designed to amplify a 131-bp fragment. The inner primer sequences were 5′-GTGAGGGTAGTTGTT-3′ (LP1) and 5′-GGTGCGAATAGTT-3′ (LP2). PCR amplification of template DNA was carried out using a thermal cycler PTC 200 (MJ Research, Bio-Rad, Munich, Germany). Cycling parameters were as follows: initial denaturation at 95°C for 15 minutes followed by 40 cycles of denaturation at 94°C for 30 seconds, annealing at 60°C for 1.5 minutes, extension at 72°C for 1.5 minutes, and final extension at 72°C for 10 minutes. PCR was performed in a 25 μL reaction mix consisting of QIAGEN Multiplex PCR (catalog number 206143), 2 μL DNA, and 200 ng each primer. LP1 and LP2 primers were used for second-round PCR amplification as described above, except that the annealing temperature was lowered to 40°C and 1 μL first-round PCR was used as DNA template. PCR product (5 μL) was added to 1.5 μL loading buffer (Sigma) and electrophoresed in UltraPure agar (Invitrogen) in 0.04 M Tris·acetate and 0.001 M ethylenediaminetetraacetic acid (EDTA) (TAE buffer). Amplified DNA was visualized by ethidium bromide staining and ultraviolet light and recorded with a Gel Doc XR System (BIO-RAD).
DNA sequencing.
PCR products were purified and sequenced using LP1 and ML1 primers by Beijing Dingguo Biotechnology Co., Ltd. The identity of the DNA sequence from the PCR products was confirmed to be M. leprae DNA sequence by Basic Local Alignment Search Tool (BLAST) search (www.ncbi.nlm.nih.gov/BLAST/).
Sensitivity and specificity of the PCR assay.
Serial 10-fold dilutions of 109 M. leprae were added to negative whole blood before DNA extraction and PCR amplification. M. leprae DNA was diluted 10-fold from 10 ng to 1 ag and PCR-amplified as described above. For determining the specificity of the PCR, first- and second-round PCR products from 10 positive patients were sent for sequencing to confirm the specificity of the PCR assay. PCR products from each of five negative control samples were also amplified after the addition of 10 fg M. leprae DNA to exclude the presence of PCR inhibitors in the template.
ELISA.
Direct detection of immunoglobulin M (IgM) with ND-O-BSA and specific IgG antibodies against LID-1 by ELISA was performed as previously described.8
Acid-fast staining.
Blood samples from four MB patients with high Bacteriological Index (BI) (≥ 4) that were positive by nested PCR were randomly selected. Blood (0.5 mL) was drawn by venipuncture of the median cubital vein with a hypodermic needle and collected in a BD vacutainer containing anticoagulant (BD Vacutainer #367884; BD). The number of AFB in blood samples was counted by the hemolysis (HL) method of Sreevatsa and others.9 Serial 10-fold dilutions of 109 M. leprae were added to negative whole blood (negative by nested PCR) and acid-fast–stained with HL.9
Statistical analysis.
Student's t test and κ-correlation analysis were performed to compare different methods of diagnosis of leprosy. P value < 0.05 was considered significant, and κ-value > 0.7 was considered to have good correlation.
Results
Acid-fast staining.
Four MB patients were randomly selected for the peripheral hemolyzed blood. Small AFB were observed by microscopy for each sample, which was also positive by nested PCR for detection of M. leprae-specific DNA. The minimum detectable concentration of M. leprae by acid-fast staining with HL using serial 10-fold dilutions in negative whole blood was 107 bacteria/mL.
Specificity of M. leprae DNA amplification.
