

Abstract
Objective
To assess the off-target effects of the histone deacetylase inhibitor (HDACi) suberoylanilide hydroxamic acid (SAHA) in human primary CD4+ T cells.
Design
A pharmacologically relevant concentration (340 nmol/l) of SAHA was shown to significantly increase histone hyperacetylation by 24 h and this length of treatment was selected to determine its impact on gene expression in primary CD4+ T cells.
Methods
Illumina Beadchips for microarray gene expression analysis were used to analyze differential gene expression between cells treated or not with SAHA with a paired analysis using multivariate permutation tests. Gene ontology, biological pathway and protein interaction network analyses were used to identify the higher order biological processes affected by SAHA treatment.
Results
Modest modulation by SAHA was observed for 1847 genes with 80% confidence level of no more than 10% false positives. A thousand genes were upregulated by SAHA and 847 downregulated. Pathways and gene ontologies overrepresented in the list of differentially expressed genes included Glycolysis/Gluconeogenesis, tRNA Modification, and the Histone Acetyltransferase Complex. Protein interaction network analysis revealed that transcription factor c-Myc, which was downregulated by SAHA treatment at the mRNA level, interacts with a number of SAHA-responsive genes.
Conclusions
The effects on transcription by SAHA were sufficiently modest to support trials to activate HIV replication as part of an eradication strategy. SAHA did not appear to modulate proliferative or apoptotic processes to a great extent, which might impact the ability of patients to eradicate the virus reservoir following activation by HDACi treatment.
Keywords: gene expression, histone deacetylase inhibitor, HIV, microarrays, primary CD4+ T cells, suberoylanilide hydroxamic acid, vorinostat
Introduction
The ability of HIV to establish persistent latent reservoirs represents the greatest obstacle to eradicating this virus in infected individuals on combination antiretroviral therapy (cART). Strategies have been proposed to coax HIV out of latency using compounds that activate viral replication, while preventing viral spread in the presence of continuing cART, so that infected cells may be recognized and killed by the host immune system [1,2]. Histone deacetylase (HDAC) inhibitors (HDACis) are one class of compounds shown to successfully activate HIV replication in cell lines [3–8]. Among HDACis, a lot of focus in HIV research has been placed on suberoylanilide hydroxamic acid (SAHA) and valproic acid (VPA). The former has already been approved by FDA for treatment of cutaneous T-cell lymphoma [9] and the latter has been used clinically as an antiepileptic drug for over four decades [10]. Recent studies demonstrated that SAHA and VPA can reactivate HIV replication ex vivo in primary resting CD4+ T cells from patients [4,11,12] and SAHA has already been successfully used to disrupt HIV latency in patients on suppressive cART [13].
HDACi treatment inhibits deacetylation and shifts the balance of the histone state toward acetylation, thus, opening chromatin structure and activating transcription from both the HIV promoter [14] and a number of host genes [15]. To investigate the potential safety risks of these drugs, it is important to identify their off-target effects in the primary cell types targeted by HIV. For example, HDACis are successful in cancer treatment because they induce growth arrest, differentiation and apoptosis of transformed cells [16–20]. However, with respect to eradicating HIV, apoptosis of uninfected bystander CD4+ T cells would be detrimental, as it may result in further reduction of CD4+ T cells in patients that are already immunologically compromised. In contrast, HDACi-induced activation and proliferation of immune cells may be detrimental by leading to an increased pool of cells susceptible to HIV infection. Previous gene expression profiling studies have mainly utilized cell lines treated with high doses of HDACis and demonstrated modulation of between 2 and 24.8% of all genes assayed [17,19–25]. As transformed cell lines were used, it was not surprising that genes and pathways involved in proliferation and apoptosis were identified [20,23,24]. Even though primary cells treated with HDACis are thought to be less prone to apoptosis compared with cell lines [26,27], a study performed by Moreira et al. [28] suggests that murine primary splenic CD4+ T cells undergo apoptosis when treated with the HDACi trichostatin A. A better understanding of the off-target effects of HDACis on gene expression in human primary immune cells relevant to HIV infection (i.e., CD4+ T cells) at concentrations achievable in HIV-infected patients is required. Therefore, gene expression changes in human primary CD4+ T cells treated with SAHA at pharmacologically achievable concentrations (340 nmol/l) were measured.
Materials and methods
Isolation of human primary CD4+ T cells
Peripheral blood from a total of nine healthy donors was collected by venipuncture according to institutional review board approved protocols into polypropylene syringes containing sodium heparin. Primary CD4+ T cells were isolated using RosetteSep CD4+ T-cell enrichment cocktail (StemCell Technologies Inc., Vancouver, Canada). All isolations had CD4+ T-cell purity greater than 95% as assessed by flow cytometry. If more than 10% of CD4+ T cells from a particular donor exhibited activation (i.e. HLA-DR+), the donor was excluded from the study. Primary CD4+ T cells were incubated overnight at 37°C, 5% CO2 in RPMI 1640 medium supplemented with 5% human AB serum and were subsequently resuspended to a concentration of 2.5 million cells per ml for treatments with HDACis (further details in the Supplemental Methods, http://links.lww.com/QAD/A271).
