

Abstract
Myelofibrosis (MF) can present as a primary disorder or evolve from polycythemia vera (PV) or essential thrombocythemia (ET) to post-PV MF or post-ET MF, respectively. MF is characterized by bone marrow fibrosis, splenomegaly, leukoerythroblastosis, extramedullary hematopoiesis, and a collection of debilitating symptoms. Until recently, the therapeutic options for patients with MF consisted of allogeneic hematopoietic stem cell transplant (alloHSCT), the use of cytoreductive agents (ie, hydroxyurea), splenectomy and splenic irradiation for treatment of splenomegaly, and management of anemia with transfusions, erythropoiesis-stimulating agents (ESAs), androgens, and immunomodulatory agents. However, with increased understanding of the pathogenesis of MF resulting from dysregulated Janus kinase (JAK) signaling, new targeted JAK inhibitor therapies, such as ruxolitinib, are now available. The purpose of this article is to review the clinical features of MF, discuss the use and future of JAK inhibitors, reassess when and how to use conventional MF treatments in the context of JAK inhibitors, and provide a perspective on the future of MF treatment.
Keywords: myelofibrosis, ruxolitinib, JAK inhibitor
Introduction
Philadelphia chromosome-negative myeloproliferative neoplasms (MPNs) are a diverse group of clonal stem cell disorders derived from hematopoietic myeloid progenitors and include polycythemia vera (PV), essential thrombocythemia (ET), and myelofibrosis (MF).1 Of the classical MPNs, MF has the worst prognosis, with a median survival of 69 months.2 MF can present as a primary (PMF) disorder or evolve from PV or ET to post-PV MF or post-ET M F, respectively. MF is characterized by bone marrow fibrosis, splenomegaly, leukoerythroblastosis, extramedullary hematopoiesis, and a collection of debilitating symptoms. These symptoms include cachexia, worsening of patient role functioning, and quality of life (QoL) – which ultimately result in increased rates of leukemic transformation – and decreased survival.3
Until recently, therapeutic options for patients with MF consisted of allogeneic hematopoietic stem cell transplant (alloHSCT), the use of cytoreductive agents (ie, hydroxyurea), splenectomy and splenic irradiation for treatment of splenomegaly, and management of anemia with transfusions, erythropoiesis-stimulating agents (ESAs), androgens, and immunomodulatory agents (IMiDs). Fortunately, our understanding of MPNs and the molecular mechanisms of the disease has been rapidly expanding. In 2005, the Janus kinase (JAK)2 V617F mutation was discovered and observed in approximately 50%–60% of patients with PMF or ET and 90%–95% of patients with PV.4–7 This discovery, along with the observation of other mutations in patients with MPNs found to activate the JAK/STAT (signal transducers and activators of transcription) pathway (JAK2 exon 12, MPL, and LNK),8–10 has established dysregulation of the JAK signaling pathway as the major contributor to the pathogenesis of MPNs. It has also translated into the development of small-molecule JAK inhibitors, the first of which, ruxolitinib, has been approved for the treatment of MF.
The purpose of this article is to review the clinical features of MF, discuss the use and future of JAK inhibitors, and reassess when and how to use conventional MF treatments in the context of JAK inhibitors.
Clinical features and prognosis of myelofibrosis (MF)
MF has an incidence of 0.5–1.5 per 100,000 individuals, and the median age of MF diagnosis is 67 years, with an equal distribution between the sexes. The clinical features of MF are similar regardless of the subtype and include anemia, leucopenia or leukocytosis, thrombocytopenia or thrombocytosis, and multi-organ extramedullary hematopoiesis – commonly causing hepatomegaly, symptomatic splenomegaly, or portal hypertension. The single most common cause of death for patients with MF is transformation to acute myeloid leukemia (20%); however, most patients die from other disease-related events, such as progression without transformation and thrombotic or cardiovascular events.2
Patients with MF often have a substantial symptom burden, including constitutional symptoms such as fatigue, cachexia, pruritus, bone pain, and fever, and symptoms related to the presence of massive splenomegaly such as pain, early satiety, splenic infarction, and dyspnea. The severity of symptoms has been found to be similar to that of advanced cancer, resulting in a diminished QoL.11 Moreover, with median survival ranging between 2 and 15 years, patients often suffer for a long period with this considerable symptom burden.
Several prognostic scoring systems have been developed for patients with MF, including the International Prognostic Scoring System (IPSS) used at the time of diagnosis2 and the dynamic IPSS (DIPSS)12,13 and DIPSS plus,14 which can be used to assess patients during the course of their disease (Table 1). It should be noted that these scoring systems have been validated only for patients with PMF. Nevertheless, they are often used for patients with post-ET MF and post-PV MF, despite potential inaccuracies associated with extrapolation to these populations. The IPSS comprises five risk factors for estimating survival from the time of diagnosis: age >65 years, hemoglobin (Hb) level <10 g/dL, leukocyte count >25 × 109/L, circulating blasts ≥1%, and presence of constitutional symptoms (eg, fever, weight loss, night sweats). Low-, intermediate-1−, intermediate-2−, and high-risk disease are defined as the presence of 0, 1, 2, and ≥3 adverse factors, respectively, with median survivals of 11.3, 7.9, 4, and 2.3 years.2 The DIPSS is a modification of the IPSS scale, in which two points are assigned for Hb level <10 g/dL. Unlike the IPSS, which is valid only at diagnosis, the DIPSS can be used to evaluate a patient’s risk category during the course of the disease.12 The DIPSS plus, a modification of the DIPSS, incorporates three additional independent risk factors: red blood cell transfusion requirements, platelet count <100 × 109/L, and unfavorable karyotype (complex karyotype or one or two abnormalities, including trisomy 8, monosomy 7/7q−, isochromosome [17q], inversion [3], deletion 5/5q−, 12p−, or 11q23 rearrangement).14 The DIPSS plus permits the identification of very low- and high-risk patients compared with the IPSS or DIPSS. For this reason, where it might alter the management, we recommend performing a karyotype for patients at diagnosis and for selected patients to follow the course of their disease.
Table 1.