In smear strongly positive (BI = 1–5+) MB blood samples, 95.92% (47/49) were positive by nested PCR compared with 70% (21/30) in smear weakly positive (BI = 0–1.8) PB samples. In 18 MB and 23 PB patients, the first-round PCR gave negative results, and the second-round PCR increased positive results by 16 and 14 in the two groups, respectively. DNA from the MB and PB patients gave the expected amplification products of 372 and 131 bp by the nested PCR (Figure 1). Positive PCR products were subjected to DNA sequencing, and the sequences were analyzed by BLAST search. The sequences were 99% homologous to M. leprae-specific DNA (FM211192.1), indicating that the PCR products were derived from M. leprae-specific DNA. Seventeen other mycobacterial species and four non-mycobacterial species were tested in the PCR to ensure the specificity of the nested-PCR assay. The 372- and 131-bp DNA fragments were only amplified by PCR with M. leprae DNA (Figure 1) and not from DNA of other mycobacterial species or bacteria belonging to other genera. Thus, these findings indicate the high specificity of the nested-PCR test (Figure 1 and Table 1).
Figure 1.

Amplification of M. Ieprae DNA from patient's blood using nested PCR.
Table 1.
Nested-PCR and ELISA tests results of all samples
| Classification R–J | No. | Nested PCR | ND-O-BSA IgM | LID-1 IgG | ||||
|---|---|---|---|---|---|---|---|---|
| 372 bp | 131 bp | Positive (%) | Mean ± SD | Positive* (%) | Mean ± SD | Positive* (%) | ||
| MB | ||||||||
| LL | 9 | 6 | 3 | 9 (100) | 1.16 ± 0.55 | 8 (88.89) | 0.78 ± 0.84 | 7 (77.78) |
| BL | 38 | 24 | 12 | 36 (94.74) | 1.37 ± 0.67 | 38 (100) | 1.49 ± 0.78 | 36 (94.74) |
| BB | 2 | 1 | 1 | 2 (100) | 1.35 ± 0.12 | 2 (100) | 0.34 ± 0.36 | 1 (50) |
| Subtotal | 49 | 31 | 16 | 47 (95.92) | 48 (97.96) | 44 (89.8) | ||
| PB | ||||||||
| BT AFB+ | 24 | 5 | 13 | 18 (75) | 0.43 ± 0.28 | 19 (79.17) | 0.46 ± 0.45 | 14 (58.33) |
| AFB− | 2 | 0 | 0 | 0 (0) | 0.24 ± 0.31 | 1 (50) | 0.10 ± 0.20 | 0 (0) |
| TT | 3 | 2 | 1 | 3 (100) | 0.20 ± 0.05 | 1 (33.33) | 0.46 ± 0.52 | 2 (66.67) |
| I | 1 | 0 | 0 | 0 (0) | 0.09 ± 0.00 | 0 (0) | 0.16 ± 0.0 | 0 (0) |
| Subtotal | 30 | 7 | 14 | 21 (70) | 21 (70) | 16 (53.33) | ||
| Contacts | 96 | 0 | 6 | 6 (6.25) | 0.21 ± 0.16 | 29 (30.21) | 0.13 ± 0.06 | 6 (6.25) |
| Tuberculosis | 18 | 0 | 0 | 0 | 0.15 ± 0.19 | 3 (16.67) | 0.08 ± 0.03 | 0 |
| Normal (Beijing) | 25 | 0 | 0 | 0 | 0.03 ± 0.00 | 0 | 0.05 ± 0.03 | 0 |
| Normal (Yunnan) | 10 | 0 | 0 | 0 | 0.12 ± 0.07 | 1 (10) | 0.07 ± 0.04 | 0 |
| Total | 228 | 38 | 39 | 77 | 102 | 66 | ||
ELISA optical density (OD) ≥ 0.2 was defined as positive.
Sensitivity of the nested-PCR assay.
M. leprae DNA (10 fg) was added to 10 negative PCR products to check for inhibition, and there was amplification of the 372-bp specific fragment at each dilution (Figure 2). The 372-bp product was successfully amplified from 1 bacterium when serial 10-fold dilutions of M. leprae (from 109 to 1 bacillus) were added to negative hemolyzed samples (Figure 3). Furthermore, the 372-bp product was amplified from as little as 1 fg purified genomic DNA (Figure 4, lane 7) when the PCR was tested against the serial 10-fold dilutions of M. leprae genomic DNA, becoming negative at 10 ag. Given that the M. leprae genome has a size of 2.2 × 109 Da,10 1 fg corresponds to a single genome for this bacterial species (Figure 4). These findings suggest that the nested-PCR test is highly sensitive for detecting M. leprae.
