

Abstract
Retinopathy of prematurity (ROP) is a potentially blinding disease affecting premature infants. ROP is characterized by pathological ocular angiogenesis or retinal neovascularization (NV). Models of ROP have yielded much of what is currently known about physiological and pathological blood vessel growth in the retina. The rat provides a particularly attractive and cost effective model of ROP. The rat model of ROP consistently produces a robust pattern of NV, similar to that seen in humans. This model has been used to study gross aspects of angiogenesis. More recently, it has been used to identify and therapeutically target specific genes and molecular mechanisms involved in the angiogenic cascade. As angiogenesis occurs as a complication of many diseases, knowledge gained from these studies has the potential to impact nonocular angiogenic conditions. This article provides historical perspective on the development and use of the rat model of ROP. Key findings generated through the use of this model are also summarized.
Keywords: Retinopathy of prematurity, Neovascularization, Angiogenesis, Rat model, Variable oxygen
ROP: Historical background
In 1942, Terry first described ROP as a disease of prematurity characterized by retinal neovascularization [1]. In the 1940s, there was an epidemic of blindness resulting from ROP, exposing the need for research focused on the identification and characterization of the pathogenesis of the disease. In 1951, Campbell proposed that the incidence of ROP was tied to the supplemental oxygen administered to premature infants with under-developed pulmonary function [2]. Subsequent studies confirmed the relationship between the clinical use of supplemental oxygen and ROP, and led to the treatment of premature infants with lower fractions of inspired oxygen (FiO2) [3–8]. However, while this new practice resulted in a decrease in the number of cases of ROP, there was a corresponding increase in the number of cases of hypoxemia-related cerebral palsy and death [9, 10]. Consequently, oxygen given to premature infants was rigorously monitored, improving morbidity and mortality outcomes. As a result of monitored oxygen regimens, the percentage of blindness attributed to ROP in certain patient groups dropped from 50% in 1950 to just 4% in 1965 [11]. The 1970s and 1980s saw a resurgence in the incidence of ROP, due to advances in neonatal intensive care, increasing the survival rates of very low-birth-weight premature infants [12–16].
It is estimated that each year 3,400 infants will suffer from ROP-related visual impairments and 650 will be blinded [17]. Thus, there exists a compelling reason to study the pathogenesis of ROP. Animal models are most commonly used for this purpose, but constant refinement of the models is necessary, taking into account emerging information about the pathogenesis of the human condition.
Human ROP: Pathogenesis
The development of retinal blood vessels is altered in infants suffering from ROP. The uterine environment has been shown to have a partial pressure of dissolved arterial oxygen (PaO2) of 30 mm Hg. This physiologic hypoxia effectively stimulates growth factor production, resulting in retinal vascular development. The exuterine environment has a PaO2 of 55–80 mm Hg [18]. This hyperoxic post-natal environment is believed initially to reduce the stimulus for growth factor production and may play a role in the retardation of developmental angiogenesis. This does not present a problem for full-term infants whose retinal vasculature has reached the retinal periphery and is fully developed. Premature infants, however, have an incompletely developed retinal vasculature at the time of birth and hyperoxia further limits vascular development, rendering them susceptible to ROP.
Retinopathy of prematurity is a biphasic disease. The first phase of ROP is characterized by vasoattenuation or the cessation of retinal vascular development. This occurs when the premature infant is born into a hyperoxic post-natal environment, and is worsened by supplemental oxygen therapy. Hyperoxia may be one factor that can slow retinal vascular development. As the vasculature is incompletely developed, it is unable to meet the increasing demands of the developing neuroretina, which leads to retinal hypoxia [19]. This “physiologic hypoxia” is made worse when oxygen therapy is terminated [20–23]. Retinal hypoxia leads to the second phase of ROP, characterized by vasoproliferation and preretinal NV. This preretinal NV predisposes the infant to intravitreal hemorrhages, retinal detachment, and subsequent vision loss.
It should be noted that fewer than 10% of infants with early stages of the disease progress to “threshold” ROP. In fact, even if the analysis is limited to infants with “prethreshold” disease (abnormal proliferation of blood vessels without rupture of the inner limiting membrane of the retina), threshold ROP develops in only 30–35% of these infants [21]. Remarkably, 65% of preterm infants receiving oxygen therapy develop threshold ROP during the course of therapy (Dale Phelps, personal communication, information derived from the STOP trial; 2004). Therefore, hypoxia resulting from the cessation of oxygen therapy is not the sole determinant of ROP pathogenesis. Developmental timing may regulate the responses of the immature retina to oxygen [24]. Human ROP involves a complex sequence of pathological events with the potential to be influenced by temporal patterns of gene expression as well as environmental factors related to clinical care.
Rat retinal vascular development
Rat retinal development follows a pattern similar to that of the human. As in the human fetus, the rat retina is one of the last tissues to be vascularized. The retinal vasculature derives from mesenchymal precursor cells of the hyaloid artery, and vascularization proceeds in a wave-like fashion, beginning at the optic disk and growing outwards until the vessels reach the retinal periphery. In the human, retinal vascularization is usually complete at the time of birth; in the rat, the process is completed at about post-natal day (P)15 [25]. The retinal vasculature of a newborn rat pup, therefore, resembles that of a preterm infant: largely avascular and highly susceptible to retinopathy. For this, and other reasons, investigators began to explore the use of the rat in modeling ROP pathogenesis.
Experimental models of ROP
In 1954, Patz first demonstrated preretinal NV in rats that had been exposed to a constant level of extreme hyperoxia [23]. Brands et al. also demonstrated abnormal vasoproliferation in rats that had been exposed to hyperoxia, followed by a period of time in room air [26]. Following their failed attempts to reproduce these findings, Ashton and Blach [27] questioned the claims of these investigators, describing their conclusions as “inadequately substantiated on the evidence provided,” due to the inconsistent nature of the vasoproliferative response. As a result, the rat model of ROP lost favor and its usefulness was not re-examined until years later.