Histone deacetylase inhibitor treatment
Two separate experiments were performed involving HDACi treatment. The first was over a time course to assess histone acetylation, and the second was to analyze gene expression by microarray analysis. Three donors were used for the time course experiment. Five and 10 million primary CD4+ T cells were isolated from each donor for RNA and protein extraction, respectively, after 6, 24, or 48 h of treatment with SAHA (340 nmol/l) or VPA (40 μmol/l). As dimethyl sulfoxide (DMSO) was used to solubilize SAHA, the untreated controls and VPA-treated cells were also exposed to this solvent to control for its presence. For microarray gene expression analysis, cells from additional six donors were treated with SAHA (solubilized in DMSO) or left untreated (but exposed to DMSO) for 24 h, and 5 million primary CD4+ T cells were collected for RNA extraction. The RNA samples from the six donors were combined with the 24 h SAHA-treated samples from the original three donors for a total of nine donors for microarray analysis.
Immunoblot analysis
Whole cell extracts were prepared using RIPA buffer (Teknova, Inc., Hollister, California, USA). Protein concentrations were determined using the bicinchoninic acid (BCA) assay (Thermo Scientific, Inc., Waltham, Massachusetts, USA). Five micrograms of protein were used for immunoblot analyses with antihistone H3 (Abcam, Inc., Cambridge, United Kingdom) and anti-acetylated histone H3 (Millipore, Inc., Billerica, Massachusetts, USA) antibodies. For fluorescence detection, Alexa fluor 680 goat antimouse (Molecular Probes, Inc., Eugene, Oregon, USA) and IRDye 800 goat antirabbit (Rockland Immunochemicals, Inc., Boyertown, Pennsylvania, USA) antibodies were used. Images were obtained on the Odyssey infrared imaging system (LiCor Biosciences, Inc., Lincoln, Nebraska, USA) and quantified using ImageJ version 1.45 according to the Gel Analysis method outlined in the ImageJ documentation (http://rsb.info.nih.gov/ij/docs/menus/analyze.html#gels). Significant differences in the levels of histone acetylation were assessed across the time course of exposure to SAHA or VPA treatment using a two-way analysis of variance (ANOVA) followed by a Tukey post-hoc test.
RNA extraction and microarrays
RNA was extracted from SAHA-treated and untreated controls for nine donors using Qiagen RNA isolation kits (Qiagen Inc., Valencia, California, USA) according to manufacturer’s instructions. RNA concentration was assessed using the NanoDrop 1000 (Thermo Scientific, Waltham, Massachusetts, USA) and RNA integrity numbers (RINs) were determined using the Agilent 2100 Bioanalyzer (Agilent Technologies Inc., Santa Clara, California, USA). The mean RIN for the 18 samples was 9.1 with a SD of ±0.47. All RINs were more than eight and deemed of sufficient quality for microarray analysis. cRNA preparations and hybridizations to the HT12 Beadchips v3 from Illumina, Inc. (San Diego, California, USA) were performed by Asuragen, Inc. (Austin, Texas, USA). Gene expression data are available at the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE41504.
Analysis of microarray gene expression data
Microarray gene expression data for SAHA-treated and untreated samples paired for nine donors was subjected to quality control assessment and normalization as described [29]. Genes differentially expressed between the SAHA-treated and untreated conditions and pathways overrepresented for the differentially expressed genes were identified using Biometric Research Branch (BRB)-Array Tools [30] developed by the BRB at the Division of Cancer Treatment and Diagnosis at the National Cancer Institute. Gene Ontology analysis was performed using the Biological Networks Gene Ontology (BiNGO) plugin version 2.42 for Cytoscape version 2.7.0 [31]. Protein–protein and protein–DNA interactions between the protein products of the differentially expressed genes were determined using MetaCore (GeneGo, St. Joseph, Michigan, USA). For further details on the statistical analyses performed during microarray data analysis please refer to the Supplemental Methods, http://links.lww.com/QAD/A271.
Validation of gene expression by real-time quantitative polymerase chain reaction
Nineteen targets were chosen for real-time quantitative PCR (RT-qPCR) confirmation of gene expression: CDH23, HLA-DMB, LGALS1, SEPT4, PDE5A, FGFRL1, DLL1, KAT5, ING3, EPC1, MYST4, ING5, BRPH3, BRD1, PHF15, PHF17, MYC, JUN, and BCL (please refer to the Supplemental Methods for primer/probe information, http://links.lww.com/QAD/A271). RNA was reverse transcribed using qScript™ cDNA SuperMix (Quanta Biosciences, Inc., Gaithersburg, Maryland, USA) and 50 ng of RNA was used for each reaction, which were performed in duplicate. The RPL27 housekeeping gene (TaqMan Gene Expression Assay Hs01652274_gH) was used to normalize input based on high expression of this gene with small variation across experimental conditions [32]. RT-qPCR reactions were performed as described [33]. Fold changes were obtained using DataAssist software version 2.0 (ABI) using the 2−ΔΔCt method. To determine significance, a one-tailed paired t-test was performed using normalized Ct values (target Ct – RPL27 Ct) between SAHA-treated and untreated samples.
