

Patients with long standing inflammatory bowel diseases (IBDs) such as ulcerative colitis and Crohn’s disease are at increased risk for colorectal cancer.1 In addition to colorectal cancer, development of hepatobiliary carcinoma and hematopoetic cancers including colonic lymphoma have also been observed in IBD patients.2 Mechanisms of chronic inflammation-associated cancers have been extensively studied; however, those linking mucosal intestinal inflammation to the sequence of dysplasia are still incompletely understood. Efforts have recently focused on the damaging effects of repair and regeneration of the epithelial barrier, dysregulated immune responses including persistently activated immune cells, cytokine profiles and inflammation-associated genotoxicity occurring in the colon. Genotoxicity has been observed as microsatellite instability in colon tumors,3 and oxidative base damage in the inflamed mouse colonic mucosa.4 Faulty DNA repair during chronic intestinal inflammation has also shown to accelerate development of colorectal cancer.4 Further research is still needed to determine the direct contribution of inflammation-associated genotoxicity to colorectal cancer development, as well as to other associated cancers such as lymphoma.
Though genotoxicity arising at the site of inflammation has been documented previously, we tested whether intestinal inflammation manifests a global effect, namely if it affects the level of systemic genotoxicity in the peripheral blood. Genotoxicity assays for DNA single- and double-strand breaks as well as oxidative base damage to peripheral leukocytes, and micronuclei formation in erythroblasts were measured in mice with dextran sulfate sodium (DSS)-induced colitis, and in Gαi2−/− and IL-10−/− mice, which represent two genetic models of immune-mediated colitis.
DSS is a non-genotoxic, non-mutagenic sulfated polysaccharide administered for multiple cycles, inducing colitis characterized by direct epithelial toxicity and activation of innate immune cells, which can lead to dysplasia followed by colorectal cancer.5–7 Interestingly, peripheral leukocytes from wild-type mice administered DSS for 3 cycles demonstrated significant increases in DNA single- and double-strand breaks as well as oxidized base damage after each cycle of treatment.8 Oxidative base damage remained elevated even during remission periods, demonstrating genotoxicity of chronic inflammation. In addition to peripheral leukocytes, erythroblasts in the bone marrow manifested clastogenic damage represented by micronucleus formation in normochromatic erythrocytes, which also increased after each cycle of DSS treatment.8 One cycle of DSS treatment caused significant increases in both 8-oxoguanine and nitrotyrosine, supporting the presence of NO-induced peroxynitrite and hydroxyl radicals potentially causative for oxidized bases and nitrative damage to proteins in the peripheral blood.
Examination of expression levels of pro-inflammatory cytokines in peripheral leukocytes of DSS-treated mice demonstrated systemic distribution and modulation throughout treatment. Particularly interesting was the expression pattern of TNFα, which mirrored patterns of DNA damage seen in peripheral leukocytes during 3 cycles of DSS treatment. Increases in pro-inflammatory cytokines may therefore orchestrate events leading to oxidative burst of inflammatory cells potentially responsible for the observed systemic genotoxicity.
We also measured DNA damage in Gαi2−/− and IL-10−/− mice with differing clinical severities of inflammation to determine whether systemic genotoxicity is in fact a general consequence of intestinal inflammation. Those with severe clinical inflammation (Gαi2−/− mice and older IL-10−/− mice) demonstrated the greatest DNA damage consisting of DNA single- and double-stranded breaks accompanied by oxidative base damage, as well as micronucleus formation in erythroblasts. DNA damage was even identified in mice with only subclinical inflammation (young IL-10−/− mice) albeit much lower than those with clinically severe colitis. As shown with DSS-treated mice, 8-oxoguanine and nitrotyrosine formation were also found to be elevated in peripheral leukocytes of IL-10−/− mice, also correlating to positive staining seen in the colonic surface epithelial cells and surrounding infiltrating inflammatory cells.
Our recent study therefore reveals systemic genotoxicity as a feature of subclinical and severe chronic intestinal inflammation. Local and systemic activation of innate immune cells in DSS-colitis as well as genetic models of immune colitis may serve as a source of oxidative stress and consequential DNA damage not only to the site of inflammation, but also systemically in the peripheral blood (Fig. 1, adapted from Macpherson et al.9). Though the specific cell types and products involved in inflammation-associated systemic genotoxicity remain to be determined, we speculate that locally activated immune cells may release reactive species damaging leukocytes that then can migrate into the peripheral circulation, or that systemically-induced inflammatory cytokines can themselves induce activation of remote leukocyte populations and therefore produce free radicals. Recent evidence for cross-talk and cross-regulation of genotoxic stress and the immune response10,11 further support the involvement of systemic genotoxicity in chronic inflammation and its role in contributing to inflammation-associated carcinogenesis. Systemic genotoxicity may therefore also serve as a sensitive biomarker of inflammation and its progression to cancer.
