

Abstract
Background & Aims
Genome-wide association studies (GWASs) have identified 140 Crohn’s disease (CD) susceptibility loci. For most loci, the variants that cause disease are not known and the genes affected by these variants have not been identified. We aimed to identify variants that cause CD through detailed sequencing, genetic association, expression, and functional studies.
Methods
We sequenced whole exomes of 42 unrelated subjects with Crohn’s disease (CD) and 5 healthy individuals (controls), and then filtered single-nucleotide variants by incorporating association results from meta-analyses of CD GWASs and in silico mutation effect prediction algorithms. We then genotyped 9348 patients with CD, 2868 with ulcerative colitis, and 14,567 controls, and associated variants analyzed in functional studies using materials from patients and controls and in vitro model systems.
Results
We identified rare missense mutations in PR domain-containing1 (PRDM1) and associated these with CD. These increased proliferation of T cells and secretion of cytokines upon activation, and increased expression of the adhesion molecule L-selectin. A common CD risk allele, identified in GWASs, correlated with reduced expression of PRDM1 in ileal biopsies and peripheral blood mononuclear cells (combined P=1.6×0−8). We identified an association between CD and a common missense variant, Val248Ala, in nuclear domain 10 protein 52 (NDP52) (P=4.83×10−9). We found that this variant impairs the regulatory functions of NDP52 to inhibit NFκB activation of genes that regulate inflammation and affect stability of proteins in toll-like receptor pathways.
Conclusions
We have extended GWAS results and provide evidence that variants in PRDM1 and NDP52 determine susceptibility to CD. PRDM1 maps adjacent to a CD interval identified in GWASs and encodes a transcription factor expressed by T and B cells. NDP52 is an adaptor protein that functions in selective autophagy of intracellular bacteria and signaling molecules, supporting the role for autophagy in pathogenesis of CD.
Keywords: inflammatory bowel disease, whole-exome sequencing, complex disease
Introduction
Crohn’s disease (CD; MIM#266600) is characterized by relapsing/remitting intestinal inflammation and results from a complex interaction of host genetic factors and environmental stimuli within the mucosal immune compartment. Meta-analyses of genome-wide association studies (GWASs) have established a total of 140 CD susceptibility loci1–3, but these account for less than 14% of the total disease variance. Most appear to exert their effect by influencing the regulation of gene transcription rather than disrupting coding sequence. A variety of approaches have been proposed to aid resolution of GWAS signals, and the need for functional interrogation is self-evident to further mechanistic understanding. However, for most GWAS loci, the causal variants remain unknown and the genes affected by these variants are yet to be identified.
The low proportion of overall heritability accounted for by GWAS studies has been a universal finding across common, complex disease4. A variety of explanations have been proposed, including over-estimation of disease heritability, epigenetic effects and a major contribution within families of low frequency and rare variants. The extent to which the latter contributes may now be amenable to direct study. Whole-exome sequencing has become state-of-the art for detecting rare coding variants in Mendelian disorders5, but the utility of this approach in dissecting the contribution of rare and low frequency coding variants to complex phenotypes is largely unknown. To further address this question we generated exome sequence data from 42 unrelated CD cases and then used a single nucleotide variant (SNV) filtering strategy that incorporated association results from a recent CD meta-analysis data set2 in combination with in silico mutation effect prediction algorithms (SNVs comprise SNPs and variants with <1% allele frequency in the general population). We carried out follow-up genotyping in eight independent case-control sets comprising 9,348 CD cases, 2,868 UC cases, and 14,567 healthy control (HC) individuals of European ancestry. Using this approach, we identified and replicated novel causative low frequency and rare missense variants associated with CD and/or ulcerative colitis (UC) and interrogated their functional impact.
Materials and Methods
Study subjects
We captured the exomes of 42 German CD patients, one HapMap trio6 and 3 German unrelated healthy control (HC) individuals by means of the NimbleGen 2.1M Human Exome Array, followed by Illumina Genome Analyzer resequencing (Supplementary Figure 1 and Supplementary Figure 2, Supplementary Table 1). For detailed information on whole-exome enrichment and sequencing process, see Supplementary Note “Whole-exome sequencing” in the Supplementary Material. For detailed information on sequence read alignment, variant calling and annotation, see Supplementary Note “Read alignment, variant calling and annotation”. Follow-up genotyping of 9,348 CD cases, 2,868 UC cases, and 14,567 healthy control (HC) individuals was carried out using Sequenom® iPlex and TaqMan® technology from Applied Biosystems. For detailed information on quality control and association testing, see Supplementary Note “Follow-up genotyping, quality control and association testing”. For detailed information on study subjects, see Supplementary Note “Study subjects”.
Expression quantitative trait locus (eQTL) analysis
We studied the correlation between PRDM1 expression and rs7746082 genotype in healthy controls on the basis of data from (i) ileal biopsies, (ii) peripheral blood mononuclear cells (PBMCs), (iii) CD4+CD45RO+ T cells, and (iv) CD4+CD45RO− T cells. Ileal biopsies were obtained from healthy study subjects undergoing colonoscopy for non-IBD indications (polyp surveillance and investigation of anaemia). For the eQTL analysis in peripheral blood subsets, we recruited 42 volunteers from the Cambridge Bioresource, 21 homozygous for each of the rs7746082 risk (C) and wildtype (G) alleles. PBMCs were isolated and from these CD4+CD45RO− and CD4+CD45RO+ T cells separated. We focused on CD4+ cells due to prior evidence predominantly implicating this cell type in IBD pathogenesis7. We extracted and reverse-transcribed mRNA and undertook qRT-PCR using Applied Biosystems 7500 Fast Real-Time PCR System. Expression analysis was undertaken blind to the genotype status. For detailed information on protocols, see Supplementary Note “eQTL analysis”.