Results
Suberoylanilide hydroxamic acid treatment of CD4+ T cells for 24 h leads to a significant increase in histone acetylation
Concentrations of SAHA (340 nmol/l) and VPA (40 μmol/l) for these experiments were chosen based on measurements of free drug concentration in tissue culture conditions to determine concentrations comparable to those achievable in patients [11]. To select a time point for microarray gene expression analysis, initial experiments were performed to identify the minimum HDACi treatment time required to detect a significant increase in histone acetylation. CD4+ T cells isolated from three healthy donors were treated with SAHA or VPA for 6, 24, or 48 h or were left untreated. Immunoblot analyses with antibodies against acetylated versus total H3 histones (Fig. 1 and S1, http://links.lww.com/QAD/A271) showed that hyperacetylation of H3 histone was significant (P <0.05) after 24–48 h but not 6 h (P =0.6) of SAHA treatment (340 nmol/l). There was no significant change in histone acetylation levels at any time point after VPA treatment (40 μmol/l) (Fig. 1), and thus this compound was not selected for microarray gene expression analysis.
Fig. 1. Suberoylanilide hydroxamic acid but not valproic acid treatment leads to a significant increase in histone acetylation.
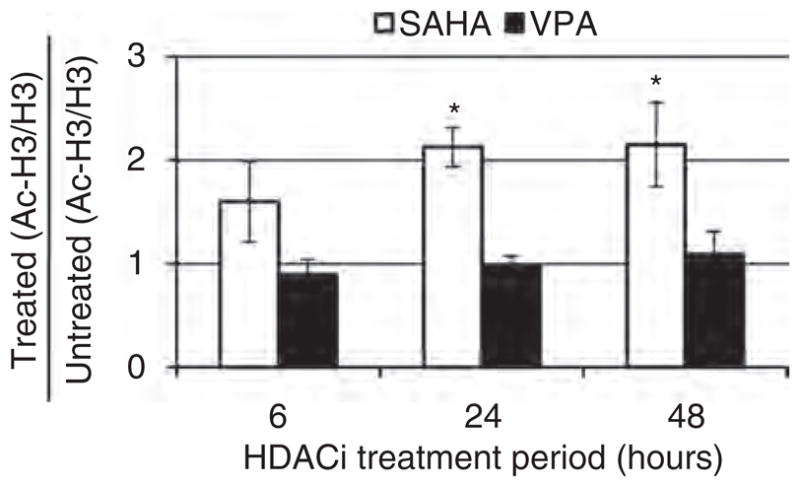
Primary CD4+ T cells from three donors were treated for 6, 24, or 48 h with suberoylanilide hydroxamic acid (SAHA) (340 nmol/l), valproic acid (VPA) (40 μmol/l), or left untreated (i.e., DMSO only, which was the solvent used to dissolve SAHA). Total protein was extracted and 5 μg used for immunoblot analysis with antiacetylated histone H3 (Ac-H3) and antitotal histone H3 (H3) antibodies. The bands were quantified using ImageJ software version 1.45. The fold changes were obtained by dividing the SAHA or VPA band intensities by the band intensities for the untreated control after normalization of the acetylated to total histone H3. The error bars represent standard error of the mean. Significant changes (P <0.05) as assessed by 2-way ANOVA with a Tukey post-hoc test are denoted by an asterisk (*).
Suberoylanilide hydroxamic acid treatment has limited effects on gene expression in CD4+ T cells
A SAHA treatment period of 24 h was selected for microarray analysis, as this was the minimum time required to induce a significant increase in histone acetylation. Gene expression in CD4+ T cells was compared between the SAHA-treated and untreated controls for nine donors. Paired multivariate permutation tests identified 2008 out of 48 803 probes (4.1%) on the microarray platform that were significantly differentially expressed, and corresponded to 1847 unique genes (1000 upregulated and 847 downregulated; Table S1, http://links.lww.com/QAD/A271). The magnitude of modulation induced by SAHA was modest, with only 38 genes displaying more than a two-fold change (Table S2, http://links.lww.com/QAD/A271). The greatest positive fold change was 3.23 for HLA-DMB and the greatest negative fold change was −2.47 for CDH23. Among genes that were upregulated more than two-fold, several were related to proliferation, differentiation and apoptosis (LGALS1, SEPT4, PDE5A, FGFRL1, and DLL1). These genes were selected for validation by RT-qPCR (Fig. 2).
Fig. 2. Validation of gene expression by real-time quantitative PCR.

Fold changes of gene expression between the SAHA-treated and untreated control groups are shown for real-time quantitative PCR (RT-qPCR) (black bars) and microarrays (grey bars). All the genes were found to be significantly modulated by SAHA treatment by microarray analysis. Genes marked with a double asterisk (**) were confirmed by RT-qPCR to be significantly modulated by SAHA (P <0.05, one-tailed paired t-test), and genes marked with a single asterisk (*) approached significance (i.e., 0.05 <P <0.1). The association of genes with functional categories indicating the criteria by which they were selected for RT-qPCR validation is shown on the left of the figure. HLA-DMB had the highest fold change and is a marker of immune activation.
