

Summary
CD19-deficient mice were used as a model to study FDC activation because these mice have normal numbers of FDC-containing primary follicles, but lack the ability to activate FDC or form GC. It was hypothesized that CD19 expression is necessary for B cell activation and upregulation of membrane-lymphotoxin (mLT) expression, which promotes FDC activation. Using VCAM-1 and FcγRII/III as FDC activation markers, it was determined that the adoptive transfer of CD19+ wild-type B cells into CD19-deficient hosts rescued GC formation and FDC activation, demonstrating that CD19 expression on B cells is required for FDC activation. In contrast, CD19+ donor B cells lacking mLT were unable to induce VCAM-1 expression on FDC and FcγRII/III upregulation was impaired. VCAM-1 expression on FDC, but not FcγRII/III, was rescued when CD19-deficient B cells expressing transgenic mLT were cotransferred into recipient mice with CD19+, mLT-deficient B cells, suggesting that FDC activation requires the CD19-dependent upregulation of mLT on activated B cells. Collectively, these data demonstrate that activated B cells are responsible for the initiation of FDC activation resulting in a microenvironment supportive of GC development and maintenance.
Keywords: Follicular Dendritic Cell, Germinal Center, CD19, Membrane Lymphotoxin
Introduction
Follicular dendritic cells (FDC) are radio-resistant stromal cells centrally located in primary B cell follicles that express high levels of complement receptors 1 (CD35) and 2 (CD21) [1, 2]. In the resting state, FDC play a critical role in the maintenance and localization of B cells within the white pulp of the spleen [3]. Once a germinal center (GC) develops, FDC polarize to the light zone where their role is greatly expanded to support high affinity antibody production and the generation of B cell memory. In addition to aiding in the segregation of lymphocytes into T and B cell areas via chemokine production [4, 5], a major role for FDC is to serve as a cellular network on which responding immune cells survey antigen bearing processes during T-dependent responses [6–8]. Opsonized antigen is actively targeted to FDC via complement and Fc receptors and can remain there for long periods of time [9–13]. BCR-mediated signaling by surveying B cells, presumably via interactions with antigen bound to FDC processes, induce changes in the integrins LFA-1 (αLβ2) and VLA-4 (α4β1) [14–16]. BCR-induced changes in clustering or affinity of these integrins for their ligands ICAM-1 and VCAM-1, respectively, facilitates interactions with the membrane-bound antigen on FDC [17]. Outside-in signaling via integrins on B cells in response to ICAM-1 and VCAM-1 on FDC has been shown to rescue GC B cells from apoptosis in vitro [18]. Additionally, neonatal inactivation of VCAM-1 in mice leads to defective humoral immune responses [19].
FDC within GC can be readily distinguished from FDC in resting follicles based on upregulation of the adhesion molecules ICAM-1 and VCAM-1. Whereas VCAM-1 is only expressed on FDC in active follicles, ICAM-1 is expressed on resting FDC at low levels and is upregulated within the GC [20, 21]. Although it is clear that FDC play an essential role to promote B cell responses, the events or signals that initiate differentiation of FDC from their resting precursors into fully functional effector cells are unclear.
The precursors of FDC are largely unknown, but may be of mesenchymal origin [22–24]; however, many signals essential for their development have been well characterized. Lymphotoxin (LT) on B cells is required for normal FDC development and is expressed as a secreted homotrimer, LTα3, or as a membrane-bound heterotrimer, LTα1β2 (mLT). LTα3 signals through TNF receptor 1, whereas mLT signals though a dedicated receptor, the lymphotoxin β receptor (LTβR) [25]. LTβR expression is restricted to a few cell types, including FDC, and blockade of LTβR signaling via an LTβR-Ig fusion protein eliminates FDC [26]. This suggests that mLT is a critical factor in FDC development and maintenance [25, 27, 28]. Membrane-LT is highly expressed on GC B cells compared to naïve B cells [29], and LTβR signaling on FDC-like cell lines and endothelial cells leads to the expression of VCAM-1 and ICAM-1 [30–33]. It has therefore been hypothesized that mLT on B cells may be responsible for regulating the expression of ICAM-1 and VCAM-1 on FDC in the GC [27, 34–37]. Confirming this hypothesis in vivo has proven difficult, as disrupting mLT signaling leads to loss of the FDC population [26]. Therefore experiments were performed to elucidate the signals that lead to FDC activation using CD19-deficient mice (CD19−/−), which have FDC in primary follicles, but lack the ability to promote FDC activation and to form GC.
CD19−/− mice were used to determine if FDC activation is indeed impaired based on examining the induction of VCAM-1 expression and FcγRII/III upregulation in response to challenge with SRBC. Following immunization, wild type (WT), but not CD19−/− mice, were observed to have activated FDC in the spleen that expressed VCAM-1 and high levels of FcγRII/III in conjunction with the formation of GC. FDC activation correlated with the earliest appearance of GC B cells in the light zone of WT mice in regions where GC B cells appeared to be in close physical contact with FDC. This observation suggested that a membrane-associated signal at the B cell:FDC interface may be responsible for FDC activation. To address the possibility that mLT signaling is involved in this process, experiments were performed in which CD19+,LTβ− B cells, which do not express mLT, were adoptively transferred into CD19−/− recipients. Whereas the transfer of CD19+,LTβ+ (i.e. WT) donor B cells resulted in activation of FDC within the GC, the adoptive transfer of CD19+LTβ− B cells failed to induce VCAM-1 expression on FDC and there was significantly less induction of FcγRII/III expression, demonstrating that mLT is necessary for optimal FDC activation. Although mLT appeared to be necessary for FDC activation, adoptive transfer of CD19−,mLT-transgenic (Tg) B cells, which express high levels of mLT equivalent to that seen on GC B cells, failed to induce FDC activation indicating that mLT is not sufficient in the absence of CD19. However, when CD19+,LTβ− B cells were cotransferred into CD19−/− mice with CD19−,mLT-Tg B cells, VCAM-1 expression was restored on FDC, but FcγRII/III expression was unaffected, confirming that mLT is required for the induction of VCAM-1 expression on FDC, whereas other signals may be necessary for full potentiation of FcγRII/III expression.
Results
CD19 expression is required for FDC activation
The induction of VCAM-1 expression and FcγRII/III (CD16/32) upregulation on FDC were determined to evaluate FDC activation. VCAM-1 is an adhesion molecule expressed on activated FDC, but not on resting FDC [20, 21]. FcγR are expressed on resting FDC, but exhibit significant upregulation when FDC are activated and mediate the uptake of Ag in the form of IC for presentation to GC B cells [38–40]. Thus, the expression of these molecules on FDC is commonly used to determine their activation status after immunization with T-dependent Ag.
To examine FDC activation C57BL/6 WT and CD19−/− mice were immunized i.p. with SRBC, a potent T-dependent antigen, to induce an immune response in the spleen. The spleens were harvested 10 days post immunization and FDC activation was visualized by fluorescence microscopy with anti-VCAM-1 and anti-FcγRII/III mAbs. First, the frequency of GC B cells, defined as PNA+, Fas (CD95)+, was determined by flow cytometry (Fig. 1A and Supplemental Fig. 1). WT mice had 4–6% GC B cells, whereas CD19−/− mice failed to generate GC B cells or GC in response to immunization with SRBC, as previously demonstrated [41]. By histology (Fig. 1B), GC B cells are distinguished by PNA binding and FDC were visualized with anti-CD35 (complement receptor 1, CR1) mAb. Tingible-body macrophages also bind PNA and can be seen throughout the GC. In addition to being expressed on activated FDC in WT mice, VCAM-1 was also expressed in the splenic red pulp and marginal zone of WT and CD19−/− mice (Fig. 1B). However, neither VCAM-1 nor FcγRII/III expression was detected on FDC in CD19−/− mice in the spleen, suggesting that CD19 is required for FDC activation and GC formation.
