

Abstract
Evidence from different laboratories using cell culture and in vivo model systems indicates that histone deacetylase-4 (HDAC4) plays an essential role in maintaining neuronal survival. Indeed, HDAC4 null knockout mice, which die within 2 weeks of birth, display cerebellar degeneration, whereas RNAi-mediated knockdown of HDAC4 expression in the retina of normal mice leads to apoptosis of retinal neurons. As a step toward analyzing the role of HDAC4 in the regulation of neuronal survival in more detail, we generated two separate lines of conditional knockout mice by breeding HDAC4-flox mice with mice expressing Cre recombinase through a Thy1 or nestin promoter. Surprisingly, both Thy1-Cre/ HDAC4−/− mice, in which HDAC4 is ablated in neurons of the cortex and hippocampus, as well as Nes-Cre/ HDAC4−/− mice, in which HDAC4 is ablated in neural progenitor cells of the CNS, appear normal at birth, have normal body weight, are fertile, and perform normally in locomotor activity assays. Histological analysis of the brains of Nes-Cre/HDAC4−/− mice revealed no obvious abnormalities in cytoarchitecture. Immunohistological analysis of tyrosine hydroxylase and calbindin also showed no discernible defects. Terminal deoxynucleotidyl transferase dUTP nick end-labeling staining showed no difference in the level of neuronal death in the cortex and cerebellum of Nes-Cre/HDAC4−/− mice compared with controls. These results indicate that neurons are less dependent on HDAC4 expression for their survival than previously believed and suggest that neuronal death observed in HDAC4 null knockout mice and after RNAi injection may result from HDAC4 deficiency
Keywords: histone deacetylase, HDAC4, neuronal cell death, neuronal survival, knockout mouse
Histone deacetylases (HDACs) are enzymes that catalyze the removal of acetyl groups from histones as well as a long and growing list of nonhistone nuclear, cytoplasmic, and mitochondrial proteins (Yang and Seto, 2008; Haberland et al., 2009). Mammals express 18 different histone deacetylases that are divided into four groups. Class I HDACs (HDAC1, –2, –3, and –8) are ubiquitously expressed and localize to the nucleus, with the exception of HDAC3, which is both nuclear and cytoplasmic. Class II HDACs (HDAC4, –5, –6, –7, –9, and –10) are specifically expressed in a variety of tissues, with highest expression in the brain and skeletal muscle. Some of the class II HDACs have been shown to translocate between the nucleus and cytoplasm in a phosphorylation-dependent manner. Class III HDACs (SIRT1–7) are structurally distinct from the other three classes of classical HDACs (I, II, and IV) and are referred to as the sirtuins. While still encompassing histone deacetylase activity, sirtuins are considered nonclassical HDACs because of their requirement of NAD+ for their activity, as opposed to the zinc-dependence of classical HDACs (Yang and Seto, 2008; Haberland et al., 2009). Finally, class IV HDACs consists of only one member, HDAC11, which shares sequence similarity with both class I and class II HDACs.
A solid body of evidence suggests that HDACs regulate neuronal viability (Morrison et al., 2007; Kazantsev and Thompson, 2008; Majdzadeh et al., 2008a; Sleiman et al., 2009; D’Mello, 2009). Whereas HDAC3 has been shown to promote neuronal death (Bardai and D’Mello, 2011), other HDACs maintain neuronal survival and protect neurons from apoptotic stimuli (Iwata et al., 2005; Morrison et al., 2006; Pandey et al., 2007; Boyault et al., 2007; Pfister et al., 2008; Ma and D’Mello, 2011); among these is HDAC4. We previously reported that elevated expression of HDAC4 protects cultured cerebellar granule neurons (CGNs) and cortical neurons from death (Majdzadeh et al., 2008b). Although HDAC4 normally resides in the cytoplasm, we found that, in neurons primed to die, HDAC4 translocates to the nucleus, where its protection was mediated (Majdzadeh et al., 2008b). This was confirmed by the inability of an HDAC4 mutant, lacking a 72-amino-acid region containing the nuclear loacalization signal, to protect neurons primed to die. A neuroprotective role for HDAC4 was also described by Chen and Cepko (2009) for the mouse retina. These authors found that RNAi-mediated knockdown of HDAC4 expression during normal retinal development led to apoptosis of rod photoreceptors and bipolar interneurons. On the other hand, retrovirus-mediated overexpression of HDAC4 reduced naturally occurring cell death of retinal neurons (Chen and Cepko, 2009). Furthermore, HDAC4 over-expression prolonged photoreceptor survival in a mouse model of retinal degeneration (Chen and Cepko, 2009). Surprisingly, and in contradiction to what was found in cultured CGNs and cortical neurons, neuroprotection by HDAC4 in the intact retina was mediated in the cytoplasm (Chen and Cepko, 2009). More recently, interaction between HDAC4 and specific HSP40 proteins was shown to be necessary for the ability of these HSP40 proteins to suppress polyglutamine protein aggregation and toxicity (Hageman et al., 2010). Together these studies strongly suggest that HDAC4 promotes neuronal survival both in vivo and in vitro.