Glucose metabolism, tRNA modification and histone acetyltransferase complexes were significantly modulated by suberoylanilide hydroxamic acid
Because some of the genes functioning in apoptosis, proliferation and immune cell activation were detected among genes with the highest fold changes, it was important to verify whether these processes were affected by SAHA on a broader scale, that is, at the pathway level. Pathway analysis performed in BRB-Array Tools and gene ontology analysis performed using BiNGO did not identify any pathway or gene ontology terms related to apoptosis, proliferation or immune activation. A single Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway, Glycolysis/Gluconeogenesis (LS P =0.011, KS P =0.016, see details on P-value definitions in the Supplemental Methods, http://links.lww.com/QAD/A271) was identified as significantly overrepresented for differentially expressed genes. The majority of the genes that mapped to this pathway were upregulated by SAHA. Significant gene ontology terms included those relating to nucleic acid metabolic processes, specifically tRNA Modification in the Biological Process category (Fig. S2A, http://links.lww.com/QAD/A271), as well as gene ontology terms related to transcriptional regulation and histone acetyltransferase complexes (HATs) in the Cellular Component category (Fig. S2B, http://links.lww.com/QAD/A271). Only genes that encode components of the MYST family of HATs (i.e. Tip60 and MOZ/MORF) and not the other HAT families were identified. The majority of the genes that mapped to gene ontology terms associated with HATs were down-regulated by SAHA and the following genes associated with HAT complexes were selected for validation by RT-qPCR: BRD1, EPC1, ING3, KAT5, PHF15, PHF17, MYST4, ING5, and BRPF3 (Fig. 2).
Suberoylanilide hydroxamic acid modulates gene expression by downregulating c-Myc
A protein interaction network was constructed using MetaCore in order to elucidate the potential mechanisms by which SAHA modulates gene expression (Fig. 3). This protein interaction network depicts those differentially expressed genes whose protein products interact at the protein–protein or protein–DNA level. Major hubs in this network were defined as those that interact with more than 10 proteins and included: c-Myc, AP-1, c-Jun, ZNF42, EZH2, BCL6, and CDK2. These hubs correspond to transcription factors modulated by SAHA treatment that regulate the expression of multiple SAHA target genes. Although the largest hub, c-Myc, was only downregulated 1.4-fold by SAHA treatment, this may have a large effect on target gene expression as even small fold changes in transcription factors can have wide ranging effects on gene expression. Specifically, c-Myc interacts with 114 of the SAHA-modulated genes (at the 1.4-fold cutoff) (see Table S3, http://links.lww.com/QAD/A271 for lists of genes that interact with each of the major hubs). The hubs, MYC, JUN and BCL6 were chosen for RT-qPCR validation (Fig. 2).
Fig. 3. Direct protein interaction network constructed from the products of suberoylanilide hydroxamic acid-modulated genes.

MetaCore was used to identify protein-protein and protein-DNA interactions between the protein products of SAHA modulated genes. For visualization purposes, only genes with a fold change <+1.4 or >−1.4 were used for constructing the protein interaction network. Each node represents a gene and nodes with multiple connections form hubs. The largest hubs are c-Myc, AP-1, c-Jun, ZNF42 (MZF1), Bcl-6, EZH2, CDK2, and NOTCH1 and correspond to transcription factors. Green and red lines refer to positive and negative regulation, respectively, whereas grey lines depict unspecified effects. Upregulated genes are depicted in red and downregulated genes in blue as indicated by the scale bar. The types of interactions are indicated with a letter code on top of the lines and can be visualized by zooming using the on-line version of the article: B, binding; C, cleavage; CM, covalent modifications; +p, phosphorylation; –p, dephosphorylation; T, transformation; TR, transcription regulation; GR, group relation; CS, complex subunit.
Validation of gene expression using RT-qPCR
The differential expression of 19 genes between SAHA-treated and untreated CD4+ T cells detected by microarray analysis was verified by RT-qPCR (Fig. 2). Differentially expressed genes selected for RT-qPCR analysis included genes with the highest negative (CDH23) and positive (HLA-DMB) fold changes, genes with fold changes greater than two that are associated with proliferation, differentiation and apoptosis (LGALS1, SEPT4, PDE5A, FGFRL1, DLL1), genes encoding components of the HAT complexes (KAT5, ING3, EPC1, MYST4, ING5, BRPH3, BRD1, PHF15, PHF17), and major hubs from the protein interaction network (MYC, JUN, BCL6). The direction of fold change from microarray analysis was confirmed by RT-qPCR for all targets, and thus a one-tailed paired t-test was used to determine significance. Expression of CDH23, HLA-DMB, SEPT4, PDE5A, KAT5, ING3, EPC1, MYST4, ING5, BRPH3, PHF17, and MYC was validated at the significance level of P <0.05, whereas LGALS1, DLL1, JUN, and BCL6 approached signifi-cance (i.e., 0.05 <P <0.1). FGFRL1, BRD1, and PHF15 were significantly differentially expressed as detected by microarray analysis but not by RT-qPCR analysis (P >0.1).
Discussion
Gene expression profiling of the entire transcriptome of human primary CD4+ T cells treated with a pharmacologically relevant concentration of SAHA (340 nmol/l) suggests that the effects of SAHA on gene expression are modest. Even though 1847 unique genes were modulated by SAHA, this number represented only 4.1% of all genes profiled. In comparison, in human lymphoid CEM cells treated with 2.5 μmol/l SAHA (not a pharmacologically achievable concentration) for 16 h, 1482-modulated genes were identified that comprised 14.9% of the 9954 genes profiled in that study [24]. Moreover, the fold changes observed here were low, compared with those observed in malignant cells treated with higher concentrations of SAHA (maximum fold change 3.23 versus 33.9 [22]). Interestingly, the degree at which the host genes were affected by low dose of SAHA was similar to the degree of transcriptional activation from HIV promoter with a comparable dose of 500 nmol/l in J89 cells [11].
