

Abstract
Purpose
Several oral multikinase inhibitors are known to interact in vitro with the human ATP-binding cassette transporter ABCC4 (MRP4), but the in vivo relevance of this interaction remains poorly understood. We hypothesized that host ABCC4 activity may influence the pharmacokinetic profile of dasatinib and subsequently affect its antitumor properties.
Experimental Design
Transport of dasatinib was studied in cells transfected with human ABCC4 or the ortholog mouse transporter, Abcc4. Pharmacokinetic studies were done in wildtype and Abcc4-null mice. The influence of Abcc4-deficiency on dasatinib efficacy was evaluated in a model of Ph+ acute lymphoblastic leukemia (ALL) by injection of luciferase-positive, p185(BCR-ABL)-expressing Arf(−/−) pre-B cells.
Results
Dasatinib accumulation was significantly changed in cells over-expressing ABCC4 or Abcc4 compared to control cells (P<0.001). Deficiency of Abcc4 in vivo was associated with a 1.75-fold decrease in systemic exposure to oral dasatinib, but had no influence on the pharmacokinetics of i.v. dasatinib. Abcc4 was found to be highly expressed in the stomach, and dasatinib efflux from isolated mouse stomachs ex vivo was impaired by Abcc4-deficiency (P<0.01), without any detectable changes in gastric pH. Abcc4-null mice receiving dasatinib had an increase in leukemic burden, based on bioluminescence imaging, and decreased overall survival compared to wildtype mice (P=0.048).
Conclusions
This study suggests that Abcc4 in the stomach facilitates the oral absorption of dasatinib, and it possibly plays a similar role for other orally-administered substrates, such as acetylsalicylic acid. This phenomenon also provides a mechanistic explanation for the malabsorption of certain drugs following gastric resection.
Keywords: Abcc4 (Mrp4), oral bioavailability, gastric absorption, dasatinib
INTRODUCTION
Dasatinib, an orally-administered inhibitor of Bcr/Abl and Src kinases, has been approved for the treatment of imatinib-resistant or imatinib-intolerant forms of chronic myeloid leukemia (CML) and Philadelphia chromosome-positive (Ph+) acute lymphoblastic leukemia (ALL) (1). A mass balance study has indicated that, following oral administration, dasatinib accounts for only less than 1% and 19% of the dose recovered in urine and feces, respectively, suggesting that dasatinib is extensively metabolized before being eliminated from the body (2). The primary pathways of dasatinib metabolism in humans include N-dealkylation (M4) and N-oxidation (M5) by CYP3A4 and flavin-containing monooxygenase 3, respectively (3).
As with other tyrosine kinase inhibitors, dasatinib is subject to extensive interindividual pharmacokinetic variability. For example, the coefficient of variation in the area under the curve (AUC) of dasatinib is >50% in adult patients (4), and as high as 46% in a pediatric patient population (5). The high degree of pharmacokinetic variability observed with dasatinib has potentially important toxicological and therapeutic ramifications. In particular, it was previously demonstrated that the steady-state trough concentration of dasatinib in patients with CML is strongly associated with treatment-related toxicities, as well as dose reductions and interruptions (6). Furthermore, pharmacokinetic studies performed in mice bearing CML xenografts have suggested that the time course and extent of inhibition of tumoral biomarkers of efficacy are correlated with the plasma levels of dasatinib (7).
The mechanistic basis underlying the unpredictable pharmacokinetic profile of dasatinib remains largely unknown. Model-based simulations have indicated that a critical determinant of dasatinib's pharmacokinetic variability is associated with absorption related processes and to a lesser extent with clearance (8). This supposition is consistent with studies indicating that deficiency of certain ATP-binding cassette (ABC) transporters normally expressed on the apical membrane of intestinal enterocytes, such as ABCB1 (P-glycoprotein), is associated with an increased AUC of dasatinib after oral (9) but not i.v. administration (10). Because dasatinib is poorly permeable (11), yet very rapidly absorbed in humans compared to other tyrosine kinase inhibitors (12), its transport across both of the enterocytic membranes after oral administration is likely transporter mediated. One candidate transporter involved in the basolateral transport process is the multidrug resistance-associated protein 4 (MRP4; ABCC4), which is known to interact with a number of tyrosine kinase inhibitors (13) and, based on functional in vivo data obtained with cefadroxil (14) and in vitro expression data in Caco-2 cells (15), may be localized on the basolateral membrane of enterocytes. In the current study, we tested the hypothesis that dasatinib is a transported substrate of ABCC4 in vitro, and that deficiency of the ortholog mouse transporter Abcc4 is associated with an altered pharmacokinetic and efficacy profile of dasatinib in vivo.
MATERIALS AND METHODS
In vitro transport studies
Dasatinib was obtained from LC Laboratories, and imatinib was kindly provided by Novartis Pharmaceuticals. [3H]Dasatinib, [3H]imatinib, [3H]-adefovir dipivoxil [bis(POM)-PMEA; hereafter referred to as PMEA], and unlabeled PMEA were purchased from Moravek Biochemicals. [3H]Estradiol-17β-D-glucuronide (E2G), [14C]tetraethylammonium bromide (TEA), and [acetyl-14C]salicylic acid (aspirin) were obtained from American Radiolabeled Chemicals, and [7-14C]salicylic acid from Perkin Elmer. Inside-out vesicles of Sf9 cells expressing ABCC3 or ABCC4 (Genomembrane) were used to determine uptake of either [3H]E2G, [3H]imatinib or [3H]dasatinib using 5-min incubations, as described (16), in the presence or absence of glutathione (GSH) (Life Technologies). The efflux transport of [3H]PMEA, [3H]imatinib, or [3H]dasatinib was also evaluated in Saos-2 cells transfected with an empty pcDNA or Abcc4 using 4-h incubations, as described (17). The uptake of [14C]TEA or [3H]dasatinib was also evaluated in HEK293 cells containing an empty PMIG II vector or a Flag-tagged cDNA of mOct1 or mOct2. The results from the in vitro transport studies were normalized to uptake values in cells transfected with an empty vector, after normalization to total protein content as measured by a Pierce BCA Protein Assay Kit (Thermo Scientific).
In vivo pharmacokinetic studies
Male mice knockout for Abcc4 [Abcc4(−/−)] were bred in-house and age-matched wildtype mice, all on a C57BL/6 background, were obtained from The Jackson Laboratory. Mice were housed in a temperature-controlled environment with a 12-hour light cycle and given a standard diet and water ad libitum. All in vivo experiments were approved by the Institutional Animal Care and Use Committee at St. Jude Children's Research Hospital.
Dasatinib was formulated in DMSO (25 mg/ml) and diluted 25-fold in 50 mM sodium acetate buffer (pH 4.6) to make a 1 mg/ml dosing solution immediately prior to drug administration by oral gavage, or in propylene glycol:deionized water (1:1, vol/vol) before i.v. administration (18). Imatinib was dissolved in sterile water to make a 10 mg/ml dosing solution. Mice were fasted for 3 h before and during the study, with unrestricted access to drinking water. Dasatinib was administered by oral gavage or by tail vein injection at a dose of 10 mg/kg (18), and imatinib was given at a dose of 50 mg/kg (19). At select time points after drug administration, blood samples (30-μl each) were taken from individual mice at 0.25, 0.5 or 0.67, and 1 h from the submandibular vein using a lancet, and at 2, and 4 h from the retro-orbital venous plexus using a heparinized capillary (Oxford Labware). A final blood draw was obtained at 6 h by a cardiac puncture after anesthetization with isoflurane using a syringe and needle. Plasma samples were analyzed by liquid chromatography-tandem mass spectrometry (LC/MS/MS), using a Waters ACQUITY separation system coupled to a TQD detector (see below). Pharmacokinetic parameters were calculated using WinNonlin 6.2 (Pharsight).