Prognostic risk assessment
| Variable | IPSS | DIPSS | DIPSS plusa |
|---|---|---|---|
| Age >65 years | ✓ | ✓ | |
| Constitutional symptoms | ✓ | ✓ | |
| Hemoglobin <10 g/dL | ✓ | ✓✓ | |
| Leukocyte count >25 × 109/L | ✓ | ✓ | |
| Circulating blasts ≥1% | ✓ | ✓ | |
| Platelet count <100 × 109/L | ✓ | ||
| RBC transfusion need | ✓ | ||
| Unfavorable karyotype: | ✓ | ||
| +8, −7/7q−, isochromosome (17q), | |||
| inversion (3), deletion −5/5q−, | |||
| 12p−, 11q23 rearrangement |
Notes: ✓ = 1 point each.
For DIPSS plus, the score is derived from the DIPSS score and additional points added as per the table.
Abbreviations: IPSS, international Prognostic Scoring System; DIPSS, dynamic IPSS; RBC, red blood cell.
Our improved molecular understanding of MF is illustrated by the recent identification of mutations in ASXL1, EZH2, IDH1/2, and SRSF2 that were associated with worse survival outcomes. If these data are validated, screening for these mutations could be used to identify patients in the IPSS groups who may have a greater likelihood of transforming to acute leukemia and could benefit from more aggressive or experimental therapies.15 However, at present, screening for such mutations is not carried out in routine practice nor is it incorporated into prognostic scores.
Janus kinase inhibitors for the treatment of MF
Ruxolitinib
As mentioned previously, discovery of the JAK2 V617F mutation and an understanding of dysregulated JAK-STAT signaling in the pathogenesis of MF have led to the development of small-molecule JAK inhibitors. Ruxolitinib (Jakavi, Novartis AG, Basel, Switzerland; Jakafi, Incyte Corporation, Wilmington, DE, USA) is the first JAK inhibitor to gain approval in the USA, Canada, and Europe.16 These approvals were based on data from two randomized Phase III trials: the COntrolled MyeloFibrosis Study With ORal JAK Inhibitor Treatment (COMFORT) trials, which were conducted in patients with primary, post-ET, or post-PV MF with intermediate-2- or high-risk disease as assessed by IPSS and platelet count >100 × 109/L.17,18 In COMFORT-I, patients (N = 309) were randomized 1:1 to ruxolitinib or placebo; in COMFORT-II, patients (N = 219) were randomized 2:1 to ruxolitinib or best available therapy (BAT). In both trials, patients received ruxolitinib 15 or 20 mg twice daily based on their baseline platelet count (100–200 or >200 × 109/L, respectively).
The primary endpoint of both trials was achieved with a proportion of patients in the ruxolitinib arms exhibiting a ≥35% reduction in spleen volume as measured by magnetic resonance imaging at 24 weeks in COMFORT-I (41.9% ruxolitinib versus [vs] 0.7% placebo; P < 0.0001) and at 48 weeks in COMFORT-II (28.5% ruxolitinib vs 0% BAT; P < 0.0001).17,18 The spleen responses in both studies were observed regardless of JAK2 V617F mutation status. Furthermore, spleen responses were durable, with 67.0% and 79.9% of responding patients in COMFORT-I and -II, respectively, maintaining their response for ≥48 weeks. With longer follow-up in both COMFORT-I and -II (median 102 and 112 weeks, respectively), the median duration of response to ruxolitinib had not been reached.19,20
The COMFORT trials also demonstrated that, in addition to the profound effects on splenomegaly, ruxolitinib provided statistically significant improvements in patients’ symptoms and QoL.17,18 Improvements in MF symptoms were rapid, with the majority of responses occurring within the first 4 weeks of ruxolitinib treatment. In COMFORT-I, there was a >50% improvement in the Myelofibrosis Symptom Assessment Form Total Symptom Score at 24 weeks in 45.9% of ruxolitinib patients compared with 5.3% of placebo patients (P < 0.001). Long-term follow-up of COMFORT-I (median 102 weeks) demonstrated that ruxolitinib treatment was associated with durable clinically significant improvements in global health status/QoL and the other functional domains of the European Organisation for Research and Treatment of Cancer QoL Questionnaire–Core 30 Items.18
Consistent with ruxolitinib’s known mechanism of action as a JAK pathway inhibitor, anemia and thrombocytopenia were the most frequently reported adverse events (AEs) overall and of grade ≥3 in the ruxolitinib arms of both studies (Table 2). In both studies, Hb levels reached a nadir at week 12 and then stabilized at an average reduction of about 1 g/dL below baseline at week 24. Anemia and thrombocytopenia rarely led to treatment discontinuation (<1% of patients in any treatment group) and were manageable with dose modi-fications and/or blood transfusions. Rates of grade 3/4 non-hematologic AEs were low in both COMFORT studies.
Table 2.
Hematologic laboratory abnormalities
| Laboratory parameter | COMFORT-I17
|
COMFORT-II18
|
||||||
|---|---|---|---|---|---|---|---|---|
| Ruxolitinib (N = 155)
|
Placebo (N = 151)
|
Ruxolitinib (N = 146)
|
BAT (N = 73)
|
|||||
| All grades, % | Grade ≥3, % | All grades, % | Grade ≥3, % | All grades, % | Grade ≥3, % | All grades, % | Grade ≥3, % | |
| Anemia | 83 | 45 | 44 | 16 | 82 | 40 | 49 | 21 |
| Thrombocytopenia | 71 | 14 | 21 | 2 | 69 | 9 | 29 | 7 |
| Neutropenia | 19 | 7 | 4 | 3 | 12 | 6 | 8 | 1 |
Note: Reproduced with permission from Novartis Pharmaceuticals Canada Inc. Jakavi (ruxolitinib) [product monograph]. Dorval, QC: 2012.58
Abbreviations: BAT, best available therapy; COMFORT, COntrolled MyeloFibrosis Study With ORal JAK Inhibitor Treatment.
A survival analysis from COMFORT-I indicated a significant survival advantage with ruxolitinib therapy compared with placebo with a median follow-up of 51 weeks (hazard ratio [HR] 0.50; 95% confidence interval [CI] 0.25–0.98; P = 0.04).18 In additional follow-up at the 2-year time point, 41 patients randomized to placebo and 27 patients randomized to ruxolitinib died, representing a continued overall survival advantage in favor of ruxolitinib (HR 0.58; 95% CI 0.36–0.95; P = 0.028; Figure 1).21 Furthermore, with a median follow-up of 112 weeks in COMFORT-II, patients randomized to ruxolitinib had longer overall survival than those randomized to BAT (HR 0.51; 95% CI 0.26–0.99; P = 0.041; the P-value from a log-rank test is provided for descriptive purposes only and was not adjusted for multiple comparisons).20 Potential reasons for this survival advantage included improved performance status, reduction of proinflammatory cytokines, improved nutritional status, and better overall physical functioning. Recent data, though only provisional, suggest that ruxolitinib therapy may lead to reduction of marrow fibrosis in a proportion of patients, while in other patients, fibrosis has remained either stable or indeed progressed.22,23 Furthermore, a modest reduction of mutant allele burden has also been reported.24
Figure 1.
