

Abstract
There have been important advances in defining effector mechanisms for several human autoimmune diseases. However, for most human autoimmune diseases, the induction stage is less well defined and there are very few clues on etiology. Our laboratory has focused on defining the molecular basis of autoantibody recognition and epitope modification in primary biliary cirrhosis (PBC). Our work has demonstrated that antibodies to mitochondria (AMA), the hallmark of disease, are directed against a very conserved site of pyruvate dehydrogenase (PDCE2). We have also demonstrated that several chemical xenobiotics, chosen based on quantitative structural activity relationship analysis and rigorous epitope analysis, when coupled to the lysine residue that normally binds the lipoic acid co-factor of PDC-E2, reacts as well or better to PBC sera than native autoantigen. In the present studies, we immunized C57BL/6 mice with one such xenobiotic, 2-octynoic acid (2OA), coupled to bovine serum albumin (BSA) and followed mice for 24 weeks. Animals were studied for appearance of histologic lesions as well as appearance of antibodies to PDC-E2, serum levels of TNF-α and IFN-γ and splenic and liver lymphoid phenotyping by flow cytometry. Mice immunized with 2OA manifest autoimmune cholangitis, typical mitochondrial autoantibodies, increased liver lymphoid cell numbers, an increase in CD8+ liver infiltrating cells, particularly CD8+ T cells that co-express CD44, and finally an elevation of serum TNF-α and IFN-γ. In conclusion, these data provide a persuasive argument in favor of an environmental origin for human PBC.
There have been important advances in defining mechanisms required for the effector stage of various human autoimmune diseases including the autoreactive populations involved in damage to the biliary epithelial cell (BEC) in primary biliary cirrhosis (PBC). On the other hand, for most autoimmune diseases, even at the induction stage, contributing genetic influences and environmental contributions are less well understood. Our laboratory has been studying the putative etiologies for the initiation of PBC for the last decade with the objective to establish a small animal model which would facilitate studies of the mechanisms that are the basis of this disease. During these studies our lab has identified several potential environmental initiators, particularly bacteria 1–3 and chemical xenobiotics 4–11 In a genetically susceptible host these initiators can disrupt immune tolerance to the dominant autoantigen of PBC, the mitochondrially localized pyruvate dehydrogenase E-2 subunit (PDC-E2), which sets in train a multi-lineage cellular and humoral immune response against this and related autoantigens. This same multi-lineage immune response then becomes the pathogenic and effector agent by virtue of the unique immunobiology of the BEC, as discussed and presented elsewhere in detail 11. The fine mapping of the epitopes recognized by the humoral and cellular autoimmune response against PDC-E2 has led to the observation that these sites are highly conserved molecular sequences flanking lipoic acid binding sites and the responses to these are among the most directed and specific responses in human autoimmunity, and includes reactivity of not only B cells, but also CD4 and CD8 T cell responses 11.
These data encouraged us to screen PBC sera against PDC-E2 molecules that carried chemical modifications of either the lysine residue (173K) or the lipoic acid co-factor that attaches to 173K. We first demonstrated that several chemical xenobiotics, when coupled via 173K to the dominant autoantigenic peptide of PDC-E2, led to the formation of a neo-antigen that surprisingly reacted at least as well or even better with PBC sera than did the native autoantigen 4, 7, 8; in other words, anti-PDC-E2 antibodies from patients with PBC could recognize xenobiotic modified PDC-E2 peptides that mimic lipoic acid, and such recognition often includes a higher titer reactivity than to the native autoantigen. Second, we have recently performed more detailed quantitative structure-activity relationship (QSAR) analysis and have identified 2-octynoic acid (2OA) as an even better xenobiotic candidate for antigenic modification of the PDC-E2 peptide 7, 8. Third, we ascertained that immunization of rabbits with a particular xenobiotic chemical, 6-bromohexanoate (6BH), coupled to bovine serum albumin (BSA), could break tolerance to PDC-E2 as judged by production of anti-mitochondrial antibodies (AMA) as seen in patients with PBC5, 6. Fourth, and finally, we recently demonstrated that immunization with the compound 6BH, coupled to BSA, led to the development of histological lesions typical of autoimmune cholangitis with the concurrent appearance of AMA in guinea pigs, albeit with a long latency of some 18 months 10.
In the present studies, we immunized mice with 2OA coupled to BSA and we observed the appearance of anti-PDC-E2 together with histological lesions typical of autoimmune cholangitis. However, of interest is our finding that immunization of mice with 2OA-BSA leads to autoimmune PBC-like disease following a latency period within just a month, rather than the latency of nearly 18 months for guinea pigs immunized with 6BH. These data illustrate the capacity of chemical xenobiotics when conjugated to a potential autoepitope site to induce a PBC-like disease model in mice. Also the data provide a persuasive argument in favor of an environmental (xenobiotic) origin for human PBC.
Materials and Methods
Murine immunization
Female C57BL/6J (B6) mice (n=13 per each group) at 8 to 9 weeks of age were obtained from The Jackson Laboratory (Bar Harbor, ME) and maintained in ventilated cages under specific pathogen-free conditions. Each mouse was immunized with a mixture of 2-octynoic acid BSA conjugate (2OA-BSA) (100μg/25μl) intraperitoneally in Complete Freund’s Adjuvant (CFA, Sigma-Aldrich, St. Louis, MO) containing 10 mg/ml of Mycobacterium tuberculosis strain H37Ra and subsequently boosted every 2 weeks with 2OA-BSA and Incomplete Freund’s Adjuvant (IFA) (Sigma-Aldrich). As controls, a comparable numbers of female B6 mice were immunized with BSA coupled to CFA (100μg/mouse) and boosted using the identical protocol. Sera were collected before immunization and thereafter every 4 weeks prior to xenobiotic immunization for definition of anti-PDC-E2. Sera were also analyzed for the cytokines, TNF-α and IFN-γ, as noted below. Animals were sacrificed 12 or 24 weeks after immunization, liver, salivary gland, thyroid, lung, kidney, small intestine and colon tissues were collected for histological evaluation. Livers and spleens were collected at the same time points and studied by flow cytometry for cellular phenotypes using the protocol below.