Figure 1.
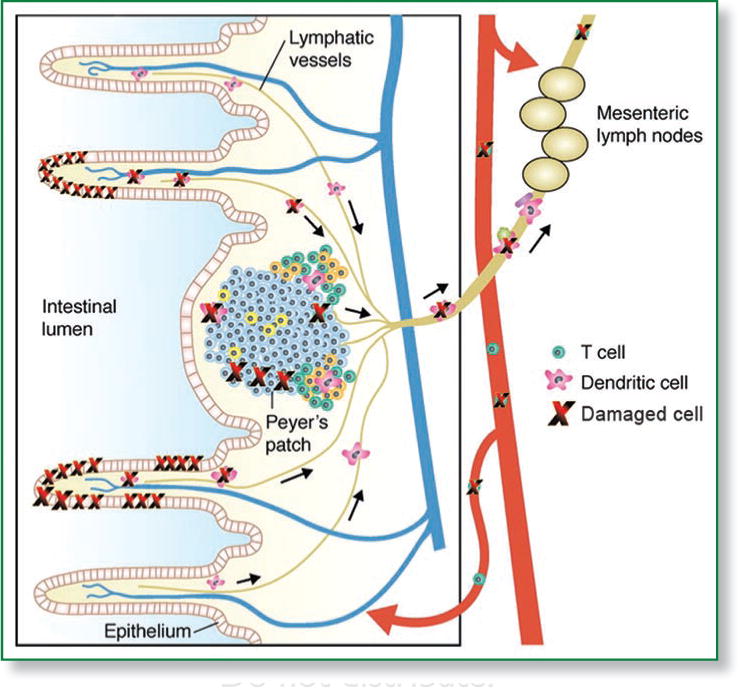
Systemic genotoxicity of intestinal inflammation. DSS-induced or immune-mediated intestinal inflammation causes DNA damage to surface epithelial cells and resident and infiltrating inflammatory cells, as well as to circulating leukocytes. Leukocytes may have been damaged that then emigrated away from the site of inflammation, or be damaged distantly by systemically activated immune responses. (Adapted from Macpherson, et al.).
Acknowledgments
Supported by NIH grants ES09519 (R.S.), DK46763 (J.B.), CA016042 (Jonsson Comprehensive Cancer Center), the Jonsson Comprehensive Cancer Center Foundation (R.S. and J.B.) and the Crohn’s and Colitis Foundation of America (J.B.).
References
- 1.Ekbom A, et al. N Engl J Med. 1990;323:1228–33. doi: 10.1056/NEJM199011013231802. [DOI] [PubMed] [Google Scholar]
- 2.Xie J, et al. World J Gastroenterol. 2008;14:378–89. doi: 10.3748/wjg.14.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kohonen-Corish MRJ, et al. Cancer Res. 2002;62:2092–7. [PubMed] [Google Scholar]
- 4.Meira LB, et al. J Clin Invest. 2008;118:2516. doi: 10.1172/JCI35073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mori H, et al. Nutr Cancer. 1984;6:92–7. doi: 10.1080/01635588509513812. [DOI] [PubMed] [Google Scholar]
- 6.Okayasu I, et al. Gastroenterology. 1990;98:694–702. doi: 10.1016/0016-5085(90)90290-h. [DOI] [PubMed] [Google Scholar]
- 7.Dieleman LA, et al. Gastroenterology. 1994;107:1643–52. doi: 10.1016/0016-5085(94)90803-6. [DOI] [PubMed] [Google Scholar]
- 8.Westbrook AM, et al. Cancer Res. 2009;69:4827–34. doi: 10.1158/0008-5472.CAN-08-4416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Macpherson AJ, et al. J Exp Med. 2006;203:497–500. doi: 10.1084/jem.20060227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coscoy L, et al. Cell. 2007;131:836–8. doi: 10.1016/j.cell.2007.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gasser S, et al. Nature. 2005;436:1186–90. doi: 10.1038/nature03884. [DOI] [PMC free article] [PubMed] [Google Scholar]