

Abstract
Peripheral immune cells and brain microglia exhibit an activated phenotype in premanifest Huntington's disease (HD) patients that persists chronically and correlates with clinical measures of neurodegeneration. However, whether activation of the immune system contributes to neurodegeneration in HD, or is a consequence thereof, remains unclear. Signaling through cannabinoid receptor 2 (CB2) dampens immune activation. Here, we show that the genetic deletion of CB2 receptors in a slowly progressing HD mouse model accelerates the onset of motor deficits and increases their severity. Treatment of mice with a CB2 receptor agonist extends life span and suppresses motor deficits, synapse loss, and CNS inflammation, while a peripherally restricted CB2 receptor antagonist blocks these effects. CB2 receptors regulate blood interleukin-6 (IL-6) levels, and IL-6 neutralizing antibodies partially rescue motor deficits and weight loss in HD mice. These findings support a causal link between CB2 receptor signaling in peripheral immune cells and the onset and severity of neurodegeneration in HD, and they provide a novel therapeutic approach to treat HD.
Introduction
Huntington's disease (HD) is an inherited and devastating neurodegenerative disease caused by a mutation in the IT-15 gene, in which an expanded CAG repeat yields a polyglutamine stretch in the huntingtin (htt) protein (The Huntington's Disease Collaborative Research Group, 1993). Mutant htt expression in the striatum is neither necessary nor sufficient for neurodegeneration, implicating other cell types in HD (Gu et al., 2005; Gu et al., 2007). Mutant htt is expressed ubiquitously (Hoogeveen et al., 1993; Li et al., 1993), and although long overlooked, its expression in non-neuronal cells might contribute to important disease phenotypes in HD. Skeletal muscle atrophy, increased cardiac failure, impaired glucose tolerance, gastrointestinal dysfunction, testicular atrophy, osteoporosis, and weight loss have all been described in HD patients (van der Burg et al., 2009).
Mutant htt is also expressed in immune cells (Moscovitch-Lopatin et al., 2010), and is associated with increased plasma levels of pro-inflammatory cytokines (Leblhuber et al., 1998; Dalrymple et al., 2007; Björkqvist et al., 2008) and chemokines (Wild et al., 2011) that are elevated years before symptom onset. Stimulated monocytes and macrophages from HD patients and mouse models produce elevated levels of these pro-inflammatory factors (Björkqvist et al., 2008). Bone marrow transplantation with wild-type cells normalizes levels of cytokines and chemokines, and partially suppresses behavioral and neuropathological deficits in HD mouse models (Kwan et al., 2012). Based on these findings, we sought to identify signaling pathways in peripheral immune cells that contribute to neurodegeneration in HD, which could be targeted by small molecules.
Activation of cannabinoid receptor 2 (CB2) receptors decreases inflammatory responses (Munro et al., 1993; Ashton and Glass, 2007), and enhanced signaling through them prevents artherosclerosis in mice by dampening cytokine secretion (Steffens et al., 2005). CB2 receptors are critical for the host response to sepsis (Tschöp et al., 2009) and colitis (Singh et al., 2012), and decrease production of pro-inflammatory cytokines (Klegeris et al., 2003; Rajesh et al., 2008; Su et al., 2012). CB2 receptor agonists are protective in mouse models of several neurodegenerative diseases (Arévalo-Martin et al., 2003; Pryce et al., 2003; Kim et al., 2006; Zhang et al., 2007; García et al., 2011; Martín-Moreno et al., 2012). A recent study showed CB2 receptor levels are increased in HD postmortem brains and mice, and genetic deletion of CB2 exacerbates disease progression in an HD mouse model (Palazuelos et al., 2009). This study, and previous ones, concluded that CB2 signaling in parenchymal microglia was important for neuroprotection by CB2.
Here we show that deletion of CB2 receptors accelerates the onset of disease phenotypes and exacerbates behavioral deficits in a slowly progressing mouse model of HD. Treatment of a mouse model of HD with a CB2-selective receptor agonist suppresses neurodegeneration, and, surprisingly, is mediated by CB2 receptor signaling not in parenchymal microglia, but rather in peripheral immune cells. Finally, enhanced CB2 receptor signaling controls IL-6 blood levels and IL-6 inhibition partially suppresses pathogenesis in a mouse model of HD.
Materials and Methods
Animals and breeding strategy.
Experiments involving mice were approved by the Institutional Animal Care and Use Committee of the University of California, San Francisco. Mice were bred and maintained in compliance with National Institutes of Health guidelines. R6/2 mice were obtained by breeding R6/2 ovarian transplants with 160 CAG repeat length (002810 Jackson) with CBA WT males. BACHD mice (Gray et al., 2008) were obtained from Dr. William Yang (University of California Los Angeles) and were maintained by breeding to WT FVB/NJ males. CB2−/− mice (Buckley et al., 2000) on C57BL/6 background were obtained from Dr. Nancy Buckley. Using speed congenics, CB2−/− mice were backcrossed for 10 generations onto a FVB/NJ background before breeding to BACHD mice. To test if loss of CB2 worsened HD progression, we crossed the CB2−/− mice to BACHD mice. Progeny from these litters that were BACHD; CB2+/− were then bred to each other to generate BACHD; CB2−/− mice and all littermate controls.