Between 1988 and 1991, there were three reports of preretinal NV generated in rat models of ROP. At this time, there was not a standard oxygen treatment protocol, and all three investigative teams used a different means of assessing NV. Ricci and Calogero [28] exposed rats to 80% oxygen for the first 5 days of life, followed by room air for 5 days, and described “marked peripheral retinal neovascularization.” However, the investigators used ink-perfused, flat-mounted retinas to assess NV. This technique is not a reliable determinant of neovascular pathology. Ink only marks patent retinal vessels. It does not mark the abnormal, nonpatent lumena of neovascular tufts. Additionally, ink can leak from, and obscure, newly developed, fragile vessels. These drawbacks underestimate the extent of vasculopathy, rendering this method unreliable [29].
Ventresca et al. exposed rats to 80% oxygen for 10 days, followed by room air for 15 days [30]. Of the 20 oxygen-exposed rat pups, 16 demonstrated preretinal NV. This group used cross sections of retinas to assess NV. Similarly, Reynaud et al. [31] presented evidence of preretinal NV in rats raised in 80% oxygen for 6 days, followed by room air for 11 days, but no mention was made of the incidence of NV in their study population. Reynaud et al. stained retinas with ADPase to determine NV. This histochemical method, developed by Flower et al., marks ADPase activity of vascular endothelium and is not dependent on vessel patency [32]. The ADPase stain is a more accurate and reliable determinant of the presence and severity of NV.
Development of the current model
Over time, investigators began to explore the role of oxygen and of factors related to the therapeutic use of oxygen, in the pathogenesis of ROP [21, 33–38]. The PaO2 of premature infants can fluctuate rapidly, leading to alternating and severe episodes of hyperoxemia and hypoxemia [39]. Frequently, this fluctuation is related to the infants’ underlying disease (e.g., patent ductus arteriosus, apnea, metabolic acidosis, and bronchopulmonary dysplasia), but it may be caused by routine medical intervention [39]. This fact may explain the inconsistencies of previous studies conducted in the rat. It is likely that investigators who meticulously monitored the oxygen level and maintained it throughout the exposure saw little to no neovascular growth [27, 40, 41], while those who were less concerned with the consistency of oxygen level were more likely to produce NV [23, 26, 28, 30, 31], due to a fluctuating PaO2. In fact, PaO2 fluctuation is now known to be associated with the development of ROP, in both clinical [42–44] and animal studies [45–47].
Penn et al. were unable to reproduce the work of previous investigators until they began to experiment with systematically varied oxygen protocols. The group exposed some rats to constant 80% oxygen, followed by a period of time in room air. Other rats were exposed to a cycle of 80% oxygen for 12 h, followed by 40% oxygen for the next 12 h, for some number of days, followed by a period of time in room air. At the time of removal from oxygen, both groups of rats demonstrated identical hyperoxia-induced vessel attenuation. None of the rats that received constant 80% oxygen, followed by a period of time in room air, developed preretinal NV. However, 66% of the rats exposed to the 80/40% oxygen exposure developed preretinal NV [48]. This study demonstrated that avascular area is not necessarily correlated with the development of retinal NV and placed in doubt the widely held theory that “more oxygen means more ROP.”
Variable oxygen protocols more closely reflect the situation in the NICU. In this setting, infants with underdeveloped and improperly functioning lungs suffer from acute pulmonary distress and associated hypoxia. Unfortunately, there is a time lag between the point at which an infant’s oxygen saturation falls below the predetermined critical level and the time at which increased oxygen delivery results in an equilibrated return to desired therapeutic levels. Despite a carefully administered oxygen protocol designed to maintain a constant saturation level [49–51], premature infants receive a highly variable course of oxygen therapy. These infants are the most likely to develop severe retinopathy, supporting the theory that variable oxygen promotes preretinal NV [35, 48].
Although variable oxygen promotes retinal NV, variations in FiO2 between 80% and 40% in intrinsically healthy animals with normal lung function do not accurately produce the levels of arterial oxygen that neonates in the NICU experience. In order to more closely mimic the clinical setting, Penn et al. altered the exposure paradigm to vary between 50% and 10% FiO2 (Fig. 1) [45], and compared it to the 80/40% paradigm. By varying the oxygen between 50% and 10%, the ΔFiO2 remained at 0.4, the same as that experienced by rats exposed to the 80/40% paradigm. This allowed for comparison of the effects of two oxygen protocols with widely divergent FiO2 values, but identical ΔFiO2. Also, the arterial blood gases measured during the 50/10% paradigm were more reflective of those experienced by a sick premature infant in the NICU. The 50/10% oxygen protocol resulted in a greater retardation of retinal blood vessel development (Fig. 2) during oxygen treatment and led to a higher incidence and severity of NV than did the 80/40% protocol. Ninety-seven percent of the 50/10%-treated rats developed retinal NV compared to only 72% of the 80/40%-treated rats. These data indicate that the development of retinopathy is not due to the amount of oxygen the subject receives. Rather, these data suggest that fluctuating oxygen treatment (FiO2) and ambient retinal hypoxia (low PaO2) contribute to the development of pathology [46].
Fig. 1.

This schematic illustrates the oxygen exposure protocol used by Penn et al. to compare the initial 40/80% treatment paradigm and the current 50/10% treatment regimen. The litters were placed in incubators within 4 h after birth and the oxygen level was adjusted every 24 h thereafter. After 14 days of variable oxygen, the rats were returned to room air to allow NV to occur. Rats were killed on day 18 and NV severity was assessed [45]
Fig. 2.