Figure 2.
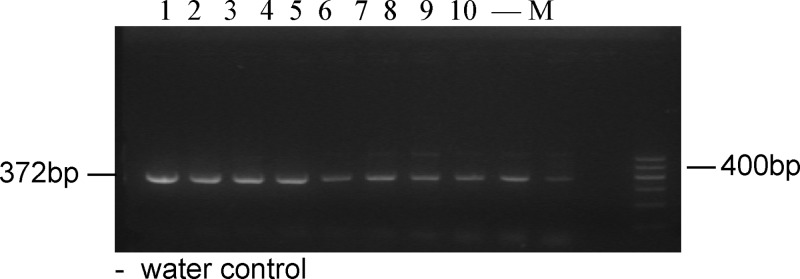
Negative PCR products (N = 10) spiked with 10 fg M. Ieprae DNA followed by nested PCR.
Figure 3.

Serial 10-fold dilutions of M. leprae in negative blood.
Figure 4.
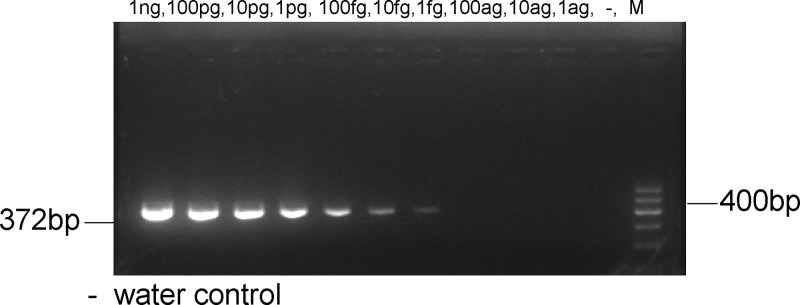
Amplification of serial 10-fold dilutions of M. leprae genomic DNA.
Comparison of the nested-PCR test and ELISA assay for diagnosis of leprosy.
We compared the detection of antibody response with M. leprae antigens (i.e., anti–ND-O-BSA IgM and anti–LID-1 IgG responses in plasma) with the nested-PCR test using whole-blood samples. The results are shown in Table 1. The highest rates of detection were ELISA, which detected anti–ND-O-BSA IgM in 48 of 49 (97.96%) MB patients, followed by nested PCR, which detected 47 of 49 (95.92%) MB patients, but there were no statistically significant differences between the two methods (P > 0.05). However, ELISA detecting anti–LID-1 IgG gave a lower sensitivity of detection in 44 of 49 (89.8%) MB patients. For PB patients, the results of ELISA detecting antibodies to ND-O-BSA and nested PCR were similar (70%), but detection of LID-1antibodies by ELISA was only 53.33%. However, ELISA test detecting antibodies to ND-O-BSA produced a significant number of positives (29/96; 30.21%) in healthy close contacts, 16.7% TB patients, and 10% normal healthy controls, suggesting some false-positive results. In contrast, nested PCR was positive in 6% of healthy close contacts and negative for TB patients and normal healthy controls as expected, indicating that the nested PCR does not have the problem of false-positive results and has a higher specificity than the ELISA tests detecting PGL-1 or LID-1. Among 228 samples, 57 samples were positive by both nested PCR and LID-1 ELISA, and 142 samples were negative by both methods. There were 20 samples positive by nested PCR but negative by LID-1 ELISA, whereas 9 samples were positive by LID-1 ELISA but negative by nested PCR. Through correlation κ-analysis, there was no statistical difference between the two methods (P > 0.05).
It is worth noting that, for HHCs, six individuals were negative for the first round of PCR (372 bp) but positive for the second round of nested PCR (131 bp). Six individuals from the HHC group were also positive for LID-1 ELISA, but the antibody titers were just above the cutoff value for positivity, indicating that the levels of anti–LID-1 antibodies were low in HHCs. These results indicate that the amounts of leprosy bacilli and specific antibody were low in the blood in six close contacts.