Importantly, SAHA-modulated genes associated with proapoptotic, proliferative, and immune activating responses were not enriched at the pathway level, which bodes well for using this compound to activate the latent HIV reservoir in infected patients. However, at the individual gene level, a few SAHA-modulated genes associated with these processes were identified. For example, the highest positive fold change was observed for HLA-DMB gene (i.e. 3.23 as detected by microarrays and 4.71 as detected by RT-qPCR), a subunit of a complex that functions in the peptide loading of MHC class II molecules. However, overall MHC class II genes were not upregulated with SAHA treatment of 340 nmol/l. The CD69 marker of early activation was upregulated slightly at the transcript level (1.7-fold), but without a corresponding increase in cell surface protein expression as assessed by mean peak antibody fluorescence intensity with flow cytometry (P =0.69, Fig. S3, http://links.lww.com/QAD/A271). A proapoptotic gene LGALS1 (galectin 1) was upregulated more than two-fold, but none of the other genes encoding galectins (LGALS2,3,7,8,9, and 12) that are able to induce apoptosis in blood cells [34] or other proapoptotic molecules such as caspases were upregulated by SAHA. On the contrary, a gene encoding an antiapoptotic molecule PDE5A [35] was also upregulated, indicating that any effect SAHA may have on apoptosis was nonspecific and bidirectional. It is possible that apoptosis can be induced without additional transcriptional responses but via activation of intrinsic or extrinsic apoptotic pathways. However, the viability of the CD4+ T cells treated with 340 nmol/l SAHA for 24 h as measured by Trypan blue staining, was similar to that of the control cells treated with DMSO (>90%, data not shown). Taken together, these results suggest that SAHA may not have any major detrimental effects in human primary CD4+ T cells, and thus represents a viable strategy for activating HIV replication in vivo to purge the latent reservoir.
Analysis of pathways and gene ontology terms over-represented in the set of genes modulated by SAHA suggests potential mechanisms that CD4+ T cells may utilize to regain control of acetylation states following exposure to inhibitors of histone acetylation such as SAHA. A reduction in the expression of genes encoding components of HAT complexes was noted following SAHA treatment. Among the four families of HATs (p300/CBP, PCAF/GCN5, MYST, and nuclear recepor coactivators), genes encoding components of the MYST family were selectively downregulated. Among the MYST family members, three out of four components (75%) of the MOZ/MORF complex and four out of 15 components (27%) of the Tip60 complex were identified. Clearly, the exposure of CD4+ T cells to HDACis such as SAHA disrupts the homeostasis of acetylation and it is plausible that the downregulation of HAT complexes represents an attempt to regain control of acetylation levels. Another group of gene ontology terms identified as significantly overrepresented for genes downregulated by SAHA were related to tRNA modification. These genes encode enzymes with a broad spectrum of functions and are required for multiple types of tRNA modification (e.g., methylation by METTL1 and WDR4, 2-thiolation by ATPBD3 and URM1, and pseudouridine modification by PUS1 and PUS3). With respect to genes upregulated by SAHA, multiple enzymes in the Glycolysis/Gluconeogenesis pathway were upregulated (PFKM, PFKP, ALDOA, ALDOC, GAPDH, ENO3). One of the downstream molecules generated as the result of glycolysis is acetyl coenzyme A (acetyl-CoA), which is the source of acetyl groups for histone acetylation. As HDACis cause a shift in the histone state towards acetylation, the demand for acetyl-CoA increases, and it is possible that CD4+ T cells replenish this molecule by upregulating components of the glycolysis pathway. Previous studies have shown that upon treatment with SAHA or VPA the soluble pool of acetyl-CoA was depleted, while more of the acetyl-CoA was found in the protein-associated fraction over time [36]. Overall, the biological importance of SAHA-modulated genes whose products constitute HAT complexes or code for enzymes capable of modifying tRNAs or facilitating glycolysis requires further investigation.
The analysis of protein-DNA interactions indicated that the transcription factor c-Myc, whose expression was downregulated by SAHA treatment (fold change −1.46 by microarray analysis and −1.72 by RT-qPCR) regulated the expression of a large number of SAHA-modulated genes (Fig. 3). It is possible that in many cases the effect of SAHA on gene expression is indirect, and occurs via transcription factors such as c-Myc. It is known that c-Myc has discordant effects on gene expression leading to either upregulation or downregulation depending on the target (Fig. 3). For example, histone acetylation and gene upregulation are facilitated through the recruitment of HAT complexes to chromatin by c-Myc and its cofactor transformation/transcription domain-associated protein [37,38]. Downregulation of gene expression is facilitated when c-Myc binds to and interferes with transcriptional activators such as Miz-1 and Sp1 [39,40] or recruits HDAC1 to promoter regions [41]. Furthermore, c-Myc inhibits transcription from the HIV LTR in resting CD4+ T cells by recruiting HDAC1 [42], and thus the downregulation of c-Myc by SAHA may serve as another mechanism contributing to HIV reactivation. Further studies are required to understand which genes are modulated directly as a result of promoter acetylation due to SAHA treatment and which genes are modulated indirectly as a result of SAHA-induced changes in the expression of transcription factors. Interestingly, a number of genes including MYC (Table S4, http://links.lww.com/QAD/A271) were modulated by a recently described bromodomain inhibitor JQ1 [43,44], a compound also capable of activating HIV transcription [45], suggesting that the two classes of compounds may have common mechanisms of action.