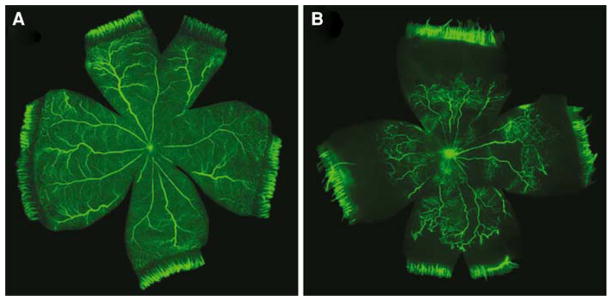
FITC-dextran infused rat retinas at P20. Exposing rats to the 50/10% model retards the development of the retinal vasculature. Room air-raised rats (a) exhibit normal retinal vascular development. 50/10% rats (b) demonstrate reduced vascular development, which leads to the development of NV. Image reproduced with permission from Springer Publishing Co. [74]
Penn et al. then performed experiments to determine what range of oxygen variability would lead to a predictable and relevant NV response [47]. The group exposed rats to alternating 24-h cycles of some level of hyperoxia and 10% oxygen. Hyperoxia treatment consisted of 50%, 40%, 30%, or 21% (room air) oxygen. This led to ΔFiO2 measures of 0.4, 0.3, 0.2, and 0.11, respectively, under conditions where absolute hypoxia remained constant. The group demonstrated a linear relationship between ΔFiO2 and the degree of retinal avascularity at the time of removal from oxygen treatment; a higher ΔFiO2 was correlated with more retinal avascularity. Also, after an appropriate post-exposure period, rats exposed to a ΔFiO2 of 0.3 or higher developed NV 100% of the time; rats exposed to a ΔFiO2 of 0.2 developed NV 33% of the time; rats exposed to a ΔFiO2 of 0.11 never developed retinopathy. This suggested that the minimum threshold for the development of NV is a ΔFiO2 of 0.2 (Fig. 3). The experiment also suggested that retinopathy could be prevented, perhaps even eliminated, by better managing the delivery of therapeutic oxygen, and that extreme oxygen fluctuations and hypoxic episodes in infants may be causal influences in ROP.
Fig. 3.

This figure demonstrates that ΔFiO2 is related to NV susceptibility. The top graph illustrates that the ΔFiO2 must be at least 0.2 for NV to develop. All animals with ΔFiO2 ≥ 0.3 developed NV. The bottom graph illustrates the importance of the extent of hypoxia during hypoxic episodes. For NV to develop, hypoxic episodes must be ≤12.5% oxygen. Image reproduced with permission from Investigative Ophthalmology and Visual Sciences [75]
The Penn group performed another set of experiments designed to determine the level of hypoxia necessary to produce NV [47]. They exposed rats to alternating 24-h cycles of 50/10%, 45/12.5%, 40/15%, and 35/21% oxygen. The ΔFiO2 of these four exposures was 0.4, 0.325, 0.25, and 0.14, respectively. The 45/12.5% exposure led to a significantly greater incidence (100%) and severity of disease than did the 40/15% treatment (4.8%), despite the modest difference in ΔFiO2. These findings demonstrated that there must be episodes of ≤12.5% oxygen for the development of NV to occur in rats [52]. This study demonstrates that relatively subtle differences in oxygen fluctuation can have large effects on the resulting NV.
The 50/10% oxygen exposure protocol reliably produces robust NV reminiscent of the NV that develops in human ROP (Fig. 4). Notably, exposure of newborn rats to the 50/10% protocol results in blood PaO2 levels that are reasonably reminiscent of those suffered by premature infants in whom ROP develops [47] and produces quantitative changes in the diameter and tortuosity of retinal arterioles that are similar to those seen in clinical ROP exams [53–55]. Exposure to the 50/10% model retards the growth of both superficial and deep retinal vessels, resulting in an avascular retinal periphery. Removal from oxygen exposure to room air causes neovascular growth at the boundary between vascular and avascular retina. Neovascular tufts can join, resulting in a ridge of preretinal vessel growth that is highly reminiscent of the mesenchymal ridge seen in human ROP. As in the human, the rat retina has been reported to detach under some experimental conditions, in a small percentage of cases [41]. Thus, the 50/10% oxygen exposure regimen developed by Penn et al. has become a significant and relevant model with which to address ROP-related questions, cementing its place in the field of ocular angiogenesis research.
Fig. 4.
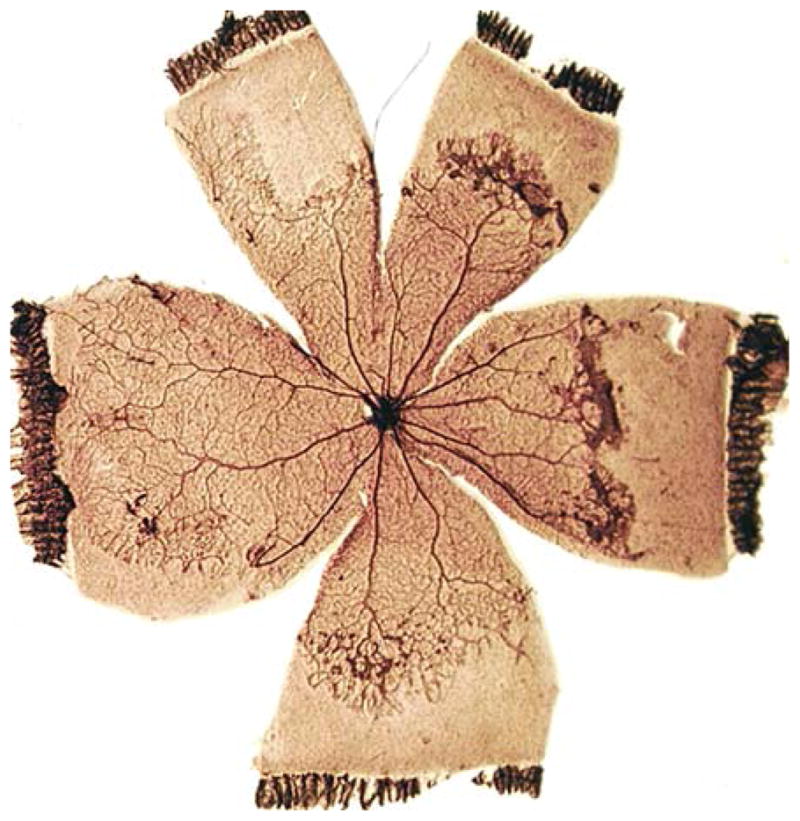
Retinal NV produced by the 50/10% rat model mimics human pathology. Retinal NV occurs at the boundary between the vascular central retina and the avascular peripheral retina, at the advancing edge of new vessel growth. Image reproduced with permission from Investigative Ophthalmology and Visual Sciences [75]
The studies by Penn et al. conclusively demonstrated that PaO2 fluctuation is an important factor related to the development of ROP. Realizing that the temporal aspect of step-wise changes in FiO2 might limit the relevance of Penn’s studies, Cunningham et al. attempted to more closely model the arterial oxygen levels in a preterm infant. The group analyzed the arterial oxygen levels in a preterm infant in whom severe ROP developed. Measurements were taken every minute for the first 14 days of life. These PaO2 values were translated to the equivalent value in the rat, and a computer-controlled delivery system was devised. This allowed the investigators to subject rats to a more clinically relevant oxygen pattern, where relatively random changes occurred minute-by-minute, and assess its effect on NV pathology [56]. Rats subject to minute-by-minute PaO2 fluctuations for the first 14 days of life had significantly larger avascular areas than room air controls. Their retinas also demonstrated more VEGF mRNA in the inner nuclear layer compared to room air controls. Importantly, however, the retinopathy produced was not as severe as that produced in rats exposed to a variable oxygen model consisting of 12-h periods alternating between 80% and 21% oxygen. The 80/21%-treated rats demonstrated a much more human-like pathology, exhibiting a larger avascular area and more VEGF mRNA staining in the inner nuclear layer, than did the minute-by-minute-treated rats. McColm et al. have exploited their minute-by-minute exposure system to better define the precise oxygen parameters that are important in ROP outcome [57].