Discussion
There are few studies that focus on the detection of M. leprae from whole-blood samples. Kaur and others11 applied the hemolytic method to samples from 54 smear-negative PB cases before MDT and found that 8 (14.8%) cases were acid-fast–positive. In the present study, four blood samples from nested PCR-positive MB patients with high BI were selected by acid-fast staining with HL. Bacteria were shown to be present in blood samples from patients before treatment. The minimum detectable concentration of M. leprae in blood by acid-fast staining with HL was 107 bacilli/mL. Therefore, a more sensitive and specific method for detecting M. leprae in blood is needed given the insensitivity of the acid-fast staining method.
With advances in molecular technology, it is now possible to improve detection of M. leprae by PCR. We selected an M. leprae-specific repetitive DNA sequence for target amplification4 because of its high copy number in the genome.6 Additional improvements in test sensitivity may be achieved by a second round of amplification.6 Thus, nested-PCR amplification may achieve greater sensitivity and specificity than the acid-fast staining method and even conventional PCR. In this study, 63.27% (31/49) of MB cases and 23.33% (7/30) of PB cases were positive after the first round of amplification. After nested PCR, the positivity increased to 95.92% (47/49) for MB and 70% (21/30) for PB. The nested PCR was also more sensitive than the LP1–LP4 primer set used in PCR by Agusni and others,12 which detected 70% of MB cases and 31% of PB cases in blood samples from 52 leprosy patients from endemic areas. It is worth noting that PB cases with low or negative BI were the highest, with a sensitivity of 70% by the nested PCR.
The 372-bp product was amplified from as little as 1 fg purified genomic DNA, indicating that the sensitivity and specificity of the nested PCR were higher than previous similar studies.4,5,13 Furthermore, PCR inhibition was not observed when negative samples were spiked with M. leprae DNA. DNA was not amplified by nested PCR of any of 17 other Mycobacterium species or 4 other bacterial species, and there was also no cross-reaction with DNA from 18 cases of TB. Truman and others14 developed a real-time PCR assay for quantifying M. leprae DNA in biological samples. The assay was specific and able to detect 10 fg purified M. leprae DNA or approximately 300 bacteria in infected tissues. The nested PCR in this study was more sensitive, although the possibility that a more efficient protocol may be found for detection of M. leprae cannot be excluded.
Nested PCR gave comparable sensitivity results to ND-O-BSA IgM and LID-1 IgG ELISA for the detection of MB patients. Although the positive rate of the ND-O-BSA IgM ELISA was slightly higher than the nested PCR, the latter seems to be more specific. It is known that HHCs of leprosy patients may have anti–PGL-I antibodies and never develop the disease.15,16 Seropositivity to PGL-I of M. leprae is relatively common in endemic areas, and currently, no evidence exists for a correlation between seroprevalence of PGL-I and the incidence of leprosy.17 It is likely that bacillimia during leprosy infection is not a continuous process and that most subclinical infections resolve spontaneously without progressing to disease. The high prevalence of seropositives among HHCs of leprosy patients shows that subclinical infection with M. leprae is common.18,19 For PB patients, both nested PCR and ND-O-BSA IgM ELISA gave higher positive rates than the LID-1 IgG. However, leprosy is an infection with a long latency period, and therefore, tests with high sensitivity and specificity are of utmost importance for tracing and follow-up of the HHCs. Our results indicate that there is no significant difference between the IgM and IgG antibodies among the different groups tested as well as no difference in nested PCR between 49 MB and 21 PB tested (P > 0.05). The combination of nested PCR with ELISA for detection of specific antibodies may, therefore, be preferred for early detection and confirming diagnosis as well as differentiation from other dermatological conditions. Additional follow-up studies are required among leprosy HHCs; also, field study for indeterminate leprosy, the very early sign of the disease that is often missed by family members and the medical personnel in the endemic area, is needed.