The present study is the first to our knowledge to identify SAHA-modulated genes in a primary immune cell (i.e., CD4+ T cells) at a pharmacologically relevant concentration (340 nmol/l). However, it would be important to investigate the effects of SAHA on other cells of the immune system, such as CD8+ T cells, whose cytotoxic capabilities are required to clear latently infected CD4+ T cells once they have been activated by HDACi treatment. In our study, 24 h of SAHA treatment was selected, as this was the shortest treatment period for which histone acetylation was significantly elevated compared with untreated controls (Fig. 1). In SAHA-treated patients, drug concentration peaks in the plasma much earlier, between 2 and 10 h following administration (SAHA package insert, Merck and Co., 2006). Furthermore, SAHA is metabolized with a half life of approximately 2 h [46] and is almost completely cleared by 24 h. Thus, future studies are required to identify genes modulated by either pulse or more sustained exposures in order to further assess the implications of using HDACis for the eradication of the latent HIV reservoir. The results of this study are very promising, because they suggest that 24 h of exposure to SAHA will not have dramatic off-target effects on host gene expression with consequent widespread effects on cell proliferation and apoptosis.
Supplementary Material
Acknowledgments
This research was supported by the grants from the National Institutes of Health (AI080193, AI096113 and AI36214) and from the Department of Veterans Affairs (BX001160). N.B.B. was supported by a T32 training grant (AI007384) during the conduct of this research.
This work was performed with the support of the Genomics and Flow Cytometry Cores at the UCSD Center for AIDS Research (AI36214), the San Diego Veterans Medical Research Foundation, and other research grants from the National Institutes of Health (AI080193 and AI096113, Collaboratory of AIDS Researchers for Eradication grant) and the Department of Veterans Affairs (BX001160). N.B.B. was supported by Ruth L. Kirschstein National Research Service Award (NRSA) Institutional Research Training Grants (2T32AI007384–21A1). This material is based upon work supported in part by the Department of Veterans Affairs (VA), Veterans Health Administration, Office of Research and Development. The views expressed in this article are those of the authors and do not necessarily reflect the position or policy of the Department of Veterans Affairs or the United States government. Merck and Co., Inc. graciously provided SAHA used in this study.
For the remaining authors, no conflicts of interest were declared.
Conflicts of interest
D.D.R. is a consultant for Merck and Co. Inc., Theraclone, Myriad, Bristol-Myers Squibb, Anadys Pharmaceuticals, Inc., Gilead Sciences, Hoffman-La Roche Inc., Monogram Biosciences, Biota, Chimerx, Idenix, and Gen-Probe, but these roles are not in conflict with the data presented in this manuscript.
C.A.S. has previously received research support from Merck Research Laboratories for an unrelated project.
Author contributions: N.B.B., D.D.R., C.A.S. and C.H.W. conceived and designed the study; J.X.Z., A.S., V.L., and V.H.T. performed the experiments; N.B.B., J.X.Z. and A.S. analyzed the data; N.B.B. and C.H.W. wrote the manuscript. All authors reviewed the manuscript and accepted it for publication
References
- 1.Demonté D, Quivy V, Colette Y, Van Lint C. Administration of HDAC inhibitors to reactivate HIV-1 expression in latent cellular reservoirs: implications for the development of therapeutic strategies. Biochem Pharmacol. 2004;68:1231–1238. doi: 10.1016/j.bcp.2004.05.040. [DOI] [PubMed] [Google Scholar]
- 2.Matalon S, Rasmussen TA, Dinarello CA. Histone deacetylase inhibitors for purging HIV-1 from latent reservoir. Mol Med. 2011;17:466–472. doi: 10.2119/molmed.2011.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yin H, Zhang Y, Zhou X, Zhu H. Histonedeacetylase inhibitor Oxamflatin increase HIV-1 transcription by inducing histone modification in latently infected cells. Mol Biol Rep. 2011;38:5071–5078. doi: 10.1007/s11033-010-0653-6. [DOI] [PubMed] [Google Scholar]
- 4.Contreras X, Schweneker M, Chen C-S, McCune JM, Deeks SG, Martin J, et al. Suberoylanilide Hydroxamic Acid Reactivates HIV from Latently Infected Cells. J Biol Chem. 2009;284:6782–6789. doi: 10.1074/jbc.M807898200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ying H, Zhang Y, Lin S, Han Y, Zhu H-Z. Histone acetylase inhibitor Scriptaid reactivates latent HIV-1 promoter by inducing histone modification in in vitro latency cell lines. Int J Mol Med. 2010;26:265–272. doi: 10.3892/ijmm_00000461. [DOI] [PubMed] [Google Scholar]