Prompted by the 1995 study by Penn et al. [47], the clinical importance of oxygen fluctuation was systematically tested in a retrospective study of premature infants [39]. Step-wise regression analysis revealed a strong correlation between PaO2 fluctuation and ROP outcome in a cohort of 231 infants with birth weights of <1500 grams. PaO2 fluctuation proved particularly important in the progression of infants from prethreshold to threshold disease. This cemented the relationship between oxygen fluctuation and neovascular ROP and underscored the value of the rat ROP model for studies of ROP pathogenesis.
In an effort to more fully characterize the rat model of ROP, Roberto et al. [58] monitored 50/10%-treated rats for the rapid, spontaneous resolution of ocular sequelae after some time in room air, as has been reported in human infants. After 7 days in room air, the group demonstrated that the retinal vasculature began to resume its peripheral outgrowth. After 14 days in room air, the retinal vasculature had reached the retinal periphery. Notably, however, the vessels that reached the periphery after 14 days in room air demonstrated abnormal vessel architecture and decreased vascular density, which improved over time, but never to control levels, even at the longest time point assessed (18 weeks post oxygen-exposure). These findings illustrate additional similarities between the rat model of ROP and the human condition, particularly regarding vascular remodeling and architecture (as shown in the report by Mintz-Hittner [59]).
Advantages and disadvantages of the rat model
The rat model of ROP confers many advantages over other models, making it an attractive model to test the efficacy of anti-angiogenic compounds, for their eventual use in the eye or in other nonocular pathologies. First, the rat model of ROP most closely approximates the human condition, producing a pattern of pathology reminiscent of human ROP. This sets the rat apart from other animal models of the disease, making it the most clinically relevant model of ROP. There are also well-defined and clinically correlated methods of quantifying retinal NV. In one method, the face of a clock is superimposed onto a flat-mounted retina with each retinal quadrant containing three clock hours. Investigators sum the number of clock hours containing pathology, obtaining a read-out of NV severity. This method is similar to the one used in the clinical setting and much less laborious than the alternative, counting endothelial cell nuclei in transverse sections of the retina. Zhang et al. compared these two methods and demonstrated a highly significant positive correlation [60]. Today, many investigators use computer-assisted image analysis techniques to increase the speed and decrease the subjectivity of assessment. The rat model was the first to use this technology [29]. It should also be noted that the rat model was the first to use fluorescein angiography to monitor the progression of the disease in real time [61–63] and the first to compare and optimize vessel staining techniques [64]. These techniques, developed and honed in the rat, have furthered our understanding of the pathogenesis of ROP and increased the value of the model as a venue for preclinical tests of efficacy.
Another advantage of the rat model is that stable dams are relatively inexpensive to purchase and maintain. Kittens and puppies are much less economical. Additionally, rat litters can be as large as 18 (approximately twice the size of mouse litters) depending upon strain. Economy aside, large litters are advantageous because they limit the amount of nutrition each pup receives. This adversely affects the health of pups, but it increases the severity of retinopathy. Holmes and Duffner studied vascular development of the retina in rats raised in litters of 10 and 18 [65]. Six days after birth, rats raised in litters of 18 have retinas with 13% less vascular area than the rats raised in litters of 10. Very low-birth-weight premature infants also have incompletely vascularized retinas, and this contributes to, and worsens, the development of retinopathy [39, 66]. Holmes and Duffner went on to demonstrate a direct correlation between increased litter size, decreased body weight, and increased severity of NV [65]. In summary, large, relatively malnourished litters not only contribute to increased NV severity, but also may more closely approximate the conditions experienced by sick premature infants.
Because of these advantages, the rat model has served as the testing ground for numerous anti-angiogenic agents, antioxidants, and anti-inflammatory drugs. The rat model has served to test numerous drug delivery methods, including: intraperitoneal, intramuscular, and oral systemic routes; intraocular injections and topical local delivery; novel methods such as the encasement of compounds in liposomes, and the introduction of chimeric proteins. Substantial pharmacokinetics data have been generated to support the use of these methods in the rat.