Preparation of immunogen
2OA was purchased from Sigma-Aldrich and conjugated to BSA as follows. Firstly, 2OA was dissolved in dry dimethyl ether. Nhydroxysuccinimide (NHS) was then added and the solution was cooled to 0°C and stirred for 20 minutes. Dicyclohexylcarbodiimide was then added and the mixture was allowed to warm to ambient temperature over night. The solution was filtered, concentrated by roto-evaporation under reduced pressure, redissolved with ethyl ether, washed with water, NaHCO3 (1M), brine, dried under magnesium sulfate, filtered and concentrated. The product was then purified using flash chromatography (30% ethyl acetate/hexane). NHS-activated 2OA was dissolved in dimethyl sulfoxide (DMSO) and then coupled to the lysine residues of BSA (EMD chemicals, Gibbstown, NJ) as previously described 5 (Fig. 1). The solution was allowed to react for 3 hours followed by dialysis with PBS.
Figure 1. Structures of lipoic acid and 2-octynoic acid-BSA.

2-octynoic acid was conjugated with BSA and used for immunization. “Z” represents any lysine residue of BSA.
Detection of anti-PDC-E2
Immunoblotting
10 μg of the purified recombinant PDC-E2 was resolved on a 10% Novex mini gel (Invitrogen, Carlsbad, CA). The proteins were electro-blotted to nitrocellulose membrane (Whatman, Dassel, Germany) and cut into 2 mm strips. Each strip was then blocked for 1 hour at room temperature (RT) with casein blocker (Piece Biotechnology, Rockford, IL). The strips were incubated for 1 hour with either mouse sera (1:200) or an AMA positive human PBC control serum (1:1,000) or a mouse anti-PDC-E2 monoclonal antibody, 2H4 12, 13 (1:50) diluted in PBS containing 0.05% Tween-20 (PBS-T) and 0.05% casein blocker (Fisher Biotech, Fair Lawn, NJ). These strips were then washed 6 times for 5 minutes in PBS-T. Secondary antibody was either horseradish peroxidase (HRP)-conjugated goat anti-mouse immunoglobulin G, A, M (IgG, A, M) or HRP-conjugated goat anti-human IgG, A, M (Zymed, San Francisco, CA) diluted 1:10,000 in the same solution as the primary antibody. These strips were exposed for 1 hour at RT to the respective secondary antibodies followed by 6 PBS-T wash cycles and exposed for 5 minutes to chemiluminescent substrate (Pierce Biotechnology). Reactive components were visualized with a Fluor Tech 8900 gel doc system (Alpha Innotech, San Leandro, CA) equipped with a chemiluminescent filter.
ELISA
This assay was performed as described 14. Purified recombinant PDC-E2 antigen at 10 μg/ml in carbonate buffer (pH 9.6) was coated onto 96-well ELISA plates at 4°C overnight, washed 5 times with PBS-T, and blocked with 3% skim milk in PBS for 1 hour. 100 μl of the diluted sera (1:500) was added to the wells and incubated for 1 hour at RT followed by PBS-T washes. 100 μl of HRP-conjugated anti-mouse IgG, IgA or IgM (1:2,000) (Zymed) was added to each well for 1 hour at RT followed by another set of PBS-T washes. Immunoreactivity was determined by measuring the optical density (O.D.) at 450 nm with an Emax precision microplate reader (Molecular Devices, Sunnyvale, CA) after incubation with 100 μl of TMB substrate (BD Biosciences, San Jose, CA) for 30 minutes.
Cytokines
Serum TNF-α and IFN-γ levels were measured using the mouse inflammatory cytometric bead array (CBA) kit (Mouse Th1/Th2 Cytokine Kit and Mouse Inflammation Kit, BD Biosciences) as previously described 15. Samples were diluted 1:5 with Assay Diluent included with the kit. Following the preparation, samples were analyzed using a FACScan flow cytometer (BD Immunocytometry System, San Jose, CA) and BD CBA Software.
Microscopy of tissue sections
Light microscopy
Immediately after sacrifice, tissue (liver, thyroid, salivary gland, lung, kidney, small intestine and colon) were harvested, fixed in 10% buffered formalin, embedded in paraffin, and cut into 5 μm sections. Liver sections were deparaffinized, stained with hematoxylin and eosin, and evaluated under light microscopy.
Immunohistochemistry
Phenotypic analysis of the intrahepatic cell infiltrates was performed utilizing rat anti-mouse CD4 and anti-mouse CD8 antibodies (1:20 dilution, BioLegend, San Diego, CA). After deparaffinization, sections were soaked in TRS buffer (pH 9.0 for CD4, pH 6.1 for CD8, Dako Cytomation, Carpinteria, CA) containing 0.1% Triton X-100 (Sigma-Aldrich) and placed in a Decloaking Chamber (Biocare Medical, Concord, CA) (SP1 at 123°C for 2 minutes, SP2 85°C for 10 seconds, SP limit 10°C) and then soaked in Universal blocking solution (BioGenex, San Roman, CA) for 15 minutes. Primary antibodies were applied overnight at 4°C in a moist chamber. After 3 washes with PBS for 5 minutes, sections were soaked in 3% H2O2 methanol solution for 5 minutes and then HRP-conjugated polyclonal rabbit anti-rat Ig (1:100, Dako Cytomation) was applied as a secondary antibody for one hour at RT in a moist chamber. After 3 washes with PBS, the sections were developed with the DAB peroxidase substrate kit (Vector Laboratories, Burlingame, CA) and counterstained with hematoxylin (Dako Cytomation).