Figure 1. FDC activation and GC formation do not take place in CD19-deficient mice.

(A) C57BL/6 WT and CD19−/− mice were immunized i.p. with SRBC and B220+, PNA+, Fas+ GC B cells were analyzed by flow cytometery 10 days later. (B) GC formation and FDC activation were visualized in the spleen by staining tissue sections with biotinylated PNA (blue), anti-CD35 mAb (red), and anti-VCAM-1 or anti-CD16/32 mAb (green). Overlay of VCAM-1 or CD16/32 with CD35 staining is visualized by yellow, whereas overlay of these markers in addition to PNA staining appears white. Scale bars are 100 μm. Images are representative of 5 independent experiments in which 3 WT and 3 CD19−/− mice were immunized per experiment.
Wild-type B cells rescue FDC activation in CD19-deficient mice
In the absence of CD19, signals delivered via the BCR complex are not potentiated and B cells fail to overcome the activation threshold necessary to form GC [41, 42]. Thus, it was hypothesized that B cell activation may be a prerequisite for FDC activation. To examine this possibility further, experiments were performed to determine whether the adoptive transfer of WT B cells would restore FDC activation and GC formation in CD19−/− mice.
CD45.2 WT, CD19+/−, or CD19−/− B cells were adoptively transferred into CD45.1 CD19−/− mice and recipient mice were immunized with SRBC 3 days post transfer. Ten days after immunization, the spleens were harvested and tissue sections were stained to visualize FDC activation (Fig. 2A). CD19−/− mice that received WT or CD19+/− B cells expressed VCAM-1 and FcγRII/III on their FDC and formed GC in response to immunization, whereas mice that received CD19−/− B cells did not contain FDC with an activated phenotype (Fig. 2B) and failed to form GC. To ensure that VCAM-1 staining was uniform across sample sections, the MFI for VCAM-1 in images of the red pulp from WT and CD19−/− mice was calculated demonstrating that overall VCAM-1 staining was indeed consistent (Supplemental Fig. 2). Both WT and CD19+/− B cells expanded in response to immunization and constituted the majority of PNA+, Fas+ GC B cells in CD19−/− recipient mice. In contrast, very few CD19−/− donor B cells survived the 2-week duration of the experiment despite the presence of Ag (Fig. 2A and data not shown). These data demonstrate that B cells expressing CD19 can rescue FDC activation and GC formation in CD19−/− mice. It is possible that CD19+ B cells indirectly contribute to FDC activation by virtue of the fact that they produce Abs that combine with Ag leading to the formation of immune complexes that in turn drive FDC activation. To test this, experiments were performed in which immune complexes were passively transferred to WT and CD19−/− mice after which mice were examined at days 3 and 10 to visualize immune complex deposition, GC formation and FDC activation based on upregulation of VCAM-1 and FcγRII/III (Supplemental Fig. 3 and 4). Although immune complexes were visible in both WT and CD19−/− mice at days 3 and 10, GC formation and FDC activation were only observed in WT mice in which GC B cells were visible. Collectively, these findings support the hypothesis that CD19 expression on B cells is required for FDC activation in a B cell intrinsic manner, and that CD19-dependent B cell activation may be crucial for acquisition of signals that in turn drive FDC activation.
Figure 2. FDC activation and GC formation requires CD19.

CD45.2+ splenic B cells from WT, CD19+/− and CD19−/− mice were adoptively transferred into CD45.1+ CD19−/− mice, after which recipient mice were immunized i.p. with SRBC. (A) Ten days post-immunization, FDC activation and GC formation were visualized by staining frozen spleen sections with PNA (blue), anti-CD35 mAb (red) anti-VCAM-1 mAb (green). CD45.2+ donor B cells were visualized using anti-CD45.2 mAb (white). Scale bars are 100 μm. Images are representative of 5 independent experiments in which 3 recipient mice per group were immunized. (B) The MFI of VCAM-1 and CD16/32 expression on FDC following adoptive transfer of WT, CD19+/−, and CD19−/− B cells into CD19−/− recipient mice was calculated using Image-J software. The student’s t-test was used to compare expression of either VCAM-1 or CD16/32 on FDC from recipients that received WT B cells versus recipients that received either CD19+/− or CD19−/− B cells. The data represent the mean ± standard error of the mean determined from 5 independent experiments in which there were 3 mice per group per experiment. **p<0.0001, *p<0.05
VCAM-1 expression on FDC occurs in conjunction with the appearance of GC B cells
PNA+ GC B cells begin to appear 2–3 days after immunization [43]. Therefore, it was of interest to determine at which point during an immune response FDC upregulate the activation markers VCAM-1 and FcγRII/III. WT mice were immunized with SRBC after which they were examined for FDC activation at days 3 and 5 (Fig. 3). A small number of PNA+ GC B cells were first seen on day 3 in the light zone, and appeared to be in contact with CD35+ FDC networks. Concurrent with the appearance of PNA+ B cells, initial VCAM-1 expression and FcγRII/III upregulation on FDC was also seen on day 3. It was noted that VCAM-1 expression on FDC was localized to regions in the follicle where it appeared that FDC and GC B cells were in close proximity to one another, suggesting that GC B cells may activate FDC through direct cell contact (Fig. 3). On day 5, VCAM-1 expression and FcγRII/III upregulation are observed throughout the FDC network as the number of GC B cells expand, once again suggesting that B cells may activate FDC through physical contact and that B cell activation via CD19 signaling is required.
Figure 3. Upregulation of VCAM-1 expression on FDC occurs at sites of FDC:GC B cell contact.
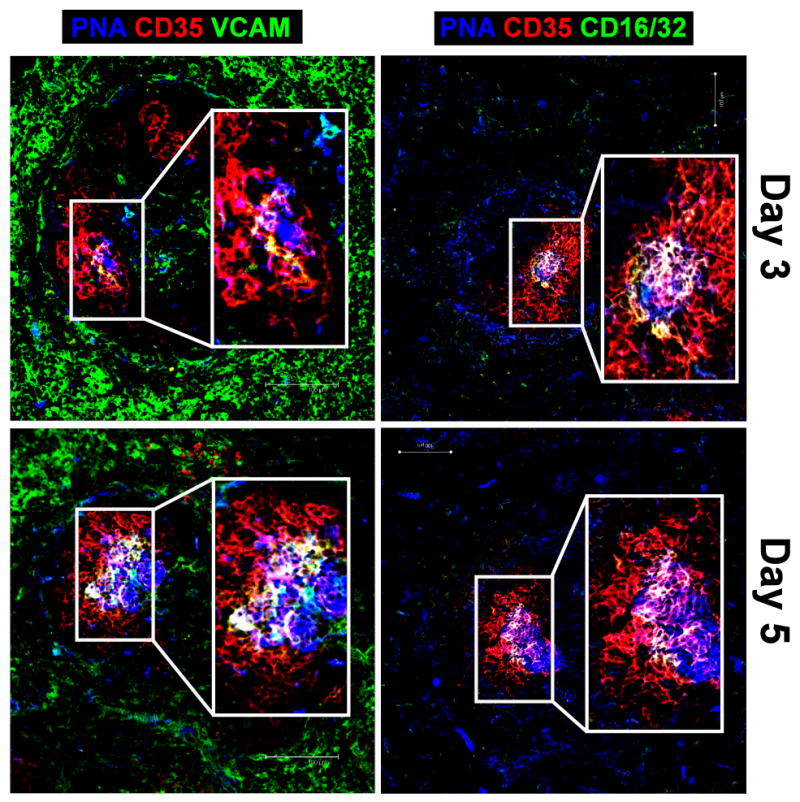
Wild-type mice were immunized i.p. with SRBC and on days 3 and 5, FDC activation and GC B cells were visualized within the GC by staining frozen spleen sections with biotinylated PNA (blue), anti-CD35 mAb (red), and either anti-VCAM-1 or anti-CD16/32 mAb (green). Overlay of VCAM-1 or CD16/32 with CD35 staining is visualized by yellow, whereas overlay of these markers in addition to PNA staining appears white. Scale bars are 100 μm. Images are representative of 3 independent experiments in which 3 WT mice were immunized per time point.