The extent of binding of dasatinib to serum proteins (20) and blood cell partitioning of dasatinib were evaluated according to published methods (18). Intrinsic intestinal permeability was evaluated by determining the recovery of D-[2-3H]mannitol (American Radiolabeled Chemicals) in plasma, kidney, and liver at 2-h after oral dosing (1 mg/kg), as described (21). In a separate experiment, the urinary bladder, gall bladder, and spleens, after perfusion, were removed from wildtype or Abcc4(−/−) mice 5 min after oral administration of dasatinib, and analyzed for the presence of dasatinib by LC/MS/MS.
Quantitative determination of dasatinib and metabolite by LC/MS/MS
Frozen samples were thawed at room temperature, and 10-μl aliquots of standard, quality control, or mouse sample were transferred to polypropylene microcentrifuge tubes containing 60 μl of a methanolic internal standards solution. The tubes were vortex-mixed for 60 s, followed by centrifugation at 10,000 rpm for 8 min at 4°C. Next, the supernatants were transferred to autosampler vials, and 3-μl volumes were injected into a Waters ACQUITY separation system coupled to a TQD triple-quadrupole MS/MS detector. Separation was achieved on a BEHC18 column (1.7 μm; 50×2.1 mm; Waters) using a column heater operating at 40°C with an in-line filter. The autosampler temperature was maintained at 15 ± 5°C. The flow rate of a gradient mobile phase, composed of 0.1% formic acid in acetonitrile and 10mM ammonium acetate in 0.5% formic acid wáter, was set at 0.7 ml/min, and separation was completed within 5 min. The instrument was equipped with an electrospray interface, and was controlled by Masslynx 4.1 software. The analysis was performed in MRM mode, as follows: m/z 488.16>401.11 for dasatinib; m/z 444.17>275.11 for N-deshydroxy dasatinib (M4), and m/z 504.17>387.12 for dasatinib N-oxide (M5). Dasatinib-d8, N-deshyhydroxy-d8 dasatinib, and dasatinib N-oxide-d8 (Toronto Research Chemicals) were used as internal standards (final concentration, 15 ng/ml). The MS/MS conditions included: capillary voltage: 0.6 kV; source temperature: 150°C; desolvation temperature: 450 °C; cone gas flow: 10 l/h; and desolvation gas flow: 900 l/h.
Calibration curves of dasatinib, M4, and M5 were created by plotting the peak area ratios of analyte to the internal standard against the analyte concentrations in the spiked matrix. The within-run and between-run precisions for dasatinib and its metabolites were less than 6.9%, and the measured concentrations were all less than 5.3% of the nominal value.
Ex vivo microsomal incubations
Mouse liver and intestinal microsomes were prepared as described (22). Assays for cytochrome P450 enzymes were performed as described by the manufacturer (BD Biosciences). Briefly, dasatinib (2 μM) was incubated with liver or intestinal microsomes (1 mg/ml) for 55 min at 37°C (3), and the reaction was terminated by 100-μL ice-cold acetonitrile. Dasatinib and its main rodent metabolites M4 and M5 (23) were determined by LC/MS/MS.
Gastric transport studies
The ex vivo mucosal-to serosal transport of dasatinib in isolated stomachs from wildtype or Abcc4(−/−) mouse was determined based on a published method (24), as was the in vivo gastric absorption of dasatinib (25). Briefly, after anesthetizing mice with isoflurance, the abdominal wall was opened longitudinally and a ligature was tightened around the cardia, in order to prevent reflux into the esophagus. Next, a tube was introduced into the duodenum and gently forced through the pylorus into the stomach cavity. To ensure tightness of the system and to avoid blood reflux into the stomach, two ligatures were fastened upstream and downstream of the pyloric sphincter. The tube was connected to a syringe to introduce dasatinib (1 mg/ml) in 50 mM sodium acetate buffer (pH 4.6) or vehicle alone into the gastric lumen. At 5 min after the administration of dasatinib, a sodium heparin solution (0.1 ml; 500 IU) was injected into the inferior cava vein and immediately thereafter, livers were obtained after cervical dislocation to ensure euthanasia. Body temperature, pulse, respiration rate, and anesthetic administration were monitored throughout the procedure. Gastric pH was determined using an IQ150 pH Meter with a PH37-SS piercing-tip microprobe (Hach).
Transporter gene and protein expression
Gene expression in the small intestine and stomach of wildtype or Abcc4(−/−) mice was performed on RNA extracted from whole organs using the RNEasy mini kit (Qiagen), and analyzed using the Mouse Transporter RT2 Profiles PCR array system (SABiosciences). Relative gene expression was determined using the ΔΔCt method, and normalized to the housekeeping gene, Gapdh.
Western blotting of Abcc4 (26) or Abcb1 and Abcg2 (27) was done according to published methods. The primary rat anti-human ABCC4 antibody, which also reacts with mouse Abcc4, was obtained from Abcam, and used at 1:50. The secondary antibody, rabbit anti-rat (Vector), was used at 1:600. Slides of 4-μm sections were cut from formalin-fixed paraffin-embedded tissues. All assay steps, including deparaffinization, rehydration, and epitope retrieval, were performed on the Bond Max with Bond wash buffer (Leica) rinses between steps. Heat-induced epitope retrieval was performed by heating slides in ER2 (Leica) for 30 min. Background Punisher (BioCare Chemical) was incubated on slides for 10 min. The Refine (Leica) detection system was used. Briefly, slides were incubated sequentially with hydrogen peroxide (5 min), primary antibody (15 min), secondary antibody (10 min), anti-rabbit horse radish peroxidase conjugated polymer (8 min), 3,3' diaminobenzidine (10 min), and hematoxylin (8 min). Normal human stomach samples were obtained from ProSci.
Activity of dasatinib against a model of Ph+ ALL
Details of the mouse model have been reported previously (28). Briefly, 2×105 luciferase-positive, p185(BCR-ABL)-expressing Arf(−/−) leukemia initiating pre-B cells (LIC) were administered by tail vein injection into cohorts of healthy, non-conditioned, immune-competent 10–12 weeks old male wildtype or Abcc4(−/−) mice on a C57BL/6 background. Dasatinib formulated in 80 mM citric acid (pH 3.1) or vehicle alone was administered by oral gavage to recipient mice at 10 mg/kg per dose once-daily, 5 days per week for 4 weeks, starting on day 10 post of LIC injection. Whole-animal luminescent imaging was performed in all mice to monitor tumor engraftment, and data analysis was performed using a Xenogen IVIS-200 system and Living Image Version 3.01 software (Caliper Life Sciences), as described (29). Mice were observed daily and the experiment was terminated when animals showed signs of terminal illness, including hind leg paralysis, inability to eat or drink, and/or were moribund.
LIC viability was determined using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) Cell Proliferation Kit I (Roche), as previously described (30). The cells were treated with dasatinib at increasing concentrations (0.001–30 μM) for 48 hours (two independent experiments were performed with eight replicates at each concentration). The concentration inhibiting cell viability by 50% compared with vehicle-treated control cells (IC50) was determined using the software program GraphPad Prism version 5.0 (GraphPad Software). In order to evaluate growth inhibitory properties of dasatinib against LIC at average concentrations observed in vivo in wildtype and Abcc4(−/−) mice, a correction factor was applied to account for anticipated differences in the fraction unbound dasatinib in plasma of wildtype mice, Abcc4(−/−) mice, and cell culture medium, as described (31).
Statistical calculations
All data are presented as mean values with standard error. Group differences as a function of cell type or mouse genotype were evaluated using a t-test. Two-tailed P values of less than 0.05 were considered as statistically significant. Overall survival was assessed by the Kaplan-Meier method, followed by a Mantel-Cox test. Statistical calculations were performed using NCSS version 2004 (Number Cruncher Statistical System).