No animal model of human disease is perfect, and the rat model of ROP has its drawbacks. There exist both strain- and vendor-related differences in susceptibility to NV. Ma et al. demonstrated strain-related differences in susceptibility to NV using Brown Norway and Sprague Dawley rats and a modified constant oxygen exposure paradigm (developed by Smith et al. for the mouse [67]). At the time of removal from oxygen, Brown Norway rats had an avascular area approximately four times greater than the Sprague Dawley rats, and 6 days post-exposure, the Brown Norway rats exhibited three times more NV than the Sprague Dawley rats [68]. These findings were confirmed by Zhang et al. who also found that Brown Norway rats exhibited increased vascular permeability relative to Sprague Dawley rats [69]. Additionally, van Wijngaarden et al. observed strain differences in a variety of rats using a 80/21% oxygen paradigm [70]. Similar to the other studies, they found Sprague Dawley rats to have an intermediate susceptibility and neovascular morphology, and also found both Hooded Wistar and Dark Agouti rats to have a 1.5-fold increase in neovascularization measured by the clock hour method. It is likely that strain-related differences in NV pathology are related to strain-related differences in pro- and anti-angiogenic factors such as VEGF and PEDF, as demonstrated in mice [68, 71]. Although informative, these studies were conducted using a model of constant extreme hyperoxia. They failed to use a clinically relevant variable oxygen regimen to investigate the presence of any strain-related differences. Using a modified variable oxygen regimen, Holmes et al. corroborated the above findings, demonstrating that 100% of the Brown Norway rats, but very few Sprague Dawley rats, developed preretinal NV [72].
There are also vendor-related differences within the same strain of rat. This phenomenon was first identified by Penn et al. (unpublished observations). He exposed Sprague Dawley rats from Charles River (Charles River Laboratories, Wilmington, MA), Zivic-Miller (Zivic Laboratories, Pittsburg, PA), Harlan (Harlan, Indianapolis, IN), and Hilltop (Hilltop Lab Animals, Scottdale, PA) to the 50/10% model. Sprague Dawley rats from Charles River produced twice as much NV as rats from Zivic-Miller. Rats purchased from Harlan and Hilltop produced intermediate susceptibilities to NV. Holmes et al. con-firmed the above findings. The group looked at NV susceptibility in Sprague Dawley rats obtained from Charles River and Harlan. As before, the rats obtained from Charles River demonstrated a 62% greater incidence and greater severity of NV [73]. Thus, the susceptibility to, and severity of, NV depend on genetic variation, environment, and oxygen treatment.
Conclusions
The current 50/10% oxygen paradigm is widely accepted as the most clinically relevant model of human ROP. This rat model of ROP has evolved over the past several decades. The development of the model has contributed much of what is currently known regarding the growth of physiological and pathological blood vessels in the retina. More specifically, this model has yielded information regarding the cellular and molecular mechanisms governing ROP pathogenesis and it offers a proven venue for testing the efficacy of anti-angiogenic agents. With technological advances, the rat genome becomes increasingly easier to manipulate, yielding the potential to test the importance of molecular targets without the limitations of pharmacologic methods. Advances in research technology, coupled with the continued refinement of the rat ROP model, will allow it to more accurately reflect the human condition. This will lead to a more thorough understanding of, and better therapeutic management of, ROP and other angiogenic conditions, both ocular and nonocular.
Acknowledgments
This study was supported by NIH EY07533, NIH EY01826, NIH AG031036, and an Unrestricted Grant from Research to Prevent Blindness, Inc., and a Research to Prevent Blindness Senior Scientific Investigator Award to JSP.
Contributor Information
Joshua M. Barnett, Department of Pharmacology, Vanderbilt University School of Medicine, Nashville, TN, USA
Susan E. Yanni, Department of Cell and Developmental Biology, Vanderbilt University School of Medicine, Nashville, TN, USA
John S. Penn, Email: john.penn@vanderbilt.edu, Department of Pharmacology, Vanderbilt University School of Medicine, Nashville, TN, USA. Department of Cell and Developmental Biology, Vanderbilt University School of Medicine, Nashville, TN, USA. Department of Ophthalmology and Visual Sciences, Vanderbilt University School of Medicine, Nashville, TN, USA. Vanderbilt Eye Institute, Vanderbilt University School of Medicine, 8000 Medical Center East, Nashville, TN 37232-8808, USA
References
- 1.Terry TL. Extreme prematurity and fibroblastic overgrowth of persistent vascular sheath behind each crystalline lens: I, preliminary report. Am J Ophthalmol. 1942;25:203–204. doi: 10.1016/j.ajo.2018.05.024. [DOI] [PubMed] [Google Scholar]
- 2.Campbell K. Intensive oxygen therapy as a possible cause of retrolental fibroplasia; a clinical approach. Med J Aust. 1951;2:48–50. [PubMed] [Google Scholar]