- 6.Archin NM, Keedy KS, Espeseth A, Dang H, Hazuda DJ, Margolis DM. Expression of latent human immunodeficiency type 1 is induced by novel and selective histone deacetylase inhibitors. AIDS. 2009;23:1799–1806. doi: 10.1097/QAD.0b013e32832ec1dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shehu-Xhilaga M, Rhodes D, Wightman F, Liu HB, Solomon A, Saleh S, et al. The novel histone deacetylase inhibitors metacept-1 and metacept-3 potently increase HIV-1 transcription in latently infected cells. AIDS. 2009;23:2047–2050. doi: 10.1097/QAD.0b013e328330342c. [DOI] [PubMed] [Google Scholar]
- 8.Van Lint C, Emiliani S, Ott M, Verdin E. Transcriptional activation and chromatin remodeling of the HIV-1 promoter in response to histone acetylation. EMBO J. 1996;15:1112–1120. [PMC free article] [PubMed] [Google Scholar]
- 9.Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous t-cell lymphoma. Oncologist. 2007;12:1247–1252. doi: 10.1634/theoncologist.12-10-1247. [DOI] [PubMed] [Google Scholar]
- 10.Perucca E. Pharmacological and therapeutic properties of valproate: a summary after 35 years of clinical experience. CNS Drugs. 2002;16:695–714. doi: 10.2165/00023210-200216100-00004. [DOI] [PubMed] [Google Scholar]
- 11.Archin NM, Espeseth A, Parker D, Cheema M, Hazuda D, Margolis DM. Expression of latent HIV induced by the potent HDAC inhibitor suberoylanilide hydroxamic acid. AIDS Res Hum Retroviruses. 2009;25:207–212. doi: 10.1089/aid.2008.0191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ylisastigui L, Archin NM, Lehrman G, Bosch RJ, Margolis DM. Coaxing HIV-1 from resting CD4 T cells: histone deacetylase inhibition allows latent viral expression. AIDS. 2004;18:1101–1108. doi: 10.1097/00002030-200405210-00003. [DOI] [PubMed] [Google Scholar]
- 13.Archin NM, Liberty AL, Kashuba AD, Choudhary SK, Kuruc JD, Crooks AM, et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature. 2012;487:482–485. doi: 10.1038/nature11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Colin L, Van Lint C. Molecular control of HIV-1 postintegration latency: implications for the development of new therapeutic strategies. Retrovirology. 2009;6:111. doi: 10.1186/1742-4690-6-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dokmanovic M, Marks PA. Prospects: histone deacetylase inhibitors. J Cell Biochem. 2005;96:293–304. doi: 10.1002/jcb.20532. [DOI] [PubMed] [Google Scholar]
- 16.Peart MJ, Tainton KM, Ruefli AA, Dear AE, Sedelies KA, O’Reilly LA, et al. Novel mechanisms of apoptosis induced by histone deacetylase inhibitors. Cancer Res. 2003;63:4460–4471. [PubMed] [Google Scholar]
- 17.He LZ, Tolentino T, Grayson P, Zhong S, Warrell RP, Jr, Rifkind RA, et al. Histone deacetylase inhibitors induce remission in transgenic models of therapy-resistant acute promyelocytic leukemia. J Clin Invest. 2001;108:1321–1330. doi: 10.1172/JCI11537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marks P, Richon VM, Breslow R, Rifkind RA. Histone deacetylase inhibitors as new cancer drugs. Curr Opin Oncol. 2001;13:477–483. doi: 10.1097/00001622-200111000-00010. [DOI] [PubMed] [Google Scholar]
- 19.Wozniak MB, Villuendas R, Bischoff JR, Aparicio CB, Martínez Leal JF, de La Cueva P, et al. Vorinostat interferes with the signaling transduction pathway of T-cell receptor and synergizes with phosphoinositide-3 kinase inhibitors in cutaneous T-cell lymphoma. Haematologica. 2010;95:613–621. doi: 10.3324/haematol.2009.013870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.LaBonte M, Wilson P, Fazzone W, Groshen S, Lenz H-J, Ladner R. DNA microarray profiling of genes differentially regulated by the histone deacetylase inhibitors vorinostat and LBH589 in colon cancer cell lines. BMC Med Genomics. 2009;2:67. doi: 10.1186/1755-8794-2-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mariadason JM, Corner GA, Augenlicht LH. Genetic reprogramming in pathways of colonic cell maturation induced by short chain fatty acids: comparison with trichostatin A, sulindac, and curcumin and implications for chemoprevention of colon cancer. Cancer Res. 2000;60:4561–4572. [PubMed] [Google Scholar]
- 22.Glaser KB, Staver MJ, Waring JF, Stender J, Ulrich RG, Davidsen SK. Gene expression profiling of multiple histone deacetylase (HDAC) inhibitors: defining a common gene set produced by HDAC inhibition in T24 and MDA carcinoma cell lines. Mol Cancer Ther. 2003;2:151–163. [PubMed] [Google Scholar]
- 23.Mitsiades CS, Mitsiades NS, McMullan CJ, Poulaki V, Shringarpure R, Hideshima T, et al. Transcriptional signature of histone deacetylase inhibition in multiple myeloma: biological and clinical implications. Proc Natl Acad Sci U S A. 2004;101:540–545. doi: 10.1073/pnas.2536759100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peart MJ, Smyth GK, van Laar RK, Bowtell DD, Richon VM, Marks PA, et al. Identification and functional significance of genes regulated by structurally different histone deacetylase inhibitors. Proc Natl Acad Sci U S A. 2005;102:3697–3702. doi: 10.1073/pnas.0500369102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Halsall J, Gupta V, O’Neill LP, Turner BM, Nightingale KP. Genes are often sheltered from the global histone hyperacetylation induced by HDAC inhibitors. PLoS One. 2012;7:e33453. doi: 10.1371/journal.pone.0033453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ungerstedt JS, Sowa Y, Xu WS, Shao Y, Dokmanovic M, Perez G, et al. Role of thioredoxin in the response of normal and transformed cells to histone deacetylase inhibitors. Proc Natl Acad Sci U S A. 2005;102:673–678. doi: 10.1073/pnas.0408732102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chang J, Varghese DS, Gillam MC, Peyton M, Modi B, Schiltz RL, et al. Differential response of cancer cells to HDAC inhibitors trichostatin A and depsipeptide. Br J Cancer. 