Flow cytometry
Immediately after sacrifice, livers and spleens were collected from B6 mice immunized with 2OA-BSA or BSA. Livers were first perfused with PBS containing 0.2% BSA, passed through a nylon mesh, and re-suspended in PBS/0.2% BSA. Hepatocytes were removed as pellets after centrifugation at 700 rpm for 1 minute and the remainder cells collected. Spleen tissue was disrupted between 2 glass slides and suspended in PBS/0.2% BSA. Lymphocytes from suspended liver and spleen cells were isolated using Accu-Paque (density: 1.086, Accurate Chemical & Scientific Corp., Westburry, NY) gradient. After centrifugation, cells at the interface were washed with PBS/0.2% BSA, and the viability of cells was confirmed by Trypan Blue dye (Invitrogen) exclusion. Cell preparations were incubated with monoclonal antibody 2.4G2 for FcR blocking (BioLegend, San Diego, CA) and then incubated at 4°C with a combination of Fluorochrome-conjugated antibodies which included anti-TCRβ FITC (eBiosciences, San Diego, CA), anti-CD4 APC-Cy7 (BioLegend), anti-CD8a PE-Cy5 (eBiosciences), anti-CD19 Alexa Fluor 647 (eBiosciences). Multi-color flow analyses were performed using a FACAcan flow cytometer upgraded by Cytec Development (Fremont, CA) to allow for 5-color analysis. Acquired data were analyzed with CELLQUEST software (BD Biosciences).
Expression of data and statistical analysis
Data for groups of mouse sera tested by ELISA for anti-PDC-E2 responses, and for levels of cytokines are cited as means ± standard error of the mean (SEM). Statistical analysis was performed using an unpaired t test to compare data for 2OA-BSA and control mice. P values less than 0.05 were considered statistically significant.
Results
2OA-BSA-immunized mice developed PDC-E2 specific IgG, IgA and IgM
Immunoblotting
Sera from 100% of mice (13/13) immunized with 2OA-BSA (Fig. 1) reacted with PDC-E2 at 8 weeks post immunization. In contrast, none of the 13 sera from the control mice immunized with BSA alone had any detectable reactivity against PDC-E2 (Fig. 2. A). ELISA. At 4 weeks, serum IgG and IgM antibodies to PDC-E2 significantly increased in mice immunized with 2OA-BSA (n=11) as compared to sera from mice immunized with BSA (n=10): O.D. (Mean ± SEM) 0.33 ± 0.12 vs. 0.04 ± 0.01, P=0.0378 for IgG and 0.08 ± 0.01 vs. 0.02 ± 0.01, P=0.0019 for IgM. At 8 weeks, IgG, IgA and IgM antibodies to PDC-E2 were all significantly increased in the sera from mice immunized with 2OA-BSA compared with mice immunized with BSA: 1.23 ± 0.23 vs. 0.03 ± 0.00, P=0.0006 for IgG, 0.55 ± 0.14 vs. 0.00 ± 0.00; P=0.0025 for IgA, and 0.27 ± 0.06 vs. 0.02 ± 0.00, P=0.0006 for IgM (Fig. 2. B).
Figure 2. Antibody to PDC-E2.

A. Immunoblot at 8 weeks after immunization with 2OA-BSA, and BSA. Recombinant PDC-E2 protein was loaded on SDS-PAGE and transferred to a nitrocellulose membrane. All 13 sera from mice immunized with 2OA-BSA versus none of 12 sera from BSA-immunized mice showed detectable reactivity with a component of ~70kDa. Sera from two PBC patients and a mouse anti-PDC-E2 monoclonal antibody (mAb), 2H4, were included as positive controls. B. Quantification of anti-PDC-E2 in the sera by ELISA at 2 weekly intervals after immunization shows significant increases in O.D. values after immunization between responder and control mice. *P<0.05, **P<0.01, ***P<0.001.
Serological TNF-α and IFN-γ were elevated in 2OA-BSA-immunized mice
TNF-α and IFN-γ were significantly increased in sera from the mice immunized with 2OA-BSA compared with levels of mice immunized with BSA from 4 weeks to 12 weeks: 79.0 ± 13.7 pg/ml vs. 19.2 ± 7.5 pg/ml, P= 0.0021 for TNF-α and 14.7 ± 3.3 pg/ml vs. 0.0 ± 0.0 pg/ml, P<0.001 for IFN-γ at 4 weeks (Fig. 3). On the other hand, there was no difference in the serum levels of Th2 prototype cytokines IL-4, IL-5 or IL-10 (data not shown), indicating Th1 cytokine bias.
Figure 3. Levels of TNF-α and IFN-γ in sera of mice after immunization.

The levels of both Th1 cytokines in sera from mice immunized with 2OA-BSA versus BSA-immunized mice were markedly increased. **P<0.01, ***P<0.001.
Presence of lymphoid infiltration in the liver and mild bile duct lesion in 2OA-BSA-immunized mice but not in BSA-immunized mice
Light microscopy
As illustrated in Figure 4, the portal tracts of mice immunized with 2OA-BSA contained a significant amount of lymphoid cell infiltrates of lymphoid cells (Fig. 4. B, C, D). Mild infiltration of lymphocytes or mononuclear cells surrounding damaged bile ducts was frequently observed in portal areas (Fig. 4. B, C). These bile duct lesions resembled those of chronic nonsuppurative destructive cholangitis (CNSDC) as seen in human PBC. Furthermore ductopenia was observed, and epithelioid granulomas were scattered within some portal tracts and also in hepatic parenchyma (Figure. 4. C, D); these pathological findings resembled those of human PBC. There were also small areas of mild focal necrosis in hepatic parenchyma. No significant change was observed in central vein areas. On the other hand, no detectable cellular infiltrates and/or bile duct destruction were present and no parenchymal histopathological change was observed in liver tissues from the control mice (Fig. 4. A). Portal infiltrates and bile duct damages were evaluated with our original scoring; 0 for no infiltrate or no bile duct damage, 1 for mild, 2 for moderate. Mild portal infiltrates and mild bile duct damages were seen in all the tested mice for 2OA-BSA (n=6) and none of them were found in all the mice for BSA (n=4) (Fig. 4. G). At 24 weeks we did not observe evidence of either steatosis, eosinophilia or cholestasis. Liver sections from mice at 24 weeks post immunization showed similar findings as mice at 12 weeks. Additional tissues, including the thyroid, salivary gland, lung, kidney, small intestine and colon were examined (data not shown). These sections were identical and interpreted as normal in both groups of animals.