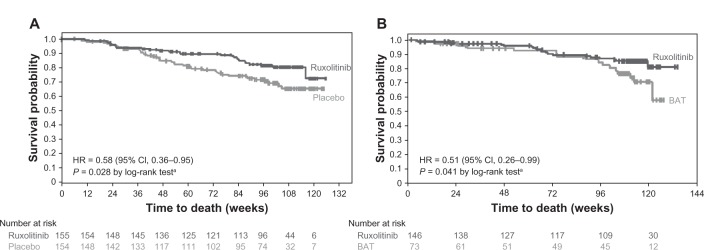
Kaplan–Meier analysis of overall survival in (A) COMFORT-I21 and (B) COMFORT-II20 with 24 months of follow-up.
Note: aP-values and CIs are unadjusted for repeat analyses.
Republished with permission of American Society of Hematology; High Wire Press, from A comprehensive review and analysis of the effect of ruxolitinib therapy on the survival of patients with myelofibrosis. Mascarenhas J, Hoffman R. Blood. 121(24):2013; permission conveyed through Copyright Clearance Center, Inc.59
Abbreviations: CI, confidence interval; HR, hazard ratio; BAT, best available therapy; COMFORT, COntrolled MyeloFibrosis Study With ORal JAK Inhibitor Treatment.
Responses to ruxolitinib are typically observed within the first 3–6 months after therapy initiation. For patients who have not had a reduction in spleen size or improvement in symptoms after this period, alternative therapies should be considered. In patients with some symptom response, symptoms returned to baseline levels within 1 week of discontinuing ruxolitinib. Therefore, dose tapering of ruxolitinib should be considered if a patient needs to discontinue ruxolitinib therapy. After 2 years of follow-up, no consistent pattern of AEs has been observed that would suggest a severe inflammatory syndrome after ruxolitinib discontinuation.
Other JAK inhibitors
Several other JAK inhibitors are in various stages of development.
SAR302503 (Sanofi; Paris, France) is a selective JAK2 inhibitor that has also shown some inhibitory activity against FLT3 and RET.25 In a Phase II study, patients were randomized to SAR302503 at 300 mg, 400 mg, or 500 mg once daily (N = 31).26 Reductions in spleen volume ≥35% at the end of cycle 3 appeared to be dose dependent (30%, 50%, and 64% for patients in the 300 mg, 400 mg, and 500 mg arms, respectively), which notably correlated with inhibition of STAT3 phosphorylation. The proportion of patients who achieved ≥50% reduction in the Myeloproliferative Neoplasm Symptom Assessment Form score was 50%, 50%, and 39% across the dosing arms, respectively. An initial report suggested that SAR302503 was associated with reduction in allele burden and improvement in bone marrow fibrosis scores;27 however, no updated results of these findings have been reported. Rates of grade 3/4 anemia were 33%, 30%, and 55%, and rates of grade 3/4 thrombocytopenia were 20%, 0%, and 9% for patients in the 300 mg, 400 mg, and 500 mg dosing arms, respectively. The most common non-hematologic AEs were gastrointestinal (rates of all-grade diarrhea were 70%, 90%, and 55%, respectively), although the incidence of diarrhea declined during the course of therapy. Currently, a Phase III study, JAK2 inhibition in a single-arm trial of SAR302503 in MF patients (JAKARTA-1), is underway comparing SAR302503 400 mg and 500 mg once daily with placebo (N = 225); the primary endpoint is spleen response. Results from this trial are anticipated soon. SAR302503 is also being evaluated in another Phase II study (JAKARTA-2) in patients previously treated with ruxolitinib (target enrolled, N = 70).
Momelotinib (CYT387; Gilead Foster City, CA, USA) is a JAK1/JAK2 inhibitor currently under evaluation in a Phase II trial of patients with MF (N = 166).28 Durable reductions in spleen length of ≥50% as assessed by palpation were observed in 37% of patients, and the median duration of spleen response was 744 days. Achievement of a complete resolution or marked improvement of constitutional symptoms was reported in the majority of patients. After a median follow-up of 16.9 months, a substantial decrease in the percentage of patients requiring transfusions during study was observed (44% at baseline vs <10% at week 40), and 13% of patients had an increase in Hb level of at least 2 g/dL. A Phase III study of CYT387 is underway, and it will be important to determine whether these encouraging results are confirmed in this setting.
Pacritinib (SB1518; Cell Technology, Inc, Mountain View, CA, USA) is a JAK2 and FLT3 inhibitor currently being evaluated at a dose of 400 mg daily in a Phase II study (N = 34) that included patients with low platelet counts (<50 × 109/L). Nearly one third of patients (32%) had a ≥35% reduction in spleen volume at 24 weeks.29 Pacritinib therapy was associated with minimal myelosuppression and no new onset of anemia or change in transfusion requirements. Furthermore, no dose reductions due to thrombocytopenia were required. A Phase III study of pacritinib in patients with low platelets and symptomatic splenomegaly is currently open (Oral Pacritinib Versus Best Available Therapy in Patients With Primary Myelofibrosis, Post-Polycythemia Vera Myelofibrosis, or Post-Essential Thrombocythemia Myelofibrosis; PERSIST).