Upregulation of LTβ expression on CD19-deficient B cells is impaired
It has been shown that signaling through the LTβR induced by an agonistic Ab on a human stromal cell line resulted in increased VCAM-1 mRNA expression [44], and that LTβR signaling leads to adhesion molecule expression on FDC-like cell lines [30]. It was therefore hypothesized that FDC activation involves the interaction of mLT on GC B cells with the LTβR on FDC and that the inability to generate GC B cells, which express high levels of mLT, might explain the block in FDC activation in CD19−/− mice.
Initially, experiments were performed to determine if mLT expression on B cells is affected by CD19 deficiency. B cells from the spleens of WT and CD19−/− mice were analyzed for mLT expression based on staining cells with anti-LTβ mAb (Fig. 4A). B cells from WT mice expressed a low basal level of mLT that was substantially upregulated on GC B cells after immunization with SRBC, which is consistent with previously published reports [29]. Although follicular B cells from CD19−/− mice expressed similar basal levels of mLT compared to WT follicular B cells, CD19−/− mice do not generate GC B cells, suggesting that in the absence of this B cell population with high level mLT expression there may be a defect in FDC development or activation, or both. Therefore, it was important to evaluate if the basal expression of mLT on B cells in CD19−/− mice is sufficient to promote FDC development and maintenance. It was determined that CD19−/− mice have normal numbers of FDC containing follicles, supporting the conclusion that basal mLT expression on resting B cells in CD19−/− mice, which is slightly reduced compared to WT mice, is indeed sufficient to support FDC development (Fig. 4B). This was further confirmed by analyzing the number of FDC-containing follicles in CD19+/− or CD19−/− mice that do not express mLT. Regardless of whether CD19 is expressed on B cells, loss of mLT expression was found to result in a significant reduction in the number of FDC-containing follicles in these mice (Fig. 4B). Thus, the failure of B cells in CD19−/− mice to overcome activation thresholds important for the generation of GC B cells, which upregulate mLT expression, may explain the block in FDC activation.
Figure 4. FDC development is not altered in CD19−/− mice.
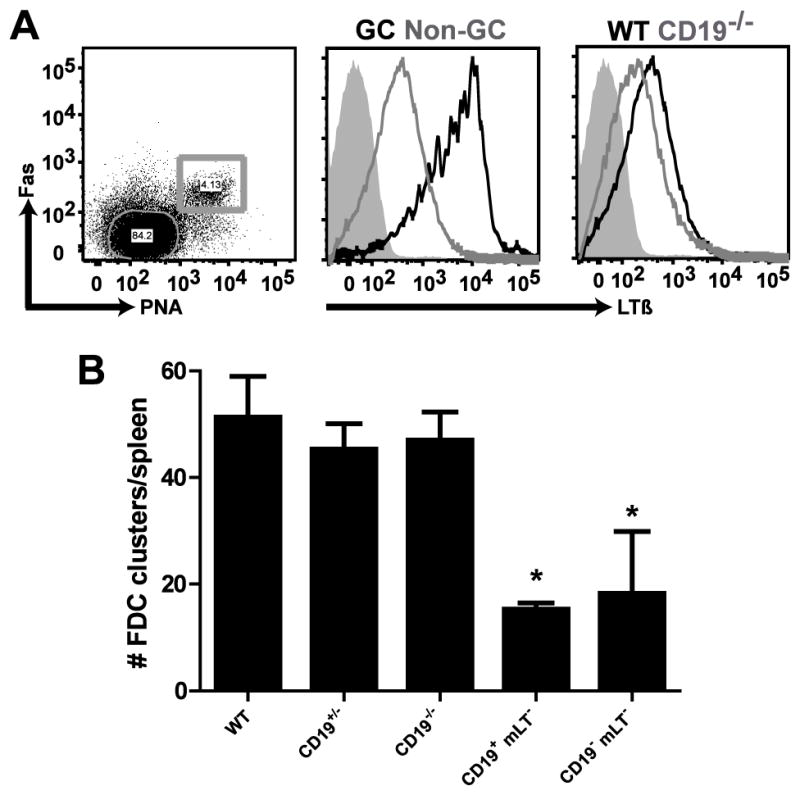
Wild-type and CD19−/− mice were immunized i.p. with SRBC. After 10 days, mLT expression on splenic B cells was analyzed by flow cytometery. (A, middle panel) Analysis of mLT expression on GC B cells in WT mice; B220+, PNA+, Fas+ GC B cells (black line), FO B cells (grey line), isotype control (filled histogram). (A, right panel) Analysis of mLT expression on FO B cells from WT versus CD19−/− mice; FO B cells isolated from WT (black line), FO B cells isolated from CD19−/− (grey line), isotype control (filled histogram). (B) FDC containing follicles in naïve, WT, CD19+/−, CD19−/−, CD19+, mLT− (CD19+/cre, LTβloxp/loxp), and CD19−, mLT− (CD19cre/cre, LTβloxp/loxp) mice were counted in frozen spleen sections using anti-CD35 mAb to visualized FDC. The data represent the average number of CD35+ FDC clusters counted in three sections per spleen and this was determined for 3 mice per genotype. The data are representative of 3 independent experiments with 3 mice per genotype per experiment. The student’s t-test was used to compare the number of FDC-containing follicles in WT versus each of the other genotypes depicted. The data represent the mean ± standard error of the mean. *p<0.01
Membrane-LT expression on GC B cells is necessary for FDC activation
The finding that adoptive transfer of WT B cells rescues FDC activation in CD19−/− mice suggests that GC B cells directly activate FDC (Fig. 2). This was further supported by the observation that in very early stages of the GC reaction the activation of FDC is restricted to those regions were FDC and GC B cells co-localize, suggesting that not only are GC B cells responsible for FDC activation, but that this process is mediated through direct cell contact between FDC and GC B cells.