Figure 4. Microscopic examination of liver tissue from immunized and control mice.

A–D. Light microscopy of stained sections of liver at 12 weeks after immunization of mice with BSA (A) shows no abnormalities, and after immunization with 2OA-BSA shows mild infiltration of lymphocytes in portal tracts (B, C, D) particularly surrounding damaged intralobular bile ducts (B, C, green *). Bile duct loss and epithelioid granulomas (yellow arrow) were observed in some portal tracts (D), and granulomas were found also in hepatic parenchyma (C). P, portal vein. (H & E ×400: scale bar, 100μm.)
E, F. Immunohistochemical analysis of liver sections from 2OA-BSA-immunized mice at 12 weeks after immunization using mAbs to CD4 (E) and CD8 (F). Both CD4+ and CD8+ T lymphocytes were distributed in portal tracts with CD8+ T cells predominating over CD4+ T cells. (×400: scale bar, 100μm.)
G. Portal infiltrates and bile duct damages were scored at 12 weeks after immunization; 0 for none, 1 for mild, 2 for moderate. Both portal infiltrates and bile duct damages were observed in all the tested mice for 2OA-BSA (n=6) while none of them were found in all the mice for BSA (n=4).
Immunohistochemistry
Portal area cellular infiltrates in the liver tissues of 2OA-BSA-immunized mice contained numerous CD4+ and CD8+ lymphocytes which were not seen in liver tissues from the control BSA-immunized mice (Fig. 4. E, F). Both CD4+ and CD8+ lymphocytes were observed in the same portal tracts and CD8+ lymphocytes predominated over CD4+ lymphocytes.
Disruption of CD4/CD8 ratio in the liver of 2OA-BSA-immunized mice
The composition of the lymphoid cell population within the liver and spleen of the immunized mice was analyzed at 12 weeks post immunization. The frequency of CD4+ cells were decreased associated with a concomitant increase in the frequency of CD8+ in liver tissues from the 2OA-BSA-immunized mice as compared with control BSA-immunized mice. The frequency of both CD4+ and CD8+ cells were on the other hand decreased in the spleen (Fig. 5. A). Therefore CD4/CD8 ratios in the 2OA-BSA-immunized mice liver were significantly decreased: 2.03 ± 0.08 vs. 12.50 ± 1.63, P=0.0015; whereas there was no difference in the spleen (Fig. 5. B).
Figure 5. Lymphoid cell population in liver and spleen from immunized mice at 12 weeks.

Cells were stained with mAbs to TCRβ, CD4 and CD8. Analysis was performed with gating of TCRβ+ cells. The CD4/CD8 ratio was significantly decreased in liver but not in spleen from mice immunized with 2OA-BSA versus control BSA-immunized mice. **P<0.01.
The total lymphoid cell number was significantly increased in liver tissues from the 2OA-BSA immunized mice as compared with control BSA-immunized mice at 12 weeks: 4.18 ± 0.24 vs. 1.88 ± 0.14 (×106), P=0.0013; whereas there was no difference in the spleen (Fig. 6. A). Although the frequency of CD4+ T cells in the liver was decreased, the absolute number of CD4+ T cells was not decreased at 12 weeks because of the increase of total cell number in the liver. On the other hand, there was no difference in either the frequency or absolute numbers of CD4+ T cells in the spleen (Fig. 6. B). Importantly, the absolute number of CD8+ T cells was markedly increased only in the liver compared with control mice: 0.468±0.053 vs. 0.063 ± 0.012 (×106), P=0.0015, and there was no difference in the spleen: 8.82 ± 1.71 vs. 11.58 ± 1.68 (×106), P=0.3114 (Fig. 6. C). Interestingly, the absolute number of CD19+ B cells in the liver was also increased: 1.29 ± 0.02 vs. 0.35 ± 0.06 (×106), P<0.0001, but there was no difference in the spleen (Fig. 6. D).
Figure 6. Numbers of lymphoid cells in liver and spleen from immunized mice.

A. Total lymphoid cells were significantly increased in the liver but not in the spleen of -BSA-immunized mice versus control BSA-immunized mice.
B–D. Cells were stained with anti-TCRβ, CD4, CD8, CD19 and enumerated by flow cytometry. Graphs show data for CD4+ T cells (B), CD8+ T cells (C) and CD19+ B cells (D). CD8+ T cells and CD19+ B cells were significantly increased in the liver but not in the spleen of 2OA-BSA-immunized mice versus control BSA-immunized mice. **P<0.01.
The frequency of CD8+ T cells that co-expressed CD44, a marker of mouse memory T cells, was increased over controls in the liver: 88.70 ± 2.04 vs. 76.47 ± 3.50, P=0.0233, but there was no difference in the spleen (Fig. 7)
Figure 7. Flow cytometry analysis of CD8 and CD44 in liver and spleen.