HDAC4 null mice have severe bone malformations resulting from chondrocyte hypertrophy and die perinatally (Vega et al., 2004). An examination of the brains of these mice revealed Purkinje cell degeneration starting within a few days of birth (Majdzadeh et al., 2008b), consistent with the conclusion that HDAC4 is required for neuronal survival. However, the early death of these mice precludes a more detailed analysis of the consequences of HDAC4 deficiency to neuronal well-being in the postnatal and adult brain. We therefore generated two separate lines of conditional knockout (cKO) mice in which HDAC4 is deleted selectively in the cortex and hippocampus or in neural progenitor cells of the CNS by breeding HDAC4-flox mice with mice expressing Cre through a Thy1 or nestin promoter, respectively. Surprisingly, both lines of HDAC4 cKO mice are normal in appearance, breeding, and performance in locomotor tests. Histological analysis of two mouse lines in which HDAC4 was deleted selectively in the cortex and hippocampus as well as the entire CNS shows no increase in cell death in the brain and no discernible abnormality in brain cytoarchitecture.
MATERIALS AND METHODS
HDAC4lox/lox mice were obtained from Dr. Eric Olson of the University of Texas Southwestern Medical Center (Backs and Olson, 2006). Thy1-Cre (Dewachter et al., 2002), nestin-Cre (Tronche et al., 1999), and Thy1-YFP (Feng et al., 2000) mice were purchased from Jackson Laboratories (Bar Harbor, ME; stock numbers are Thy1-Cre 006143, Nes-Cre 003771, and Thy1-YFP 003782).
Crossing of Nes-Cre/HDAC4−/− with Thy1-YFP
Nes-Cre/HDAC4 cKO mice were bred with Thy1-YFP+/+, and their offspring were then bred to obtain Nes-Cre/HDAC4 cKO/Thy1-YFP+/+ mice.
Genotyping
Tail tips of mice were collected, DNA was extracted using the REDExtract-N-Amp Tissue PCR Kit from Sigma Aldrich (St. Louis, MO), and the DNA was used in PCRs to determine genotype (HDAC4 fwd: ATCTGCCCACCAGAG TATGTG, HDAC4 rev: CTTGTTGAGAACAAACTCCTG CAGCT, nLacZ rev: GATTGACCGTAATGGGATAGGT TACG; Thy1-YFP fwd: TCTGAGTGGCAAAGGACCTT AG, Thy1-YFP rev: TGAACTTGTGGCCGTTTACGl; Cre fwd: GCGGTCTGGCAGTAAAAACTATC, Cre rev: GTG AAACAGCATTGCTGTCACTT).
Western Blot
Mice were euthanized by carbon dioxide inhalation prior to dissection of the various brain parts analyzed in this study. Brain tissue was homogenized in RIPA buffer and centrifuged at 14,000 rpm for 10 min. Protein concentrations were taken using the Bradford reagent from Bio-Rad (Hercules, CA); 100 μg of protein was loaded on a 10% SDS-PAGE gel, transferred to a PVDF membrane, and probed with the following antibodies: anti-HDAC4 (Sigma; 1:250), anti-Total Erk (Cell Signaling, Beverly, MA; 1:1,000), anti-HDAC3 (Santa Cruz Biotechnology, Santa Cruz, CA; 1:500), and anti-c-Jun (Santa Cruz Biotechnology; 1:200). After incubation in the appropriate secondary antibody, membranes were subjected to enhanced chemiluminescence (GE Healthcare, Piscataway, NJ). Densitometric analysis of films was performed in Kodak imaging software.