Thus it was hypothesized that FDC activation is dependent on upregulation of mLT by GC B cells, which in turn signals through the LTβR on FDC. To test the hypothesis that B cells activate FDC through mLT-LTβR interactions, LTβloxp/loxp mice [45] were obtained and were crossed with CD19cre/cre mice. B cells from CD45.2 CD19+/+,LTβloxp/loxp; CD19+/cre,LTβloxp/loxp and CD19cre/cre,LTβloxp/loxp mice were adoptively transferred into CD45.1 CD19−/− recipient mice. In these experiments, donor B cells are CD19+,mLT+; CD19+,mLT− or CD19−,mLT−, respectively. The CD19−/− hosts express mLT, but lack the ability to generate GC B cells that upregulate mLT. Three days after adoptive transfer of B cells, recipient mice were immunized with SRBC and the spleens were harvested 10 days post-immunization. CD19−/− mice that received B cells from CD19+/+,LTβloxp/loxp mice, which express CD19 and mLT, formed GC in which FDC were observed to express VCAM-1 and FcγRII/III (Fig. 5, top row). In contrast, CD19−/− mice that received B cells from CD19cre/cre,LTβloxp/loxp mice, which lack CD19 and mLT failed to generate GC, and FDC from these mice did not exhibit phenotypic changes associated with activation (Fig. 5, bottom row). CD19−/− mice that received B cells from CD19+/cre,LTβloxp/loxp, that is B cells that express CD19 but lack mLT, were able to form GC, but VCAM-1 expression was not detectable on FDC (Fig. 5, middle row). Additionally, upregulation of FcγRII/III expression was significantly reduced in the absence of mLT (Fig. 5). Thus, although not required for GC formation, mLT on B cells was necessary for optimal FDC activation.
Figure 5. Membrane-LT expression on B cells is necessary for FDC activation.

Splenic B cells from CD45.2+, CD19+, mLT+ (CD19+/+, LTβflox/flox), CD19+, mLT− (CD19+/cre, LTβflox/flox) or CD19−, mLT− (CD19cre/cre, LTβflox/flox) were adoptively transferred into CD45.1+, CD19−/− mice, after which the mice were immunized i.p. with SRBC. Ten days post-immunization, splenic FDC activation and GC formation were visualized with biotinylated PNA (blue), anti-CD35 mAb (red), anti-VCAM-1 mAb (green) or anti-CD16/32 mAb (green). CD45.2+ donor B cells were detected using anti-CD45.2 mAb (white). Overlay of VCAM-1 or CD16/32 with CD35 staining is visualized by yellow, whereas overlay of these markers in addition to PNA staining appears white. Scale bars are 100 μm. Images are representative of 3 independent experiments in which 3 mice per experimental group were immunized.
Because mLT expression on B cells is necessary for optimal FDC activation, it was of interest to determine if enforced mLT expression in the absence of CD19 would be sufficient to promote GC formation and FDC activation. To address this question, mice were obtained from Dr. Jason Cyster [46], which contain a Tg that specifically and constitutively overexpresses a membrane-bound LTα chain, leading to enhanced expression of mLT on B cells. These κLTα-Tg mice were crossed with various CD19 Cre lines to generate B cells that differ in their expression of CD19 and mLT that were subsequently transferred into CD19−/− recipients. Following the adoptive transfer of CD19−/−, κLTα-Tg B cells (i.e. B cells that are CD19−,mLT-Tg) into CD19−/− mice and immunization with SRBC, VCAM-1 and FcγRII/III expression were monitored on FDC 10 days later. CD19−/− mice receiving CD19−,mLT-Tg B cells did not form GC in response to immunization, and were unable to activate FDC, as neither VCAM-1 nor FcγRII/III were expressed on the surface of FDC (Fig. 6A and B). Thus, it was apparent that enforced expression of mLT is not sufficient to drive a GC response and FDC activation. It was hypothesized that the failure of B cells that express elevated mLT to induce FDC activation was due to the deficiency in CD19 expression resulting in an inability of those B cells to overcome the activation threshold required for normal B cell responses or survival, or both. In support of this hypothesis, it was not possible to detect the adoptively transferred CD19−,mLT-Tg donor B cells 10 days post-transfer, indicating that they did not survive in the host. However, these cells could be detected in the spleen 24 h post transfer demonstrating that there was no defect in their ability to home to the splenic microenvironment (data not shown). This finding was not unexpected as earlier adoptive transfer experiments with CD19−/− B cells into CD19−/− recipient mice yielded few donor B cells after 10 days (Fig 2A).
Figure 6. Membrane-LT expression on B cells is not sufficient for induction of VCAM-1 expression on FDC in the absence of CD19.

(A) Splenic B cells from CD45.2+, CD19−, mLT-Tg (CD19−/−, κLTα-Tg) mice alone, or in a 4:1 mixture with CD45.2+, CD19+, mLT− (CD19+/cre, LTβflox/flox) and CD19- mLT-Tg (CD19−/− κLTα-Tg) were adoptively transferred into CD45.1+, CD19−/− recipient mice and subsequently immunized i.p. with SRBC. Ten days post-immunization, FDC activation and GC formation were visualized in the spleen based on staining of tissue sections with biotinylated PNA (blue), anti-CD35 mAb (red), anti-VCAM-1 mAb (green) or anti-CD16/32 mAb (green). CD45.2+, donor B cells were visualized using anti-CD45.2 mAb (white). Scale bars are 100 μm. Images are representative of 4 independent experiments in which 3 mice were immunized per experimental group. (B) The MFI of VCAM-1 and CD16/32 expression was calculated on FDC from frozen spleen sections analyzed in Figures 5 and 6. The data represent the average MFI calculated from analyzing 3 spleen sections per mouse with 3 mice per experimental group over a series of 3 independent experiments. The student’s t-test was used to compare the MFI for VCAM-1 or CD16/32 expression on FDC in WT mice with that calculated on FDC from each of the other experimental groups. The data represent the mean ± standard error of the mean. *p<0.01 **p<0.001 (C) Analysis of Ab affinity maturation based on calculating the ratio of NP8:NP30 binding activity detected by ELISA in serum collected on days 7 and 14 after immunization of mice i.p. with NP-CGG in incomplete Freund’s adjuvant. The data represent the mean ratio calculated for 3 mice per experimental group for 3 independent experiments. (D) The relative change in affinity was calculated by dividing the mean for the ratio of NP8:NP30 binding activity on day 14 by the mean on day 7.
Based on the earlier observation that CD19+,mLT− B cells were able to form GC, but did not promote FDC activation and the fact that mLT expression on B cells is necessary for optimal FDC activation, it was of interest to determine if CD19 and mLT work in concert to provide the necessary signals to drive both GC formation and FDC activation. For these experiments, a mixture of CD19+,mLT− B cells and CD19−,mLT-Tg B cells in a 5:1 ratio was adoptively transferred into CD19−/− recipient mice. The rationale was that the CD19+,mLT− B cells would initiate and sustain a GC response, in which the CD19−,mLT-Tg B cells could then participate by providing signals to FDC via binding of mLT on their surface to the LTβR on FDC thereby promoting FDC activation. Ten days after adoptive transfer and immunization with SRBC, FDC were examined to quantitate VCAM-1 and FcγRII/III expression. FDC in mice receiving CD19+,mLT− B cells with the addition of CD19−,mLT-Tg B cells, but not CD19+,mLT− B cells alone, exhibited VCAM-1 expression on their surface (Fig 6A and B), indicating that mLT expression on B cells is required for induction of VCAM-1 expression on FDC. However, FcγRII/III upregulation on FDC was unaffected by the addition of CD19−,mLT-Tg B cells (Fig. 6B), suggesting that even though mLT plays a role in potentiation of FDC activation, other signals may be required to drive FcγRII/III upregulation.