In each of the four analyses, we computed the standardized mean differential expression T between individuals homozygous for the CD risk-associated rs7746082 allele “C”, and individuals homozygous for the wildtype allele “G”. For detailed information on protocols, see Supplementary Note “Statistical analysis of the correlation between PRDM1 expression and rs7746082 genotype”.
Functional studies on rare PRDM1 variants and allelic variants of NDP52
We studied peripheral blood lymphocyte (PBL) subsets in CD patients with and without PRDM1 variants chr6:106659789 (Ser354Asn) and chr6:10666076 (Leu450Phe) to investigate whether these variants are associated with alterations in the phenotype and function of PBLs. Furthermore, we analysed the impact of the allelic NDP52 variants on protein function and NF-κB activation. For detailed information on protocols, see Supplementary Note “Functional PRDM1 studies” and “Functional studies on CALCOCO2/NDP52”.
In silico analyses of PRDM1, NDP52 and CYB561D2 protein sequences and protein structure modeling of the NDP52 coiled coil domain
We performed comprehensive sequence analysis and protein secondary structure predictions. For detailed information on methods, see Supplementary Note “In silico analyses of PRDM1, NDP52 and CYB561D2 protein sequences” and “protein structure modeling of the NDP52 coiled coil domain”.
Results
Discovery of low frequency variants using whole-exome sequencing
In total, we identified 117,957 SNVs in our 48 individuals, including 59,076 coding and splice site SNVs (Supplementary Table 2 and 3). The overall SNV analysis workflow of our study is displayed in Supplementary Figure 3. In order to pinpoint a small subset of functionally important SNVs from the large amount of genetic variation, we developed a software tool for SNV annotation, categorization, and filtering named snpActs (Supplementary Table 4, see also Supplementary Note “SNV filtering and validation phase”).
Results of association testing
Two missense SNPs, chr6:106659789 (Ser354Asn) and chr6:106660076 (Leu450Phe) at PRDM1 (MIM#603423) on chromosome 6q21, were significantly associated with CD (PSer354Asn=1.18×10−3, odds ratio [OR] of 1.23 (95% CI=1.07–1.40); risk variant) and UC (PLeu450Phe=5.88×10−6, OR=0.37 (95% CI [0.23–0.59]); protective variant), respectively (Table 1, see also Supplementary Table 5, 6 and 7 for full association results). We validated TaqMan® genotype calls by Sanger sequencing (see Supplementary Note “Detailed results of association testing”). Conditional analyses on the three common and two low-frequency markers indicated that common and low-frequency alleles contribute independently to risk at the PRDM1 gene locus (Supplementary Table 8).
Table 1.
Association results of genotyping in a large European panel comprising 9,348 CD cases, 2,868 UC cases and 14,567 control individuals (panel C, see also description in Supplementary Table 5c) in combination with additional GWAS meta-analysis data on CD (6,333 CD cases and 15,056 control individuals; panel D, Supplementary Table 5d) and UC (6,687 UC cases and 19,718 control individuals; panel F, Supplementary Table 5f), respectively. The variants chr6:106659789A>G and chr106660076T>C correspond to Ser354Asn (CD-risk) and Leu450Phe (UC-protection), respectively.
| (a) Association with Crohn’s disease
| |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNV | Locus | A1min. | A2maj. | CD Cases | Controls | A1freq.cases | A1freq.controls | Stage 2 genotyping (9,348/14,567) | Stage 2 genotyping & CD GWAS meta-analysis (15,691/29,623) | ||||
| A1Obs. | Alleles Total | A1Obs. | AllelesTotal | P | OR (95% CI) | Pcombined | OR (95% CI) | ||||||
| chr2:27291700 | C2orf28 | T | C | 38 | 19356 | 66 | 28598 | 0.00196 | 0.00231 | 4.09×10−1 | 0.95 (0.63–1.45) | NA | NA |
| chr3:48613476 | UQCRC1 | C | T | 120 | 19348 | 131 | 28596 | 0.00620 | 0.00458 | 1.77×10−2 | 1.32 (1.02–1.72) | NA | NA |
| chr3:50365760 | CYB561D2 | T | C | 5 | 19360 | 0 | 28614 | 0.00026 | 0 | 3.57×10−3 | NA | NA | NA |
| chr6:106659789 | PRDM1 | A | G | 427 | 19358 | 525 | 28614 | 0.02206 | 0.01835 | 1.18×10−3 | 1.23 (1.07–1.41) | NA | NA |
| chr6:106660076 | PRDM1 | T | C | 89 | 19266 | 247 | 28590 | 0.00462 | 0.00864 | 8.50×10−3 | 0.73 (0.56–0.94) | NA | NA |
| rs2303015 | NDP52 | G | A | 992 | 19358 | 1244 | 28612 | 0.05124 | 0.04348 | 3.27×10−6 | 1.24 (1.13–1.36) | 4.83×10−9 | 1.23 (1.15–1.32) |
|
| |||||||||||||