RT-PCR Analysis of Gene Expression
Mice were euthanized in the same fashion as for protein collection. Selected brain parts were dissected and homogenized in Trizol reagent from Life Technologies (Carlsbad, CA), and cDNA was obtained from extracted RNA with the Superscript-II RT-PCR kit, also from Life Technologies. PCR was carried out with primers for amplification of HDAC4 and c-Jun; actin served as a loading control.
Open-Field Locomotor Testing
To assess possible differences in locomotion in conditional knockout mice, the TruScan system from Coulbourn Instruments (Whitehall, PA) was utilized. Mice were analyzed for 15 min, and six different measurements were taken. Five measurements corresponded to movements in the floor plane, and one measurement was taken for vertical movements. Data shown are an average of cKO mice and wild-type littermates.
Histology
Brains were fixed with 4% paraformaldehyde in 0.1 M phosphate buffer. Samples were allowed to sink in 20% sucrose solution in phosphate-buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, 1.47 mM KH2PO4, pH 7.4) overnight following fixation. Whole brains were then frozen in cryopreservative solution (Cryo-STAT2; StatLab Medial Products, Lewisville, TX) on dry ice, and then sectioned sagitally at 20 μm (Nes-Cre/HDAC4 cKO/Thy1-YFP brains were sectioned at 40 μm) on a cryostat. Sections were either stained with cresyl violet for structural studies or subjected to immunohistochemistry. Primary antibodies used on sections were as follows: Calbindin and tubulin (Sigma-Aldrich; 1:1,000), GFAP (Dako Cytomation, Glostrup, Denmark; 1:500), and tyrosine hydroxylase (Calbiochem, Sunnyvale, CA; 1:500). Sections were incubated with primary antibody at 4°C overnight, followed by secondary antibody incubation as suggested in the manufacturer’s protocol (Avidin/Biotinylated Enzyme Complex [ABC] Kit; Vector, Burlingame, CA). This was followed by 3,3′-diaminobenzidine (DAB; Vector) staining of the sections. For fluorescence imaging, Texas red secondary antibody (Jackson Immunore-search, West Grove, PA; 1:200) was used for 2 hr at room temperature, and 4′,6-diamidino-2-phenylindole (DAPI) was used to stain cell nuclei. The numbers of Thy1-YFP-expressing cells were counted blindly, and an average was taken from five different fields of both cortex and hippocampus for wild type (WT) and conditional knockout (cKO). DAPI counts were performed to analyze the cell density of the cortex; data displayed are an average from five fields for both WT and cKO. For both experiments, cKO was normalized to WT control (100%).
TUNEL Assay
TUNEL assays were performed using a commercially available apoptosis detection kit from Promega (Madison, WI). Briefly, frozen brain sections were fixed in 4% paraformaldehyde. After washing with PBS, the sections were permeabilized with 0.2% Triton X-100 solution and equilibrated in the provided buffer before labeling with fluorescein-conjugated dNTP. After reaction termination, the sections were washed in PBS and counterstained with DAPI. The sections were then mounted with Fluoromount-G and kept at 4°C before viewing under a fluorescent microscope. TUNEL staining was quantified from comparable sagittal brain sections from the cortex, hippocampus, and cerebellum of 4-day-old Nes-Cre/HDAC4 cKO and WT mice. The number of TUNEL-positive cells was counted blindly, and an average was taken from six different fields of each brain region. TUNEL-positive cells for cKO mice were normalized to the number of TUNEL-positive cells in WT mice for each brain region (100%).
RESULTS
Deletion of HDAC4 in Postnatal Cortical and Hippocampal Neurons Causes No Major Abnormality
HDAC4 null mice are severely abnormal and die within 2 weeks of birth (Vega et al., 2004). To understand better the role of HDAC4 in regulating neuronal survival, we generated cKO mice in which HDAC4 was selectively deleted only in specific regions of the brain. We first crossed HDAC4lox/lox mice with Thy1-Cre mice; the Thy1 promoter in these mice is most active in the cortex and hippocampus but is also expressed in other brain regions (Dewachter et al., 2002). No overt abnormality in appearance was observed in cKO mice homozygous for HDAC4 deletion (Thy1-Cre/ HDAC4−/−). In contrast to the HDAC4 null knockout mice, which are substantially smaller at birth, the cKO mice were of normal size at birth and throughout adulthood (data not shown). Although litters containing Thy1-Cre/HDAC4−/− mice were of normal sizes, typical Mendelian ratios were not observed in the offspring, which can be attributed to an inability of the genotyping primers for Cre expression to distinguish between homozygosity and heteroxygosity (Fig. 1). Results from Western blot and RT-PCR experiments confirmed that HDAC4 expression is reduced at both the protein and the RNA levels in Thy1-Cre/HDAC4−/− mice compared with their wild-type littermates (Fig. 2). Because increased c-jun is generally associated with neuronal death, we looked at its expression in the mutant. Interestingly, a small increase in c-jun expression was observed in the cortex of Thy1-Cre/HDAC4 cKO mice. In contrast, however, expression of HDAC3, an HDAC associated with neuronal death, showed no change. To assess behavioral performance, we conducted an open-field locomotor assay. As shown in Figure 3, no difference in locomotor performance was observed in Thy1-Cre/HDAC4−/− mice.