RESULTS
In vitro transport of dasatinib by ABCC4
Results from ABCC4-overexpressing inside-out vesicles indicated that dasatinib was actively transported by ABCC4 (Fig. 1A). When compared to control vesicles, dasatinib accumulation in the ABCC4-overexpressing vesicles increased by 3.0-fold (P = 0.0014). Similarly, the accumulation of the prototypical ABCC4 substrate E2G was increased by 3.2-fold (P = 0.0016), although this was not observed for imatinib (1.3-fold; P = 0.098). The transport of dasatinib into vesicles expressing ABCC4 did not reach a plateau within a clinically relevant range of concentrations (Supplementary Fig. S1). This observation is consistent with the relatively high Michaelis-Menten constants (Km) reported for several other xenobiotic substrates of ABCC4, including 220–1300 μM for MTX, 640 μM for leucovorin, and >1000 μM for adefovir and tenofovir (32). Since several compounds undergo transport by ABCC4 only in the presence of physiological concentrations of glutathione (33), we also evaluated ATP-dependent co-transport of reduced glutathione (GSH). While GSH (2mM) increased ABCC4-mediated transport of E2G by about 2-fold, this phenomenon was not observed for dasatinib (Supplementary Figure 2A). Under the same experimental conditions, dasatinib uptake was also facilitated by the related transporter ABCC3 (MRP3), although the extent of accumulation (1.3-fold; P = 0.028) was lower compared with that observed for ABCC4 (Supplementary Fig. S2B).
Figure 1.
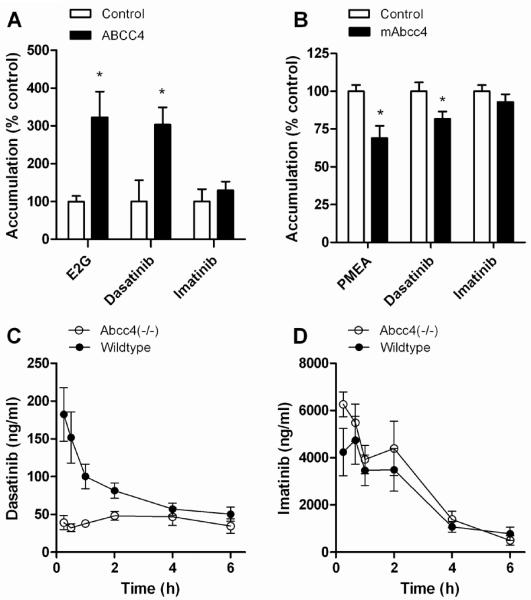
Influence of ABCC4 on dasatinib transport and pharmacokinetics. (A) Uptake of [3H]estradiol-17β-D-glucuronide (E2G) (10 μM), a positive control substrate, [3H]dasatinib (1 μM), or [3H]imatinib (1 μM) in control inside-out vesicles of Sf9 cells or vesicles expressing human ABCC4. (B) Accumulation of [3H]PMEA (10 μM), a positive control substrate, [3H]dasatinib (100 μM), or [3H]imatinib (100 μM) in Saos-2 cells transfected with an empty pcDNA (control) or mouse Abcc4. Data represent the mean 2 to 4 independent experiments, and are expressed as the average percent of uptake values in control cells. Error bars represent the standard error. The star (*) denotes a significant difference from control (P < 0.05). (C) Plasma concentration-time profile of dasatinib (oral dose, 10 mg/kg) in wildtype mice and Abcc4(−/−) mice. (D) Plasma concentration-time profile of imatinib (oral dose, 50 mg/kg) in wildtype mice and Abcc4(−/−) mice. Data represent the mean of 6 (imatinib) or 8 (dasatinib) observations per time point, and error bars represent the standard error.
Dasatinib was also found to be transported by mouse Abcc4, as exemplified by a significantly reduced accumulation in Saos-2 cells overexpressing Abcc4 compared to control cells (Fig. 1B). In this model system, no noticeable efflux transport by Abcc4 was noted with imatinib (Fig. 1B).
Dasatinib pharmacokinetics in Abcc4-knockout mice
We next evaluated the possible importance of this transporter for dasatinib in mice with a genetic deletion of Abcc4. The AUC for dasatinib in these animals after oral administration was significantly decreased by 1.75-fold compared with that observed in wildtype mice (Table 1). The respective concentration-time profiles of dasatinib suggest that the low systemic exposure in the Abcc4(−/−) mice is possibly due to an absorption defect rather than an event occurring in the terminal phase (Fig. 1C). This is consistent with the notion that the absorption rate constant is dramatically reduced in Abcc4(−/−) mice, leading to a delayed time to peak concentration, whereas the terminal half-lives of dasatinib were not significantly different between mouse genotypes (Table 1). As anticipated based on in vitro transport data, the pharmacokinetic profile of oral imatinib was not substantially affected by Abcc4-deficiency (Fig. 1D).
Table 1.
Dasatinib pharmacokinetic parameters in wildtype mice and Abcc4(−/−) mice after oral or i.v. administration of dasatinib (10 mg/kg).
| Parameter | Wildtype | Abcc4(−/−) | P |
|---|---|---|---|
| Oral | |||
| Ka(1/h) | 34.3 ± 6.85 | 2.49 ± 0.92 | 0.022 |
| Tmax (h) | 0.306 ± 0.223 | 2.19 ± 1.06 | <0.0001 |
| Cmax (ng/ml) | 182 ± 93.8 | 48.1 ± 16.5 | 0.0014 |
| AUC (ng.h/ml) | 442 ± 152 | 253 ± 86.8 | 0.0041 |
| T1/2 (h) | 4.05 ± 2.44 | 5.10 ± 2.34 | 0.54 |
| F (%) | 13.2 | 6.36 | |
| Intravenous | |||
| Cmax (ng/ml) | 3,387 ± 305 | 3,458 ± 323 | 0.73 |
| AUC (ng.h/ml) | 3,361 ± 314 | 3,975 ± 371 | 0.93 |
| T1/2 (h) | 3.96 ± 2.33 | 3.81 ± 2.49 | 0.90 |
| CL (ml/min/kg) | 49.6 | 41.9 | |
| Cblood/Cplasma | 0.896 ± 0.013 | 0.940 ± 0.04 | 0.17 |
| Blood cell partitioning (%) | 33.0 ± 1.04 | 36.1 ± 2.86 | 0.16 |
| Fraction unbound (%) | 8.70 ± 0.440 | 8.12 ± 0.493 | 0.13 |
Abbreviations: Ka, absorption rate constant; Tmax, time to peak plasma concentration; Cmax, peak plasma concentration; AUC, area under the plasma-concentration time curve extrapolated to infinity; T1/2, half-life of the terminal phase; F, apparent oral bioavailability; Cblood/Cplasma, ratio of concentration in blood to concentration in plasma.
To obtain further insights into the mechanism underlying the pharmacokinetic changes observed with dasatinib in the Abcc4(−/−) mice, the drug was also administered via i.v. bolus injection. The resulting concentration-time profiles (Supplementary Fig. S3) and pharmacokinetic parameter estimates (Table 1) were not significantly affected by Abcc4-deficiency. Moreover, the binding of dasatinib to serum proteins and its extent of blood cell partitioning (18) were independent of mouse genotype (Table 1).