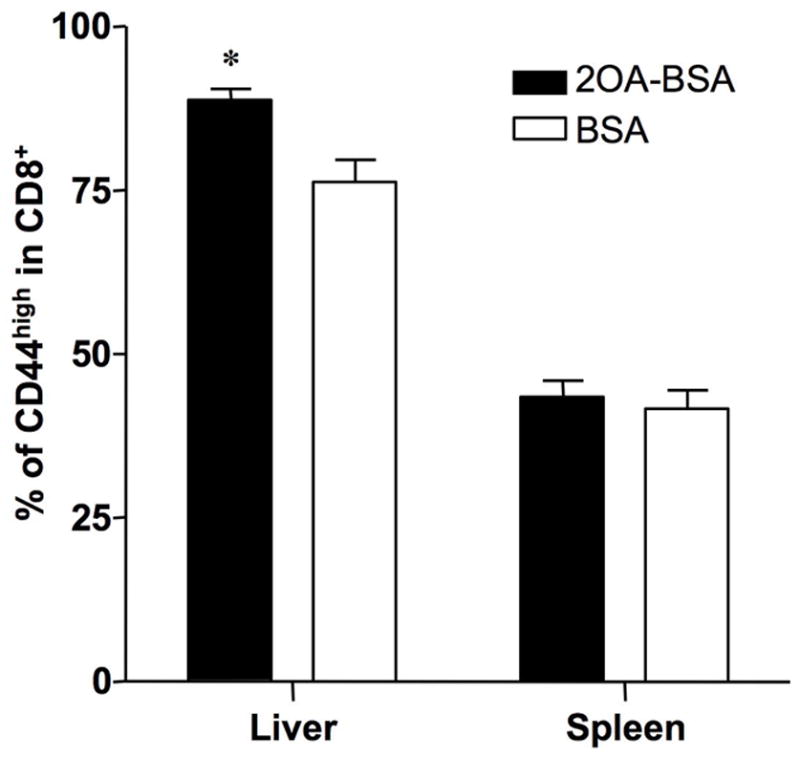
Analysis was performed with gating of TCRβ+ and CD8+ cells. Frequency of CD44high memory T cells in CD8+ T cells were increased in 2OA-BSA-immunized mice liver but not in the spleen compared with BSA-immunized mice. *P<0.05
Discussion
The data reported herein has several major advances since our earlier work on induction, using xenobiotics, of AMAs and biliary pathology in older guinea pigs 10. Firstly, our use of QSAR has allowed us to choose 2OA instead of 6BH as a more likely candidate for disease induction. Secondly, the latency time for appearance of AMAs and biliary pathology is now measured in weeks, rather than a year or longer. Third, the use of a murine model, with the ability to track and follow infiltrates with mAbs, is much greater than with guinea pigs. However, the model still has disadvantages, including the failure to see fibrosis and the inability in mice to measure cholestatic enzymes. We suspect that longer periods of observation, and likely additional host factors, are required to eventually demonstrate destructive cholangitis. These observations notwithstanding, these data are strikingly important for studies of tolerance and study of the early events of autoimmune cholangitis. Finally, and before putting these data into the context of human PBC, we should emphasize that we believe that many chemicals may be involved in similar induction of disease and the use of 2OA has proven to be a convenient method to break tolerance. This is discussed in more detail in a recent review11.
PBC is an autoimmune disease with a long latency period during which there is a progressive loss of self-tolerance and seropositivity for the signature antibody, anti-PDC-E2. Valid animals models of PBC would greatly facilitate investigation of this loss of self-tolerance. Those recently described include genetically manipulated mouse strains, including NOD.c3c4 mice 16, 17, TGF-β receptor II dominant-negative mice 18–20, and IL-2 receptor α−/− mice 15. Such mouse models spontaneously develop selective features of PBC. However, to date, there has been no accepted murine model of human PBC based on experimental immunization. Claimed models are described, including B6 mice injected with either poly I:C 21, or with purified PDC-E2 in lipopolysaccharide (LPS) or recombinant polypeptides of PDC-E2 22, but these models lack essential diagnostic criteria for human PBC. Further, in a graft versus host disease (GVHD) model 23, there was exemplary histopathology but the characteristic AMA reactivity was not reliably demonstrated. Finally, in SJL/J mice immunized with PDC-E2 emulsified in CFA, there developed an apparent autoimmune cholangitis associated with T cells that in vitro displayed a Th1/Th2 mixed cytokine profile after stimulation with the autoantigen 24, 25, but unfortunately a similar histopathology was observed in mice immunized with control protein (α-casein) emulsified in CFA 26, putting into question the significance of this finding. Particular features of the presently described mouse model, generated by immunization of B6 mice with 2OA conjugated with BSA were that no inflammation was found in organs other than liver, no analogous findings were seen in control B6 mice immunized with BSA in CFA, and antibodies to PDC-E2 were regularly demonstrable by immunoblot and ELISA. The mouse strain used in the present study, C57BL/6J (B6), does not spontaneously develop biliary disease, but does so only after immunization with 2OA-BSA with an anti-PDC-E2 response.
As regard to genetics of PBC, a number of genetic factors have been suggested in population and family studies. However, no definitive genetic association with disease susceptibility or onset has been found 27. The relative risk for siblings and the concordance rate in monozygotic twins in PBC are among the highest reported in autoimmune conditions. However, discordant pairs were identified. Therefore not only the role of genetics but also emphasize that either epigenetic factors and/or environment plays critical role in the induction of PBC 28.
Results from our attempts to develop an animal model for human PBC indicate that there are several variables that contribute to induction of a “pathological” autoimmune response versus an autoimmune response with low to undetectable levels of tissue pathology. These variables include the species of the animal being utilized, the nature of the auto-antigenic mimic, and the requirement for an adjuvant. Various clonotypes of self-reactive T cells do escape negative selection within the thymus and, whilst present in the periphery, do not induce any pathology in the host 29. Antigenic ignorance (likely based on a naïve phenotype), peripheral deletion, induction of anergy and influences of regulatory cells have all been reasoned as contributors to the failure of such clones of T cells to cause immunopathology. It is also becoming clear that 3 signals could be required for these inert self reactive T cells to initiate a pathological response against an autoantigen: these include the nature of the antigenic epitope that leads to the engagement of the appropriate clones of T cells via the cognate TCR; the co-stimulatory signals available; and a third signal which will vary according to the lineage and organ within which the autoimmune response is generated. For example, in selected cases, the presence of IL-12 can efficiently induce such clones of auto-reactive T cells to become activated and initiate autoimmune pathology 30.