| rs7746082 | PRDM1 | C | G | 5967 | 19258 | 8112 | 28024 | 0.3098 | 0.2895 | 5.11×10−6 | 1.10 (1.06–1.15) | 7.19×10−12 | 1.12 (1.08–1.15) |
| rs6568421 | PRDM1 | G | A | 6243 | 19924 | 8531 | 29174 | 0.3133 | 0.2924 | 4.21×10−6 | 1.10 (1.06–1.15) | 5.72×10−10 | 1.11 (1.07–1.15) |
| rs6911490 | PRDM1 | T | C | 4231 | 19258 | 5652 | 28024 | 0.2197 | 0.2017 | 5.55×10−7 | 1.12 (1.07–1.18) | 7.13×10−11 | 1.13 (1.09–1.17) |
| (b) Association with ulcerative colitis
| |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNV | Locus | A1min. | A2maj. | UC Cases | Controls | A1freq.cases | A1freq.controls | Stage 2 genotyping (2,868/14,567) | Stage 2 genotyping & UC GWAS meta- analysis (9,555/34,285) | ||||
| A1Obs. | AllelesTotal | A1Obs. | AllelesTotal | P | OR (95% CI) | Pcombined | OR (95% CI) | ||||||
| chr2:27291700 | C2orf28 | T | C | 15 | 5052 | 55 | 20266 | 0.00297 | 0.00271 | 3.03×10−1 | 1.17 (0.65–2.10) | NA | NA |
| chr3:48613476 | UQCRC1 | C | T | 25 | 5052 | 95 | 20264 | 0.00495 | 0.00469 | 3.70×10−1 | 1.08 (0.69–1.70) | NA | NA |
| chr3:50365760 | CYB561D2 | T | C | 2 | 5052 | 0 | 20282 | 0.00030 | 0 | 4.31×10−3 | NA | NA | NA |
| chr6:106659789 | PRDM1 | A | G | 95 | 5052 | 336 | 20282 | 0.01880 | 0.01657 | 1.93×10−1 | 1.11 (0.88–1.41) | NA | NA |
| chr6:106660076 | PRDM1 | T | C | 19 | 5052 | 230 | 20258 | 0.00376 | 0.01135 | 5.77×10−6 | 0.37 (0.23–0.59) | NA | NA |
| rs2303015 | NDP52 | G | A | 240 | 5052 | 919 | 20280 | 0.04751 | 0.04532 | 2.03×10−1 | 1.07 (0.92–1.24) | 2.32×10−2 | 1.11 (1.01–1.21) |
|
| |||||||||||||
| rs7746082 | PRDM1 | C | G | 1604 | 5110 | 5701 | 19724 | 0.3139 | 0.2890 | 1.99×10−4 | 1.13 (1.06–1.21) | 8.03×10−5 | 1.10 (1.05–1.15) |
| rs6568421 | PRDM1 | G | A | 1627 | 5176 | 6024 | 20586 | 0.3143 | 0.2926 | 1.13×10−3 | 1.11 (1.04–1.19) | 7.92×10−6 | 1.09 (1.05–1.13) |
| rs6911490 | PRDM1 | T | C | 1071 | 5110 | 3898 | 19724 | 0.2096 | 0.1976 | 3.72×10−2 | 1.07 (1.00–1.14) | 1.16×10−8 | 1.12 (1.08–1.16) |
The first six SNVs are low-frequency variants identified by the heuristic SNV filtering strategy that used whole-exome sequence data. For case/control allele frequencies in the individual genotyping panels see Supplementary Table 7. Additionally genotyped SNP markers listed below the double line represent best common association signals that were found previously in independent GWAS meta-analyses on CD and UC1, 2, 32. Marker rs7746082 represents the best association signal at 6q21 (PRDM1) in the scan of the first GWAS meta-analysis on CD1. rs6568421 represents the most significant associated SNP at 6q21 (PRDM1) in the scan of the second GWAS meta-analysis on CD2. rs6911490 is the best associational signal at 6q21 (PRDM1) from the scan of the GWAS meta-analysis on UC32. Markers, which remain significant after Bonferroni correction, are shown in bold type. At locus 6q21 (PRDM1), we observed significant associations of rare missense variants with CD (SNP chr6:106659789/) and UC (SNP chr6:106660076). At 17p12 (NDP52), a highly significant association of the missense SNP rs230315 with CD but not UC was observed.
CD: Crohn’s disease; UC: ulcerative colitis; SNV: SNP rs-id (if available from dbSNP130) or chromosome and position (NCBI build 36 coordinates) of marker; Locus: the affected gene; A1: minor allele; A2: major allele; A1Obs.: number of minor alleles observed in stage 1&2 genotyping; AllelesTotal: number of overall alleles in stage 1&2 genotyping; A1freq.cases: average allele frequency of minor allele in cases in stage 1&2 genotyping; A1freq.controls: average allele frequency of minor allele in controlsm in stage 1&2 genotyping; P: P value from permutation association testing (mega-analysis of rare variants); OR (95% CI): Cochran-Mantel-Haenszel (CMH) odds ratio and 95% confidence interval with respect to minor allele A1; Pcombined: Combined analysis P value of the European panel (panel C, Supplementary Table 5c) and the scan from the independent GWAS meta-analysis on CD2 (panel D, Supplementary Table 5d) and UC32 (panel F, Supplementary Table 5f), respectively. For each panel, numbers of cases/controls in total are displayed in parentheses.
The missense SNP rs2303015 (Val248Ala) in the novel CD candidate gene NDP52 (MIM#604587; also known as CALCOCO2) was associated with CD (P=3.27×10−6) with an odds ratio of 1.24 (95% CI=1.13–1.36) in panel C (Table 1, Supplementary Table 5). In the combined analysis of panels C and D, SNP rs2303015 attained classical genome-wide significance for CD (P=4.83×10−9). We observed no association with UC at the NDP52 locus.