Perspective: we would recommend the use of ruxolitinib first line for patients with burdening symptoms and/or splenomegaly. As with any new therapy, the long-term benefits and safety profile of ruxolitinib and the other JAK inhibitors (in time) will need to be evaluated as more patients receive treatment over longer periods. There has been some evidence of an increased incidence of herpes zoster and tuberculosis reactivation in the ruxolitinib arms of the COMFORT studies,17,18 and it will be important to assess the risk of increased viral reactivation in patients treated with JAK inhibitors as well as other concerns of immunosuppression (eg, opportunistic infections and secondary malignancies). We also believe that it will be important to understand how disease progression may present in patients receiving JAK inhibitors because increases in spleen size and worsening of constitutional symptoms may be masked by treatment. Additionally, as more JAK inhibitors are approved for MF and become commercially available, it will be necessary to determine which patients may benefit the most from a particular agent. For example, momelotinib may be a better choice for patients with severe anemia, while ruxolitinib may be better for those who experience non-hematologic AEs with other JAK inhibitors such as SAR302503. At present, our understanding of the reasons for these apparent differences in anemia is unclear but has been related to differing binding affinities for the various members of the JAK family. It will also be important to determine if any biomarkers exist to help select the most appropriate inhibitors for a particular patient. Furthermore, we need to be able to assess long-term benefits in comparing these agents with each other and with future therapeutic strategies (eg, combination studies) that may be developed.
Non–JAK inhibitor therapies for MF
Allogeneic hematopoietic stem cell transplant
AlloHSCT is currently the only curative treatment for patients with M F. However, as a large proportion of patients are not in the transplant age group at the time of diagnosis, alloHSCT has a limited role in the overall disease management of patients with M F. Significant regimen-related toxicities, graft failure, and graft-versus-host disease are major barriers to the success of alloHSCT in MF. However, it remains a valid option for patients in the transplant age group with adequate performance status and without any prohibitive comorbidities; among these typically younger patients, suitable donors are found in approximately 40%–50% of cases.30 Data from the most recent studies suggest that the expected progression-free survival rate after alloHSCT is in the range of 40%–50% at 3 years.30
Perspective: the recommended indications for transplant in our clinic are expected survival <5 years, transfusion dependency, and/or an increased risk of leukemic transformation, perhaps using the DIPSS plus or novel molecular markers as discussed earlier. The availability of a fully matched sibling donor would lead us to consider conducting a transplant earlier (for patients with intermediate-2 or intermediate-1 risk with anemia, transfusion, or a rising blast count). While splenectomy is not routinely recommended prior to alloHSCT, it is reasonable to explore the safety and efficacy of novel drugs that can provide rapid spleen shrinkage and improvement of constitutional symptoms in the immediate pretransplant period.30 Along these lines, a clinical trial (study MPD-RC114) to explore the safety of ruxolitinib in the pretransplant setting is underway.31
Splenomegaly and extramedullary hematopoiesis
Cytoreductive agents
Cytoreductive agents have been the treatment of choice for most patients with symptomatic splenomegaly. Hydroxycarbamide (hydroxyurea, HC) is the most commonly used cytoreductive agent, which usually produces modest responses at higher doses (1–2 g). However, HC can often exacerbate cytopenias and therefore is often not well tolerated. Reductions in spleen size >25% and 50% have been reported in up to 35% and 17%, respectively, of the patients treated with HC.32 In patients who do not respond to HC, busulfan or melphalan can be used, especially in older patients, since there is evidence that these agents can increase the frequency of leukemic transformation. Spleen responses with low-dose thalidomide (50 mg daily) are infrequent (<20%). However, lenalidomide has been shown to result in a 33% response rate in a study that included some patients who had failed prior thalidomide therapy. In cases of massive refractory splenomegaly, monthly intravenous cladribine courses have produced responses up to 50%, with severe but reversible cytopenias being the primary toxicity.33 Interferonalfa (standard and PEGylated versions) has demonstrated minimal clinical effect in reducing splenomegaly, and therefore its use is not generally recommended.34 Ruxolitinib has proven superior to BAT in the COMFORT-II study and thus we would use this agent first line to control symptomatic or progressive splenomegaly.
Splenectomy and radiotherapy
While the management of MF-associated splenomegaly with splenectomy is well established, the procedure is associated with morbidity and mortality rates of approximately 31% and 9%, respectively.35 Hepatic extramedullary hematopoiesis, which sometimes leads to rapid hepatic enlargement, is an unusual but well recognized complication following splenectomy, as is the increased thrombotic risk. As a result, splenectomy should be restricted to selected patients with refractory hemolysis or anemia, symptomatic splenomegaly, significant splenic infarction, severe portal hypertension, and/or severe hypercatabolic symptoms. Furthermore, patients undergoing splenectomy need to be made well aware of the risks and provided with meticulous preoperative assessment and postoperative follow-up care.
Radiotherapy can be an alternative to splenectomy in patients with symptomatic splenomegaly and an adequate platelet count (>50 × 109/L). In a report from the Mayo Clinic, a median radiation dose of 277 cGy administered in a median of 7.5 fractions reduced spleen size in the majority of cases for a median of 6 months. However, 44% of patients experienced cytopenias, of which 13% were fatal.36 Our preference is to use even lower dosing fractions with caution. Low-dose radiotherapy remains a preferred treatment for nonsplenic extramedullary hematopoiesis, including involvement of the peritoneum and pleura with resultant ascites and pleural effusions.
Perspective: cytoreductive agents and/or surgical intervention have been the main approaches to treat symptomatic splenomegaly. JAK inhibitors, primarily ruxolitinib at present, will radically alter the way this aspect of disease is managed. In the COMFORT-II study, none of the 73 patients in the BAT arm, of whom 60% received HC, achieved a sustained >35% reduction in spleen volume.17 Ruxolitinib is likely to surpass HC as first-line treatment of symptomatic splenomegaly and will also be a valuable option in the management of extramedullary hematopoiesis at other sites.
Anemia management
The management of anemia can be one of the most challenging aspects of treating patients with MF. Blood transfusion is a standard therapy for symptomatically anemic patients, and the transfusion target should be assessed individually. Since regular transfusions will ultimately result in iron overload, iron chelation is frequently required; therefore, it is useful to also consider other treatment options.
Erythropoiesis-stimulating agents
Responses to ESAs are more likely in transfusion-independent patients with higher baseline Hb. In an analysis of 20 anemic patients with MF treated with ESAs, responses were seen in 45% of cases but were maintained long-term in only 20%.37 A pooled analysis that combined these 20 patients with 31 patients from the literature suggested an overall response rate of 55% (31% complete responses), with a median response duration of 12 months.37
Perspective: the use of ESAs in combination with ruxolitinib therapy has also been reported.38 In an analysis of 13 patients enrolled in COMFORT-II, concomitant ESA and ruxolitinib therapy was well tolerated, with an observed safety profile similar to that of ruxolitinib therapy alone. Furthermore, the combination did not appear to affect the efficacy of ruxolitinib regarding reductions in spleen size. Further analysis is required to determine if ESAs provided any substantial alleviation of anemia in these patients. Unless a patient has chronic kidney disease or an endogenous erythropoietin level ,125 IU/L, we generally do not use ESAs to treat patients in our clinic, although a short therapeutic trial may be useful.