Fig. 1.

Genotyping of HDAC4 cKO mouse crosses. A: Ratio of observed genotypes compared with expected Mendelian ratio. The expected ratios of both WT and cKO are different from traditional genetic expectancies of a cross between mice heterozygous for the two transgenes. The number for expected cKO mice was obtained by combining the genotypes for homozygous inheritance of both genes (Cre and HDAC4) and mice that were heterozygous for Cre and homozygous for HDAC4; this number was adjusted to compensate for the incapability of Cre primers to distinguish between heterozygous and homozygous gene inheritance. B: Representative figure showing identification of genotypes. The leftmost lane represents cKO, and the rightmost lane represents WT. For Cre PCR results, “+” denotes presence of Cre and “−” denotes absence of the gene. C: Body and brain weight analysis of Thy1-Cre-HDAC4 mice (n = 3 litters) and Nes-Cre-HDAC4 cKO mice (n = 4 litters). The body weight of male cKO and WT mice was measured at 6 weeks, the average of four WT and five cKO mice for both lines are represented here. Brains of male mice from both cKO lines were weighed at the time of dissection prior to their use in other experiments in this study. The numbers shown are averaged from the following sample sizes (number of individual mice analyzed): Thy1-Cre-HDAC4 line WT n = 4 and cKO n = 5; Nes-Cre-HDAC4 line WT n = 3 and cKO n = 3.
Fig. 2.

RT-PCR and Western blot analysis confirm knockdown of HDAC4 in the brains of cKO mice. RNA or protein lysates were prepared from the cortex, cerebellum, and other brain parts excluding cortex and cerebellum (OBP) of 6-week-old Thy1-Cre/ HDAC4−/− and Nes-Cre/HDAC4−/− mice (cKO) and wild-type (WT) littermates. Results are from both Thy1-Cre/HDAC4 (Thy1) and Nes-Cre/HDAC4 cKO (Nes) mice. A: RT-PCR analysis. HDAC4 mRNA expression was analyzed in brain regions of HDAC4 cKO and WT littermates. Actin RNA was also amplified as a control. In both lines, HDAC4 expression is substantially reduced, with a more robust knockdown observed in the Nes-Cre/HDAC4 cKO mice because of the more widespread ablation of HDAC4 in this line. B: Western blot analysis results. Tissue lysates were subjected to Western blot analysis and probed with HDAC4 antibody. An additional blot was run and probed for total ERK to demonstrate equal loading. The lysates were also analyzed for expression of c-Jun and HDAC3 using antibodies to these proteins. C: Densitometric analysis of c-jun expression in the cortex of both Thy1 and Nes cKO mice reveals a twofold increase in expression compared with the expression of their WT littermates.
Fig. 3.

Open-field locomotor testing of cKO mice. A–F: Data from the six different measurements collected from the TruScan system are shown for both the Thy1-Cre/HDAC4 (n = 3 litters) and the Nes-Cre/HDAC4 cKO (n = 4 litters) lines. The graphs represent averages of males tested at 6 weeks of age for both lines; number of individual mice analyzed: WT n = 4 and cKO n = 5.
No Major Abnormalities Are Observed When HDAC4 Is Selectively Deleted in the CNS
Because no abnormality was seen in Thy1-Cre/ HDAC4 cKO mice, we proceeded to achieve a more widespread knockout of HDAC4 in the brain. This was accomplished by crossing HDAC4lox/lox mice with the Nes-Cre line, which expresses Cre in neural progenitor cells of the CNS (Tronche et al., 1999). Crossing of Nes-Cre mice ablates target floxed genes in all neurons and glia of the brain (Graus-Porta et al., 2001). RT-PCR and Western blot analyses confirmed that HDAC4 expression is severely reduced in the cortex and cerebellum, as well as the remaining areas of the brain (OBP; Fig. 2). Residual expression is likely due to contributions from nonneural cell types such as blood vessel cells. Interestingly, and as observed in Thy1-Cre/HDAC4 cKO mice, c-jun expression was higher in the cortex as well as in lysates from other brain parts (Fig. 2). Also consistent with Thy1-Cre/HDAC4 cKO mice, there was no change in the expression of HDAC3 in Nes-Cre/HDAC4 cKO mice.