Results of eQTL analysis
A locus on chromosome 6q21 adjacent to PRDM1 has been associated with Crohn’s disease in three GWAS meta-analyses1–3. To explore possible eQTL effects relating to this locus we directly interrogated correlation between PRDM1 expression and genotype at rs7746082, the common lead SNP identified by the first CD GWAS meta-analysis1. SNP rs7746082 maps to a gene desert and PRDM1 represents one of only two genes within 500 kb – the other being ATG5 (MIM#604261). Both are strong positional candidate genes for IBD: PRDM1 (maps 100 kb downstream of rs7746082) encodes a master transcriptional regulator in B and T cells. ATG5 (197 kb downstream) encodes a critical component of autophagy, a pathway required for cellular innate immunity, which has previously been strongly implicated in CD pathogenesis8–12. Ileal biopsies from the 13 individuals homozygous for the CD risk-associated “C” allele showed significantly lower expression of PRDM1 when compared to the 39 individuals homozygous for the wildtype allele “G” (P=1.15×10−6 – Figure 1a). No such eQTL effect was observed for ATG5. Using immunohistochemistry, we observed PRDM1 to be expressed predominantly in mucosal T cells and plasma cells, and not epithelial cells (Supplementary Figure 4).
Figure 1. Correlation of expression of PRDM1 with genotype at CD GWAS-associated lead SNP rs7746082 in healthy controls.

Panel (a) expression analysis by qRT-PCR in terminal ileal biopsies from 13 controls homozygous for rs7746082 risk (C) allele vs. 39 homozygous wildtype (G) allele showed (i): PRDM1 expression was significantly lower in the CC than in the GG genotype group (P=1.15×10−6) and (ii) ATG5 expression did not differ significantly between the two genotype groups (P=0.4229). Panel (b) (i) and (ii): PBMC and CD4+CD45RO+ T cell populations from an independent panel of controls demonstrated a significant difference in PRDM1 expression between CC and GG genotypes at rs7746082 (P=0.0006 and 0.0125, respectively). (iii): PRDM1 was expressed at lower levels in the CD4+CD45RO− population and did not correlate with genotype at rs7746082 (P=0.449). CC: homozygous Crohn’s disease risk genotype at rs7746082; GG: homozygous wildtype genotype at rs7746082.
Given potential confounding by the mixed cell populations present in ileal mucosal biopsies, we additionally studied this eQTL effect in peripheral blood subsets from an independent panel of healthy volunteers. Using qRT-PCR, we observed the same direction of eQTL effect as seen in ileal biopsies in both PBMCs and CD4+CD45RO+ T cells, with significantly lower expression in CC vs. GG homozygotes (P=0.0006 and 0.0125 for respective cell populations – Figure 1b; combined P with ileal data: 1.6×10−8). The CD4+CD45RO− sub-population showed much lower PRDM1 expression and no eQTL effect. No eQTL effect was seen for ATG5 expression in the PBMCs or subsets.
Functional characterization of rare PRDM1 variants
To investigate whether the rare PRDM1 variants we had identified alter the phenotype and function of peripheral blood lymphocytes (PBLs), we studied PBL subsets in CD patients with and without PRDM1 variants chr6:106659789 (Ser354Asn) and chr6:10666076 (Leu450Phe). In accordance with murine studies on B cell-specific Prdm1 deletion13, CD patients heterozygous for Ser354Asn and Leu450Phe exhibited significantly decreased levels of plasma cells and a non-significant trend towards reduction in memory and overall B cell levels (Figure 2a). In contrast, absolute and relative numbers of CD4+, CD8+ and double negative T cells and baseline expression levels of activation markers CD69, CD44, and CD25 were similar in all groups of CD patients (Supplementary Figure 5a–c and data not shown). However, while numbers of naive, central memory and effector memory populations did not differ depending on the PRDM1 genotype (Supplementary Figure 5d–e), CD patients heterozygous for the risk allele “A” of Ser354Asn but not the protective allele “T” of Leu450Phe exhibited increased expression of L selectin (CD62L), an adhesion molecule critical for migration of PBLs into mesenteric lymph nodes, Peyer’s patches and the lamina propria (Figure 2b)14–16. In addition, despite similar baseline characteristics in expression of activation markers, both CD4+ and CD8+ T cells obtained from CD patients heterozygous for the risk allele Ser354Asn but not the protective allele of Leu450Phe exhibited dramatically increased proliferation, IFN-γ secretion, and upregulation of activation markers upon stimulation, even under conditions of minimal T cell receptor engagement (Figure 2c–e; flow cytometry histograms for 2e are displayed in Supplementary Figure 6). To investigate whether ectopic expression of the risk allele Ser354Asn in T cells from healthy donors is associated with increased activation and cytokine secretion, we transduced PBLs from healthy controls expressing wildtype PRDM1 with lentiviruses expressing wildtype, Ser354Asn, and Leu450Phe PRDM1. In accordance with data obtained in CD patients, T cells from healthy controls expressing Ser354Asn but not Leu450Phe exhibited increased proliferation, IFN-γ secretion, and upregulation of activation markers upon stimulation (Figure 3a–b, Supplementary Figure 5f). Transduction rates were similar in all conditions (Figure 3c). Thus, PRDM1 Ser354Asn is associated with increased activation, cytokine secretion and proliferation of CD4+ and CD8+ T cells.
Figure 2. Phenotypic and functional characterization of PBL populations in CD patients with and without the indicated PRDM1 variants.