- 3.Patz A, Hoech LE, Cruz ED. Studies on the effect of high oxygen administration in retrolental fibroplasia. I. Nursery observations. Am J Ophthalmol. 1953;35:1245–1253. doi: 10.1016/0002-9394(52)91140-9. [DOI] [PubMed] [Google Scholar]
- 4.Kinsey VE. Retrolental fibroplasia; cooperative study of retrolental fibroplasia and the use of oxygen. AMA Arch Ophthalmol. 1956;56:481–543. [PubMed] [Google Scholar]
- 5.Lucey JF, Dangman B. A reexamination of the role of oxygen in retrolental fibroplasia. Pediatrics. 1984;73:82–96. [PubMed] [Google Scholar]
- 6.Patz A, Hoeck LE, De La Cruz E. Studies on the effect of high oxygen administration in retrolental fibroplasia. I. Nursery observations. Am J Ophthalmol. 1952;35:1248–1253. doi: 10.1016/0002-9394(52)91140-9. [DOI] [PubMed] [Google Scholar]
- 7.Gyllensten LJ, Hellstrom BE. Retrolental fibroplasia; animal experiments: the effect of intermittingly administered oxygen on the postnatal development of the eyes of fullterm mice. Acta Paediatr. 1952;41:577–582. doi: 10.1111/j.1651-2227.1952.tb17854.x. [DOI] [PubMed] [Google Scholar]
- 8.Ashton N, Ward B, Serpell G. Role of oxygen in the genesis of retrolental fibroplasia: a preliminary report. Br J Ophthalmol. 1953;37:513–520. doi: 10.1136/bjo.37.9.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Forrester RM. Oxygen, cerebral palsy and retrolental fibroplasia. Dev Med Child Neurol. 1964;6:648–650. doi: 10.1111/j.1469-8749.1964.tb02817.x. [DOI] [PubMed] [Google Scholar]
- 10.McDonald AD. Oxygen treatment of premature babies and cerebral palsy. Dev Med Child Neurol. 1964;6:313–314. doi: 10.1111/j.1469-8749.1964.tb10800.x. [DOI] [PubMed] [Google Scholar]
- 11.Hatfield EM. Blindness in infants and young children. Sight Sav Rev. 1972;42:69–89. [PubMed] [Google Scholar]
- 12.Gibson DL, Sheps SB, Schechter MT, Wiggins S, McCormick AQ. Retinopathy of prematurity: a new epidemic? Pediatrics. 1989;83:486–492. [PubMed] [Google Scholar]
- 13.Phelps DL. Retinopathy of prematurity: an estimate of vision loss in the United States—1979. Pediatrics. 1981;67:924–925. [PubMed] [Google Scholar]
- 14.Curley MA, Thompson JE, Arnold JH. The effects of early and repeated prone positioning in pediatric patients with acute lung injury. Chest. 2000;118:156–163. doi: 10.1378/chest.118.1.156. [DOI] [PubMed] [Google Scholar]
- 15.Calkovska A, Sun B, Curstedt T, Renheim G, Robertson B. Combined effects of high-frequency ventilation and surfactant treatment in experimental meconium aspiration syndrome. Acta Anaesthesiol Scand. 1999;43:135–145. doi: 10.1034/j.1399-6576.1999.430204.x. [DOI] [PubMed] [Google Scholar]
- 16.Tang SF, Sherwood MC, Miller OI. Randomised trial of three doses of inhaled nitric oxide in acute respiratory distress syndrome. Arch Dis Child. 1998;79:415–418. doi: 10.1136/adc.79.5.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gibson DL, Sheps SB, Uh SH, Schechter MT, McCormick AQ. Retinopathy of prematurity-induced blindness: birth weight-specific survival and the new epidemic. Pediatrics. 1990;86:405–412. [PubMed] [Google Scholar]
- 18.Payne JW, Patz A. Fluorescein angiography in retrolental fibroplasia. Int Ophthalmol Clin. 1977;17:121–135. doi: 10.1097/00004397-197701720-00011. [DOI] [PubMed] [Google Scholar]
- 19.Chan-Ling T, Stone J. Retinopathy of prematurity: origins of the architecture of the retina. Prog Retin Eye Res. 1993;12:155–178. [Google Scholar]
- 20.Hardy P, Dumont I, Bhattacharya M, Hou X, Lachapelle P, Varma DR, Chemtob S. Oxidants, nitric oxide and prostanoids in the developing ocular vasculature: a basis for ischemic retinopathy. Cardiovasc Res. 2000;47:489–509. doi: 10.1016/s0008-6363(00)00084-5. [DOI] [PubMed] [Google Scholar]
- 21.Schaffer DB, Palmer EA, Plotsky DF, Metz HS, Flynn JT, Tung B, Hardy RJ. Prognostic factors in the natural course of retinopathy of prematurity. The Cryotherapy for Retinopathy of Prematurity Cooperative Group. Ophthalmology. 1993;100:230–237. doi: 10.1016/s0161-6420(93)31665-9. [DOI] [PubMed] [Google Scholar]
- 22.Foos RY. Chronic retinopathy of prematurity. Ophthalmology. 1985;92:563–574. doi: 10.1016/s0161-6420(85)34007-1. [DOI] [PubMed] [Google Scholar]
- 23.Patz A. Oxygen studies in retrolental fibroplasia. IV. Clinical and experimental observations. Am J Ophthalmol. 1954;38:291–308. doi: 10.1016/0002-9394(54)90845-4. [DOI] [PubMed] [Google Scholar]
- 24.Coats DK, Paysse EA, Steinkuller PG. Threshold retinopathy of prematurity in neonates less than 25 weeks’ estimated gestational age. J AAPOS. 2000;4:183–185. [PubMed] [Google Scholar]
- 25.Fruttiger M. Development of the mouse retinal vasculature: angiogenesis versus vasculogenesis. Invest Ophthalmol Vis Sci. 2002;43:522–527. [PubMed] [Google Scholar]
- 26.Brands KH, Hofmann H, Klees E. Die retrolentale fibroplasie. Geburtshilfe Frauenheilkd. 1958;18:805–814. [PubMed] [Google Scholar]
- 27.Ashton N, Blach R. Studies on developing retinal vessels Viii. Effect of oxygen on the retinal vessels of the ratling. Br J Ophthalmol. 1961;45:321–340. doi: 10.1136/bjo.45.5.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ricci B, Calogero G. Oxygen-induced retinopathy in newborn rats: effects of prolonged normobaric and hyper-baric oxygen supplementation. Pediatrics. 1988;82:193–198. [PubMed] [Google Scholar]
- 29.Penn JS, Tolman BL, Lowery LA, Koutz CA. Oxygen-induced retinopathy in the rat: hemorrhages and dysplasias may lead to retinal detachment. Curr Eye Res. 1992;11:939–953. doi: 10.3109/02713689209033492. [DOI] [PubMed] [Google Scholar]