In a previous study on PBC, we reported a ten fold increase in the relative frequency of PDC-E2-specific CD8+ T cells in liver compared with that in peripheral blood 31, and the CD8+ T cell frequency in the liver correlated directly with biliary ductular damage 20, 32. Comparable observations were made for liver tissue from 2OA-BSA-immunized B6 mice in which there were significant numbers of intrahepatic CD8+ T cells (Fig. 6) with a decrease in the CD4/CD8 ratio (Fig. 5) and with a increase of CD44high memory T cells (Fig. 7). These data suggest that CD8+ T cells contribute to the immunopathogenesis of cholangiolitis in 2OA-BSA immunized B6 mice, but this possibility was not addressed directly in the present study. In future work, the effects of transfer of distinct auto-antigen primed lymphoid cell populations within the B6 strain should prove informative. Further characterization of cell population in the liver, hepatic T cell responses to self antigen and MHC expression on the bile duct epithelial cells should also be examined and these works are in progress.
With regards to the mechanism of disease induction in 2OA-BSA immunized mice, we hypothesize that a molecular mimicry between the self-antigen, lipoylated PDC-E2, and xenobiotically modified 2OA-PDC-E2 can break tolerance, particularly under the additional influence of CFA in the immunizing inoculum; tolerance failure to PDC-E2 then leads on to PBC-like bile duct damage. This hypothesis is well supported by the observation that anti-PDC-E2 from patients with PBC can recognize certain xenobiotically modified PDC-E2 peptides that mimic lipoic acid 4, and that PDC-E2 can be expressed on the surface of biliary epithelial cells in the course of apoptosis 12, 13, 33. We should note that immunization with PDC-E2 alone leads to an antimitochondrial response that reacts to mitochondria of other species, but not to self 34. Moreover rabbits and guinea pigs immunized with the lipoic acid mimic 6BH, conjugated to BSA in CFA did produce AMA, albeit after a long interval 5, 6, 10. We selected 2OA-BSA for the present study because the inhibition study using human PBC patients’ sera demonstrated that 2OA coupled to the lysine residue (173K) of the PDC-E2 peptide inner lipoyl domain is unique among the tested compounds in that it did not cross-react with the lipoylated PDC-E2 peptide 7. Importantly this xenobiotic compound is not found in nature but can be chemically synthesized and the methyl and ethyl esters are widely used in cosmetic products such as perfume, lipstick, soap, detergents, cream, lotion and many common food flavorings 7, 8, 35. BSA-conjugated 6BH was also investigated as an immunogen in the present study but anti-PDC-E2 were not evident in mice immunized with 6BH-BSA by 6 weeks (data not shown), whereas in mice immunized with 2OA-BSA, seropositivity was evident as early as four weeks post immunization. For the purposes of this study, 2OA-BSA was used to immunize the mice, but obviously another, or potentially many, chemically equivalent environmental xenobiotics may well be involved with the initiation of human PBC.
Acknowledgments
Funded by National Institutes of Health grant, DK067003.
The authers thank Thomas P. Kenny for helpful advice and Willy M. Hsu for maintaining our mitochondrial clones, and Nikki Phipps for manuscript preparation.
Abbreviations
- 2OA
2-octynoic acid
- 6BH
6-bromohexanoate
- AMA
anti-mitochondrial antibody
- BEC
biliary epithelial cell
- BSA
bovine serum albumin
- CFA
Complete Freund’s Adjuvant
- CNSDC
chronic nonsuppurative destructive cholangitis
- IFA
Incomplete Freund’s Adjuvant
- Ig
immunoglobulin
- PBC
primary biliary cirrhosis
- PDC-E2
E2 subunit of pyruvate dehydrogenase
- TCRβ
T cell receptor β
Footnotes
No conflicts of interest exist
References
- 1.Selmi C, Balkwill DL, Invernizzi P, Ansari AA, Coppel RL, Podda M, Leung PS, Kenny TP, Van De Water J, Nantz MH, Kurth MJ, Gershwin ME. Patients with primary biliary cirrhosis react against a ubiquitous xenobiotic-metabolizing bacterium. Hepatology. 2003;38:1250–7. doi: 10.1053/jhep.2003.50446. [DOI] [PubMed] [Google Scholar]
- 2.Leung PS, Park O, Matsumura S, Ansari AA, Coppel RL, Gershwin ME. Is there a relation between Chlamydia infection and primary biliary cirrhosis? Clin Dev Immunol. 2003;10:227–33. doi: 10.1080/10446670310001642429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Padgett KA, Selmi C, Kenny TP, Leung PS, Balkwill DL, Ansari AA, Coppel RL, Gershwin ME. Phylogenetic and immunological definition of four lipoylated proteins from Novosphingobium aromaticivorans, implications for primary biliary cirrhosis. J Autoimmun. 2005;24:209–19. doi: 10.1016/j.jaut.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 4.Long SA, Quan C, Van de Water J, Nantz MH, Kurth MJ, Barsky D, Colvin ME, Lam KS, Coppel RL, Ansari A, Gershwin ME. Immunoreactivity of organic mimeotopes of the E2 component of pyruvate dehydrogenase: connecting xenobiotics with primary biliary cirrhosis. J Immunol. 2001;167:2956–63. doi: 10.4049/jimmunol.167.5.2956. [DOI] [PubMed] [Google Scholar]