(a) Percentages of indicated B cell populations among PBLs of CD patients with and without PRDM1 variants. (b) Percentage of CD62L+ cells among T cells and mean fluorescence intensity of CD62L on T cells of CD patients with and without PRDM1 variants. (c–e) PBLs of the indicated groups of CD patients were stimulated with plate-bound anti-CD3 and soluble anti-CD28 antibodies and proliferation (c, day 5), IFNγ secretion (d, day 3), and upregulation of activation markers (e, day 2) were investigated. Flow cytometry histograms for e are displayed in Supplementary Figure 6). Results from studies shown in Figure 2a–e are based on five CD patients with wildtype PRDM1 expression, six CD patients with PRDM1 Ser354Asn and two CD patients with PRDM1 Leu450Phe. *P<0.05, **P<0.01, ***P<0.001. Mean ± sem of 4 to 6 CD patients per group (a–e) are shown.
Figure 3. Phenotypic and functional characterization of healthy control PBLs transduced with PRDM1 lentiviruses.
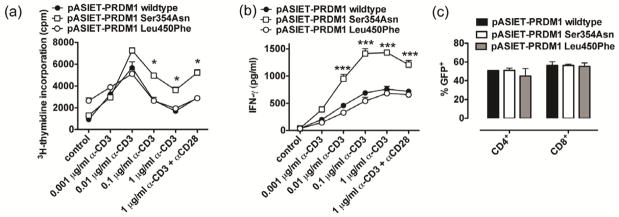
(a–c) PBLs of healthy controls with PRDM1 wildtype expression were transduced with the indicated PRDM1 lentiviruses and proliferation (a, day 5) and IFN-γ secretion (b, day 3) were determined. Tranduction rates as detected by lentivirus-derived GFP are shown in (c). *P<0.05, **P<0.01, ***P<0.001. Mean ± sem of 4 to 6 CD patients per triplicates (a–c) are shown.
Functional assessment of allelic variants of NDP52
It has recently been shown that NDP52 negatively regulates toll-like receptor (TLR)-triggered NF-κB activation17. We therefore analysed the impact of the two allelic variants 248Val (wildtype; WT) and 248Ala (risk) on NF-κB activation using the specific TLR stimulus poly(I:C), which is affected by NDP52 regulatory activity. As expected, the 248Val WT variant decreased NF-κB activation in response to poly(I:C) treatment. However, the 248Ala risk variant failed to decrease NF-κB activation (Figure 4a). We further confirmed the impaired capacity of the risk allele to dampen proinflammatory signalling by assessing nuclear localization of NF-κB subunit p65 after poly(I:C) treatment (Figure 4b). It is estimated that the suppressive function of NDP52 for proinflammatory signalling is mediated by ubiquitination events and a complex interplay between the proteasomal and the autophagic degradation systems17. We therefore analyzed structural features of the respective coiled coil region surrounding amino acid 248. As depicted in Figure 4c, this structure prediction analysis of the allelic variants indicates a possible role of the affected amino acid position 248 for binding of ubiquitin chains. Since NDP52 itself is supposed to be post-translationally modified and subject to selective degradation in the course of TLR activation17, we next characterized the stability of NDP52 protein complexes under poly(I:C)-triggered TLR-activating conditions. As demonstrated in Figure 4d, TLR activation resulted in time-dependent degradation of NDP52 of the 248Val WT allele. Intriguingly, this effect was impaired for the 248Ala risk variant as documented by the increased stability of the NDP52 (248Ala) protein in a time- and dose-dependent fashion (Figure 4d and Supplementary Figure 7). We constructed an NDP52 protein structure model (Figure 4c) in complex with Lys63-linked di-ubiquitin (PDB ID 3jsv) to visualize the position of the variant in the predicted coiled coil domain (for details see also Supplementary Note “Protein Structure Model and Analysis of NDP52”, Supplementary Figure 8).
Figure 4. Role of NDP52 variants in modulation of proinflammatory signalling and protein stability.

(a) Dual-Luciferase reporter assay for NF-κB activation after stimulation of HeLa cells with poly(I:C) for 6 h. (b) Analysis of nuclear localisation of p65/NF-κB in poly(I:C) stimulated HeLa cells expressing NDP52 variants. Ratio of nuclear/cytoplasmic presence of p65/NF-κB was quantified in NDP52 expressing cells using automated image analysis. Exemplary images and results of the automated analysis are given at the right side. Green lines indicate cell boundaries, blue lines indicate nucleus boundaries as identified by the software. (c) Structure-based alignment and model of the NDP52 coiled coil domain region (”CACOCO2 human”; amino acids 225–267) based on the structure of the NEMO UBAN motif positions 293–335 (“Nemo mouse”) in complex with Lys63-linked di-ubiquitin. Coiled coil helices are colored yellow. The variant position Val248 is visualized by red spheres in the 3D model and marked in red in the sequence lineup. Ubiquitin is shown in blue with surface representation. (d) Western Blot analysis of NDP52 stability. HeLa cells were transfected with plasmids encoding NDP52 WT and risk variants and stimulated using 50 μg/ml of poly(I:C) for 0, 3 or 6 h. While NDP52 WT is effectively degraded after 6h, the risk variant exhibits increased stability. RFP alone (lower band on the left) is not affected.
Discussion
Outcomes of the present study highlight the importance of detailed multi-faceted analyses to extend genetic insights beyond those derived from GWAS alone. By undertaking whole exome sequencing followed by rare variant analysis, and detailed expression and functional studies, we have substantially advanced understanding of a previously defined GWAS locus and have identified a new CD susceptibility gene which further emphasizes the contribution of autophagy and NFkB pathways to CD pathogenesis.