- 30.Ventresca MR, Gonder JR, Tanswell AK. Oxygen-induced proliferative retinopathy in the newborn rat. Can J Ophthalmol. 1990;25:186–189. [PubMed] [Google Scholar]
- 31.Reynaud X, Vallat M, Vincent D, Dorey CK. Protective effect of the ginkgo biloba extract in the rat model of retinopathy of prematurity. Invest Ophthalmol Vis Sci. 1991;32(suppl):1147. [Google Scholar]
- 32.Flower RW, McLeod DS, Lutty GA, Goldberg B, Wajer SD. Postnatal retinal vascular development of the puppy. Invest Ophthalmol Vis Sci. 1985;26:957–968. [PubMed] [Google Scholar]
- 33.Biglan AW, Brown DR, Reynolds JD, Milley JR. Risk factors associated with retrolental fibroplasia. Ophthalmology. 1984;91:1504–1511. doi: 10.1016/s0161-6420(84)34104-5. [DOI] [PubMed] [Google Scholar]
- 34.Brown BA, Thach AB, Song JC, Marx JL, Kwun RC, Frambach DA. Retinopathy of prematurity: evaluation of risk factors. Int Ophthalmol. 1998;22:279–283. doi: 10.1023/a:1006326008909. [DOI] [PubMed] [Google Scholar]
- 35.Gunn TR, Easdown J, Outerbridge EW, Aranda JV. Risk factors in retrolental fibroplasia. Pediatrics. 1980;65:1096–1100. [PubMed] [Google Scholar]
- 36.Holmstrom G. Retinopathy of prematurity. BMJ. 1993;307:694–695. doi: 10.1136/bmj.307.6906.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hammer ME, Mullen PW, Ferguson JG, Pai S, Cosby C, Jackson KL. Logistic analysis of risk factors in acute retinopathy of prematurity. Am J Ophthalmol. 1986;102:1–6. doi: 10.1016/0002-9394(86)90200-x. [DOI] [PubMed] [Google Scholar]
- 38.Purohit DM, Ellison RC, Zierler S, Miettinen OS, Nadas AS. Risk factors for retrolental fibroplasia: experience with 3,025 premature infants. National Collaborative Study on Patent Ductus Arteriosus in Premature Infants. Pediatrics. 1985;76:339–344. [PubMed] [Google Scholar]
- 39.York JR, Landers S, Kirby RS, Arbogast PG, Penn JS. Arterial oxygen fluctuation and retinopathy of prematurity in very-low-birth-weight infants. J Perinatol. 2004;24:82–87. doi: 10.1038/sj.jp.7211040. [DOI] [PubMed] [Google Scholar]
- 40.Penn JS, Thum LA. The rat as an animal model for retinopathy of prematurity. Prog Clin Biol Res. 1989;314:623–642. [PubMed] [Google Scholar]
- 41.Penn JS, Thum LA, Naash MI. Oxygen-induced retinopathy in the rat. Vitamins C and E as potential therapies. Invest Ophthalmol Vis Sci. 1992;33:1836–1845. [PubMed] [Google Scholar]
- 42.Cunningham S, Fleck BW, Elton RA, McIntosh N. Transcutaneous oxygen levels in retinopathy of prematurity. Lancet. 1995;346:1464–1465. doi: 10.1016/s0140-6736(95)92475-2. [DOI] [PubMed] [Google Scholar]
- 43.Gaynon MW, Stevenson DK. What can we learn from STOP-ROP and earlier studies? Pediatrics. 2000;105:420–421. doi: 10.1542/peds.105.2.420. [DOI] [PubMed] [Google Scholar]
- 44.Saito Y, Omoto T, Cho Y, Hatsukawa Y, Fujimura M, Takeuchi T. The progression of retinopathy of prematurity and fluctuation in blood gas tension. Graefes Arch Clin Exp Ophthalmol. 1993;231:151–156. doi: 10.1007/BF00920938. [DOI] [PubMed] [Google Scholar]
- 45.Penn JS, Tolman BL, Lowery LA. Variable oxygen exposure causes preretinal neovascularization in the newborn rat. Invest Ophthalmol Vis Sci. 1993;34:576–585. [PubMed] [Google Scholar]
- 46.Penn JS, Henry MM, Tolman BL. Exposure to alternating hypoxia and hyperoxia causes severe proliferative retinopathy in the newborn rat. Pediatr Res. 1994;36:724–731. doi: 10.1203/00006450-199412000-00007. [DOI] [PubMed] [Google Scholar]
- 47.Penn JS, Henry MM, Wall PT, Tolman BL. The range of PaO2 variation determines the severity of oxygen-induced retinopathy in newborn rats. Invest Ophthalmol Vis Sci. 1995;36:2063–2070. [PubMed] [Google Scholar]
- 48.Shahinian L, Jr, Malachowski N. Retrolental fibroplasia: a new analysis of risk factors based on recent cases. Arch Ophthalmol. 1978;96:70–74. doi: 10.1001/archopht.1978.03910050034008. [DOI] [PubMed] [Google Scholar]
- 49.Askie LM, Henderson-Smart DJ, Irwig L, Simpson JM. Oxygen-saturation targets and outcomes in extremely preterm infants. N Engl J Med. 2003;349:959–967. doi: 10.1056/NEJMoa023080. [DOI] [PubMed] [Google Scholar]
- 50.Leach CL, Greenspan JS, Rubenstein SD, Shaffer TH, Wolfson MR, Jackson JC, DeLemos R, Fuhrman BP. Partial liquid ventilation with perflubron in premature infants with severe respiratory distress syndrome. The LiquiVent Study Group. N Engl J Med. 1996;335:761–767. doi: 10.1056/NEJM199609123351101. [DOI] [PubMed] [Google Scholar]
- 51.Courtney SE, Durand DJ, Asselin JM, Hudak ML, Aschner JL, Shoemaker CT. High-frequency oscillatory ventilation versus conventional mechanical ventilation for very-low-birth-weight infants. N Engl J Med. 2002;347:643–652. doi: 10.1056/NEJMoa012750. [DOI] [PubMed] [Google Scholar]
- 52.Werdich XQ, McCollum GW, Rajaratnam VS, Penn JS. Variable oxygen and retinal VEGF levels: correlation with incidence and severity of pathology in a rat model of oxygen-induced retinopathy. Exp Eye Res. 2004;79:623–630. doi: 10.1016/j.exer.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 53.Liu K, Akula JD, Falk C, Hansen RM, Fulton AB. The retinal vasculature and function of the neural retina in a rat model of retinopathy of prematurity. Invest Ophthalmol Vis Sci. 2006;47:2639–2647. doi: 10.1167/iovs.06-0016. [DOI] [PubMed] [Google Scholar]