To examine the functional importance of VCAM-1 and FcγRII/III expression on FDC for the humoral response, experiments were performed to measure affinity maturation in recipient mice following immunization with NP-CGG. B cells from CD45.2 CD19+/+,LTβloxp/loxp; CD19+/cre,LTβloxp/loxp, or CD19cre/cre,LTβloxp/loxp mice or a mixture of CD19+,mLT− B cells and CD19−,mLT-Tg B cells were adoptively transferred into CD45.1 CD19−/− recipient mice followed by i.p. immunization with 50μg NP-CGG in incomplete Freund’s adjuvant. The affinity of serum anti-NP antibodies from day 7 and 14 post-immunization was determined by calculating the ratio of binding to NP8 versus NP30. Whereas the binding ratio was not statistically different among the groups at either day 7 or 14 (Fig. 6C), the relative change in affinity between day 7 and day 14 was increased almost 2-fold in mice receiving CD19+/+,LTβloxp/loxp B cells compared to all other groups (Fig. 6D). As these mice are the only mice to have VCAM-1 and fully potentiated FcγRII/III expression on FDC, these data suggest that VCAM-1 and FcγRII/III are required for normal affinity maturation. In conclusion, these results demonstrate that mLT expressed on GC B cells is required for induction of VCAM-1 expression on FDC, and that fully potentiated FDC activation is required for normal affinity maturation.
Discussion
Using CD19-deficient mice, experiments demonstrated that mLT on B cells is required for the activation of FDC. Studying the function of FDC in conclusive detail has proven difficult because of the technical challenges associated with isolation of FDC for analysis in vitro. Additionally, many of the signals thought to be involved in FDC activation have been shown to play an indispensable role in their development in vivo. Consequently, the disruption of LT signaling abrogates the development of FDC, rendering results from these experiments difficult to interpret with respect to FDC activation [34]. CD19−/− mice have normal numbers of FDC-containing primary follicles, but due to a B cell intrinsic defect, lack the ability to form GC or to activate FDC in response to immunization, providing a model to study the mechanism of FDC activation. In this system, FDC activation required CD19-dependent B cell activation. Activated B cells participating in the GC response upregulate mLT [29], which is dependent on growth-factor receptor-bound protein-2 (Grb2) signaling [47]. In the absence of CD19, B cell activation and the resulting upregulation of mLT are impaired, presumably because these cells exhibit attenuated BCR-mediated signaling and are unable to overcome the activation threshold necessary for upregulation of mLT expression. This conclusion was supported by the finding that adoptive transfer of WT B cells into CD19−/− recipients restored GC B cell production, the GC response and FDC activation.
The importance of mLT expression on B cells for FDC activation was further supported by the deletion of mLT on CD19+ donor B cells. Although adoptive transfer of mLT− B cells into CD19−/− mice did not abrogate the generation of GC, the induction of VCAM-1 expression on FDC within these GC was not observed, suggesting that mLT does indeed play an important role in FDC activation. This was confirmed by complementation of CD19+,mLT− B cells with CD19− B cells that express high levels of mLT via expression of the κLTα transgene. Adoptive transfer of this mixed population of B cells restored VCAM-1 expression on FDC in response to immunization with SRBC. Collectively, these results indicate that mLT expression on B cells is required for VCAM-1 expression on FDC, and that the defect in FDC activation in CD19−/− mice is caused by the inability of B cells to undergo optimal activation leading to upregulation of mLT expression. Although upregulation of FcγRII/III expression on FDC was attenuated in the absence of mLT on GC B cells, it was not potentiated in the presence of CD19−,mLT-Tg B cells, indicating that other CD19-dependent B cell intrinsic signals are important for modulating expression of these receptors.
The complex interplay between TNF, LTα, and LTβ in FDC development has been extensively examined [26, 48], but questions remain as to the role of these molecules in FDC activation associated with the induction of a GC response. Studies have shown that once FDC networks are established, which is TNF and LT dependent, LTα and TNF are no longer necessary for the maintenance of FDC or for GC formation after immunization [49]. However, little is known about how mLT, which is highly expressed on GC B cells, may influence FDC function. Membrane-LT upregulation is induced by CXCL13-dependent signaling through CXCR5 on B cells within the light zone of the GC [29]. It is in the light zone, 2–3 days after immunization that PNA+ GC B cells initially interact with FDC, resulting in changes in VCAM-1 and FcγRII/III expression within this microenvironment of the GC. Several lines of evidence suggest that a direct link exists between LT signaling and regulation of adhesion molecule expression on FDC. Treatment of FDC-like or endothelial cell lines with LT induces VCAM-1 and ICAM-1 expression [30, 31]. Additionally, blocking mLT signaling by the injection of a LTβR-Ig fusion protein eliminates VCAM-1 staining in the marginal zone [50]. The LTβR signals through both the canonical and non-canonical NF-κB pathways, but it is the classical pathway that is responsible for adhesion molecule expression [31, 51]. Genetically altered mice with a FDC-specific deficiency in IKKβ did not exhibit changes in VCAM-1 and ICAM-1 expression on FDC following SRBC immunization, further suggesting that mLT-LTβR signaling plays a critical role in the activation of FDC [21].
Although the adoptive transfer of CD19+,mLT− B cells into CD19−/− mice supports GC formation in response to immunization, VCAM-1 expression on FDC was not detectable. However, complementation with CD19−,mLT-Tg B cells in a mixed adoptive transfer rescued VCAM-1 expression on FDC. In these mice, the majority of GC B cells were CD19+,mLT−, suggesting that the CD19−,mLT-Tg B cells provided the required signal for VCAM-1 expression, but did not participate in the GC reaction as they could not be detected 10 days after immunization. The GC represents an open and dynamic microenvironment [8] and as such, it is plausible that while surveying the FDC surface for antigen, CD19−,mLT-Tg B cells provided signals to FDC via the LTβR resulting in upregulation of VCAM-1 expression. In the CD19−,mLT-Tg B cells, mLT upregulation is constitutive and therefore is decoupled from B cell activation, which depends on CD19 expression. Because the CD19−,mLT-Tg B cell population was unable to overcome the requisite activation threshold in the absence of CD19 [52], these cells may not have been retained in the GC, explaining why they could not be detected in the GC 10 days after adoptive transfer. This may also explain why CD19−,mLT-Tg B cells fail to induce FcγRII/III upregulation on FDC; possibly due to interrupted mLT signaling because of their inability to remain within the GC. Alternatively, FcγRII/III upregulation may not occur in this system because of a requirement for CD19- and mLT-mediated signaling to occur on the same B cell for optimal FDC activation to take place. Regardless, enforced expression of mLT on B cells was sufficient to induce VCAM-1 expression on FDC despite the transient survival of the CD19−,mLT-Tg B cells, as long as B cells were present in the GC that expressed CD19 on their surface.
In summary, these experiments demonstrate that mLT expressed on GC B cells is required for FDC activation. Our data also suggest that GC B cells activate FDC directly, resulting in upregulation of integrins. This in turn results in reciprocal signal transduction to B cells engaged with FDC via the newly expressed adhesion molecules. VCAM-1 and ICAM-1 on FDC provide anti-apoptotic signals to GC B cells through their respective integrins on the surface of the B cell [18], suggesting that upregulation of mLT on B cells is indirectly involved in promoting their survival. From a functional perspective, optimal expression of both VCAM-1 and FcγRII/III are required for affinity maturation of GC B cells. These results provide insight into the functional role that crosstalk between B cells and FDC within the GC plays in promoting an optimal humoral response.