To rule out potentially altered, compensatory changes in the expression of enzymes in the liver and intestine of Abcc4(−/−) mice at baseline, differential expression profiles of Cyp1a1, Cyp1b1, and Cyp3a11, the three isoforms with the highest dasatinib turnover (18), were evaluated. Compared to levels in liver and intestine of wildtype mice, transcripts of these enzymes were not increased in the Abcc4(−/−) mice (Fig. 2A–B). Furthermore, there were no potentially compensatory changes in enzyme activity, since Abcc4-knockout had no influence on the hepatic or intestinal microsomal metabolism of dasatinib to M4 (Fig. 2C) or M5 (Fig. 2D). This finding is consistent with recent data indicating that hepatic Cyp3a11 protein expression is not altered in adult male Abcc4(−/−) mice (34).
Figure 2.
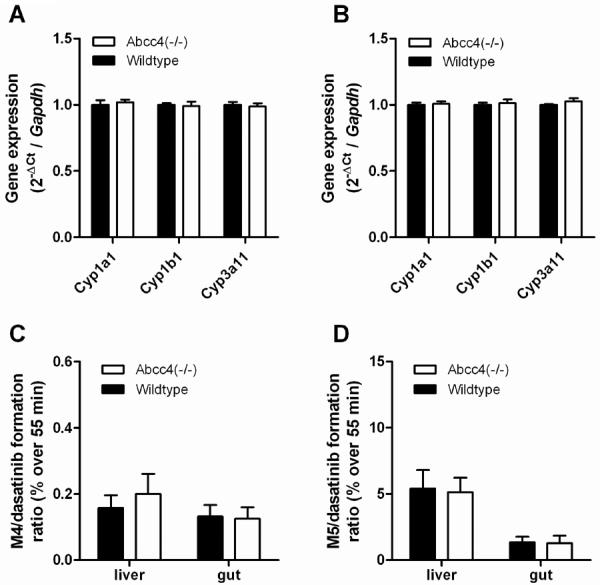
Influence of Abcc4-knockout on dasatinib metabolism. (A, B) Comparative expression of Cyp1a1, Cyp1b1, and Cyp3a11 at baseline in liver (A) and small intestine (B) of wildtype mice and Abcc4(−/−) mice. (C, D) Comparative metabolism of dasatinib to M4 (C) or M5 (D) in liver or small intestinal microsomes of wildtype mice and Abcc4(−/−) mice. Data represent the mean of 4–8 independent observations per group and are expressed as the mean (bars) and the standard error (error bars).
Since dasatinib is a substrate for Abcb1 and Abcg2 (35) and these transporters have been implicated in restricting dasatinib absorption (9), we confirmed that their protein expression levels in the small intestine were not altered in Abcc4(−/−) mice (Supplementary Fig. S4A–B). In addition, there were no differences in intrinsic intestinal permeability between wildtype and Abcc4(−/−) mice, as determined from the recovery of mannitol in plasma, kidney, and liver after oral mannitol dosing (Supplementary Fig. S4C). Likewise, the altered systemic levels of dasatinib in Abcc4(−/−) mice cannot be explained by an intrinsically altered ability to excrete dasatinib, since drug levels remained undetectable (<10 ng/ml) in the urinary bladder and gall bladder at 5 min after oral administration.
Gastric transport as a mechanism for dasatinib absorption
The notion that, in wildtype mice receiving oral dasatinib, peak concentrations in plasma were already observed at the first collection time point, prompted us to consider the possibility that dasatinib absorption occurs through the stomach. Consistent with this hypothesis, the Abcc4 protein was readily detectable in stomach samples from wildtype mice (Fig. 3A). In stomachs of wildtype mice, Abcc4 was detected in the mucosa of the and fundus (Fig. 3B) with basal labeling of gastric pit mucosal cells and basolateral labeling of the parietal and chief cells; in the parietal cells, the basal labeling was faint to weak and the lateral labeling was strong. Labeling was absent in Abcc4 (−/−) mice (Fig. 3C). In the human stomach, ABCC4 was detected in the gastric pit mucous cells, and basolateral labeling was present in parietal and chief cells (Fig. 3D).
Figure 3.

Expression of Abcc4 in mouse and human stomach. (A) Protein expression of Abcc4 normalized to β-actin in stomachs of wildtype mice and Abcc4(−/−) mice. (B–D) Localization of Abcc4 by immunohistochemistry in the stomach of a wildtype mouse (B), its absence in the stomach of an Abcc4(−/−) mouse (C), and the presence of ABCC4 in stomach of a human (D). Magnifications are 20×.
We next evaluated the ex vivo transport of dasatinib in sacs prepared from mouse stomachs, and found that Abcc4-deficiency was associated with up to 3.9-fold (P = 0.0025) reduced drug transport to the serosal side (Fig. 4A). To test whether the stomach is a site of absorption of dasatinib in vivo, a surgical procedure was implemented on anesthetized mice, allowing us to introduce a dasatinib dosing solution directly into the stomach, ligated at both the cardia and the pylorus. Analysis of liver samples, as a surrogate for portal and systemic plasma, obtained 5 min after introducing the drug confirmed that dasatinib can permeate the gastric mucosa through an Abcc4-mediated mechanism (Fig. 4B). During this experiment, the gastric pH remained on average 4.36 ± 0.32 and 4.31 ± 0.23 (P = 0.81) in wildtype and Abcc4(−/−) mice, respectively. This suggests that Abcc4-deficiency is unlikely to cause changes in gastric pH that could subsequently cause altered dasatinib solubility and lead to impaired absorption (36).
Figure 4.
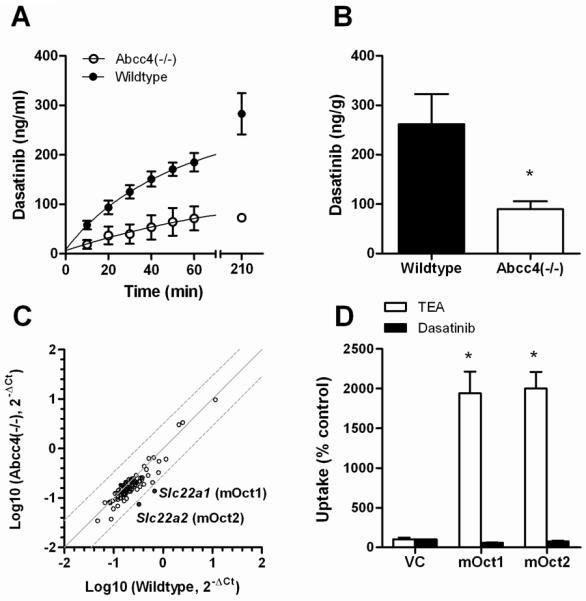
Influence of Abcc4 on gastric transport of dasatinib. (A) Time course of the ex vivo mucosal-to serosal transport of dasatinib in isolated sacs from wildtype mice or Abcc4(−/−) mouse stomachs. Data represent the mean of 4 independent observations per group and are expressed as the mean (bars) and the standard error (error bars). (B) The in vivo gastric absorption of dasatinib in wildtype or Abcc4(−/−) mice with ligated stomachs. See Methods for details. Data represent the mean of 4 observations per group and are expressed as the mean (bars) and the standard error (error bars). The star (*) denotes a significant difference from wildtype (P < 0.05). (C) Pairwise comparison of gene expression in stomachs of wildtype mice and Abcc4(−/−) mice of 84 genes encoding ATP-binding cassette transporters or solute carriers. Each symbol represents a single gene, the solid line is the line of identity, and the dotted lines are the 95% confidence intervals. Expression of the genes highlighted in red is decreased in the Abcc4(−/−) mice. (D) Transport of [14C]tetraethyl-ammonium bromide (TEA) (1 μM), a positive control substrate, or [3H]dasatinib (1 μM) in HEK293 cells containing an empty PMIG II vector (VC) or a Flag-tagged cDNA of mOct1 or mOct2 for the organic cation transporters mOct1 and mOct2. Data represent the mean of 6–12 observations, and are expressed as the average percent of uptake values (bars) with standard error (error bars) in VC cells. The star (*) denotes a significant difference from VC (P < 0.05).