Androgens
Androgenic hormones have been shown to stimulate erythropoiesis in patients with refractory anemia, leading to increased Hb level, reticulocytosis, and decreased need for blood transfusions.39 Danazol, a synthetic attenuated androgen, has demonstrated efficacy in treating anemic patients with MF and even reduced spleen size in a proportion of patients.40 Dosages are dependent on body weight (600 mg daily for patients weighing up to 80 kg and 800 mg daily for those weighing >80 kg) and should be continued for a period of 6 months. Patients achieving favorable responses can be maintained on danazol at a reduced dose of 400 mg daily for 6 months and then titrated down to the minimum dose required to maintain a response (generally 200 mg daily).
Side effects of androgen therapy include fluid retention, increased libido, hirsutism, abnormal liver function tests, and hepatic tumors. Therefore, all patients receiving danazol should be monitored using monthly liver function tests during initial therapy and a periodic liver ultrasound to detect any hepatic malignancy. Males should be screened for prostate cancer before and during treatment.
Perspective: in our clinic, we have had limited but successful experiences of combining androgens with ruxolitinib. However, it should be noted that these patients require careful liver function monitoring during androgen therapy.
Immunomodulatory agents
IMiDs, such as thalidomide, lenalidomide, and pomalidomide, are agents that inhibit neoangiogenesis by downregulating vascular endothelial growth factor, basic fibroblast growth factor, and tumor necrosis factor. IMiDs have shown some efficacy in managing anemia, with some responses in patients with thrombocytopenia and splenomegaly (reviewed in the study by Thapaliya et al41). In our clinic, thalidomide is used in combination with prednisolone. However, due to the side-effect profile of thalidomide, it would not be selected for first-line management of anemia.42 Lenalidomide is the recommended first-line therapy in rare cases of del(5q31)-associated anemia, because significant improvement – with resolution of anemia and evidence of occasional molecular remission – has been described.43 There is great interest in the potential for pomalidomide to treat anemia, and the benefits of combining pomalidomide with prednisolone have recently been reported.44 A number of Phase II studies are underway, 45–47 and results from a Phase III study (Pomalidomide in Persons With Myeloproliferative- Neoplasm-Associated Myelofibrosis and RBC [red blood cell]-Transfusion-Depen-dence Myelofibrosis and RBC-Transfusion-Dependence; RESUME) are anticipated.
Perspective: in our clinic, IMiDs are rarely used as monotherapy for treating anemia in patients with M F, so other agents, such as androgens, are often selected instead. It will be interesting to see if the combination of IMiDs with a JAK inhibitor, such as ruxolitinib, can ameliorate therapy-induced anemia. However, we are doubtful that the combination will yield a benefit in this challenging scenario and would rather focus efforts on deepening disease response. The results of the RESUME trial are likely to be critical here, although we would note that anemia response is notoriously difficult to assess.
Other experimental strategies
Everolimus
In addition to JAK/STAT, other related pathways, such as the phosphatidylinositol 3-kinase/mammalian target of rapamycin (PI3K/mTOR) pathway, have been found to be dysregulated in M F.48 In studies in vitro, the proliferation of JAK2 V617F–positive cells decreased when treated with the mTOR inhibitor everolimus.49–51 Results from a Phase I/II study of 39 high- or intermediate-risk patients with PMF or post-PV/ET MF treated with everolimus have also been reported.49 Of 30 evaluable patients, 69% and 80% experienced complete resolution of systemic symptoms and pruritus, respectively. The response rate was 60% when European Myelofibrosis Network criteria were applied (eight major, seven moderate, and three minor responses) or 23% when International Working Group for Myelofibrosis Research and Treatment criteria were used (one partial response, six clinical improvements). These results provide proof of concept that targeting the mTOR pathway may be clinically relevant in patients with M F.
Panobinostat
Deacetylases (DACs) are enzymes that modify the acetylation of both non-histone and histone proteins. The inhibition of DACs has been shown to influence a number of cellular events involved in cancer initiation and progression.52,53 Panobinostat (LBH589) is a novel pan-DAC inhibitor that has demonstrated clinical activity in Phase I/II studies in patients with MF. At 16 months, one patient achieved a near-complete response, with resolution of palpable splenomegaly.54 This patient also achieved elimination of peripheral blood dacrocytes and leukoerythroblastosis, a 4 g/dL increase in Hb from baseline, and improvement in overall marrow cellularity and megakaryocyte atypia, with an increase in erythroid precursors and a significant reduction in reticulin/collagen fibrosis. In a Phase I/II study of panobinostat, reversible thrombocytopenia was observed to be the dose-limiting toxicity.54,55 Overall, low doses of panobinostat administered for >6 months improved symptoms and clinical features and reversed pathologic marrow changes in patients with MF.
Combination approaches
There is significant interest in improving patient outcomes using the combination of JAK inhibitors and traditional and/or experimental agents.56 Studies assessing the JAK1/2 inhibitor ruxolitinib combined with IMiDs, androgens, and DAC, PI3K, and smoothened inhibitors, or administered prior to HSCT have already been initiated (Table 3). Preliminary results in a small number of patients have been reported for panobinostat combined with ruxolitinib.57 The combination has been well tolerated, and reductions in spleen size and improvement of MF-related symptoms have been observed, even at doses below the therapeutic dosage of each agent as monotherapy.
Table 3.
Ruxolitinib combination therapy – clinical studies
| Combination agent | Study title | Trial identifier |
|---|---|---|
| Lenalidomide | Ruxolitinib and Lenalidomide for Patients with Myelofibrosis | NCT01375140 |
| Pomalidomide | Ruxolitinib and Pomalidomide Combination Therapy in Patients with Primary and Secondary MF (POMINC) | NCT01644110 |
| Danazol | Ruxolitinib Phosphate and Danazol in Treating Anemia in Patients with Myelofibrosis | NCT01732445 |
| Panobinostat | Panobinostat and Ruxolitinib in Myelofibrosis (PRIME Trial) | NCT01693601 |
| Buparlisib (BKM120) | A Study to Find the Maximum Tolerated Dose of the Experimental Combination of the Drugs INC424 and BKM120 in Patients with Primary or Secondary Myelofibrosis | NCT01730248 |
| LDE225 | A Phase Ib/II Dose-finding Study to Assess the Safety and Efficacy of LDE225 + INC424 in Patients with MF | NCT01787552 |
| Stem cell transplantation | Ruxolitinib Prior to Transplant in Patients with Myelofibrosis | NCT01790295 |
Abbreviation: MF, myelofibrosis.