2012;106:116–125. doi: 10.1038/bjc.2011.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moreira JM, Scheipers P, Sorensen P. The histone deacetylase inhibitor Trichostatin A modulates CD4+ T cell responses. BMC Cancer. 2003;3:30. doi: 10.1186/1471-2407-3-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Woelk CH, Beliakova-Bethell N, Goicoechea M, Zhao Y, Du P, Rought S, et al. Gene expression before HAART initiation predicts HIV-infected individuals at risk of poor CD4+ T cell recovery. AIDS. 2010;24:217–222. doi: 10.1097/QAD.0b013e328334f1f0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Simon R, Lam A, Li MC, Ngan M, Menenzes S, Zhao Y. Analysis of gene expression data using BRB-array tools. Cancer Informatics. 2007;3:11–17. [PMC free article] [PubMed] [Google Scholar]
- 31.Maere S, Heymans K, Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of Gene Ontology categories in Biological Networks. Bioinformatics. 2005;21:3448–3449. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
- 32.De Jonge HJ, Fehrmann RS, de Bont ES, Hofstra RM, Gerbens F, Kamps WA, et al. Evidence based selection of housekeeping genes. PLoS One. 2007;2:e898. doi: 10.1371/journal.pone.0000898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chana G, Lucero G, Salaria S, Lozach J, Du P, Woelk C, et al. Upregulation of NRG-1 and VAMP-1 in human brain aggregates exposed to clozapine. Schizophrenia Res. 2009;113:273–276. doi: 10.1016/j.schres.2009.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cummings RD, Liu F-T. In: Essentials of glycobiology. Varki A, Cummings RD, Esko JD, et al., editors. New York: Cold Spring Harbor Laboratory Press; 2009. [PubMed] [Google Scholar]
- 35.Serafini P, Meckel K, Kelso M, Noonan K, Califano J, Koch W, et al. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J Exp Med. 2006;203:2691–2702. doi: 10.1084/jem.20061104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wardell SE, Ilkayeva OR, Wieman HL, Frigo DE, Rathmell JC, Newgard CB, et al. Glucose metabolism as a target of histone deacetylase inhibitors. Mol Endocrinol. 2009;23:388–401. doi: 10.1210/me.2008-0179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frank SR, Parisi T, Taubert S, Fernandez P, Fuchs M, Chan H-M, et al. MYC recruits the TIP60 histone acetyltransferase complex to chromatin. EMBO. 2003;4:575–580. doi: 10.1038/sj.embor.embor861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McMahon SB, Wood MA, Cole MD. The essential cofactor TRRAP recruits the histone acetyltransferase hGCN5 to c-Myc. Mol Cell Biol. 2000;20:556–562. doi: 10.1128/mcb.20.2.556-562.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Staller P, Peukert K, Kiermaier A, Seoane J, Lukas J, Karsunky H, et al. Repression of p15INK4b expression by Myc through association with Miz-1. Nat Cell Biol. 2001;3:392–399. doi: 10.1038/35070076. [DOI] [PubMed] [Google Scholar]
- 40.Gartel AL, Ye X, Goufman E, Shianov P, Hay N, Najmabadi F, et al. Myc represses the p21(WAF1/CIP1) promoter and interacts with Sp1/Sp3. Proc Natl Acad Sci U S A. 2001;98:4510–4515. doi: 10.1073/pnas.081074898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Satou A, Taira T, Iguchi-Ariga SMM, Ariga H. A novel transrepression pathway of c-Myc. J Biol Chem. 2001;276:46562–46567. doi: 10.1074/jbc.M104937200. [DOI] [PubMed] [Google Scholar]
- 42.Jiang G, Espeseth A, Hazuda DJ, Margolis DM. c-Myc and Sp1 contribute to proviral latency by recruiting histone deacetylase 1 to the human immunodeficiency virus type 1 promoter. J Virol. 2007;81:10914–10923. doi: 10.1128/JVI.01208-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Delmore Jake E, Issa Ghayas C, Lemieux Madeleine E, Rahl Peter B, Shi J, Jacobs Hannah M, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, et al. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci. 2011;108:16669–16674. doi: 10.1073/pnas.1108190108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Banerjee C, Archin N, Michaels D, Belkina AC, Denis GV, Bradner J, et al. BET bromodomain inhibition as a novel strategy for reactivation of HIV-1. J Leukocyte Biol. 2012 doi: 10.1189/jlb.0312165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mann BS, Johnson JR, He K, Sridhara R, Abraham S, Booth BP, et al. Vorinostat for treatment of cutaneous manifestations of advanced primary cutaneous t-cell lymphoma. Clin Cancer Res. 2007;13:2318–2322. doi: 10.1158/1078-0432.CCR-06-2672. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.