Analysis of stomachs from Abcc4(−/−) mice revealed that the expression of 84 ATP-binding cassette transporter and solute carrier genes was not substantially changed compared with stomachs obtained from wildtype animals, with the exception of a downregulation of Slc22a1 (4.85-fold) and Slc22a2 (4.35-fold) (Fig. 4C). These genes encode the organic cation transporters Oct1 and Oct2 that have been implicated in xenobiotic transport (37). However, the altered expression of Slc22a1 and Slc22a2 in the stomach of Abcc4(−/−) mice is unlikely to affect the pharmacokinetic profile of dasatinib since overexpression of Oct1 or Oct2 in mammalian cells did not alter intracellular accumulation of dasatinib (Fig. 4D), which suggests that the drug is not a substrate of these transporters.
Since the stomach drains either directly or indirectly into the portal vein through short gastric veins from the fundus to the splenic vein, we also measured dasatinib concentrations in the spleen, an organ that expresses high levels of Abcc4, which in turn can restrict splenic accumulation of substrates such as topotecan (26) and PMEA (38). At 5 min after oral dasatinib administration, however, the drug levels were still higher in the spleen of wildtype mice (46.5 ± 11.8 ng/g) than in Abcc4(−/−) mice (26.7 ± 7.51 ng/g). This suggests that splenic trapping of dasatinib after oral administration is not substantially contributing to the low systemic plasma levels in Abcc4(−/−) mice.
Influence of Abcc4-deficiency on dasatinib efficacy
In view of the altered systemic exposure caused by Abcc4-deficiency and the established pharmacokinetic-pharmacodynamic relationships for dasatinib (7), we hypothesized that the antileukemic properties of dasatinib are dependent on host Abcc4 function. This hypothesis was evaluated in paired wildtype and Abcc4(−/−) mice receiving daily oral dasatinib starting on day 10 following the injection of luciferase-positive, p185(BCR-ABL)-expressing Arf(−/−) leukemia initiating pre-B cells (28). We found that these cells express low levels of Abcc4, as detected by real time RT-PCR, with an Abcc4 to Gapdh expression ratio of 0.002653. However, despite detectable transcripts, the intracellular accumulation or in vitro cell growth inhibitory properties of dasatinib in p185(BCR-ABL)-expressing Arf(−/−) leukemia initiating pre-B cells were not influenced by co-incubation with the ABC transporter inhibitor, MK571 (Supplementary Fig. S5A–B). Additional in vitro studies indicated that exposure of these cells to dasatinib at a concentration equivalent to the average in vivo levels of dasatinib in plasma of wildtype mice was associated with a 5.5-fold increase (P < 0.001) in cell growth inhibition compared to levels of dasatinib in plasma of Abcc4(−/−) mice (Supplementary Fig. S5C).
In subsequent in vivo experiments, whole-animal luminescent imaging was performed to monitor tumor engraftment (Fig. 5A). Consistent with the prediction from the in vitro experiments, two weeks after start of treatment with dasatinib (day 24), the luminescent signal was increased by 1.6-fold in wildtype mice and by more than 5-fold in Abcc4(−/−) mice (P = 0.016) (Fig. 5B). This suggests that the initial antileukemic activity of dasatinib given at the applied dose and schedule against this p185(BCR-ABL)-expressing Arf(−/−) leukemia is dependent on host Abcc4 genotype. Whereas all mice receiving vehicle only died within 2 weeks irrespective of genotype, there was a statistically significant survival advantage for wildtype mice receiving dasatinib compared to Abcc4(−/−) mice (median survival, 35 versus 31 days; P = 0.048) (Fig. 5C). Importantly, the observed systemic concentrations of dasatinib in these tumor-bearing mice receiving multiple daily doses of the drug were similarly dependent on Abcc4 genotype compared to results obtained in non tumor-bearing mice receiving a single dose (Fig. 5D).
Figure 5.
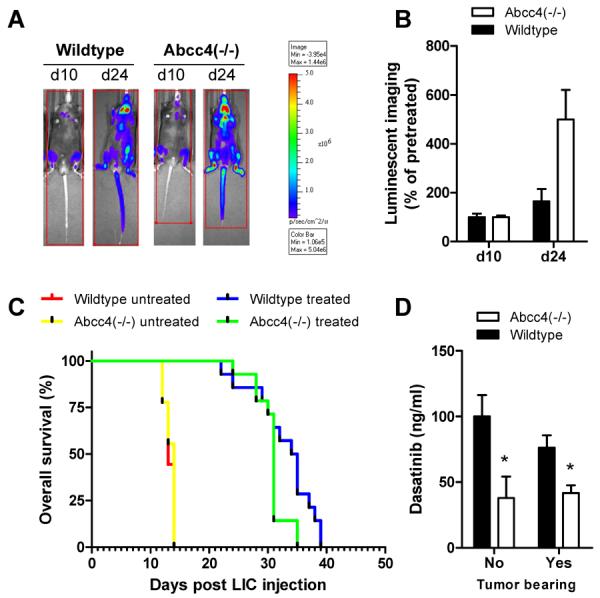
Influence of Abcc4 on antileukemic efficacy of dasatinib. (A, B) Noninvasive bioluminescence imaging immediately before drug administration (day 10) and two weeks after start of a daily oral dose (10 mg/kg) of dasatinib (day 24) in mice injected with luciferase-positive, p185(BCRABL)-expressing Arf(−/−) leukemia initiating pre-B cells (LIC). Data represent the mean (bars) and standard error (error bars) of 14 animals per group. (C) Overall survival probability for wildtype mice and Abcc4(−/−) mice receiving no treatment or dasatinib at a daily oral dose (10 mg/kg) relative to the time of LIC injection. (D) Observed systemic concentrations of dasatinib in plasma of wildtype mice or Abcc4(−/−) without leukemia (1 h after a single dose) or with leukemia (1 hour after the 8th daily oral dose). Data represent the mean (bars) and standard error (error bars) of 8–14 animals per group. The star (*) denotes a significant difference from wildtype (P < 0.05).
DISCUSSION
In the current study we found that dasatinib is a substrate for the ATP-binding cassette transporter ABCC4, that the oral absorption of dasatinib is at least partially dependent on ABCC4-mediated gastric transport, and that this process influences the drug's antitumor efficacy. The current data complement previous knowledge on the interaction of tyrosine kinase inhibitors with ATP-binding cassette transporters, and may have important practical implications for their optimal use.
The oral absorption of small molecules is believed to be primarily occurring in the small intestine of the gastrointestinal tract, with the stomach playing a supporting role in the solubilization of weakly basic compounds (39). Indeed, only a handful of molecules, including ethanol, acetylsalicylic acid (aspirin), and certain anthocyanin-flavonoids in the Vitis vinifera grape (Cabernet Sauvignon), have been demonstrated to be absorbed from the gastric compartment (25). Conventionally, for small molecules to cross cellular membranes by simple diffusion they are thought to possess certain physicochemical properties, most notably those defined by Lipinski's rule of five (40). However, mounting recent evidence points to drug transport proteins playing a more significant role in the transfer of small molecules across cellular membranes than held previously (41).
In the case of aspirin, for example, the drug is rapidly absorbed from the stomach by crossing the luminal membrane in a non-ionized form, which is favored by the highly acidic environment (42). The resulting change in pH caused by crossing the gastric mucosa would switch aspirin to a predominately ionized form that requires the assistance of a transporter to facilitate its movement across the basal side of the lumen before entering the bloodstream. Interestingly, aspirin was recently identified as a transported substrate of human ABCC4 (43), and we found that both aspirin and its metabolite salicylic acid are also substrates of mouse Abcc4 (Supplementary Fig. S6). This finding supports the possibility that aspirin, like dasatinib, may be entering the circulation through a process that is dependent, at least in part, on a gastric ATP-binding cassette transporter.