Despite the widespread knockout of expression, Nes-Cre/HDAC4−/− mice appear normal and have a normal brain weight (Fig. 1C) compared with wild-type littermates. Locomotor performance in these mice was also not different from that of wild-type littermates (Fig. 3). Ablation of HDAC4 occurs in neurons and glia throughout the CNS of these mice, so we predicted that some abnormalities in cellular organization would be detected. However, based on cresyl violet staining, the overall cytoarchitecture of the brain appeared normal (Fig. 4). Tubulin immunostaining showed a normal staining pattern in Nes-Cre/HDAC4−/− mice (Fig. 5). We also performed an immunohistochemical analysis using antibodies against tyrosine hydroxylase (TH) and calbindin. As shown in Figure 6, the staining of Purkinje neurons in the cerebellum with calbindin is normal in Nes-Cre/HDAC4−/− mice, as is TH staining in the striatum. The overall staining pattern of TH and calbindin in other regions of the brain was also normal. Although histological and behavioral analyses of aged Nes-Cre/HDAC4−/− mice have not been performed, the mice grow to adulthood normally and are fertile. A sensitive approach to detect abnormalities in the appearance, distribution, and number of specific neuronal populations is to cross mutant mice with mice expressing green or yellow fluorescent protein (GFP or YFP, respectively) under the control of promoters that are active in those neurons (Feng et al., 2000; Oliva et al., 2000). We conducted such an approach using Thy1-YFP mice. In these mice, YFP is expressed in large pyramidal neurons of layer 5 within the cerebral cortex as well as pyramidal neurons within the hippocampus (Feng et al., 2000; Oliva et al., 2000). Histological analysis of HDAC4 cKO/Thy1-YFP+/+ mice showed a normal pattern of expression, further confirming that the absence of HDAC4 has no effect on neuronal cytoarchitecture (Fig. 7A). Quantification of neuronal cells expressing YFP was performed for both cortex and hippocampus (Fig. 7B); there was no difference in number of YFP-expressing cells in cKO mice compared with WT littermate.
Fig. 4.

Histological analysis of Nes-Cre/HDAC4 cKO mice. Sagittal sections stained with cresyl violet are shown from a 9-week-old Nes-Cre/HDAC4−/− (cKO) and wild-type (WT) littermate. A: Pictures from three brain regions are displayed at low magnification, from left to right: cortex, hippocampus, and cerebellum. B: Higher magnifications of the same brain regions from A. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Fig. 5.

Tubulin immunoreactivity in Nes-Cre/HDAC4 cKO mice. Sections from 9-week-old Nes-Cre/HDAC4−/− (cKO) and wild-type (WT) littermate were stained with an antibody against tubulin. Tubulin staining reveals no disruption in cell appearance in either the cortex or the internal granule layer of the cerebellum of HDAC4 cKO mice. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Fig. 6.
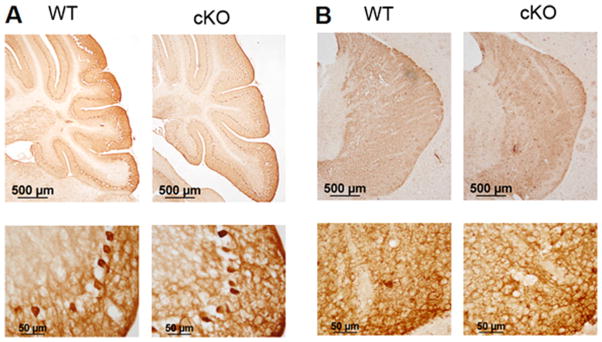
Immunohistological analysis of calbindin and tyrosine hydroxylase expression in Nes-Cre/HDAC4 cKO mice. Sections of 9-week-old Nes-Cre/HDAC4− − (cKO) and wild-type (WT) littermate were subjected to immunohistological analysis for calbindinn and tyrosine hydroxylase. A: Calbindin immunostaining of the cerebellum showing Purkinje cell layer at low magnification (top) and higher magnification (bottom). B: Tyrosine hydroxylase immunostaining of striatium at low magnification (top) and higher magnification (bottom). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Fig. 7.