- 54.Akula JD, Hansen RM, Martinez-Perez ME, Fulton AB. Rod photoreceptor function predicts blood vessel abnormality in retinopathy of prematurity. Invest Ophthalmol Vis Sci. 2007;48:4351–4359. doi: 10.1167/iovs.07-0204. [DOI] [PubMed] [Google Scholar]
- 55.Fulton AB, Akula JD, Mocko JA, Hansen RM, Benador IY, Beck SC, Fahl E, Seeliger MW, Moskowitz A, Harris ME. Retinal degenerative and hypoxic ischemic disease. Doc Ophthalmol. 2009;118:55–61. doi: 10.1007/s10633-008-9127-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cunningham S, McColm JR, Wade J, Sedowofia K, McIntosh N, Fleck B. A novel model of retinopathy of prematurity simulating preterm oxygen variability in the rat. Invest Ophthalmol Vis Sci. 2000;41:4275–4280. [PubMed] [Google Scholar]
- 57.McColm JR, Cunningham S, Wade J, Sedowofia K, Gellen B, Sharma T, McIntosh N, Fleck BW. Hypoxic oxygen fluctuations produce less severe retinopathy than hyperoxic fluctuations in a rat model of retinopathy of prematurity. Pediatr Res. 2004;55:107–113. doi: 10.1203/01.PDR.0000099772.66376.02. [DOI] [PubMed] [Google Scholar]
- 58.Roberto KA, Tolman BL, Penn JS. Long-term retinal vascular abnormalities in an animal model of retinopathy of prematurity. Curr Eye Res. 1996;15:932–937. doi: 10.3109/02713689609017637. [DOI] [PubMed] [Google Scholar]
- 59.Mintz-Hittner HA, Prager TC, Kretzer FL. Visual acuity correlates with severity of retinopathy of prematurity in untreated infants weighing 750 g or less at birth. Arch Ophthalmol. 1992;110:1087–1091. doi: 10.1001/archopht.1992.01080200067026. [DOI] [PubMed] [Google Scholar]
- 60.Zhang S, Leske DA, Holmes JM. Neovascularization grading methods in a rat model of retinopathy of prematurity. Invest Ophthalmol Vis Sci. 2000;41:887–891. [PubMed] [Google Scholar]
- 61.Larrazabal LI, Penn JS. Study of ocular vasculature in the newborn rat by fluorescein angiography. J Ophthal Phot. 1989;11:49–52. [Google Scholar]
- 62.Larrazabal LI, Penn JS. Fluorescein angiography of the newborn rat. Implications in oxygen-induced retinopathy. Invest Ophthalmol Vis Sci. 1990;31:810–818. [PubMed] [Google Scholar]
- 63.Penn JS, Johnson BD. Fluorescein angiography as a means of assessing retinal vascular pathology in oxygen-exposed newborn rats. Curr Eye Res. 1993;12:561–570. doi: 10.3109/02713689309001834. [DOI] [PubMed] [Google Scholar]
- 64.Penn JS, Henry MM. Assessing retinal neovascularization in an animal model of proliferative retinopathy. Microvasc Res. 1996;51:126–130. doi: 10.1006/mvre.1996.0014. [DOI] [PubMed] [Google Scholar]
- 65.Holmes JM, Duffner LA. The effect of postnatal growth retardation on abnormal neovascularization in the oxygen exposed neonatal rat. Curr Eye Res. 1996;15:403–409. doi: 10.3109/02713689608995831. [DOI] [PubMed] [Google Scholar]
- 66.Shohat M, Reisner SH, Krikler R, Nissenkorn I, Yassur Y, Ben-Sira I. Retinopathy of prematurity: incidence and risk factors. Pediatrics. 1983;72:159–163. [PubMed] [Google Scholar]
- 67.Smith LE, Wesolowski E, McLellan A, Kostyk SK, D’Amato R, Sullivan R, D’Amore PA. Oxygen-induced retinopathy in the mouse. Invest Ophthalmol Vis Sci. 1994;35:101–111. [PubMed] [Google Scholar]
- 68.Gao G, Li Y, Fant J, Crosson CE, Becerra SP, Ma JX. Difference in ischemic regulation of vascular endothelial growth factor and pigment epithelium-derived factor in brown norway and sprague dawley rats contributing to different susceptibilities to retinal neovascularization. Diabetes. 2002;51:1218–1225. doi: 10.2337/diabetes.51.4.1218. [DOI] [PubMed] [Google Scholar]
- 69.Zhang SX, Ma JX, Sima J, Chen Y, Hu MS, Ottlecz A, Lambrou GN. Genetic difference in susceptibility to the blood-retina barrier breakdown in diabetes and oxygen-induced retinopathy. Am J Pathol. 2005;166:313–321. doi: 10.1016/S0002-9440(10)62255-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.van Wijngaarden P, Coster DJ, Brereton HM, Gibbins IL, Williams KA. Strain-dependent differences in oxygen-induced retinopathy in the inbred rat. Invest Ophthalmol Vis Sci. 2005;46:1445–1452. doi: 10.1167/iovs.04-0708. [DOI] [PubMed] [Google Scholar]
- 71.Chan CK, Pham LN, Zhou J, Spee C, Ryan SJ, Hinton DR. Differential expression of pro- and antiangiogenic factors in mouse strain-dependent hypoxia-induced retinal neovascularization. Lab Invest. 2005;85:721–733. doi: 10.1038/labinvest.3700277. [DOI] [PubMed] [Google Scholar]
- 72.Floyd BN, Leske DA, Wren SM, Mookadam M, Fautsch MP, Holmes JM. Differences between rat strains in models of retinopathy of prematurity. Mol Vis. 2005;11:524–530. [PubMed] [Google Scholar]
- 73.Kitzmann A, Leske D, Chen Y, Kendall A, Lanier W, Holmes J. Incidence and severity of neovascularization in oxygen- and metabolic acidosis-induced retinopathy depend on rat source. Curr Eye Res. 2002;25:215–220. doi: 10.1076/ceyr.25.4.215.13483. [DOI] [PubMed] [Google Scholar]
- 74.Yanni SE, McCollum GW, Penn JS. Rodent models of oxygen-induced retinopathy. In: Penn JS, editor. Retinal and choroidal angiogenesis. Springer; Dordrecht: 2008. pp. 57–80. [Google Scholar]
- 75.Penn JS, Rajaratnam VS. Inhibition of retinal neovascularization by intravitreal injection of human rPAI-1 in a rat model of retinopathy of prematurity. Invest Ophthalmol Vis Sci. 2003;44:5423–5429. doi: 10.1167/iovs.02-0804. [DOI] [PubMed] [Google Scholar]