Perspective: it is our practice to offer patients trials exploring combination therapy if transplant is not an option or if ruxolitinib has either failed or may not be suitable. Other patients for consideration in combination studies are those seeking to delay disease progression who do not yet require transplant (eg, a young patient with leukocytosis but no other risk factors). Such patients are lower risk than those in the former categories and need to be very carefully counseled and monitored in experimental trials. Combination studies offer patients the opportunity to move toward ultimately attaining a possible cure or at least deeper/more meaningful responses. However, challenges lie ahead in understanding how to assess the benefits of combination approaches since observation of a survival benefit or leukemia-free survival would require very large trials conducted over a long period. Spleen volume response has almost become the standard endpoint in MF and may be selected as a primary endpoint in these studies, but other measures may be more appropriate.
Conclusion
It is encouraging to witness the recent developments in the understanding and treatment of MF and observe the benefits that these new options can provide to patients. However, it will be important to assess the long-term safety and efficacy of new treatments, such as JAK inhibitors, and establish the role of combination therapies in MF treatment.
A challenge in the introduction of novel therapies is determining both the current prevalence of disease and the true societal cost – this is poorly understood for M F, and efforts should focus here. If we could better understand the events surrounding either progression in MF or transformation in post-ET/post-PV MF, we might unlock even more biologically significant targets. As such, it will be important to assess whether early JAK inhibitor intervention in patients with PV and ET has an impact on reducing the transformation rate to post-PV MF and post-ET M F. Furthermore, such efforts may help us to identify surrogate markers of response that could enable more rapid advancement in the selection of novel therapies or combinations: the presence of splenomegaly and degree of spleen response seem to be of utility in identifying patients who may have a survival benefit, but other markers could be more relevant (eg, allele burden, cluster of differentiation molecule [CD]34+, fibrosis grade, and/or genetic signature).
Acknowledgments
Financial support for medical editorial assistance was provided by Novartis Pharmaceuticals Corporation. The authors thank Daniel Hutta, PhD, for medical editorial assistance with this manuscript.
Footnotes
Disclosure
Dr Clodagh Keohane has received research funding from Novartis. Dr Deepti Radia has received honoraria from Novartis, Shire, Pfizer, and Vifor Pharma. Dr Claire Harrison has received honoraria from Novartis, Sanofi-Aventis, Celgene, and Shire; research funding from Novartis and Shire; and acted as a consultant to YM BioSciences, S*BIO, Sanofi-Aventis, and Novartis.
References
- 1.Mesa RA, Green A, Barosi G, Verstovsek S, Vardiman J, Gale RP. MPN-associated myelofibrosis (MPN-MF) Leuk Res. 2011;35(1):12–13. doi: 10.1016/j.leukres.2010.07.019. [DOI] [PubMed] [Google Scholar]
- 2.Cervantes F, Dupriez B, Pereira A, et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood. 2009;113(13):2895–2901. doi: 10.1182/blood-2008-07-170449. [DOI] [PubMed] [Google Scholar]
- 3.Barosi G, Rosti V, Vannucchi AM. Therapeutic approaches in myelofibrosis. Expert Opin Pharmacother. 2011;12(10):1597–1611. doi: 10.1517/14656566.2011.568939. [DOI] [PubMed] [Google Scholar]
- 4.James C, Ugo V, Le Couédic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144–1148. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- 5.Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779–1790. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- 6.Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365(9464):1054–1061. doi: 10.1016/S0140-6736(05)71142-9. [DOI] [PubMed] [Google Scholar]
- 7.Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7(4):387–397. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 8.Scott LM, Tong W, Levine RL, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med. 2007;356(5):459–468. doi: 10.1056/NEJMoa065202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oh ST, Simonds EF, Jones C, et al. Novel mutations in the inhibitory adaptor protein LNK drive JAK-STAT signaling in patients with myeloproliferative neoplasms. Blood. 2010;116(6):988–992. doi: 10.1182/blood-2010-02-270108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pikman Y, Lee BH, Mercher T, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006;3(7):e270. doi: 10.1371/journal.pmed.0030270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mesa RA, Niblack J, Wadleigh M, et al. The burden of fatigue and quality of life in myeloproliferative disorders (MPDs): an international Internet-based survey of 1179 MPD patients. Cancer. 2007;109(1):68–76. doi: 10.1002/cncr.22365. [DOI] [PubMed] [Google Scholar]
- 12.Passamonti F, Cervantes F, Vannucchi AM, et al. Dynamic International Prognostic Scoring System (DIPSS) predicts progression to acute myeloid leukemia in primary myelofibrosis. Blood. 2010;116(15):2857–2858. doi: 10.1182/blood-2010-06-293415. [DOI] [PubMed] [Google Scholar]
- 13.Passamonti F, Cervantes F, Vannucchi AM, et al. A dynamic prognostic model to predict survival in primary myelofibrosis: a study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment) Blood. 2010;115(9):1703–1708. doi: 10.1182/blood-2009-09-245837. [DOI] [PubMed] [Google Scholar]