- 5.Leung PS, Quan C, Park O, Van de Water J, Kurth MJ, Nantz MH, Ansari AA, Coppel RL, Lam KS, Gershwin ME. Immunization with a xenobiotic 6-bromohexanoate bovine serum albumin conjugate induces antimitochondrial antibodies. J Immunol. 2003;170:5326–32. doi: 10.4049/jimmunol.170.10.5326. [DOI] [PubMed] [Google Scholar]
- 6.Amano K, Leung PS, Xu Q, Marik J, Quan C, Kurth MJ, Nantz MH, Ansari AA, Lam KS, Zeniya M, Coppel RL, Gershwin ME. Xenobiotic-induced loss of tolerance in rabbits to the mitochondrial autoantigen of primary biliary cirrhosis is reversible. J Immunol. 2004;172:6444–52. doi: 10.4049/jimmunol.172.10.6444. [DOI] [PubMed] [Google Scholar]
- 7.Amano K, Leung PS, Rieger R, Quan C, Wang X, Marik J, Suen YF, Kurth MJ, Nantz MH, Ansari AA, Lam KS, Zeniya M, Matsuura E, Coppel RL, Gershwin ME. Chemical xenobiotics and mitochondrial autoantigens in primary biliary cirrhosis: identification of antibodies against a common environmental, cosmetic, and food additive, 2-octynoic acid. J Immunol. 2005;174:5874–83. doi: 10.4049/jimmunol.174.9.5874. [DOI] [PubMed] [Google Scholar]
- 8.Rieger R, Leung PS, Jeddeloh MR, Kurth MJ, Nantz MH, Lam KS, Barsky D, Ansari AA, Coppel RL, Mackay IR, Gershwin ME. Identification of 2-nonynoic acid, a cosmetic component, as a potential trigger of primary biliary cirrhosis. J Autoimmun. 2006;27:7–16. doi: 10.1016/j.jaut.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 9.Rieger R, Gershwin ME. The X and why of xenobiotics in primary biliary cirrhosis. J Autoimmun. 2007;28:76–84. doi: 10.1016/j.jaut.2007.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leung PS, Park O, Tsuneyama K, Kurth MJ, Lam KS, Ansari AA, Coppel RL, Gershwin ME. Induction of Primary Biliary Cirrhosis in Guinea Pigs following Chemical Xenobiotic Immunization. J Immunol. 2007;179:2651–7. doi: 10.4049/jimmunol.179.4.2651. [DOI] [PubMed] [Google Scholar]
- 11.Gershwin ME, Mackay IR. The causes of primary biliary cirrhosis: Convenient and inconvenient truths. Hepatology. 2007 doi: 10.1002/hep.22042. [DOI] [PubMed] [Google Scholar]
- 12.Migliaccio C, Nishio A, Van de Water J, Ansari AA, Leung PS, Nakanuma Y, Coppel RL, Gershwin ME. Monoclonal antibodies to mitochondrial E2 components define autoepitopes in primary biliary cirrhosis. J Immunol. 1998;161:5157–63. [PubMed] [Google Scholar]
- 13.Migliaccio C, Van de Water J, Ansari AA, Kaplan MM, Coppel RL, Lam KS, Thompson RK, Stevenson F, Gershwin ME. Heterogeneous response of antimitochondrial autoantibodies and bile duct apical staining monoclonal antibodies to pyruvate dehydrogenase complex E2: the molecule versus the mimic. Hepatology. 2001;33:792–801. doi: 10.1053/jhep.2001.23783. [DOI] [PubMed] [Google Scholar]
- 14.Moteki S, Leung PS, Coppel RL, Dickson ER, Kaplan MM, Munoz S, Gershwin ME. Use of a designer triple expression hybrid clone for three different lipoyl domain for the detection of antimitochondrial autoantibodies. Hepatology. 1996;24:97–103. doi: 10.1002/hep.510240117. [DOI] [PubMed] [Google Scholar]
- 15.Wakabayashi K, Lian ZX, Moritoki Y, Lan RY, Tsuneyama K, Chuang YH, Yang GX, Ridgway W, Ueno Y, Ansari AA, Coppel RL, Mackay IR, Gershwin ME. IL-2 receptor alpha(−/−) mice and the development of primary biliary cirrhosis. Hepatology. 2006;44:1240–9. doi: 10.1002/hep.21385. [DOI] [PubMed] [Google Scholar]
- 16.Koarada S, Wu Y, Fertig N, Sass DA, Nalesnik M, Todd JA, Lyons PA, Fenyk-Melody J, Rainbow DB, Wicker LS, Peterson LB, Ridgway WM. Genetic control of autoimmunity: protection from diabetes, but spontaneous autoimmune biliary disease in a nonobese diabetic congenic strain. J Immunol. 2004;173:2315–23. doi: 10.4049/jimmunol.173.4.2315. [DOI] [PubMed] [Google Scholar]
- 17.Irie J, Wu Y, Wicker LS, Rainbow D, Nalesnik MA, Hirsch R, Peterson LB, Leung PS, Cheng C, Mackay IR, Gershwin ME, Ridgway WM. NOD. c3c4 congenic mice develop autoimmune biliary disease that serologically and pathogenetically models human primary biliary cirrhosis. J Exp Med. 2006;203:1209–19. doi: 10.1084/jem.20051911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oertelt S, Lian ZX, Cheng CM, Chuang YH, Padgett KA, He XS, Ridgway WM, Ansari AA, Coppel RL, Li MO, Flavell RA, Kronenberg M, Mackay IR, Gershwin ME. Anti-mitochondrial antibodies and primary biliary cirrhosis in TGF-beta receptor II dominant-negative mice. J Immunol. 2006;177:1655–60. doi: 10.4049/jimmunol.177.3.1655. [DOI] [PubMed] [Google Scholar]