Materials and Methods
Mice and Immunizations
C57BL/6 mice were obtained from The Jackson Laboratory (Bangor, ME). CD19−/− mice were obtained from Dr. Robert Rickert [41] and backcrossed onto the C57BL/6 background for greater than 10 generations. LTβ floxed mice, a gift from Dr. Sergei A. Nedospasov [45], and κLTα-Tg mice, a gift from Dr. Jason Cyster [46], were crossed with CD19cre/cre mice. Mice used in experiments were 8–12 weeks of age. All mice were housed in the University of Alabama at Birmingham animal housing facility in accordance with the UAB Institutional Animal Care and Use Committee policies and procedures. Mice were immunized with 100 μL SRBC in Alsever’s solution or 50 μg NP-CGG via the i.p. route.
Flow Cytometry
Splenocytes were stained as described previously [29] with FITC-labeled peanut agglutinin (PNA) (Vector Laboratories, Burlingame, CA), phycoerythrin (PE) anti-Fas (Jo2), and allophycocyanin-Cy7 anti-B220 (RA3-6B2) (BD Biosciences, San Diego CA). LTβ expression was quantitated with anti-LTβ mAb (BBF6, a gift from Dr. Jeffery L. Browning) [53], followed by anti-hamster PE. Flow cytometry was performed on a LSR II (BD Biosciences, San Jose, CA) and the data were analyzed with FlowJo version 8.2 (Tree Star Inc, Ashland, OR).
Histology
Spleens were harvested from immunized mice, embedded in OCT tissue media (Tissue-Tek), and frozen on dry ice. Ten micrometer-thick frozen sections were fixed to slides in ice-cold acetone for 15 min and air dried for 30 min. The sections were blocked with 10% horse serum for 30 min at room temperature and then stained for 30 min at room temperature in a humidified chamber with fluorescently-labeled antibody cocktails and biotinylated PNA (Vector Laboratories). The following conjugations with Alexa Flour dyes (Invitrogen, Carlsbad, CA) were performed according to the manufacturer’s instructions: anti-CD16/CD32 (2.4G2, BD Biosciences) to Alexa 488, anti-CD35 (8C12, BD Biosciences) to Alexa 555, IgD (11-26c.2a, BD Biosciences) and anti-CD45.2 (104, BD Biosciences) to Alexa 647. Biotinylated PNA was detected using strepavidin-Alexa 350 (Invitrogen). VCAM-1 on FDC was visualized with anti-VCAM-1 (CD106) conjugated to Alexa 488 (429, Serotec, Raleigh NC). Sections were mounted with Gel/Mount (Biomeda, Foster City CA). Stained tissue sections were examined with a Leica/Leitz DMRB microscope and the data were processed using Adobe Photoshop (Adobe Systems Inc., San Jose, CA). FDC containing follicles in naïve, WT, CD19+/−, CD19−/−, CD19+/cre LTβloxp/loxp, and CD19cre/cre LTβloxp/loxp were visually counted using a Zeiss Axio fluorescence microscope. The number of FDC containing follicles was averaged over three sections per spleen from three independent experiments.
Determination of Mean Fluorescence Intensity and Statistical Analysis
The MFIs for VCAM-1 and FcγRII/III were determined from 2–3 unprocessed raw image files per mouse from each of 3–5 experimental repetitions in which there were 3 mice per experimental group. Briefly, using Image J software (NIH, http://rsbweb.nih.gov/ij/), VCAM-1 and FcγRII/III on FDC was determined by, first tightly defining the area of CD35+ FDC on the anti-CD35 single color image. This gate was then copied to the anti-VCAM-1 or anti-CD16/32 single color image and the MFI of VCAM-1 or FcγRII/III was measured. To determine the VCAM-1 MFI in the red pulp, first B cell follicles were removed from the anti-VCAM-1 single color image, and then the MFI was measured over the entire remaining image. The student’s t-test was used for statistical analysis.
Adoptive Transfer Experiments
Spleens from CD45.2 WT, CD19+/−, CD19−/−, CD19+/+,LTβflox/flox; CD19+/cre,LTβflox/flox; CD19cre/cre,LTβflox/flox, or CD19−/−,κLTα-Tg mice were harvested and a single cell suspension was obtained by passing the cells through a 70 micrometer cell strainer. After incubation of the single cell suspension in ACK lysis buffer (0.15 M NH4Cl, 1 mM KHCO3, 0.1 mM Na2EDTA in diH2O) to lyse RBCs, splenocytes were washed twice in PBS. B cells were negatively selected on LS columns via the manufacture’s protocol with anti-CD11b and anti-CD43 conjugated to microbeads (Miltenyi Biotech, Auburn CA). For each adoptive transfer, 1.5×107 cells were injected intravenously in 100 μL PBS and the mice were immunized three days after adoptive transfer.
ELISA
ELISA plates were coated with NP8-BSA (bovine serum albumin) or NP30-BSA (Biosearch Technologies, Novato, CA) overnight at 4°C. The plates were blocked with 10% BSA for 1 hour at 37°C, followed by diluted serum for 1 hour at 37°C. Anti-NP antibodies were detected using anti-mouse IgG conjugated to horseradish peroxidase (HRP) (Southern Biotech, Birmingham, AL). The ratio of NP8:NP30 O.D. values were used to determine relative affinity.
Acknowledgments
We would like to thank Jason G. Cyster for κLTα-Tg mice, Sergei A. Nedospasov and David D. Chaplin for LTβ floxed mice, Jeffery Browning for anti-LTβ, the UAB High Resolution Imaging facility, and the UAB Arthritis and Musculoskeletal Flow Core facility. This work was supported by the National Institutes of Health (grant AI042265-13 to L.B. Justement).
Abbreviations used in this paper
- CR
complement receptor
- FDC
follicular dendritic cell
- FO
follicular
- LTβR
lymphotoxin β receptor
- mLT
membrane-lymphotoxin
- NRS
normal rabbit serum
- PNA
peanut agglutinin
Footnotes
Conflict of Interest
The authors declare no financial or commercial conflict of interest.