Genotyping.
Mouse-tail DNA was analyzed by PCR to determine the genotypes. The BACHD transgene was identified as described previously (Gray et al., 2008). CB2−/− mice were identified as described previously (Buckley et al., 2000). The R6/2 transgene was identified as described previously (Hockly et al., 2003).
RNA extraction, reverse-transcription, real-time PCR analysis.
Brains from BACHD and R6/2 mice were collected after perfusing with 0.9% saline transcardially followed by snap freezing in dry ice and storage at −80°C until use. On the day of RNA isolation, striatal tissue was collected and placed in 1 ml of Qiazol reagent. Brains were homogenized and 200 μl chloroform was added and centrifuged at 4°C for 15 min. An equal volume of 70% EtOH was added to the supernatant and mixed together. Immediately after this step, samples were isolated with the Qiagen RNA Lipid Isolation Kit and their standard protocol and then treated with DNase (Qiagen) for 15 min and subsequent reverse transcription was performed with RT Superscript III (Invitrogen). Quantitative PCR (qPCR) were performed on the ABI Prism Sequence Detector 7700HT with TAQMAN PCR master mix (Applied Biosystems). For qPCR data we used the delta CT method (Schmittgen and Livak, 2008) and each reaction was normalized against the expression of the ATP5B (4331182; Applied Biosystems). The primer sequences from 5′ to 3′ for CB2 are forward primer, AAGGTCCTCGGTTACAGAAACA; reverse primer, GGAGTGAACTGAACGGACTTCT; and taqman probe, 6FAM-TCCAGAACTCCAGGCTGCTCCAAC-TAMRA (Applied Biosystems).
Drug studies.
For the first experiment, R6/2 mice received either vehicle (10% EtOH, 20% Cremaphor EL (Sigma), 70% saline), SR2 antagonist (2.1 mg/kg), GW405833 (20 mg/kg), or SR2 and GW405833 (2.1 mg/kg and 20 mg/kg, respectively), once daily intraperitoneally. Treatment began at ∼4 weeks of age and continued throughout the experiment until mice either died or were killed. The methods for the second experiment were identical, except that dosing was initiated at ∼8 weeks of age. All compounds were dissolved in vehicle. Briefly, ethanol was added and each mixture was incubated for 5 min at 55°C. This was then followed by the addition of Cremaphor EL and another incubation at 55°C for 15–20 min. Saline was added once all compounds were dissolved. Drugs were formulated for 7 d at a time and stored at −20°C until use.
Behavioral analysis.
An accelerating rotarod was used to analyze motor coordination and balance. Baseline behavior was performed at ∼4 weeks, and mice were placed into balanced cohorts based on behavior, sex, and genotype. For R6/2 studies, mice were trained three times at a constant speed of 16 rpm for maximum of 300 s. During testing, mice were subjected to beam acceleration of 4–40 rpm for a maximum of 300 s, three times per session for a total of three sessions. Performance was quantified by measuring the latency to fall off of the rotarod apparatus.
The balance beam was also used to assess motor coordination and balance. This test consisted of three sessions with three trials in each session: one session of training, one session of testing on large diameter beam, one session of testing on medium diameter beam. Motor performance was assessed by measuring the time it takes for the mouse to traverse the beam and the number of hindpaw slips that occur in this process.
Rearing activity was assessed in an automated Flex-Field/Open-Field Photobeam Activity System (San Diego Instruments). Before testing, mice were acclimated in the testing room for 1 h. Mice were then placed in open-field chambers with two 16 × 16 photobeam arrays to detect horizontal and vertical movements. Rearing behavior (the number of times the mouse stood erect on its hindlegs) was measured over 30 min. Chambers were cleaned with 70% ethanol before and after each mouse.
Survival in R6/2 mice was evaluated as the time when the animals either died spontaneously or had lost >15% of their peak body weight. For all studies the experimenter was blind to the genotype and/or treatment group.
Neuropathology.