Our findings implicate PRDM1 as the causal gene at the 6q21 CD/UC GWAS locus, combining genetic association with rare coding variants, functional interrogation and an eQTL effect in two disease-relevant tissues18, 19. PRDM1 encodes PR domain containing 1, also known as B-lymphocyte-induced maturation protein (Blimp-1). It is a zinc finger-containing transcriptional repressor, now known to be a master regulator of terminal B and T cell differentiation.20 The rare Ser354Asn CD risk allele that we identified in PRDM1 led to increased PBL expression of adhesion molecule L selectin, critical for PBL migration to the sites of intestinal inflammation, and increased CD4+ and CD8+ T cell proliferation, IFN-γ secretion, and upregulation of activation markers upon stimulation. Each of these factors may contribute to its pathogenic role.
Our observed ‘eQTL’ signal, correlating expression with genotype for the common PRDM1 SNP rs7746082, has not yet been reported in publicly available data sets21. This highlights that genotype-expression correlations can be highly tissue-specific and must be studied in relevant cell types22. Intriguingly, conditional T cell-specific deletion of Prdm1 in mice has been associated with increased numbers of activated effector CD4+ and CD8+ T cells, hyperproliferation, increased production of inflammatory cytokines, and the development of spontaneous colitis23, 24. In accordance with these observations in mice, we found that PRDM1 expression in the human intestinal mucosa was largely limited to T cells and plasma cells (Supplementary Figure 4). In addition, PRDM1 was predominantly expressed by effector memory cells within T cell compartments, characterized by expression of CD45RO (Figure 1b). Importantly, ileal biopsies of healthy individuals homozygous for the CD risk-associated allele at rs7746082 showed significantly lower expression of PRDM1 than individuals homozygous for the wildtype allele (Figure 1a). Similar observations were made for purified PBMCs and effector memory T cells (Figure 1b). It may therefore be hypothesized that lower PRDM1 expression of rs7746082 risk-allele carriers leads to the same pro-inflammatory effects as in the afore-described Prdm1 conditional knock-out mice.
We also identify novel genome-wide significant association of the genetic marker rs2303015 with susceptibility to CD. This SNP represents a missense mutation in the gene NDP52 (nuclear dot protein 52 kDa). Initially described as a nuclear protein25, NDP52 was later recognized as a cytosolic protein with a crucial role in immunity and as an adaptor for selective autophagy26–29. Besides the detection of ubiquitin-decorated cytosolic bacteria, NDP52 is also essentially involved in an entirely different type of selective autophagy. Inomata et al. reported that NDP52 selectively degrades TLR signalling adaptors leading to attenuated NF-κB activation17. In line with this, our data demonstrate a role of the missense mutation Val248Ala for differential regulation of NF-κB signalling. Our observation that the risk variant displays an almost complete loss of its ability to restrict NF-κB activation downstream of TLR pathways could be potentially causative for the chronically increased (hyper-) activation of NF-κB in Crohn’s disease patients30. Inomata and colleagues have shown that proinflammatory gene expression is only suppressed under the condition of A20 silencing and that the coiled coil domain of NDP52 is subjected to polyubiquitination events mediated by the ubiquitin-modifying enzyme A2017 – itself encoded by positional candidate gene TNFAIP3 within a confirmed CD GWAS locus2. The missense mutation described here affects the amino acid Val248, which is located within the coiled coil domain. Since our data additionally argue for an allele-specific effect on NDP52 protein stability (Figure 4d), it is tempting to speculate that the same molecular mechanism is causative for both observations (i.e. NDP52 degradation and NF-κB inhibition). In summary, we identified NDP52 as an additional autophagy-related risk factor for CD (see pathway details in Supplementary Figure 9) and provide evidence for a functional role of the disease-associated missense mutation in the context of protein stability and differences in its ability to restrict proinflammatory signalling.
The new genetic associations that we have identified emphasize the contribution of risk alleles from across the frequency spectrum to CD pathogenesis. Low frequency and rare variant associations have been relatively poorly studied to date as they are not generally interrogated by GWAS microarrays, and require different study designs and analytic methods. Hence their contribution to the ‘missing heritability’ of complex disease is largely unknown at present. There is an expectation that many hundreds or thousands of such associations might exist, and that the effect sizes might be significantly larger than is typical for common variants. Cumulatively these might contribute a significant fraction of the current ‘missing heritability’; and their detection, particularly in the context of large effect sizes, might significantly impact personalised medicine options in the future.
As with the study reported by Nejentsev et al. in type 1 diabetes31 our detailed sequencing efforts here, in identifying rare variants in PRDM1 which are associated with Crohn’s disease, have helped to pinpoint this as a causal gene at a GWAS locus (or in our case, immediately adjacent to a GWAS locus). This corroborates the genotype-expression analysis described above, which suggests that the common IBD-predisposing mechanism is by an effect on transcriptional regulation. Another, broader, conclusion that can be drawn is that, after a detailed exome sequencing study, we have found only modest evidence that coding variation across the genome makes a major contribution to Crohn’s disease susceptibility. While our study was underpowered to detect rare variants of small effect size, one would have expected that, if this made a major contribution to missing heritability, we would have found much more.
Here we focused sequencing efforts on exomes but future studies will encompass whole genomes and, as sequencing costs continue to fall dramatically, will interrogate panels comprising thousands of Crohn’s disease patients. Although our current study suggests that rare coding variation does not make a big contribution to IBD susceptibility this does not rule out an effect of rare variants in non-coding sequence. Indeed, one of the key lessons from the studies of common variation through GWAS is that such regulatory polymorphisms make a much bigger contribution than do coding variants. Only once whole genome sequencing data have been analysed will we have a true appreciation of the relative contribution of common vs. low frequency vs. rare variants to disease heritability. This will aid understanding of the heterogeneity of genetic effects in Crohn’s disease and the extent to which specific disease strata might benefit from individualised treatment approaches.