The ability of dasatinib to permeate the gastric mucosa through an ABCC4-mediated mechanism could be at the basis of the fast absorption kinetics of dasatinib in rodents as well as in humans (12). Although absorption is likely to also occur in the upper intestine, the relative contribution of the stomach in the process of dasatinib absorption remains to be determined. In this context, it is worthwhile pointing out that the fate of dasatinib in lower segments of the digestive tract is severely complicated by its strong pH-dependent aqueous solubility, which ranges from 18.4 mg/ml at pH 2.6 to only 0.008 mg/ml at pH 6.0 (36). Since the intraluminal pH in humans acutely changes from highly acidic in the stomach to about pH 6 in the duodenum, and then gradually to pH 7.4 in the terminal ileum (44), it is very likely that the stomach contributes substantially to the absorption of oral dasatinib in patients. This possibility is consistent with previous studies indicating that dasatinib is exquisitely sensitive to agents that modulate the luminal pH of the stomach by suppressing gastric acid secretion, such as H2-receptor antagonists and proton pump inhibitors (45, 46).
It is possible that gastric transport also contributes to the oral absorption of other clinically important tyrosine kinase inhibitors. Although direct evidence of this is lacking, some recent studies have demonstrated that major gastrectomy, but not small bowel resection, is associated with significantly decreased plasma levels of imatinib in patients with gastrointestinal stromal tumors (47, 48). Similar findings have been reported for nilotinib (49), and in some patients major gastrectomy was resulting in rapid disease progression. The observed decreases in exposure of these drugs have been attributed to impaired pH-dependent drug dissolution and solubility and/or an altered gastric motility resulting from the surgery. However, these explanations seem inadequate in view of the fact that the pharmacokinetics of imatinib (50) and nilotinib (51) are not substantially affected by omeprazole or its S-enantiomer esomeprazole, agents that both increase the pH of the gut as well as delay gastric emptying (52).
It is interesting to note that previous studies demonstrated that, after oral administration, the pharmacokinetic profile of the ABCC4 substrates methotrexate (24) and cefadroxil (14) was unchanged in Abcc4(−/−) mice. The reasons underlying the apparent differences in outcome of these prior studies and our current results for dasatinib are not entirely clear, but it is likely that differential mechanisms regulating drug entry on the apical side of the gut lumen contribute. In addition, unlike dasatinib, methotrexate and cefadroxil are relatively high affinity substrates of Abcc3, and this transporter may compensate for the loss of Abcc4 in the knockout mice. Indeed, concentrations of cefadroxil in portal and peripheral blood, after injection of the drug directly into a ligated jejunum, were significantly decreased in mice deficient for both Abcc3 and Abcc4, compared to wildtype mice or animals lacking only one of the transporters (14). Regardless of the exact mechanism, our current findings provide further evidence that ABCC4 can affect the pharmacokinetic properties of a remarkably broad range of substrates.
Collectively, our results indicate that in humans and mice ABCC4 is abundantly expressed in the stomach, where it facilitates the gastric absorption of dasatinib. Moreover, Abcc4-deficiency in mice was associated with reduced activity of dasatinib against a model of Ph+-ALL. These results identified ABCC4 as an important, previously unrecognized, contributor to the oral absorption of dasatinib, and this process may be relevant for other substrate drugs, such as aspirin. In view of the established exposure-efficacy relationships for dasatinib, we suggest that caution is warranted if dasatinib has to be administered together with agents that potently inhibit ABCC4.
Supplementary Material
Translational Relevance.
Dasatinib is an inhibitor of the Bcr/Abl and Src kinases, and is used in the treatment of chronic myeloid leukemia and Philadelphia chromosome-positive (Ph+) acute lymphoblastic leukemia (ALL). The interindividual pharmacokinetic variability for dasatinib treatment is extensive and largely unexplained, and this phenomenon may have important ramification for the agent's clinical activity. We speculated that differential expression of the ATP-binding cassette transporter ABCC4 (MRP4) may play a role in explaining this pharmacologic variability. We investigated the contribution of ABCC4 to the pharmacokinetics and efficacy of dasatinib using an array of in vitro and in vivo model systems. Our results indicate that ABCC4 is abundantly expressed in the stomach, where it facilitates the gastric absorption of dasatinib. Moreover, Abcc4-deficiency in mice was associated with reduced activity of dasatinib against a model of Ph+-ALL. These results identified ABCC4 as an important, previously unrecognized, contributor to the oral absorption of dasatinib and this process may be relevant for other substrate drugs, such as acetylsalicylic acid.
Acknowledgments
The authors thank Sarah Colvin, Chaoxin Hu, Markos Leggas, Shelley Orwick, Praveen Potukuchi, Laura Ramsey, and Samantha Wilcox for their various contributions to this work.
Grant Support This work was supported in part by the American Lebanese Syrian Associated Charities (ALSAC), USPHS CCSG grant 3P30CA021765 to S. D. B., and NIH grant 2R01 GM060904 to J. D. S.
Footnotes
Disclosure of Potential Conflicts of Interest No potential conflicts of interests were disclosed.
Authors' Contributions Conception and design: A. Sparreboom, S.D. Baker
Development of methodology: B.D. Furmanski, S. Hu, L. Li, L.J. Janke, R.T. Williams, J.D. Schuetz
Acquisition of data: B.D. Furmanski, S.Hu, K.-i. Fujita, L. Li, A.A. Gibson, L.J. Janke
Analysis and interpretation of data: B.D. Furmanski, L.J. Janke, R.T. Williams, J.D. Schuetz, A. Sparreboom, S.D. Baker
Writing, review, and/or revision of manuscript: B.D. Furmanski, S. Hu, K.-i. Fujita, L. Li, L.J. Janke, R.T. Williams, J.D. Schuetz, A. Sparreboom, S.D. Baker
Administrative, technical, or material support: B.D. Furmanski, S. Hu, A.A. Gibson
Study supersivion: A. Sparreboom, S.D. Baker
References
- 1.Talpaz M, Shah NP, Kantarjian H, Donato N, Nicoll J, Paquette R, et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med. 2006;354:2531–41. doi: 10.1056/NEJMoa055229. [DOI] [PubMed] [Google Scholar]
- 2.Christopher LJ, Cui D, Wu C, Luo R, Manning JA, Bonacorsi SJ, et al. Metabolism and disposition of dasatinib after oral administration to humans. Drug Metab Dispos. 2008;36:1357–64. doi: 10.1124/dmd.107.018267. [DOI] [PubMed] [Google Scholar]