Mice were anesthetized with avertin (tribromoethanol, 250 mg/kg) and perfused with saline. Hemi-brains were then drop fixed in 4% paraformaldehyde for 48 h and sectioned into 40 μm sections with a Leica Vibratome. Free-floating sections were incubated in either 10% horse (synaptophysin) or goat serum (Iba-1) in 0.1% Triton X-100 for 1 h before incubation with primary antibodies at 4°C for 24 h. To investigate the effects of the CB2 receptor agonist on microglial activation/accumulation, free-floating sections were immunostained with a mouse monoclonal antibody against Iba-1 (1:1000; Dako Cytomation), followed by biotinylated secondary antibody, avidin coupled to horseradish peroxidase, and reacted with diaminobenzidine. The number of microglia per unit area of 0.1 mm2 was assessed using ImageJ. For synaptophysin staining, sections were immunostained with anti-synaptophysin (1:200; Roche Diagnostics) and FITC-conjugated horse anti-mouse IgG (1:500; Vector Laboratories), and fluorescence images were obtained on the Spectral Confocal C1Si at the NIKON imaging center, University of California San Francisco. The percentage of fluorescent immunostaining was evaluated using the ImageJ software. For all neuropathological analysis, at least three sections were analyzed per mouse and for each section at least three fields of view were imaged randomly. For comparison across genotypes we selected sections with similar neuroanatomical features, such as the hippocampus or striatum. For image quantification each image was assessed using similar microscopy settings and thresholding in ImageJ. For all studies the experimenter was blind to the genotype and/or treatment group.
Cytokine analysis.
Blood samples were collected from R6/2 mice at 12–13 weeks and BACHD mice at ∼9–10 months of age. Interleukin-6 (IL-6) plasma levels were assessed with the Mouse IL-6 Quantikine kit (R & D Systems) for R6/2 studies, and with the mouse chemokine 5-plex panel (Invitrogen) and the Luminex SD xMAP machine for BACHD studies.
IL-6 antibody treatment.
R6/2 and WT mice were weaned and genotyped at ∼3 weeks. Baseline behavior was performed at ∼4 weeks, and mice were placed into balanced cohorts based on behavior, sex, and genotype. R6/2 and WT mice were given intraperitoneal injections of a neutralizing antibody against IL-6 (anti-IL-6; rat IgG) (R & D Systems) dissolved in saline. Control mice were given injections of the rat IgG control antibody (R & D Systems), which is the same isotype as the anti-IL6 neutralizing antibody used in this study. Mice were injected with IL-6 neutralizing antibodies or control antibodies (1.5 mg/kg, i.p.) every other day starting at ∼4 weeks and continued until mice were killed at ∼9.5 weeks. Behavioral performance was assessed at ∼5.5 and ∼9.5 weeks of age using the rotarod assay. Mice were weighed biweekly.
Statistics.
All data are expressed as the mean ± SEM. Unless otherwise indicated, a one-way ANOVA with Bonferroni's post hoc test was performed to determine levels of significance between experimental groups at each time point.
Results
Genetic deletion of CB2 receptors accelerate disease onset and exacerbate severity in a slowly progressing HD mouse model
To determine whether loss of CB2 receptors influences disease progression in a slowly progressing HD mouse model, we crossed CB2−/− mice with BACHD mice (Gray et al., 2008), which express full-length htt under the control of its endogenous promoter and regulatory sequences, and generated BACHD; CB2−/− mice and littermate controls. We measured their motor behaviors on the balance beam and rotarod, two measures of motor coordination, and in open-field assays, a measure of locomotor activity. BACHD mice took significantly longer than WT mice to traverse the balance beam at 6 months, but not 3 months of age, consistent with reported deficits of these mice in a rotarod assay (Gray et al., 2008; Menalled et al., 2009).
Remarkably, at 3 months of age, behavioral deficits were clearly detectable in BACHD mice lacking CB2. Indeed, BACHD; CB2−/− mice took significantly longer to traverse the beam than WT or BACHD mice. These deficits were even greater at 6 and 15 months (Fig. 1A). Note that BACHD; CB2−/− mice also had significantly more slips, and these deficits were also detected at 3 months of age, when BACHD mice are phenotypically normal (Fig. 1B). Loss of CB2 alone had no effect on latency or slips at any time, indicating a specific genetic interaction between mutant htt and CB2 that mediates the onset and severity of motor deficits in BACHD mice. BACHD;CB2−/− mice performed worse on the rotarod when compared with WT and BACHD mice, but this difference did not reach statistical significance after accounting for differences in animal weight (data not shown).
Figure 1.

Genetic deletion of CB2 receptors accelerates the onset of motor symptoms and increases their severity in a slowly progressing mouse model of HD. A, B, Deletion of CB2 receptors increases the latency to cross and the number of slips of BACHD mice in a balance beam traversal assay at 3, 6, and 15 months. C, Deletion of CB2 receptors decreases rearing activity in BACHD mice. Spontaneous locomotor activity was quantified in an automated open field and the numbers of rearings were scored for 30 min. Data represent the sum of total rearings in 30 min. A two-way ANOVA (p < 0.05) was performed in conjunction with post hoc t tests (significant p value adjusted for multiple comparisons with Bonferroni correction) to investigate the effects of time and genotype on rearing activity. D, Quantification of synaptophysin levels in the striatum at 15 months of age. One-way ANOVA was performed in conjunction with Dunnett's multiple-comparisons test. Values are means ± SEM; n = 25–30/genotype. *p < 0.05, **p < 0.01, ***p < 0.001.