- 19.Chuang YH, Lian ZX, Yang GX, Shu SA, Moritoki Y, Ridgway WM, Ansari AA, Kronenberg M, Flavell RA, Gao B, Gershwin ME. Natural killer T cells exacerbate liver injury in a transforming growth factor beta receptor II dominant-negative mouse model of primary biliary cirrhosis. Hepatology. 2008;47:571–80. doi: 10.1002/hep.22052. [DOI] [PubMed] [Google Scholar]
- 20.Yang GX, Lian ZX, Chuang YH, Moritoki Y, Lan RY, Wakabayashi K, Ansari AA, Flavel RA, Ridgway WM, Coppel RL, Tsuneyama K, Mackay IR, Gershwin ME. Adoptive transfer of CD8+ T cells from dnTGFβRII mice induces autoimmune cholangitis into Rag1(−/−) mice. Hepatology. 2008 doi: 10.1002/hep.22226. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Okada C, Akbar SM, Horiike N, Onji M. Early development of primary biliary cirrhosis in female C57BL/6 mice because of poly I:C administration. Liver Int. 2005;25:595–603. doi: 10.1111/j.1478-3231.2005.01043.x. [DOI] [PubMed] [Google Scholar]
- 22.Ide T, Sata M, Suzuki H, Uchimura Y, Murashima S, Shirachi M, Tanikawa K. An experimental animal model of primary biliary cirrhosis induced by lipopolysaccharide and pyruvate dehydrogenase. Kurume Med J. 1996;43:185–8. doi: 10.2739/kurumemedj.43.185. [DOI] [PubMed] [Google Scholar]
- 23.Kimura T, Suzuki K, Inada S, Hayashi A, Isobe M, Matsuzaki Y, Tanaka N, Osuga T, Fujiwara M. Monoclonal antibody against lymphocyte function-associated antigen 1 inhibits the formation of primary biliary cirrhosis-like lesions induced by murine graft-versus-host reaction. Hepatology. 1996;24:888–94. doi: 10.1053/jhep.1996.v24.pm0008855193. [DOI] [PubMed] [Google Scholar]
- 24.Jones DE, Palmer JM, Yeaman SJ, Kirby JA, Bassendine MF. Breakdown of tolerance to pyruvate dehydrogenase complex in experimental autoimmune cholangitis: a mouse model of primary biliary cirrhosis. Hepatology. 1999;30:65–70. doi: 10.1002/hep.510300123. [DOI] [PubMed] [Google Scholar]
- 25.Jones DE, Palmer JM, Kirby JA, De Cruz DJ, McCaughan GW, Sedgwick JD, Yeaman SJ, Burt AD, Bassendine MF. Experimental autoimmune cholangitis: a mouse model of immune-mediated cholangiopathy. Liver. 2000;20:351–6. doi: 10.1034/j.1600-0676.2000.020005351.x. [DOI] [PubMed] [Google Scholar]
- 26.Sasaki M, Long SA, Van De Water J, He XS, Shultz L, Coppel RL, Ansari A, Nakanuma Y, Gershwin ME. The SJL/J mouse is not a model for PBC. Hepatology. 2002;35:1284–6. doi: 10.1053/jhep.2002.32540. [DOI] [PubMed] [Google Scholar]
- 27.Tanaka A, Borchers AT, Ishibashi H, Ansari AA, Keen CL, Gershwin ME. Genetic and familial considerations of primary biliary cirrhosis. Am J Gastroenterol. 2001;96:8–15. doi: 10.1111/j.1572-0241.2001.03446.x. [DOI] [PubMed] [Google Scholar]
- 28.Selmi C, Mayo MJ, Bach N, Ishibashi H, Invernizzi P, Gish RG, Gordon SC, Wright HI, Zweiban B, Podda M, Gershwin ME. Primary biliary cirrhosis in monozygotic and dizygotic twins: genetics, epigenetics, and environment. Gastroenterology. 2004;127:485–92. doi: 10.1053/j.gastro.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 29.Sohn SJ, Thompson J, Winoto A. Apoptosis during negative selection of autoreactive thymocytes. Curr Opin Immunol. 2007;19:510–5. doi: 10.1016/j.coi.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 30.Segal R, Dayan M, Zinger H, Habut B, Shearer GM, Mozes E. The effect of IL-12 on clinical and laboratory aspects of experimental SLE in young and aging mice. Exp Gerontol. 2003;38:661–8. doi: 10.1016/s0531-5565(03)00060-3. [DOI] [PubMed] [Google Scholar]
- 31.Kita H, Matsumura S, He XS, Ansari AA, Lian ZX, Van de Water J, Coppel RL, Kaplan MM, Gershwin ME. Quantitative and functional analysis of PDC-E2-specific autoreactive cytotoxic T lymphocytes in primary biliary cirrhosis. J Clin Invest. 2002;109:1231–40. doi: 10.1172/JCI14698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lan RY, Cheng C, Lian ZX, Tsuneyama K, Yang GX, Moritoki Y, Chuang YH, Nakamura T, Saito S, Shimoda S, Tanaka A, Bowlus CL, Takano Y, Ansari AA, Coppel RL, Gershwin ME. Liver-targeted and peripheral blood alterations of regulatory T cells in rimary biliary cirrhosis. Hepatology. 2006;43:729–37. doi: 10.1002/hep.21123. [DOI] [PubMed] [Google Scholar]
- 33.Van de Water J, Turchany J, Leung PS, Lake J, Munoz S, Surh CD, Coppel R, Ansari A, Nakanuma Y, Gershwin ME. Molecular mimicry in primary biliary cirrhosis. Evidence for biliary epithelial expression of a molecule cross-reactive with pyruvate dehydrogenase complex-E2. J Clin Invest. 1993;91:2653–64. doi: 10.1172/JCI116504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krams SM, Surh CD, Coppel RL, Ansari A, Ruebner B, Gershwin ME. Immunization of experimental animals with dihydrolipoamide acetyltransferase, as a purified recombinant polypeptide, generates mitochondrial antibodies but not primary biliary cirrhosis. Hepatology. 1989;9:411–6. doi: 10.1002/hep.1840090311. [DOI] [PubMed] [Google Scholar]
- 35.Opdyke DL. Monographs on fragrance raw materials. Food Cosmet Toxicol. 1979;17:357–90. doi: 10.1016/0015-6264(79)90330-4. [DOI] [PubMed] [Google Scholar]