- 3.Wang L, Christopher LJ, Cui D, Li W, Iyer R, Humphreys WG, et al. Identification of the human enzymes involved in the oxidative metabolism of dasatinib: an effective approach for determining metabolite formation kinetics. Drug Metab Dispos. 2008;36:1828–39. doi: 10.1124/dmd.107.020255. [DOI] [PubMed] [Google Scholar]
- 4.Demetri GD, Lo Russo P, MacPherson IRJ, Wang D, Morgan JA, Brunton VG, et al. Phase I Dose-Escalation and Pharmacokinetic Study of Dasatinib in Patients with Advanced Solid Tumors. Clinical Cancer Research. 2009;15:6232–40. doi: 10.1158/1078-0432.CCR-09-0224. [DOI] [PubMed] [Google Scholar]
- 5.Aplenc R, Blaney SM, Strauss LC, Balis FM, Shusterman S, Ingle AM, et al. Pediatric phase I trial and pharmacokinetic study of dasatinib: a report from the children's oncology group phase I consortium. J Clin Oncol. 2011;29:839–44. doi: 10.1200/JCO.2010.30.7231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gao B, Yeap S, Clements A, Balakrishnar B, Wong M, Gurney H. Evidence for therapeutic drug monitoring of targeted anticancer therapies. J Clin Oncol. 2012;30:4017–25. doi: 10.1200/JCO.2012.43.5362. [DOI] [PubMed] [Google Scholar]
- 7.Luo FR, Yang Z, Camuso A, Smykla R, McGlinchey K, Fager K, et al. Dasatinib (BMS-354825) pharmacokinetics and pharmacodynamic biomarkers in animal models predict optimal clinical exposure. Clin Cancer Res. 2006;12:7180–6. doi: 10.1158/1078-0432.CCR-06-1112. [DOI] [PubMed] [Google Scholar]
- 8.Dai G, Pfister M, Blackwood-Chirchir A, Roy A. Importance of characterizing determinants of variability in exposure: application to dasatinib in subjects with chronic myeloid leukemia. J Clin Pharmacol. 2008;48:1254–69. doi: 10.1177/0091270008320604. [DOI] [PubMed] [Google Scholar]
- 9.Lagas JS, van Waterschoot RA, van Tilburg VA, Hillebrand MJ, Lankheet N, Rosing H, et al. Brain accumulation of dasatinib is restricted by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) and can be enhanced by elacridar treatment. Clin Cancer Res. 2009;15:2344–51. doi: 10.1158/1078-0432.CCR-08-2253. [DOI] [PubMed] [Google Scholar]
- 10.Chen Y, Agarwal S, Shaik NM, Chen C, Yang Z, Elmquist WF. P-glycoprotein and breast cancer resistance protein influence brain distribution of dasatinib. J Pharmacol Exp Ther. 2009;330:956–63. doi: 10.1124/jpet.109.154781. [DOI] [PubMed] [Google Scholar]
- 11.Zimmerman EI, Hu S, Roberts JL, Gibson AA, Orwick S, Li L, et al. Contribution of OATP1B1 and OATP1B3 to the Disposition of Sorafenib and Sorafenib-Glucuronide. Clin Cancer Res. 2013;19:1458–1466. doi: 10.1158/1078-0432.CCR-12-3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Erp NP, Gelderblom H, Guchelaar HJ. Clinical pharmacokinetics of tyrosine kinase inhibitors. Cancer Treat Rev. 2009;35:692–706. doi: 10.1016/j.ctrv.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 13.Hu S, Chen Z, Franke R, Orwick S, Zhao M, Rudek MA, et al. Interaction of the multikinase inhibitors sorafenib and sunitinib with solute carriers and ATP-binding cassette transporters. Clin Cancer Res. 2009;15:6062–9. doi: 10.1158/1078-0432.CCR-09-0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Waart DR, van de Wetering K, Kunne C, Duijst S, Paulusma CC, Oude Elferink RP. Oral availability of cefadroxil depends on ABCC3 and ABCC4. Drug Metab Dispos. 2012;40:515–21. doi: 10.1124/dmd.111.041731. [DOI] [PubMed] [Google Scholar]
- 15.Ming X, Thakker DR. Role of basolateral efflux transporter MRP4 in the intestinal absorption of the antiviral drug adefovir dipivoxil. Biochem Pharmacol. 2010;79:455–62. doi: 10.1016/j.bcp.2009.08.029. [DOI] [PubMed] [Google Scholar]
- 16.Franke RM, Lancaster CS, Peer CJ, Gibson AA, Kosloske AM, Orwick SJ, et al. Effect of ABCC2 (MRP2) transport function on erythromycin metabolism. Clin Pharmacol Ther. 2011;89:693–701. doi: 10.1038/clpt.2011.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu S, Franke RM, Filipski KK, Hu C, Orwick SJ, de Bruijn EA, et al. Interaction of imatinib with human organic ion carriers. Clin Cancer Res. 2008;14:3141–8. doi: 10.1158/1078-0432.CCR-07-4913. [DOI] [PubMed] [Google Scholar]
- 18.Kamath AV, Wang J, Lee FY, Marathe PH. Preclinical pharmacokinetics and in vitro metabolism of dasatinib (BMS-354825): a potent oral multi-targeted kinase inhibitor against SRC and BCR-ABL. Cancer Chemother Pharmacol. 2008;61:365–76. doi: 10.1007/s00280-007-0478-8. [DOI] [PubMed] [Google Scholar]
- 19.Gardner ER, Smith NF, Figg WD, Sparreboom A. Influence of the dual ABCB1 and ABCG2 inhibitor tariquidar on the disposition of oral imatinib in mice. J Exp Clin Cancer Res. 2009;28:99. doi: 10.1186/1756-9966-28-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li J, Brahmer J, Messersmith W, Hidalgo M, Baker SD. Binding of gefitinib, an inhibitor of epidermal growth factor receptor-tyrosine kinase, to plasma proteins and blood cells: in vitro and in cancer patients. Invest New Drugs. 2006;24:291–7. doi: 10.1007/s10637-006-5269-2. [DOI] [PubMed] [Google Scholar]
- 21.Bijlsma PB, Peeters RA, Groot JA, Dekker PR, Taminiau JA, Van Der Meer R. Differential in vivo and in vitro intestinal permeability to lactulose and mannitol in animals and humans: a hypothesis. Gastroenterology. 1995;108:687–96. doi: 10.1016/0016-5085(95)90440-9. [DOI] [PubMed] [Google Scholar]
- 22.Emoto C, Yamazaki H, Yamasaki S, Shimada N, Nakajima M, Yokoi T. Characterization of cytochrome P450 enzymes involved in drug oxidations in mouse intestinal microsomes. Xenobiotica. 2000;30:943–53. doi: 10.1080/00498250050200104. [DOI] [PubMed] [Google Scholar]
- 23.Christopher LJ, Cui D, Li W, Barros A, Jr, Arora VK, Zhang H, et al. Biotransformation of [14C]dasatinib: in vitro studies in rat, monkey, and human and disposition after administration to rats and monkeys. Drug Metab Dispos. 2008;36:1341–56. doi: 10.1124/dmd.107.018234. [DOI] [PubMed] [Google Scholar]
- 24.Kitamura Y, Hirouchi M, Kusuhara H, Schuetz JD, Sugiyama Y. Increasing systemic exposure of methotrexate by active efflux mediated by multidrug resistance-associated protein 3 (mrp3/abcc3) J Pharmacol Exp Ther. 2008;327:465–73. doi: 10.1124/jpet.108.140475. [DOI] [PubMed] [Google Scholar]
- 25.Passamonti S, Vrhovsek U, Vanzo A, Mattivi F. The stomach as a site for anthocyanins absorption from food. FEBS Lett. 2003;544:210–3. doi: 10.1016/s0014-5793(03)00504-0. [DOI] [PubMed] [Google Scholar]
- 26.Leggas M, Adachi M, Scheffer GL, Sun D, Wielinga P, Du G, et al. Mrp4 confers resistance to topotecan and protects the brain from chemotherapy. Mol Cell Biol. 2004;24:7612–21. doi: 10.1128/MCB.24.17.7612-7621.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yasuda K, Lan LB, Sanglard D, Furuya K, Schuetz JD, Schuetz EG. Interaction of cytochrome P450 3A inhibitors with P-glycoprotein. J Pharmacol Exp Ther. 2002;303:323–32. doi: 10.1124/jpet.102.037549. [DOI] [PubMed] [Google Scholar]