Monitoring of leprosy bacillus DNA in the blood of HHCs of MB leprosy cases is particularly important for early detection, because about 30% may develop leprosy themselves. Banerjee and others20 found that multiplex PCR (M-PCR) was a promising technique for early detection among contacts of leprosy cases. Nasal swab samples from a total of 110 MB patients and 72 PB contacts were tested by M-PCR; 10.9% were found to be positive among MB contacts, and 1.3% were found to be positive among PB contacts. Two contacts of M-PCR–positive MB cases developed leprosy during the next 2 years, but PCR-positive nasal swabs indicated the presence of M. leprae DNA only and not infection. The positivity was 6.25% in HHCs by nested PCR. Although M. leprae DNA in the blood may indicate infection, the relationship between positive blood PCR and development of the disease requires additional analysis. The six HHCs who were nested PCR-positive should be followed up closely for clinical signs as well as SSSs and ELISA serology.
Although our experimental protocol requires additional refinement, the comparable sensitivity of the nested PCR but better specificity compared with anti–PGL-1 ELISA test for detecting MB and PB patients and the successful use of nested PCR for detection of M. leprae DNA from blood samples rather than skin biopsies are most encouraging. Future studies are needed to validate the use of the nested-PCR test along with the ELISA test to detect the PGL-1 for early diagnosis of leprosy in more patients.
ACKNOWLEDGMENTS
We are grateful to Prof. P. J. Brennan for providing M. leprae DNA (NHDP63) and antigen ND-O-BSA, Dr. M. S. Duthie for providing the recombinant antigen LID-1, and Dr. T. P. Gillis for providing bacterial strains. The authors thank Dr. Sen Wang for help with the statistical analysis.
Footnotes
Authors' addresses: Yan Wen, Capital University of Medicine Affiliated Beijing Friendship Hospital, Beijing Tropical Medicine Research Institute, Beijing, China, E-mail: weny8@163.com. Yan Xing, Lian-Chao Yuan, Jian Liu, and Huan-Ying Li, Department of Leprosy Research, Beijing Tropical Medicine Research Institute, Beijing, China, E-mails: xingyan@163.com, yuanlianchao@126.com, liujian@sina.com, and lihybj@163.com. Ying Zhang, Department of Molecular Microbiology and Immunology, Bloomberg School of Public Health, Johns Hopkins University, Baltimore, MD, E-mail: yzhang@jhsph.edu.
References
- 1.Duthie MS, Goto W, Ireton GC, Reece ST, Cardoso LP, Martelli CM, Stefani MM, Nakatani M, de Jesus RC, Netto EM, Balagon MV, Tan E, Gelber RH, Maeda Y, Makino M, Hoft D, Reed SG. Use of protein antigens for early serological diagnosis of leprosy. Clin Vaccine Immunol. 2007;14:1400–1408. doi: 10.1128/CVI.00299-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Douglas JT, Worth RM. Field evaluation of an ELISA to detect antibody in leprosy patients and their contacts. Int J Lepr Other Mycobact Dis. 1984;52:26–33. [PubMed] [Google Scholar]
- 3.De Wit MYL, Faber WR, Krieg SR, Douglas JT, Lucas SB, Montreewasuwat N, Pattyn SR, Hussain R, Ponnighaus JM, Hartskeerl RA. Application of a polymerase chain reaction for the detection of Mycobacterium leprae in skin tissues. J Clin Microbiol. 1991;29:906–910. doi: 10.1128/jcm.29.5.906-910.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Woods SA, Cole ST. A rapid method for the detection of potentially viable Mycobacterium leprae in human biopsies: a novel application of PCR. FEMS Microbiol Lett. 1989;65:305–310. doi: 10.1016/0378-1097(89)90235-8. [DOI] [PubMed] [Google Scholar]