In the open-field test, BACHD mice lacking CB2 reared 30% less by 6 months and nearly 70% less by 15 months than WT and BACHD mice (Fig. 1C). We could not detect an effect in BACHD or CB2−/− mice alone, indicating that the loss of CB2 in BACHD mice influences the onset and severity of spontaneous motor activity in the open field. These results demonstrate that loss of CB2 receptor signaling accelerates behavioral phenotypes of disease progression in BACHD mice.
We next quantified levels of the presynaptic marker synaptophysin in our experimental cohorts. In agreement with a previous study (Kwan et al., 2012), we observed that synaptophysin levels in 15-month-old BACHD striata were significantly lower than in WT controls (Fig. 1D). Synaptophysin levels were further decreased in BACHD mice lacking CB2; however, this decrease did not reach statistical significance. Similar results were observed in the cortex (data not shown).
CB2 receptor signaling in peripheral immune cells suppresses neurodegeneration in HD mouse models
We next sought to determine in which cells CB2 receptor signaling is important for modulating behavioral phenotypes in BACHD mice. CB2 receptors are expressed at high levels in peripheral immune cells (Munro et al., 1993; Galiègue et al., 1995; Schatz et al., 1997; Griffin et al., 1999) and under healthy conditions are thought to be expressed in the CNS only by specific neurons on the brainstem (Van Sickle et al., 2005). However, in some neurological diseases associated with strong inflammatory responses and immune cell infiltration into the CNS, such as multiple sclerosis, CB2 receptor expression is dramatically increased (Maresz et al., 2005; Yiangou et al., 2006). Using qPCR, we measured the levels of CB2 mRNA transcripts in isolated striatal tissue from BACHD and WT littermates. In the striatum, CB2 transcript levels were near the limit of detection in our assay (data not shown). In contrast, levels of the transcript were ∼200-fold higher in the spleen (Fig. 2A,B). We also quantified CB2 mRNA in R6/2 mice, which express the first exon of human htt driven by the human htt promoter and display a more rapid and severe disease progression than BACHD mice (Mangiarini et al., 1996; Menalled et al., 2009). Similar to BACHD mice, R6/2 mice had very low levels of CB2 in striatal tissue and ∼300-fold higher levels in the spleen (Fig. 2C,D). These studies are consistent with previous studies showing that CB2 mRNA cannot be detected in rodent brains by in situ hybridization, Northern blot analyses, or qPCR, but is readily detected by these methods in immune cells (Munro et al., 1993; Galiègue et al., 1995; Schatz et al., 1997; Griffin et al., 1999). Unfortunately, we could not use antibodies to quantify CB2 protein levels in HD mouse models, since we found that several commercially available CB2 antibodies stained brain sections from CB2−/− mice, indicating a lack of specificity for CB2 (data not shown).
Figure 2.

CB2 mRNA and the CB2 receptor antagonist SR2 are detected at low levels in brain tissue from HD mouse models. A, B, Relative levels of CB2 mRNA in striatum from ∼15- to 18-month-old BACHD mice (n = 13) and WT littermates (n = 13), normalized to WT levels as determined by qPCR. C, D, Relative levels of CB2 mRNA in striatum from ∼10- to 12-week-old R6/2 mice (n = 6) and WT littermates (n = 6), normalized to CB2−/− levels as determined by qPCR. E, Chemical structure and pharmacological properties of GW, a CB2 receptor partial agonist. F, Chemical structure and pharmacological properties of SR2, a CB2 receptor antagonist. G, The CB2 receptor agonist GW accumulates at high levels in plasma and brains from mice dosed with GW alone (20 mg/kg, i.p.; n = 11) or GW coadministered with SR2 (2.1 mg/kg, i.p.; n = 6). Blood and brains were harvested 1 h after intraperitoneal administration, and GW levels were quantified by LC MS/MS. H, The CB2 receptor antagonist SR2 accumulates at high levels in plasma but not in brain tissues isolated from mice dosed with SR2 alone (2.1 mg/kg; n = 5) or SR2 coadministered with GW (20 mg/kg; n = 6). Blood and brains were harvested 1 h after intraperitoneal administration, and SR2 levels were quantified by LC MS/MS. I, Concentration of SR2 detected in brains of mice expressed as a percentage of plasma levels in mice dosed with SR2 alone (2.1 mg/kg; n = 5) or SR2 coadministered with GW (20 mg/kg; n = 6). ns, not significant. Values are means ± SEM.