Histological analysis of Nes-Cre/HDAC4 cKO/Thy1-YFP+ mice. A: Sagittal sections from the cortex (left) and hippocampus (right) of 4-week-old Nes-Cre/HDAC4 cKO/Thy1-YFP+/+ and WT/Thy1-YFP+/+ mice are shown; YFP expression of cKO is similar to that of WT littermate, confirming that there is no obvious abnormality in cell arrangement in mutant mice. B: Quantification of cells positive for YFP expression in the cortex and hippocampus; number of positive cells for cKO was normalized to WT (100%). [Color figure can be viewed in the online issue, which is available at wiley onlinelibrary.com.]
It was possible that a small level of neuronal death does occur in Nes-Cre/HDAC4−/− mice that could not be detected using the histological and immunohistological analyses we performed. To examine this possibility, we performed terminal deoxynucleotidyl transferase dUTP nick end-labeling (TUNEL) staining to label apoptotic cells. Although TUNEL-positive cells were observed in the cortex and cerebellum of Nes-Cre/ HDAC4−/− mice, the numbers of dying cells were similar to what was seen in the corresponding regions of WT littermates (Fig. 8A–C). To rule out the possibility that neuronal death occurred prior to the time at which TUNEL analysis was conducted, we performed immunohistochemistry with a glial fibrillary acidic protein (GFAP) antibody. Consistent with the lack of neuronal death, there was no change in cell density in the HDAC4 cKO mice (Fig. 8D). It is known that neuronal death is often followed by activation of reactive astrocytes. However, no sign of glial activation was evident in HDAC4 cKO brains, again arguing against a significant amount of cell death. Moreover, the normal pattern of GFAP staining suggests that HDAC4 deletion has no major effect on astroglial cytoarchitecture (Fig. 8E).
Fig. 8.

Cell death in Nes-Cre/HDAC4 cKO mice. TUNEL staining was performed on brain sections of 4-day-old Nes-Cre/HDAC4−/− (cKO) and wild-type (WT) littermates. The 4-day time point was chosen because naturally occurring cell death is observed at this point in development. Furthermore, our previous analysis of HDAC4 null knockout mice showed commencement of Purkinje cell degeneration starting 3–4 days after birth. TUNEL staining in the cortex (A) and cerebellum (B) from Nes-Cre/HDAC4 cKO and WT mice. C: Quantification of TUNEL-positive cells for cortex (Ctx), hippocampus (Hip), and cerebellum (Cere). Numbers are normalized to WT, which is adjusted to 100%. D: Quantification of cortical cell density by presence of DAPI stained nuclei; cKO numbers were normalized to WT (100%). E: GFAP immunostaining was performed on brain sections of 9-week-old HDAC4 cKO and WT littermate. Top: Representative cortical sections from WT and Nes-Cre/HDAC4 cKO littermates reveal no pronounced increase in GFAP staining in cKO mice. Bottom: As a control for detection of gliosis, cortical brain sections from the CaMK-p25-GFP/CDK5 double-transgenic model of tauopathic neurodegeneration (Cruz et al., 2003) were also subjected to GFAP staining. In this mouse, cortical and hippocampal degeneration is followed by gliosis. The panel shows GFAP staining within a portion of the cortex. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
DISCUSSION
Compelling evidence from studies performed in different laboratories using tissue culture and in vivo systems suggests that HDAC4 plays an essential role in promoting neuronal survival and protecting neurons from death. Indeed, HDAC4 has been shown to protect neurons from death in tissue culture paradigms, whereas the complete knockout of HDAC4 in mice results in perinatal Purkinje cell degeneration (Majdzadeh et al., 2008b). Similarly, shRNA-mediated knockdown of HDAC4 in the retina induces neuronal death, whereas the overexpression of HDAC4 has been shown to provide strong protective effects in a mouse model of retinal neurodegeneration (Chen and Cepko, 2009). HDAC4 has also been shown to be necessary for the neuroprotective effect of HSP40 proteins against toxic protein aggregates in cell culture models (Hageman et al., 2010). Surprisingly, analyses of two separate HDAC4 cKO lines, including one in which the HDAC4 gene is ablated in all neural cells of the CNS, revealed no gross abnormalities that could be detected based on basic histological and immunohistological analyses. Locomotor tests also failed to reveal motor abnormalities in either of the two HDAC4 cKO lines. Further analysis is necessary to determine whether the cKO mice suffer any cognitive or memory impairment. These analyses were not performed in the present study. Additionally, it is possible that more detailed immunohistological analyses will reveal minor abnormalities in the HDAC4 cKO mice.