- 5.Plikaytis BB, Gelber RH, Shinnick TM. Rapid and sensitive detection of Mycobacterium leprae using a nested-primer gene amplification assay. J Clin Microbiol. 1990;28:1913–1917. doi: 10.1128/jcm.28.9.1913-1917.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang HB, Weng XM, Wen Y, Li HY. Improved detection of Mycobacterium leprae rifampin resistance with nested PCR. Chin J Lab Med. 2005;28:1015–1017. [Google Scholar]
- 7.Ridley DS, Jopling WH. Classification of leprosy according to immunity—a five group system. Int J Lepr. 1966;34:255–273. [PubMed] [Google Scholar]
- 8.Wen Y, Vissa VD, Li W, Li HY. Antigenicity of Mycobacterium leprae recombinant proteins. J Trop Med. 2010;10:1153–1157. [Google Scholar]
- 9.Sreevatsa RG, Sengupta U, Ramu G, Desikan KV. Evaluation of bacteraemia in leprosy patients. Lepr India. 1978;50:381–387. [PubMed] [Google Scholar]
- 10.Santos AR, De Miranda AB, Sarno EN, Suffys PN, Degrave WM. Use of PCR-mediated amplification of Mycobacterium leprae DNA in different types of clinical samples for the diagnosis of leprosy. J Med Microbiol. 1993;39:298–304. doi: 10.1099/00222615-39-4-298. [DOI] [PubMed] [Google Scholar]
- 11.Kaur I, Kaur S, Sharma VK, Agnihotri N, Vaishnavi C, Ganguly NK. Bacillaemia and Mycobacterium leprae cell wall antigen in paucibacillary leprosy. Indian J Lepr. 1993;65:283–288. [PubMed] [Google Scholar]
- 12.Agusni I, Prakoeswa CR, Adriaty D. Detection of M. leprae in the blood of leprosy patients and healthy inhabitants live in endemic leprosy area. Proceedings of the 17th International Leprosy Congress; Hyderabad, India. 30th January–4th February 2008.2008. [Google Scholar]
- 13.Hartskeerl RA, De Wit MYL, Klatser PR. Polymerase chain reaction for the detection of Mycobacterium leprae. J Gen Microbiol. 1989;135:2357–2364. doi: 10.1099/00221287-135-9-2357. [DOI] [PubMed] [Google Scholar]
- 14.Truman RW, Andrews PK, Robbins NY, Adams LB, Krahenbuhl JL, Gillis TP. Enumeration of Mycobacterium leprae using real-time PCR. PLoS Negl Trop Dis. 2008;2:e328. doi: 10.1371/journal.pntd.0000328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buhrer-Sekula S, Smits HL, Gussenhoven GC, van Leeuwen J, Amador S, Fujiwara T, Klatser PR, Oskam L. Simple and fast lateral flow test for classification of leprosy patients and identification of contacts with high risk of developing leprosy. J Clin Microbiol. 2003;41:1991–1995. doi: 10.1128/JCM.41.5.1991-1995.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oskam L, Slim E, Buhrer-Se'kula S. Serology: recent developments, strengths, limitations and prospects: a state of the art overview. Lepr Rev. 2003;74:196–205. [PubMed] [Google Scholar]
- 17.Klatser PR, Cho SN, Brennan PJ. The contribution of serological tests to leprosy control. Int J Lepr Other Mycobact Dis. 1996;64((Suppl 4)):S63–S66. [PubMed] [Google Scholar]
- 18.Menzel S, Harboe M, Bergsvik H, Brennan PJ. Antibodies to a synthetic analog of phenolic glycolipid-I of Mycobacterium leprae in healthy household contacts of patients with leprosy. Int J Lepr Other Mycobact Dis. 1987;55:617–625. [PubMed] [Google Scholar]
- 19.Saad MH, Medeiros MA, Gallo ME, Gontijo PP, Fonseca LS. IgM immunoglobulins reacting with the phenolic glycolipid-1 antigen from Mycobacterium leprae in sera of leprosy patients and their contacts. Mem Inst Oswaldo Cruz. 1990;85:191–194. doi: 10.1590/s0074-02761990000200008. [DOI] [PubMed] [Google Scholar]
- 20.Banerjee S, Sarkar K, Gupta S, Mahapatra PS, Gupta S, Guha S, Bandhopadhayay D, Ghosal C, Paine SK, Dutta RN, Biswas N, Bhattacharya B. Multiplex PCR technique could be an alternative approach for early detection of leprosy among close contacts—a pilot study from India. BMC Infect Dis. 2010;10:252. doi: 10.1186/1471-2334-10-252. [DOI] [PMC free article] [PubMed] [Google Scholar]