Based on these results, we hypothesized that the action of CB2 receptor signaling on BACHD progression was likely mediated through peripheral immune cells. If this hypothesis is correct, then treating HD mice with a CB2 receptor-selective agonist should protect against HD phenotypes, and critically, coadministration with a CB2 receptor antagonist that does not cross the blood–brain barrier (BBB) should block this protective effect. For these drug studies, we used R6/2 mice (Mangiarini et al., 1996), a well characterized and widely used genetic model of HD. In this mouse model, the first exon of the htt gene (IT-15) with a large CAG repeat expansion is expressed under the control of the 5′ end of human IT-15. R6/2 mice reliably develop progressive neurological phenotypes, including motor deficits, weight loss, and premature death. These features, along with the rapid progression of symptoms and the relatively short life span of the mice, have contributed to their popularity and utility for preclinical studies.
We treated R6/2 mice with GW405833 (GW) (Fig. 2E), a high-affinity (Ki = 4 nm) and highly selective CB2 receptor partial agonist with a half-life of ∼4 h (Valenzano et al., 2005). To block this effect, we coadministered GW with SR144528 (SR2) (Rinaldi-Carmona et al., 1998) (Fig. 2F), a highly selective CB2 receptor antagonist that displays subnanomolar affinity (Ki = 0.6 nm). A previous study showed that oral administration of SR2 as a single bolus readily displaces ex vivo [3H]-CP 55,940 binding to mouse spleen membranes (ED50 0.35 mg/kg) for up to 20 h, but does not prevent binding to cannabinoid receptors expressed in mouse brain membranes (Rinaldi-Carmona et al., 1998), suggesting that SR2 does not cross the BBB. To confirm that SR2 does not cross the BBB, starting at 4 weeks of age, we treated R6/2 mice daily with GW alone (20 mg/kg/d, i.p.), SR2 alone (2.1 mg/kg/d, i.p.), or GW together with SR2 (20 mg/kg/d, i.p. and 2.1 mg/kg/d, i.p., respectively). At ∼12 weeks of age, plasma and brains samples were isolated from R6/2 mice 1 h after the daily intraperitoneal injections to quantify GW and SR2 levels. When administered alone, we found that GW accumulates at a high concentration in plasma (3.8 ± 0.6 μm) and brain (5.0 ± 0.7 μm) (Fig. 2G). In contrast, although SR2 was also present at a high concentration in plasma when administered alone (1.5 ± 0.2 μm), only very low levels (34.8 ± 6.9 nm, or ∼2% of plasma levels) were found in the brain (Fig. 2H,I). Similar findings were observed when GW was coadministered with SR2. Low concentrations of SR2 in the brain might reflect small amounts of residual blood that remained in brain preparations despite the saline perfusion. These results indicate that, while GW readily crosses the BBB, CNS penetration of SR2 is negligible.
Using the agonist/antagonist strategy described above, we tested the hypothesis that the CB2 receptor acts via the peripheral immune system. Confirming our hypothesis, administration of the CB2 receptor partial agonist GW starting at 4 weeks of age, an early symptomatic stage in these mice (Menalled et al., 2009), ameliorated behavioral and neuropathological deficits in R6/2 mice, and this protective effect was negated when GW was coadministered with the peripherally restricted CB2 receptor antagonist SR2 (Fig. 3). Indeed, after only ∼1.5 weeks of treatment with GW (at ∼5.5 weeks of age), GW significantly improved R6/2 mice performance on both the rotarod and balance beam, indicating that CB2 receptor signaling in peripheral immune cells is important at early disease stages in R6/2 mice. On the rotarod, mice treated with GW performed better during training than vehicle-treated R6/2 mice (data not shown) and stayed on the accelerating rotarod longer during each individual testing session. The protective effect of GW in R6/2 mice in the rotarod assay was observed at all time points tested (5, 9, and 11 weeks) (Fig. 3A). GW-treated mice also took significantly less time to traverse a balance beam and slipped less than controls (Fig. 3B,C). Notably, the protective effect of GW in these assays was not observed in mice treated with GW and SR2. Although genetic deletion of CB2 receptors accelerated the onset and exacerbated the severity of behavioral phenotypes in BACHD mice (Fig. 1), the CB2 antagonist SR2 alone did not worsen phenotypes in R6/2 mice (Fig. 3). This result is not unexpected, since SR2, similar to other receptor antagonists, does not provoke a biological response itself upon binding to CB2 receptors, but only blocks or dampens agonist-mediated responses (Rinaldi-Carmona et al., 1998). Kaplan–Meier survival analysis revealed that R6/2 mice treated with GW lived significantly longer than mice treated with vehicle alone (Fig. 3D). Importantly, the survival curves of R6/2 mice treated with the combination of GW and SR2 were no longer distinguishable from vehicle-treated mice, as were the endpoint survival rates. These results support the two main tenets of our hypothesis: enhanced CB2 receptor signaling is protective in mouse models of HD, and this effect is mediated by CB2 receptor signaling in peripheral immune cells.