It is difficult to reconcile the results in this study with those obtained previously by our own group and other laboratories (Chen and Cepko, 2009; Hageman et al., 2010; Majdzadeh et al., 2008b). It is possible that, although HDAC4 has a dispensable role in the maintenance of neuronal viability under physiological conditions, it might contribute to the protection of neurons when subjected to extreme stress or under neuropathological conditions; overexpression could enhance this neuroprotective function. The degeneration of Purkinje neurons in the HDAC4 null knockout mice could, at least in part, be due to secondary effects resulting from gross abnormalities of the skull observed in these mice. The death of retinal neurons induced by HDAC4 shRNA might be due to inhibitory effects on the expression of other HDAC proteins or even unrelated proteins. Alternatively, the neuronal survival-promoting effect of HDAC4 may depend on its expression in other nonneuronal cell types in the brain. The Thy1 promoter is expressed only in neurons, and, although nestin is expressed in progenitors of both neurons and glial cells, it is not expressed in nonneural such as cells of the vasculature.
In summary, analyses of two separate HDAC4 cKO mice in which HDAC4 is deleted in overlapping and nonoverlapping neuronal populations has failed to reveal any major effect on brain cytoarchitecture. These results argue against an essential role for HDAC4 in the maintenance of neuronal survival, at least under normal physiological conditions. Analyses were performed on young adult mice, so the consequences of HDAC4 deficiency during aging remain to be studied. It also remains to be determined whether the absence of HDAC4 might increase vulnerability to neurodegenerative disease or severity of the disease pathology and progression. Crossing these lines to already established mouse models of neurodegenerative diseases should provide information on this issue. We hope to resolve these questions in future studies. It deserves mention that c-jun expression, which is often associated with neuronal death, is elevated in HDAC4-deficient mice. It is possible that sustained elevation of c-jun could render the brains of HDAC4-deficient mice more vulnerable to degeneration either during normal aging or under pathological conditions.
Acknowledgments
Contract grant sponsor: National Institutes of Health; Contract grant number: NS040408.
We are grateful to Drs. Eric Olsen and Rhonda Bassel-Duby for the HDAC4-flox mice.
References
- Backs J, Olson EN. Control of cardiac growth by histone cetylation/deacetylation. Circ Res. 2006;98:15–24. doi: 10.1161/01.RES.0000197782.21444.8f. [DOI] [PubMed] [Google Scholar]
- Bardai FH, D’Mello SR. Selective toxicity by HDAC3 in neurons: regulation by Akt and GSK3beta. J Neurosci. 2011;31:1746–1751. doi: 10.1523/JNEUROSCI.5704-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyault C, Zhang Y, Fritah S, Caron C, Gilquin B, Kwon SH, Garrido C, Yao T, Pourc’h C, Matthias P, Khochbin S. HDAC6 controls major cell response pathways to cytotoxic accumulation of protein aggregates. Genes Dev. 2007;21:2172–2181. doi: 10.1101/gad.436407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Cepko CL. HDAC4 regulates neuronal survival in normal and diseased retinas. Science. 2009;323:256–259. doi: 10.1126/science.1166226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz JC, Tseng HC, Goldman JA, Shih H, Tsai LH. Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron. 2003;40:471–483. doi: 10.1016/s0896-6273(03)00627-5. [DOI] [PubMed] [Google Scholar]
- D’Mello SR. Histone deacetylases as targets for the treatment of human neurodegenerative diseases. Drug News Perspect. 2009;22:513–524. doi: 10.1358/dnp.2009.9.1428871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewachter I, Reverse D, Caluwaerts N, Ris L, Kuiperi C, Van den Haute C, Spittaels K, Umans L, Serneels L, Thiry E, Moechars D, Mercken M, Godaux E, Van Leuven F. Neuronal deficiency of presenilin 1 inhibits amyloid plaque formation and corrects hippocampal long-term potentiation but not a cognitive defect of amyloid precursor protein [V717I] transgenic mice. J Neurosci. 2002;22:3445–3453. doi: 10.1523/JNEUROSCI.22-09-03445.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Graus-Porta D, Blaess S, Senften M, Littlewood-Evans A, Damsky C, Huang Z, Orban P, Klein R, Schittny JC, Muller U. Beta1-class integrins regulate the development of laminae and folia in the cerebral and cerebellar cortex. Neuron. 2001;31:367–379. doi: 10.1016/s0896-6273(01)00374-9. [DOI] [PubMed] [Google Scholar]
- Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hageman J, Rujano MA, van Waarde MA, Kakkar V, Dirks RP, Govorukhina N, Oosterveld-Hut HM, Lubsen NH, Kampinga HH. A DNAJB chaperone subfamily with HDAC-dependent activities suppresses toxic protein aggregation. Mol Cell. 2010;37:355–369. doi: 10.1016/j.molcel.2010.01.001. [DOI] [PubMed] [Google Scholar]
- Iwata A, Riley BE, Johnston JA, Kopito RR. HDAC6 and micro-tubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem. 2005;280:40282–40292. doi: 10.1074/jbc.M508786200. [DOI] [PubMed] [Google Scholar]
- Kazantsev AG, Thompson LM. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat Rev Drug Discov. 2008;7:854–868. doi: 10.1038/nrd2681. [DOI] [PubMed] [Google Scholar]
- Ma C, D’Mello SR. Neuroprotection by histone deacetylase-7 (HDAC7) occurs by inhibition of c-jun expression through a deacetylase-independent mechanism. J Biol Chem. 2011;286:4819–4828. doi: 10.1074/jbc.M110.146860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majdzadeh N, Morrison BE, D’Mello SR. Class IIA HDACs in the regulation of neurodegeneration. Front Biosci. 2008a;13:1072–1082. doi: 10.2741/2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majdzadeh N, Wang L, Morrison BE, Bassel-Duby R, Olson EN, D’Mello SR. HDAC4 inhibits cell-cycle progression and protects neurons from cell death. Dev Neurobiol. 2008b;68:1076–1092. doi: 10.1002/dneu.20637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison BE, Majdzadeh N, Zhang X, Lyles A, Bassel-Duby R, Olson EN, D’Mello SR. Neuroprotection by histone deacetylase-related protein. Mol Cell Biol. 2006;26:3550–3564. doi: 10.1128/MCB.26.9.3550-3564.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison BE, Majdzadeh N, D’Mello SR. Histone deacetylases: focus on the nervous system. Cell Mol Life Sci. 2007;64:2258–2269. doi: 10.1007/s00018-007-7035-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva AA, Jr, Jiang M, Lam T, Smith KL, Swann JW. Novel hippocampal interneuronal subtypes identified using transgenic mice that express green fluorescent protein in GABAergic interneurons. J Neurosci. 2000;20:3354–3368. doi: 10.1523/JNEUROSCI.20-09-03354.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, Schwartz SL, DiProspero NA, Knight MA, Schuldiner O, Padmanabhan R, Hild M, Berry DL, Garza D, Hubbert CC, Yao TP, Baehrecke EH, Taylor JP. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–863. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- Pfister JA, Ma C, Morrison BE, D’Mello SR. Opposing effects of sirtuins on neuronal survival: SIRT1-mediated neuroprotection is independent of its deacetylase activity. PLoS ONE. 2008;3:e4090. doi: 10.1371/journal.pone.0004090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleiman SF, Basso M, Mahishi L, Kozikowski AP, Donohoe ME, Langley B, Ratan RR. Putting the “HAT” back on survival signalling: the promises and challenges of HDAC inhibition in the treatment of neurological conditions. Expert Opin Invest Drugs. 2009;18:573–584. doi: 10.1517/13543780902810345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Schutz G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet. 1999;23:99–103. doi: 10.1038/12703. [DOI] [PubMed] [Google Scholar]
- Vega RB, Matsuda K, Oh J, Barbosa AC, Yang X, Meadows E, McA-nally J, Pomajzl C, Shelton JM, Richardson JA, Karsenty G, Olson EN. Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell. 2004;119:555–566. doi: 10.1016/j.cell.2004.10.024. [DOI] [PubMed] [Google Scholar]
- Yang XJ, Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol. 2008;9:206–218. doi: 10.1038/nrm2346. [DOI] [PMC free article] [PubMed] [Google Scholar]