- 14.Gangat N, Caramazza D, Vaidya R, et al. DIPSS plus: a refined Dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol. 2011;29(4):392–397. doi: 10.1200/JCO.2010.32.2446. [DOI] [PubMed] [Google Scholar]
- 15.Vannucchi AM, Lasho TL, Guglielmelli P, et al. Mutations and prognosis in primary myelofibrosis. Leukemia. 2013 Apr 26; doi: 10.1038/leu.2013.119. Epub. [DOI] [PubMed] [Google Scholar]
- 16.Jakafi (ruxolitinib) [package insert] Wilmington, DE: Incyte Corporation; 2011. [Google Scholar]
- 17.Harrison C, Kiladjian JJ, Al-Ali HK, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366(9):787–798. doi: 10.1056/NEJMoa1110556. [DOI] [PubMed] [Google Scholar]
- 18.Verstovsek S, Mesa RA, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366(9):799–807. doi: 10.1056/NEJMoa1110557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Verstovsek S, Passamonti F, Rambaldi A, et al. Long-term efficacy and safety results from a phase II study of ruxolitinib in patients with polycythemia vera. Blood (ASH Annual Meeting Abstracts) 2012;120(21):804. [Google Scholar]
- 20.Cervantes F, Kiladjian J, Niederwieser D, et al. Long-term safety, efficacy, and survival findings from COMFORT-II, a phase 3 study comparing ruxolitinib with best available therapy (BAT) for the treatment of myelofibrosis (MF) Blood (ASH Annual Meeting Abstracts) 2012;120(21):801. [Google Scholar]
- 21.Verstovsek S, Mesa R, Gotlib J, et al. Long-term outcome of ruxolitinib treatment in patients with myelofibrosis: durable reductions in spleen volume, improvements in quality of life, and overall survival advantage in COMFORT-I. Blood (ASH Annual Meeting Abstracts) 2012;120(21):800. [Google Scholar]
- 22.Kvasnicka HM, Thiele J, Bueso-Ramos C, et al. Long-term intervention effects on bone marrow morphology in myelofibrosis: Patients treated with ruxolitinib and best available therapy. Haematologica. 2013;98(Suppl 1):249. [abstract S591] [Google Scholar]
- 23.Kvasnicka HM, Thiele J, Bueso-Ramos C, et al. Exploratory analysis of the effect of ruxolitinib on bone marrow morphology in patients with myelofibrosis. J Clin Oncol, 2013 Annual Meeting Proc (Post-Meeting Edition) 2013;31(Suppl 15) (May 20 Suppl)[abstract 7030] [Google Scholar]
- 24.Vannucchi A, Kiladjian JJ, Gisslinger H, et al. Reductions in JAK2 V617F allele burden with ruxolitinib treatment in COMFORT-II, a phase 3 study comparing the safety and efficacy of ruxolitinib to best available therapy (BAT) Haematologica. 2012;97(Suppl 1):151. [abstract 0373] [Google Scholar]
- 25.Wernig G, Kharas MG, Okabe R, et al. Efficacy of TG101348, a selective JAK2 inhibitor, in treatment of a murine model of JAK2V617F-induced polycythemia vera. Cancer Cell. 2008;13(4):311–320. doi: 10.1016/j.ccr.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 26.Talpaz M, Jamieson C, Gabrail N, et al. A phase II randomized doseranging study of the JAK2-selective inhibitor SAR302503 in patients with intermediate-2 or high-risk primary myelofibrosis (MF), post-polycythemia vera (PV) MF, or post-essential thrombocythemia (ET) M F. Blood (ASH Annual Meeting Abstracts) 2012;120(21):2837. [Google Scholar]
- 27.Pardanani A, Gotlib JR, Jamieson C, et al. Safety and efficacy of TG101348, a selective JAK2 inhibitor, in myelofibrosis. J Clin Oncol. 2011;29(7):789–796. doi: 10.1200/JCO.2010.32.8021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pardanani A, Gotlib J, Gupta V, et al. Phase I/II study of CYT387, a JAK1/JAK2 inhibitor for the treatment of myelofibrosis. Blood (ASH Annual Meeting Abstracts) 2012;120(21):178. [Google Scholar]
- 29.Komrokji RS, Wadleigh M, Seymour JF, et al. Results of a phase 2 study of pacritinib (SB1518), a novel oral JAK2 inhibitor, in patients with primary, post-polycythemia vera, and post-essential thrombocythemia myelofibrosis. Blood (ASH Annual Meeting Abstracts) 2011;118(21):282. [Google Scholar]
- 30.McLornan DP, Mead AJ, Jackson G, Harrison CN. Allogeneic stem cell transplantation for myelofibrosis in 2012. Br J Haematol. 2012;157(4):413–425. doi: 10.1111/j.1365-2141.2012.09107.x. [DOI] [PubMed] [Google Scholar]
- 31.Mascarenhas J.Ruxolitinib Prior to Transplant in Patients With Myelofibrosis Available from: http://clinicaltrials.gov/show/NCT01790295Accessed July 29, 2013
- 32.Martínez-Trillos A, Gaya A, Maffioli M, et al. Efficacy and tolerability of hydroxyurea in the treatment of the hyperproliferative manifestations of myelofibrosis: results in 40 patients. Ann Hematol. 2010;89(12):1233–1237. doi: 10.1007/s00277-010-1019-9. [DOI] [PubMed] [Google Scholar]
- 33.Faoro LN, Tefferi A, Mesa RA. Long-term analysis of the palliative benefit of 2-chlorodeoxyadenosine for myelofibrosis with myeloid metaplasia. Eur J Haematol. 2005;74(2):117–120. doi: 10.1111/j.1600-0609.2004.00370.x. [DOI] [PubMed] [Google Scholar]
- 34.Ianotto JC, Kiladjian JJ, Demory JL, et al. PEG-IFN-alpha-2a therapy in patients with myelofibrosis: a study of the French Groupe d’Etudes des Myelofibroses (GEM) and France Intergroupe des syndromes Myéloprolifératifs (FIM) Br J Haematol. 2009;146(2):223–225. doi: 10.1111/j.1365-2141.2009.07745.x. [DOI] [PubMed] [Google Scholar]
- 35.Mesa RA. How I treat symptomatic splenomegaly in patients with myelofibrosis. Blood. 2009;113(22):5394–5400. doi: 10.1182/blood-2009-02-195974. [DOI] [PubMed] [Google Scholar]
- 36.Elliott MA, Tefferi A. Splenic irradiation in myelofibrosis with myeloid metaplasia: a review. Blood Rev. 1999;13(3):163–170. doi: 10.1054/blre.1999.0110. [DOI] [PubMed] [Google Scholar]