Figure 3.

Enhanced CB2 receptor signaling in peripheral immune cells suppresses behavioral deficits and increases survival in R6/2 mice. A, A CB2 receptor agonist (GW; 20 mg/kg/d, i.p. starting at ∼4 weeks of age) improves motor deficits in R6/2 mice on the rotarod, while coadministration of a peripherally restricted CB2 receptor antagonist (SR2; 2.1 mg/kg/d, i.p.) prevents this effect. B, C, GW improves the latency of R6/2 mice to cross and the number of slips in a balance beam traversal assay, while coadministration of SR2 prevents this effect. D, Kaplan–Meier survival analysis shows that GW increases survival in R6/2 mice, while coadministration of SR2 prevents this effect. Vehicle and GW-treated R6/2 mice were subjected to log-rank (Mantel–Cox) test, p = 0.041; all groups compared, p = 0.17; n = 10–11 mice/group at beginning of study. The number of mice in each group when taken down for neuropathology at ∼13-weeks of age: GW-treated mice, n = 11; all other groups; n = 5–7. Values are means ± SEM; n = 10–11/treatment group for time points 5 and 9 weeks and n = 5–11/treatment group at 11 weeks. *p < 0.05; **p < 0.01; ***p < 0.001; ns, not significant.
We next asked whether there were neuropathological correlates of improvement in R6/2 mice treated with GW. R6/2 mice show marked neurodegeneration as measured by loss of the presynaptic marker synaptophysin in the cortex and striatum by 11–15 weeks (Zwilling et al., 2011), analogous to BACHD mice (Kwan et al., 2012) at later times. Consistent with our behavioral results, synaptophysin levels were higher in the striatum (Fig. 4A,B) and cortex (data not shown) of ∼12-week-old R6/2 mice treated with GW than in those treated with vehicle control. As observed in behavioral assays, coadministration of SR2 prevented the protective effect of GW against synapse loss. Therefore, CB2 receptor signaling in peripheral immune cells protects against synapse loss in the brains of R6/2 mice. Previous studies in R6/2 mice and HD brains showed increased Iba-1expression (Simmons et al., 2007; Zwilling et al., 2011), consistent with microglial activation and/or proliferation. R6/2 mice treated with GW had significantly fewer Iba-1-positive cells than vehicle-treated controls (Fig. 4C,D). Surprisingly, this effect was not prevented by coadministration of GW with SR2. As the protective effects of CB2 receptor signaling on behavioral deficits and synapse loss in R6/2 mice are antagonized by SR2, increased Iba-1 expression in the CNS per se probably does not contribute to neurodegeneration, at least under these experimental conditions. However, immunohistochemical levels of Iba-1 are not necessarily indicative of the functional state of microglia, and one cannot rule out the possibility that GW/SR2-treated microglia in R6/2 mice may still secrete higher levels of pro-inflammatory cytokines/chemokines or other mediators of neuronal toxicity. Nevertheless, this result provides further support that the protective effects of enhanced CB2 receptor signaling are mediated primarily through peripheral immune cells and not through changes in Iba-1 levels in parenchymal microglia.
Figure 4.
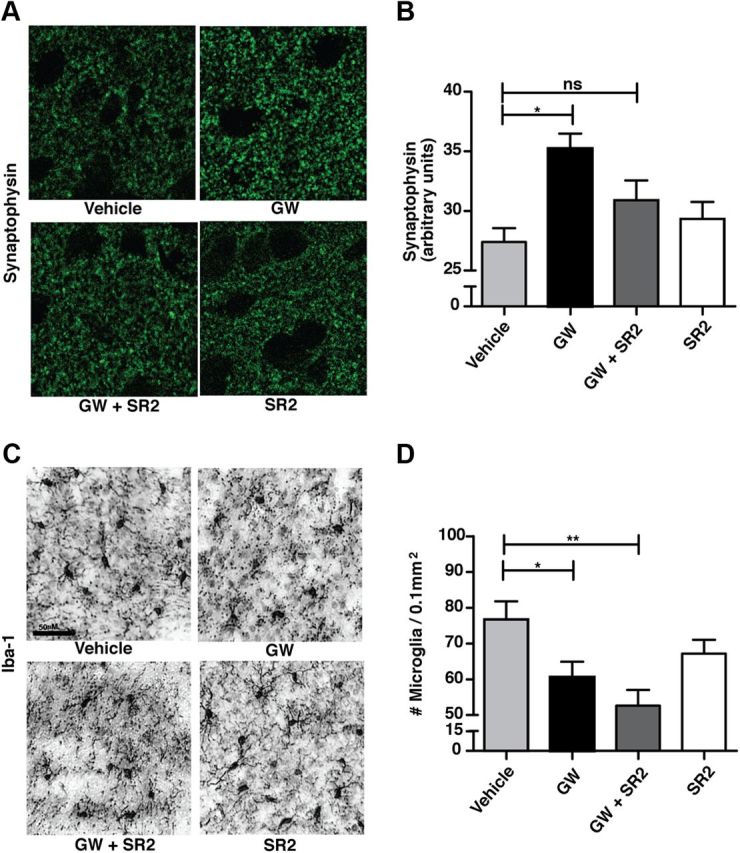
Enhanced CB2 receptor signaling in peripheral immune cells suppresses neurodegeneration and reduces CNS inflammation in R6/2 mice. A, B, A CB2 receptor agonist (GW; 20 mg/kg/d, i.p.) prevents synapse loss in the striatum of ∼12-week-old R6/2 mice, while coadministration of a peripherally restricted CB2 receptor antagonist (SR2; 2.1 mg/kg/d, i.p.) prevents this effect. C, D, GW reduces Iba-1 immunostaining in the striatum of ∼12-week-old R6/2 mice, but SR2 does not prevent this effect. Values are means ± SEM; n = 5–10/treatment group. *p < 0.05; **p < 0.01; ns, not significant.