Supplementary Material
Acknowledgments
Grant support
We thank all individuals with CD or UC, their families, control individuals and clinicians for their participation. We wish to thank Tanja Wesse, Tanja Henke, Sanaz Sedghpour Sabet, Sandra Greve and Ilona Urbach for expert technical help. The study was supported by the Deutsche Forschungsgemeinschaft (DFG), grant no. FR 2821/2-1 and research fellowship Ti 640 1-1, the SFB 877 subproject B9, the German Ministry of Education and Research (BMBF) through the National Genome Research Network (NGFN), the PopGen biobank (http://www.popgen.de), and the NIH (grant GM069373). The project received infrastructure support through the DFG Clusters of Excellence “Inflammation at Interfaces” and “Multimodal Computing and Interaction”.
Stephan Brand is supported by the DFG grant BR1912/6-1 and by the Else-Kröner-Fresenius-Stiftung (stipend 2010_EKES.32). We acknowledge use of material from the British 1958 Birth Cohort DNA collection, funded by the Medical Research Council grant G0000934 and the Wellcome Trust grant 068545/Z/02 (http://www.b58cgene.sgul.ac.uk/). We also gratefully acknowledge the participation of all Cambridge BioResource (CBR) subjects and staff (http://www.cambridgebioresource.org.uk/); the National Institute of Health Research (NIHR) Biomedical Research Centre awards to Guy’s & St. Thomas’ National Health Service Trust/King’s College London and to Addenbrooke’s Hospital/University of Cambridge School of Clinical Medicine; the Wellcome Trust Case Control Consortium (WTCCC), a seed grant (to A.Till and S. Subramani) from the San Diego Center for Systems Biology (SDCSB, grant #GM085764), one of the NIGMS-funded P50 National Centers for Systems Biology, the National Institutes of Health grant 1R01CA141743-01 (PI: Richard H. Duerr), the University of Pittsburgh Genomics and Proteomics Core Laboratories for the Immunochip genotyping services, and the DFG grants ZE 814/4-1 and ZE 814/5-1 (SZ). Mauro D’Amato received support from The Swedish Research Council (VR). Finally, we would like to thank the International IBD Genetics Consortium (IIBDGC) for providing us with summary statistics of the CD and UC meta-analyses through the IIBDGC webpage (http://www.ibdgenetics.org).
Footnotes
Author names in bold designate shared co-first authorship.
Competing financial interests
The authors declare no competing financial interests.
Author contributions
- Design of genetic experiments: D Ellinghaus, A Franke.
- Performed the experiments: D Ellinghaus, H Zhang, T Jiang, J Wang, E Ellinghaus, X Liu, Q Liu, F Jiang, R. Häsler, P Rosenstiel, S Zeissig, S Lipinski, A Till, TH Karlsen.
- Analyzed the data: D Ellinghaus. H Zhang, T Jiang, S Zeissig, S Lipinski, A Till, B Stade, E Ellinghaus, Y Bromberg, A Keller, G Mayr, M Albrecht, NT Doncheva, R Häsler, A Franke, A Keller, MA Rivas, M Daly, CR Berzuini.
- Contributed sample collections: S Nikolaus and S Schreiber provided the German CD and UC samples. T Balschun helped providing German CD and UC samples. BO Boehm provided German control samples from the EMIL-study. V Annese and A Latiano provided Italian samples. V Andersen and U Vogel provided Danish samples. J Skieceviciene and L Kupcinskas provided Baltic samples. RK Weersma and CY Ponsioen provided Dutch samples. C Wijmenga provided control samples from The Netherlands. S Vermeire and P Rutgeerts provided samples from Belgium. M Sans provided Spanish samples. D Strachan, WL McArdle, JD Sanderson, CG Mathew, J Lee and M Parkes provided samples from the United Kingdom. M D’Amato and J Halfvarson provided Swedish samples. MH Vatn, TH Karlsen and the IBSEN study group collected and provided the Norwegian samples. MM Nöthen, J Winkelmann and T Illig provided German control samples. S Brand, J Glas and C Büning provided German control samples which were genotyped at the University of Pittsburgh Genomics and Proteomics Core Laboratories (PI: RH Duerr).
- M Kayser and M Krawzcak helped with principal component analysis and provided summary statistics for the European reference panel.
- Supervised the experiments: A Franke, M Parkes, P Rosenstiel, S Subramani, S Schreiber.