- 28.Boulos N, Mulder HL, Calabrese CR, Morrison JB, Rehg JE, Relling MV, et al. Chemotherapeutic agents circumvent emergence of dasatinib-resistant BCR-ABL kinase mutations in a precise mouse model of Philadelphia chromosome-positive acute lymphoblastic leukemia. Blood. 2011;117:3585–95. doi: 10.1182/blood-2010-08-301267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu S, Niu H, Inaba H, Orwick S, Rose C, Panetta JC, et al. Activity of the multikinase inhibitor sorafenib in combination with cytarabine in acute myeloid leukemia. J Natl Cancer Inst. 2011;103:893–905. doi: 10.1093/jnci/djr107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu S, Niu H, Minkin P, Orwick S, Shimada A, Inaba H, et al. Comparison of antitumor effects of multitargeted tyrosine kinase inhibitors in acute myelogenous leukemia. Mol Cancer Ther. 2008;7:1110–20. doi: 10.1158/1535-7163.MCT-07-2218. [DOI] [PubMed] [Google Scholar]
- 31.Baker SD, Hu S. Pharmacokinetic considerations for new targeted therapies. Clin Pharmacol Ther. 2009;85:208–11. doi: 10.1038/clpt.2008.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheepala SB, Sukthankar M, Schuetz JD. MRP4 (ABCC4) In: Ishikawa T, Kim RB, Konig J, editors. Pharmacogenomics of Human Drug Transporters: Clinical Impacts. John Wiley & Sons, Inc.; Hoboken, NJ: 2013. pp. 365–85. [Google Scholar]
- 33.Rius M, Nies AT, Hummel-Eisenbeiss J, Jedlitschky G, Keppler D. Cotransport of reduced glutathione with bile salts by MRP4 (ABCC4) localized to the basolateral hepatocyte membrane. Hepatology. 2003;38:374–84. doi: 10.1053/jhep.2003.50331. [DOI] [PubMed] [Google Scholar]
- 34.Morgan JA, Cheepala SB, Wang Y, Neale G, Adachi M, Nachagari D, et al. Deregulated hepatic metabolism exacerbates impaired testosterone production in Mrp4-deficient mice. J Biol Chem. 2012;287:14456–66. doi: 10.1074/jbc.M111.319681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hiwase DK, Saunders V, Hewett D, Frede A, Zrim S, Dang P, et al. Dasatinib cellular uptake and efflux in chronic myeloid leukemia cells: therapeutic implications. Clin Cancer Res. 2008;14:3881–8. doi: 10.1158/1078-0432.CCR-07-5095. [DOI] [PubMed] [Google Scholar]
- 36.Eley T, Luo FR, Agrawal S, Sanil A, Manning J, Li T, et al. Phase I study of the effect of gastric acid pH modulators on the bioavailability of oral dasatinib in healthy subjects. J Clin Pharmacol. 2009;49:700–9. doi: 10.1177/0091270009333854. [DOI] [PubMed] [Google Scholar]
- 37.Nies AT, Koepsell H, Damme K, Schwab M. Organic cation transporters (OCTs, MATEs), in vitro and in vivo evidence for the importance in drug therapy. Handb Exp Pharmacol. 2011:105–67. doi: 10.1007/978-3-642-14541-4_3. [DOI] [PubMed] [Google Scholar]
- 38.Takenaka K, Morgan JA, Scheffer GL, Adachi M, Stewart CF, Sun D, et al. Substrate overlap between Mrp4 and Abcg2/Bcrp affects purine analogue drug cytotoxicity and tissue distribution. Cancer Res. 2007;67:6965–72. doi: 10.1158/0008-5472.CAN-06-4720. [DOI] [PubMed] [Google Scholar]
- 39.van de Waterbeemd H, Jones BC. Predicting oral absorption and bioavailability. Prog Med Chem. 2003;41:1–59. doi: 10.1016/s0079-6468(02)41001-6. [DOI] [PubMed] [Google Scholar]
- 40.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced Drug Delivery Reviews. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 41.Dobson PD, Kell DB. Carrier-mediated cellular uptake of pharmaceutical drugs: an exception or the rule? Nat Rev Drug Discov. 2008;7:205–20. doi: 10.1038/nrd2438. [DOI] [PubMed] [Google Scholar]
- 42.Hollander D DV, Fairchild PA. Intestinal absorption of aspirin. Influence of pH, taurocholate, ascorbate, and ethanol. J Lab Clin Med. 1981;4:591–8. [PubMed] [Google Scholar]
- 43.Mattiello T, Guerriero R, Lotti LV, Trifiro E, Felli MP, Barbarulo A, et al. Aspirin extrusion from human platelets through multidrug resistance protein-4-mediated transport: evidence of a reduced drug action in patients after coronary artery bypass grafting. J Am Coll Cardiol. 2011;58:752–61. doi: 10.1016/j.jacc.2011.03.049. [DOI] [PubMed] [Google Scholar]
- 44.Fallingborg J. Intraluminal pH of the human gastrointestinal tract. Dan Med Bull. 1999;46:183–96. [PubMed] [Google Scholar]
- 45.Matsuoka A, Takahashi N, Miura M, Niioka T, Kawakami K, Matsunaga T, et al. H2-receptor antagonist influences dasatinib pharmacokinetics in a patient with Philadelphia-positive acute lymphoblastic leukemia. Cancer Chemother Pharmacol. 2012;70:351–2. doi: 10.1007/s00280-012-1900-4. [DOI] [PubMed] [Google Scholar]
- 46.Takahashi N, Miura M, Niioka T, Sawada K. Influence of H2-receptor antagonists and proton pump inhibitors on dasatinib pharmacokinetics in Japanese leukemia patients. Cancer Chemother Pharmacol. 2012;69:999–1004. doi: 10.1007/s00280-011-1797-3. [DOI] [PubMed] [Google Scholar]
- 47.Pavlovsky C, Egorin MJ, Shah DD, Beumer JH, Rogel S, Pavlovsky S. Imatinib mesylate pharmacokinetics before and after sleeve gastrectomy in a morbidly obese patient with chronic myeloid leukemia. Pharmacotherapy. 2009;29:1152–6. doi: 10.1592/phco.29.9.1152. [DOI] [PubMed] [Google Scholar]
- 48.Yoo C, Ryu MH, Kang BW, Yoon SK, Ryoo BY, Chang HM, et al. Cross-sectional study of imatinib plasma trough levels in patients with advanced gastrointestinal stromal tumors: impact of gastrointestinal resection on exposure to imatinib. J Clin Oncol. 2010;28:1554–9. doi: 10.1200/JCO.2009.26.5785. [DOI] [PubMed] [Google Scholar]
- 49.Kim KP, Ryu MH, Yoo C, Ryoo BY, Choi DR, Chang HM, et al. Nilotinib in patients with GIST who failed imatinib and sunitinib: importance of prior surgery on drug bioavailability. Cancer Chemother Pharmacol. 2011;68:285–91. doi: 10.1007/s00280-010-1479-6. [DOI] [PubMed] [Google Scholar]
- 50.Egorin MJ, Shah DD, Christner SM, Yerk MA, Komazec KA, Appleman LR, et al. Effect of a proton pump inhibitor on the pharmacokinetics of imatinib. Br J Clin Pharmacol. 2009;68:370–4. doi: 10.1111/j.1365-2125.2009.03466.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yin OQ, Gallagher N, Fischer D, Demirhan E, Zhou W, Golor G, et al. Effect of the proton pump inhibitor esomeprazole on the oral absorption and pharmacokinetics of nilotinib. J Clin Pharmacol. 2010;50:960–7. doi: 10.1177/0091270009346061. [DOI] [PubMed] [Google Scholar]
- 52.Tougas G, Earnest DL, Chen Y, Vanderkoy C, Rojavin M. Omeprazole delays gastric emptying in healthy volunteers: an effect prevented by tegaserod. Aliment Pharmacol Ther. 2005;22:59–65. doi: 10.1111/j.1365-2036.2005.02528.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.