CB2 receptor signaling in peripheral immune cells is protective even when dosing is initiated at late disease stages in HD mice
Given that treatment with GW prevented behavioral and neuropathological deficits in R6/2 mice when administered starting at an early symptomatic stage (∼4 weeks), we next wanted to determine whether this effect would still be observed if mice were treated at a late symptomatic stage (when the mice are severely impaired in behavioral assays) (Menalled et al., 2009). Here, treatment began at ∼8 weeks of age (20 mg/kg/d, i.p.) and continued daily until mice were ∼13 weeks when neuropathological indices were measured. When measuring the rotarod performance of these cohorts at 9 and 11 weeks of age, we found that GW treatment later in disease progression still confers significant benefits (Fig. 5A). Here also, GW partially prevented the loss of synaptophysin immunoreactivity and decreased Iba-1 levels in R6/2 mice (Fig. 5B–E), indicating that increasing CB2 receptor signaling in peripheral immune cells even at late stages of disease is still beneficial in R6/2 mice.
Figure 5.

CB2 receptor signaling in peripheral immune cells suppresses neurodegeneration and CNS inflammation in late stage R6/2 mice. A, A CB2 receptor agonist (GW; 20 mg/kg/d, i.p. starting at ∼8 weeks of age) improves motor deficits in late-stage R6/2 mice (rotarod). B, C, GW (20 mg/kg/d, i.p. starting at ∼8 weeks of age) prevents synapse loss in the striatum of ∼12-week-old R6/2 mice. D, E, GW (20 mg/kg/d, i.p. starting at ∼8 weeks of age) reduces Iba-1 immunostaining in the striatum of ∼12-week-old R6/2 mice. Values are means ± SEM; n = 12–17/treatment group for A, and 5–10 for B–E. *p < 0.05; ***p < 0.001; ns, not significant.
CB2 receptors control IL-6 levels that contribute to disease phenotypes in HD mice
Our results indicate that the beneficial effects of treatment induced by a CB2 receptor agonist on behavioral deficits were elicited rapidly (within ∼12 d of dosing), suggesting that perhaps soluble factors secreted from peripheral immune cells, such as cytokines and chemokines, might contribute to neurodegeneration. In an effort to identify potential factors downstream from CB2 that might contribute to its effects on neurodegeneration, we next quantified levels of the pro-inflammatory cytokines IL-6 and TNF-α in the blood of HD mice from our studies, since CB2 receptors regulate the production of these cytokines (Klegeris et al., 2003; Rajesh et al., 2008; Su et al., 2012) and that these immune mediators are elevated in HD patients and mouse models (Björkqvist et al., 2008). Genetic deletion of CB2 receptors in BACHD mice led to increased blood levels of IL-6 (Fig. 6A). Conversely, blood IL-6 levels were markedly lower in R6/2 mice treated with GW than in those treated with a vehicle control (Fig. 6B). Importantly, coadministration of SR2 with GW prevented the GW-mediated decrease in IL-6 levels, suggesting that CB2 receptor signaling in peripheral immune cells is the major source of increased IL-6 production in R6/2 mouse blood. Similar results were found with IL-1β (data not shown), which increases in R6/2 mice (Björkqvist et al., 2008). Levels of TNF-α were not significantly increased in R6/2 or BACHD mice and were not changed in R6/2 mice by GW treatment (data not shown).
Figure 6.