References
- 1.Allen CD, Cyster JG. Follicular dendritic cell networks of primary follicles and germinal centers: phenotype and function. Semin Immunol. 2008;20:14–25. doi: 10.1016/j.smim.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Imal Y, Yamakawa M. Morphology, function and pathology of follicular dendritic cells. Pathol Int. 1996;46:807–833. doi: 10.1111/j.1440-1827.1996.tb03555.x. [DOI] [PubMed] [Google Scholar]
- 3.Cyster JG, Ansel KM, Reif K, Ekland EH, Hyman PL, Tang HL, Luther SA, Ngo VN. Follicular stromal cells and lymphocyte homing to follicles. Immunol Rev. 2000;176:181–193. doi: 10.1034/j.1600-065x.2000.00618.x. [DOI] [PubMed] [Google Scholar]
- 4.Wang X, Cho B, Suzuki K, Xu Y, Green JA, An J, Cyster JG. Follicular dendritic cells help establish follicle identity and promote B cell retention in germinal centers. J Exp Med. 2011;208:2497–2510. doi: 10.1084/jem.20111449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allen CD, Ansel KM, Low C, Lesley R, Tamamura H, Fujii N, Cyster JG. Germinal center dark and light zone organization is mediated by CXCR4 and CXCR5. Nat Immunol. 2004;5:943–952. doi: 10.1038/ni1100. [DOI] [PubMed] [Google Scholar]
- 6.Suzuki K, Grigorova I, Phan TG, Kelly LM, Cyster JG. Visualizing B cell capture of cognate antigen from follicular dendritic cells. J Exp Med. 2009;206:1485–1493. doi: 10.1084/jem.20090209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Allen CD, Okada T, Tang HL, Cyster JG. Imaging of germinal center selection events during affinity maturation. Science. 2007;315:528–531. doi: 10.1126/science.1136736. [DOI] [PubMed] [Google Scholar]
- 8.Schwickert TA, Lindquist RL, Shakhar G, Livshits G, Skokos D, Kosco-Vilbois MH, Dustin ML, Nussenzweig MC. In vivo imaging of germinal centres reveals a dynamic open structure. Nature. 2007;446:83–87. doi: 10.1038/nature05573. [DOI] [PubMed] [Google Scholar]
- 9.Link A, Zabel F, Schnetzler Y, Titz A, Brombacher F, Bachmann MF. Innate immunity mediates follicular transport of particulate but not soluble protein antigen. J Immunol. 2012;188:3724–3733. doi: 10.4049/jimmunol.1103312. [DOI] [PubMed] [Google Scholar]
- 10.Carroll MC. The role of complement and complement receptors in induction and regulation of immunity. Annu Rev Immunol. 1998;16:545–568. doi: 10.1146/annurev.immunol.16.1.545. [DOI] [PubMed] [Google Scholar]
- 11.Yoshida K, van den Berg TK, Dijkstra CD. Two functionally different follicular dendritic cells in secondary lymphoid follicles of mouse spleen, as revealed by CR1/2 and FcR gamma II-mediated immune-complex trapping. Immunology. 1993;80:34–39. [PMC free article] [PubMed] [Google Scholar]
- 12.Cinamon G, Zachariah MA, Lam OM, Foss FW, Jr, Cyster JG. Follicular shuttling of marginal zone B cells facilitates antigen transport. Nat Immunol. 2008;9:54–62. doi: 10.1038/ni1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mattsson J, Yrlid U, Stensson A, Schon K, Karlsson MC, Ravetch JV, Lycke NY. Complement activation and complement receptors on follicular dendritic cells are critical for the function of a targeted adjuvant. J Immunol. 2011;187:3641–3652. doi: 10.4049/jimmunol.1101107. [DOI] [PubMed] [Google Scholar]
- 14.Carrasco YR, Fleire SJ, Cameron T, Dustin ML, Batista FD. LFA-1/ICAM-1 interaction lowers the threshold of B cell activation by facilitating B cell adhesion and synapse formation. Immunity. 2004;20:589–599. doi: 10.1016/s1074-7613(04)00105-0. [DOI] [PubMed] [Google Scholar]
- 15.Spaargaren M, Beuling EA, Rurup ML, Meijer HP, Klok MD, Middendorp S, Hendriks RW, Pals ST. The B cell antigen receptor controls integrin activity through Btk and PLCgamma2. J Exp Med. 2003;198:1539–1550. doi: 10.1084/jem.20011866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arana E, Harwood NE, Batista FD. Regulation of integrin activation through the B-cell receptor. J Cell Sci. 2008;121:2279–2286. doi: 10.1242/jcs.017905. [DOI] [PubMed] [Google Scholar]
- 17.Koopman G, Parmentier HK, Schuurman HJ, Newman W, Meijer CJ, Pals ST. Adhesion of human B cells to follicular dendritic cells involves both the lymphocyte function-associated antigen 1/intercellular adhesion molecule 1 and very late antigen 4/vascular cell adhesion molecule 1 pathways. J Exp Med. 1991;173:1297–1304. doi: 10.1084/jem.173.6.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koopman G, Keehnen RM, Lindhout E, Newman W, Shimizu Y, van Seventer GA, de Groot C, Pals ST. Adhesion through the LFA-1 (CD11a/CD18)-ICAM-1 (CD54) and the VLA-4 (CD49d)-VCAM-1 (CD106) pathways prevents apoptosis of germinal center B cells. J Immunol. 1994;152:3760–3767. [PubMed] [Google Scholar]
- 19.Leuker CE, Labow M, Muller W, Wagner N. Neonatally induced inactivation of the vascular cell adhesion molecule 1 gene impairs B cell localization and T cell-dependent humoral immune response. J Exp Med. 2001;193:755–768. doi: 10.1084/jem.193.6.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Balogh P, Aydar Y, Tew JG, Szakal AK. Appearance and phenotype of murine follicular dendritic cells expressing VCAM-1. Anat Rec. 2002;268:160–168. doi: 10.1002/ar.10148. [DOI] [PubMed] [Google Scholar]
- 21.Victoratos P, Lagnel J, Tzima S, Alimzhanov MB, Rajewsky K, Pasparakis M, Kollias G. FDC-specific functions of p55TNFR and IKK2 in the development of FDC networks and of antibody responses. Immunity. 2006;24:65–77. doi: 10.1016/j.immuni.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 22.Mabbott NA, Kenneth Baillie J, Kobayashi A, Donaldson DS, Ohmori H, Yoon SO, Freedman AS, Freeman TC, Summers KM. Expression of mesenchyme-specific gene signatures by follicular dendritic cells: insights from the meta-analysis of microarray data from multiple mouse cell populations. Immunology. 2011;133:482–98. doi: 10.1111/j.1365-2567.2011.03461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aguzzi A, Krautler NJ. Characterizing follicular dendritic cells: A progress report. Eur J Immunol. 2010;40:2134–2138. doi: 10.1002/eji.201040765. [DOI] [PubMed] [Google Scholar]
- 24.Pasparakis M, Kousteni S, Peschon J, Kollias G. Tumor necrosis factor and the p55TNF receptor are required for optimal development of the marginal sinus and for migration of follicular dendritic cell precursors into splenic follicles. Cell Immunol. 2000;201:33–41. doi: 10.1006/cimm.2000.1636. [DOI] [PubMed] [Google Scholar]
- 25.Fu YX, Chaplin DD. Development and maturation of secondary lymphoid tissues. Annu Rev Immunol. 1999;17:399–433. doi: 10.1146/annurev.immunol.17.1.399. [DOI] [PubMed] [Google Scholar]
- 26.Mackay F, Browning JL. Turning off follicular dendritic cells. Nature. 1998;395:26–27. doi: 10.1038/25630. [DOI] [PubMed] [Google Scholar]
- 27.Gonzalez M, Mackay F, Browning JL, Kosco-Vilbois MH, Noelle RJ. The sequential role of lymphotoxin and B cells in the development of splenic follicles. J Exp Med. 1998;187:997–1007. doi: 10.1084/jem.187.7.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fu YX, Huang G, Wang Y, Chaplin DD. B lymphocytes induce the formation of follicular dendritic cell clusters in a lymphotoxin alpha-dependent fashion. J Exp Med. 1998;187:1009–1018. doi: 10.1084/jem.187.7.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ansel KM, Ngo VN, Hyman PL, Luther SA, Forster R, Sedgwick JD, Browning JL, Lipp M, Cyster JG. A chemokine-driven positive feedback loop organizes lymphoid follicles. Nature. 2000;406:309–314. doi: 10.1038/35018581. [DOI] [PubMed] [Google Scholar]