- 37.Cervantes F, Alvarez-Larrán A, Hernández-Boluda JC, Sureda A, Torrebadell M, Montserrat E. Erythropoietin treatment of the anaemia of myelofibrosis with myeloid metaplasia: results in 20 patients and review of the literature. Br J Haematol. 2004;127(4):399–403. doi: 10.1111/j.1365-2141.2004.05229.x. [DOI] [PubMed] [Google Scholar]
- 38.McMullin MF, Harrison CN, Niederwieser D, et al. The use of erythropoietic-stimulating agents (ESAs) with ruxolitinib in patients with primary myelofibrosis (PMF), post-polycythemia vera myelofibrosis (PPV-MF), and post-essential thrombocythemia myelofibrosis (PET-MF) Blood (ASH Annual Meeting Abstracts) 2012;120(21):2838. [Google Scholar]
- 39.Kennedy BJ. Effect of androgenic hormone in myelofibrosis. JAMA. 1962;182:116–119. doi: 10.1001/jama.1962.03050410012003. [DOI] [PubMed] [Google Scholar]
- 40.Cervantes F, Alvarez-Larrán A, Domingo A, Arellano-Rodrigo E, Montserrat E. Efficacy and tolerability of danazol as a treatment for the anaemia of myelofibrosis with myeloid metaplasia: long-term results in 30 patients. Br J Haematol. 2005;129(6):771–775. doi: 10.1111/j.1365-2141.2005.05524.x. [DOI] [PubMed] [Google Scholar]
- 41.Thapaliya P, Tefferi A, Pardanani A, et al. International working group for myelofibrosis research and treatment response assessment and long-term follow-up of 50 myelofibrosis patients treated with thalidomide-prednisone based regimens. Am J Hematol. 2011;86(1):96–98. doi: 10.1002/ajh.21892. [DOI] [PubMed] [Google Scholar]
- 42.Weinkove R, Reilly JT, McMullin MF, Curtin NJ, Radia D, Harrison CN. Low-dose thalidomide in myelofibrosis. Haematologica. 2008;93(7):1100–1101. doi: 10.3324/haematol.12416. [DOI] [PubMed] [Google Scholar]
- 43.Tefferi A, Lasho TL, Mesa RA, Pardanani A, Ketterling RP, Hanson CA. Lenalidomide therapy in del(5)(q31)-associated myelofibrosis: cytogenetic and JAK2V617F molecular remissions. Leukemia. 2007;21(8):1827–1828. doi: 10.1038/sj.leu.2404711. [DOI] [PubMed] [Google Scholar]
- 44.Shastri A, Cortes JE, Jabbour EJ, et al. A phase II study of low-dose pomalidomide (0.5 mg/day) and prednisone combination therapy in patients with myelofibrosis and significant anaemia. Blood (ASH Annual Meeting Abstracts) 2012;120(21):1728. [Google Scholar]
- 45.Begna KH, Mesa RA, Pardanani A, et al. A phase-2 trial of low-dose pomalidomide in myelofibrosis. Leukemia. 2011;25(2):301–304. doi: 10.1038/leu.2010.254. [DOI] [PubMed] [Google Scholar]
- 46.Mesa RA, Pardanani AD, Hussein K, et al. Phase 1/-2 study of pomalidomide in myelofibrosis. Am J Hematol. 2010;85(2):129–130. doi: 10.1002/ajh.21598. [DOI] [PubMed] [Google Scholar]
- 47.Tefferi A, Verstovsek S, Barosi G, et al. Pomalidomide is active in the treatment of anaemia associated with myelofibrosis. J Clin Oncol. 2009;27(27):4563–4569. doi: 10.1200/JCO.2008.21.7356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tefferi A. Primary myelofibrosis: 2012 update on diagnosis, risk stratification, and management. Am J Hematol. 2011;86(12):1017–1026. doi: 10.1002/ajh.22210. [DOI] [PubMed] [Google Scholar]
- 49.Guglielmelli P, Barosi G, Rambaldi A, et al. Safety and efficacy of everolimus, a mTOR inhibitor, as single agent in a phase 1/2 study in patients with myelofibrosis. Blood. 2011;118(8):2069–2076. doi: 10.1182/blood-2011-01-330563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vannucchi AM, Bogani C, Bartalucci N, et al. The mTOR inhibitor, RAD001, inhibits the growth of cells from patients with myeloproliferative neoplasms. Blood (ASH Annual Meeting Abstracts) 2009;114(22):2914. [Google Scholar]
- 51.Vannucchi AM, Guglielmelli P, Lupo L, et al. A phase 1/2 study of RAD001, a mTOR inhibitor, in patients with myelofibrosis: final results. Blood (ASH Annual Meeting Abstracts) 2010;116(21):314. [Google Scholar]
- 52.Marks PA, Xu WS. Histone deacetylase inhibitors: potential in cancer therapy. J Cell Biochem. 2009;107(4):600–608. doi: 10.1002/jcb.22185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5(9):769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 54.Mascarenhas J, Lu M, Li T, et al. A phase I study of panobinostat (LBH589) in patients with primary myelofibrosis (PMF) and post-polycythaemia vera/essential thrombocythaemia myelofibrosis (post-PV/ET MF) Br J Haematol. 2013;161(1):68–75. doi: 10.1111/bjh.12220. [DOI] [PubMed] [Google Scholar]
- 55.DeAngelo DJ, Tefferi A, Fiskus W, et al. A phase II trial of panobinostat, an orally available deacetylase inhibitor, in patients with primary myelofibrosis, post-essential thrombocythemia myelofibrosis, and post-polycythemia vera myelofibrosis. Blood (ASH Annual Meeting Abstracts) 2010;116(21):630. [Google Scholar]
- 56.Harrison C, Verstovsek S, McMullin MF, Mesa R. Janus kinase inhibition and its effect upon the therapeutic landscape for myelofibrosis: from palliation to cure? Br J Haematol. 2012;157(4):426–437. doi: 10.1111/j.1365-2141.2012.09108.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Harrison CN, Kiladjian J, Passamonti F, et al. A phase 1b dose-finding study of ruxolitinib plus panobinostat in patients with primary myelofibrosis, post-polycythemia vera myelofibrosis, or post-essential thrombocythemia myelofibrosis. Haematologica. 2012;97(Suppl 1):146. [abstract 0364] [Google Scholar]
- 58.Jakavi (ruxolitinib) [product monograph] Dorval, QC: Novartis Pharmaceuticals Canada, Inc; 2012. [Google Scholar]
- 59.Mascarenhas J, Hoffman R. A comprehensive review and analysis of the effect of ruxolitinib therapy on the survival of patients with myelofibrosis. Blood. 2013;121(24):4832–4837. doi: 10.1182/blood-2013-02-482232. [DOI] [PubMed] [Google Scholar]