- Wrote the first draft paper: D Ellinghaus.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Barrett JC, Hansoul S, Nicolae DL, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet. 2008;40:955–62. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Franke A, McGovern DP, Barrett JC, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 2010;42:1118–25. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jostins L, Ripke S, Weersma RK, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–24. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–53. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bamshad MJ, Ng SB, Bigham AW, et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 2011;12:745–55. doi: 10.1038/nrg3031. [DOI] [PubMed] [Google Scholar]
- 6.A haplotype map of the human genome. Nature. 2005;437:1299–320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zenewicz LA, Antov A, Flavell RA. CD4 T-cell differentiation and inflammatory bowel disease. Trends Mol Med. 2009;15:199–207. doi: 10.1016/j.molmed.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 8.Hampe J, Franke A, Rosenstiel P, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39:207–211. doi: 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- 9.Rioux JD, Xavier RJ, Taylor KD, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39:596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prescott NJ, Fisher SA, Franke A, et al. A Nonsynonymous SNP in ATG16L1 Predisposes to Ileal Crohn’s Disease and Is Independent of CARD15 and IBD5. Gastroenterology. 2007;132:1665–1671. doi: 10.1053/j.gastro.2007.03.034. [DOI] [PubMed] [Google Scholar]
- 11.Parkes M, Barrett JC, Prescott NJ, et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility. Nat Genet. 2007;39:830–2. doi: 10.1038/ng2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Henckaerts L, Cleynen I, Brinar M, et al. Genetic variation in the autophagy gene ULK1 and risk of Crohn’s disease. Inflamm Bowel Dis. 2011;17:1392–7. doi: 10.1002/ibd.21486. [DOI] [PubMed] [Google Scholar]
- 13.Shapiro-Shelef M, Lin KI, McHeyzer-Williams LJ, et al. Blimp-1 is required for the formation of immunoglobulin secreting plasma cells and pre-plasma memory B cells. Immunity. 2003;19:607–20. doi: 10.1016/s1074-7613(03)00267-x. [DOI] [PubMed] [Google Scholar]
- 14.Steeber DA, Tang ML, Zhang XQ, et al. Efficient lymphocyte migration across high endothelial venules of mouse Peyer’s patches requires overlapping expression of L-selectin and beta7 integrin. J Immunol. 1998;161:6638–47. [PubMed] [Google Scholar]
- 15.Wagner N, Lohler J, Tedder TF, et al. L-selectin and beta7 integrin synergistically mediate lymphocyte migration to mesenteric lymph nodes. Eur J Immunol. 1998;28:3832–9. doi: 10.1002/(SICI)1521-4141(199811)28:11<3832::AID-IMMU3832>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 16.Salmi M, Granfors K, MacDermott R, et al. Aberrant binding of lamina propria lymphocytes to vascular endothelium in inflammatory bowel diseases. Gastroenterology. 1994;106:596–605. doi: 10.1016/0016-5085(94)90691-2. [DOI] [PubMed] [Google Scholar]
- 17.Inomata M, Niida S, Shibata KI, et al. Regulation of Toll-like receptor signaling by NDP52-mediated selective autophagy is normally inactivated by A20. Cell Mol Life Sci. 2011 doi: 10.1007/s00018-011-0819-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dotan I, Allez M, Nakazawa A, et al. Intestinal epithelial cells from inflammatory bowel disease patients preferentially stimulate CD4+ T cells to proliferate and secrete interferon-gamma. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1630–40. doi: 10.1152/ajpgi.00294.2006. [DOI] [PubMed] [Google Scholar]
- 19.Fuss IJ, Heller F, Boirivant M, et al. Nonclassical CD1d-restricted NK T cells that produce IL-13 characterize an atypical Th2 response in ulcerative colitis. J Clin Invest. 2004;113:1490–7. doi: 10.1172/JCI19836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crotty S, Johnston RJ, Schoenberger SP. Effectors and memories: Bcl-6 and Blimp-1 in T and B lymphocyte differentiation. Nat Immunol. 2010;11:114–20. doi: 10.1038/ni.1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dixon AL, Liang L, Moffatt MF, et al. A genome-wide association study of global gene expression. Nat Genet. 2007;39:1202–7. doi: 10.1038/ng2109. [DOI] [PubMed] [Google Scholar]
- 22.McCarroll SA, Huett A, Kuballa P, et al. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn’s disease. Nat Genet. 2008;40:1107–12. doi: 10.1038/ng.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martins GA, Cimmino L, Shapiro-Shelef M, et al. Transcriptional repressor Blimp-1 regulates T cell homeostasis and function. Nat Immunol. 2006;7:457–65. doi: 10.1038/ni1320. [DOI] [PubMed] [Google Scholar]
- 24.Kallies A, Hawkins ED, Belz GT, et al. Transcriptional repressor Blimp-1 is essential for T cell homeostasis and self-tolerance. Nat Immunol. 2006;7:466–74. doi: 10.1038/ni1321. [DOI] [PubMed] [Google Scholar]
- 25.Korioth F, Gieffers C, Maul GG, et al. Molecular characterization of NDP52, a novel protein of the nuclear domain 10, which is redistributed upon virus infection and interferon treatment. Journal of Cell Biology. 1995;130:1–13. doi: 10.1083/jcb.130.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thurston TL, Ryzhakov G, Bloor S, et al. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol. 2009;10:1215–21. doi: 10.1038/ni.1800. [DOI] [PubMed] [Google Scholar]
- 27.Morriswood B, Ryzhakov G, Puri C, et al. T6BP and NDP52 are myosin VI binding partners with potential roles in cytokine signalling and cell adhesion. Journal of Cell Science. 2007;120:2574–85. doi: 10.1242/jcs.007005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ivanov S, Roy CR. NDP52: the missing link between ubiquitinated bacteria and autophagy. Nat Immunol. 2009;10:1137–9. doi: 10.1038/ni1109-1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.von Muhlinen N, Thurston T, Ryzhakov G, et al. NDP52, a novel autophagy receptor for ubiquitin-decorated cytosolic bacteria. Autophagy. 2010;6:288–9. doi: 10.4161/auto.6.2.11118. [DOI] [PubMed] [Google Scholar]
- 30.Schreiber S, Rosenstiel P, Albrecht M, et al. Genetics of Crohn disease, an archetypal inflammatory barrier disease. Nat Rev Genet. 2005;6:376–88. doi: 10.1038/nrg1607. [DOI] [PubMed] [Google Scholar]
- 31.Nejentsev S, Walker N, Riches D, et al. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science. 2009;324:387–9. doi: 10.1126/science.1167728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anderson CA, Boucher G, Lees CW, et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat Genet. 2011;43:246–52. doi: 10.1038/ng.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.