- 30.Husson H, Lugli SM, Ghia P, Cardoso A, Roth A, Brohmi K, Carideo EG, Choi YS, Browning J, Freedman AS. Functional effects of TNF and lymphotoxin alpha1beta2 on FDC-like cells. Cell Immunol. 2000;203:134–143. doi: 10.1006/cimm.2000.1688. [DOI] [PubMed] [Google Scholar]
- 31.Madge LA, Kluger MS, Orange JS, May MJ. Lymphotoxin-alpha 1 beta 2 and LIGHT induce classical and noncanonical NF-kappa B-dependent proinflammatory gene expression in vascular endothelial cells. J Immunol. 2008;180:3467–3477. doi: 10.4049/jimmunol.180.5.3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matsumoto M, Iwamasa K, Rennert PD, Yamada T, Suzuki R, Matsushima A, Okabe M, Fujita S, Yokoyama M. Involvement of distinct cellular compartments in the abnormal lymphoid organogenesis in lymphotoxin-alpha-deficient mice and alymphoplasia (aly) mice defined by the chimeric analysis. J Immunol. 1999;163:1584–1591. [PubMed] [Google Scholar]
- 33.Zindl CL, Kim TH, Zeng M, Archambault AS, Grayson MH, Choi K, Schreiber RD, Chaplin DD. The lymphotoxin LTalpha(1)beta(2) controls postnatal and adult spleen marginal sinus vascular structure and function. Immunity. 2009;30 :408–420. doi: 10.1016/j.immuni.2009.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gommerman JL, Browning JL. Lymphotoxin/light, lymphoid microenvironments and autoimmune disease. Nat Rev Immunol. 2003;3:642–655. doi: 10.1038/nri1151. [DOI] [PubMed] [Google Scholar]
- 35.Endres R, Alimzhanov MB, Plitz T, Futterer A, Kosco-Vilbois MH, Nedospasov SA, Rajewsky K, Pfeffer K. Mature follicular dendritic cell networks depend on expression of lymphotoxin beta receptor by radioresistant stromal cells and of lymphotoxin beta and tumor necrosis factor by B cells. J Exp Med. 1999;189:159–168. doi: 10.1084/jem.189.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gommerman JL, Mackay F, Donskoy E, Meier W, Martin P, Browning JL. Manipulation of lymphoid microenvironments in nonhuman primates by an inhibitor of the lymphotoxin pathway. J Clin Invest. 2002;110:1359–1369. doi: 10.1172/JCI15975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matsumoto M, Fu YX, Molina H, Huang G, Kim J, Thomas DA, Nahm MH, Chaplin DD. Distinct roles of lymphotoxin alpha and the type I tumor necrosis factor (TNF) receptor in the establishment of follicular dendritic cells from non-bone marrow-derived cells. J Exp Med. 1997;186:1997–2004. doi: 10.1084/jem.186.12.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aydar Y, Balogh P, Tew JG, Szakal AK. Altered regulation of Fc gamma RII on aged follicular dendritic cells correlates with immunoreceptor tyrosine-based inhibition motif signaling in B cells and reduced germinal center formation. J Immunol. 2003;171:5975–5987. doi: 10.4049/jimmunol.171.11.5975. [DOI] [PubMed] [Google Scholar]
- 39.Aydar Y, Wu J, Song J, Szakal AK, Tew JG. FcgammaRII expression on follicular dendritic cells and immunoreceptor tyrosine-based inhibition motif signaling in B cells. Eur J Immunol. 2004;34:98–107. doi: 10.1002/eji.200324147. [DOI] [PubMed] [Google Scholar]
- 40.El Shikh ME, El Sayed R, Szakal AK, Tew JG. Follicular dendritic cell (FDC)-FcgammaRIIB engagement via immune complexes induces the activated FDC phenotype associated with secondary follicle development. Eur J Immunol. 2006;36:2715–2724. doi: 10.1002/eji.200636122. [DOI] [PubMed] [Google Scholar]
- 41.Rickert RC, Rajewsky K, Roes J. Impairment of T-cell-dependent B-cell responses and B-1 cell development in CD19-deficient mice. Nature. 1995;376:352–355. doi: 10.1038/376352a0. [DOI] [PubMed] [Google Scholar]
- 42.Carter RH, Tuveson DA, Park DJ, Rhee SG, Fearon DT. The CD19 complex of B lymphocytes. Activation of phospholipase C by a protein tyrosine kinase-dependent pathway that can be enhanced by the membrane IgM complex. J Immunol. 1991;147:3663–3671. [PubMed] [Google Scholar]
- 43.Chan TD, Gatto D, Wood K, Camidge T, Basten A, Brink R. Antigen affinity controls rapid T-dependent antibody production by driving the expansion rather than the differentiation or extrafollicular migration of early plasmablasts. J Immunol. 2009;183:3139–3149. doi: 10.4049/jimmunol.0901690. [DOI] [PubMed] [Google Scholar]
- 44.Bonizzi G, Bebien M, Otero DC, Johnson-Vroom KE, Cao Y, Vu D, Jegga AG, Aronow BJ, Ghosh G, Rickert RC, Karin M. Activation of IKKalpha target genes depends on recognition of specific kappaB binding sites by RelB:p52 dimers. Embo J. 2004;23:4202–4210. doi: 10.1038/sj.emboj.7600391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tumanov A, Kuprash D, Lagarkova M, Grivennikov S, Abe K, Shakhov A, Drutskaya L, Stewart C, Chervonsky A, Nedospasov S. Distinct role of surface lymphotoxin expressed by B cells in the organization of secondary lymphoid tissues. Immunity. 2002;17:239–250. doi: 10.1016/s1074-7613(02)00397-7. [DOI] [PubMed] [Google Scholar]
- 46.Ngo VN, Cornall RJ, Cyster JG. Splenic T zone development is B cell dependent. J Exp Med. 2001;194:1649–1660. doi: 10.1084/jem.194.11.1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jang IK, Cronshaw DG, Xie LK, Fang G, Zhang J, Oh H, Fu YX, Gu H, Zou Y. Growth-factor receptor-bound protein-2 (Grb2) signaling in B cells controls lymphoid follicle organization and germinal center reaction. Proc Natl Acad Sci U S A. 2011;108:7926–7931. doi: 10.1073/pnas.1016451108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Matsumoto M, Fu YX, Molina H, Chaplin DD. Lymphotoxin-alpha-deficient and TNF receptor-I-deficient mice define developmental and functional characteristics of germinal centers. Immunol Rev. 1997;156:137–144. doi: 10.1111/j.1600-065x.1997.tb00965.x. [DOI] [PubMed] [Google Scholar]
- 49.Mackay F, Majeau GR, Lawton P, Hochman PS, Browning JL. Lymphotoxin but not tumor necrosis factor functions to maintain splenic architecture and humoral responsiveness in adult mice. Eur J Immunol. 1997;27:2033–2042. doi: 10.1002/eji.1830270830. [DOI] [PubMed] [Google Scholar]
- 50.Lu TT, Cyster JG. Integrin-mediated long-term B cell retention in the splenic marginal zone. Science. 2002;297:409–412. doi: 10.1126/science.1071632. [DOI] [PubMed] [Google Scholar]
- 51.Dejardin E, Droin NM, Delhase M, Haas E, Cao Y, Makris C, Li ZW, Karin M, Ware CF, Green DR. The lymphotoxin-beta receptor induces different patterns of gene expression via two NF-kappaB pathways. Immunity. 2002;17:525–535. doi: 10.1016/s1074-7613(02)00423-5. [DOI] [PubMed] [Google Scholar]
- 52.Carter RH, Fearon DT. CD19: lowering the threshold for antigen receptor stimulation of B lymphocytes. Science. 1992;256:105–107. [PubMed] [Google Scholar]
- 53.Browning JL, Sizing ID, Lawton P, Bourdon PR, Rennert PD, Majeau GR, Ambrose CM, Hession C, Miatkowski K, Griffiths DA, Ngam-ek A, Meier W, Benjamin CD, Hochman PS. Characterization of lymphotoxin-alpha beta complexes on the surface of mouse lymphocytes. J Immunol. 1997;159:3288–3298. [PubMed] [Google Scholar]