CB2 receptors control IL-6 levels, which contribute to disease phenotypes in R6/2 mice. A, IL-6 serum levels are increased in ∼9- to 10-month-old BACHD mice lacking CB2. Data were subjected to one-way ANOVA (p < 0.05) followed by Tukey's post hoc analysis; n = 8–10/genotype. B, A CB2 receptor agonist (GW) decreases IL-6 levels in ∼12-week-old R6/2 mice, and this effect is prevented by coadministration of a CB2 receptor antagonist (SR2). *p < 0.05 (Student's t test, vehicle vs GW); ns, not significant; n = 6–11 mice/cohort. C, Treatment with a neutralizing IL-6 antibody partially rescues motor deficits in R6/2 mice on the rotarod. *p < 0.05 (Student's t test); n = 12–15 mice/group. D, Treatment with a neutralizing IL-6 antibody prevents weight loss at late stages in R6/2 mice. *p < 0.05 (Student's t test); n = 12–15 mice/group. Values are means ± SEM. *p < 0.05.
These findings led us to hypothesize that IL-6 produced by peripheral immune cells might contribute to pathogenesis in R6/2 mice. To test this hypothesis, we administered an IL-6 neutralizing antibody (1.5 mg/kg, i.p.) or a control IgG antibody (1.5 mg/kg, i.p.) every other day for 30 d beginning at ∼4 weeks of age to R6/2 and WT littermate controls. R6/2 mice treated with the IL-6 antibody performed significantly better than IgG-treated mice in a rotarod assay at each time point investigated (Fig. 6C). Moreover, treatment with the IL-6 antibody prevented weight loss at 9 weeks of age in R6/2 mice (Fig. 6D). Treatment with the IL-6 antibody had no effect on rotarod performance or body weight in WT mice.
Discussion
In HD, immune system activation is detected many years before the onset of clinical symptoms, persists chronically, and correlates with clinical severity scores (Björkqvist et al., 2008). Here we show that genetic deletion of CB2 receptors, which are expressed predominantly in peripheral immune cells, increases the severity of behavioral deficits in BACHD mice and dramatically accelerates their onset. Previous work has shown that the age of onset in HD patients with similar CAG repeat lengths varies widely, suggesting that environmental factors may influence disease onset (Wexler et al., 2004). It is therefore tempting to speculate that immune activation may be one important factor that influences disease onset and/or progression. For instance, polymorphisms in CB2 or other immune-related molecules may be associated with variation in the age of onset. Moreover, our results suggest that immune system activation by bacterial and viral infections early during HD pathogenesis might be sufficient to trigger the onset of behavioral symptoms and/or accelerate/exacerbate neurodegeneration, hypotheses we are currently testing in HD mouse models.
Our results confirm a previous study in which genetic deletion of CB2 receptors exacerbated behavioral and neuropathological deficits in R6/2 mice (Palazuelos et al., 2009). However, in this previous study it was argued that CB2 receptor signaling in parenchymal microglia mediated these effects. Our data from the present study clearly show that a peripherally restricted CB2 receptor antagonist (SR2) fully blocks all beneficial effects of a CB2 receptor agonist (GW). Although low nanomolar concentrations of SR2 that were detected in R6/2 brains could conceivably mediate a local neuroprotective effect on microglia if CB2 receptors were indeed expressed at functional levels on these cells, converging lines of evidence argue against this possibility. First, using qPCR we found that CB2 mRNA is present at 300× higher levels in peripheral immune tissues relative to striatal tissue in R6/2 and BACHD mice, questioning the physiological relevance of the low levels of CB2 in the brain. Second, the ED50 of SR2 after oral administration required to displace ex vivo [3H]-CP 55,940 binding to mouse spleen membranes being 0.35 mg/kg is most likely associated with peripheral drug concentrations >100 nm (∼350 nm, assuming a perfect dose-dependent PK linearity) (i.e., far above the brain concentration achieved after SR2 administration to R6/2 mice at 2.1 mg/kg, which is 35 nm in our experiment; Fig. 2H). Third, while treatment with a CB2 receptor agonist (GW) decreased apparent microglial activation in R6/2 brains (as determined by Iba-1 immunostaining), coadministration of GW with SR2 did not block this effect (i.e., although the number of Iba-1+ microglia in CNS was reduced, behavioral and neuropathological deficits still persisted). Together, these data strongly argue that the effect of CB2 receptor activity in HD mouse models is primarily mediated by cells in the peripheral immune system, not on microglia in the CNS.
Previous genetic and pharmacological studies that report beneficial effects of CB2 receptor signaling in animal models of neurodegeneration have interpreted these results as being mediated by parenchymal microglia (Arévalo-Martin et al., 2003; Pryce et al., 2003; Kim et al., 2006; Shoemaker et al., 2007; Zhang et al., 2007; Palazuelos et al., 2009; Murikinati et al., 2010; García et al., 2011; Martín-Moreno et al., 2012). However, data from this study combined with methodological issues in these previous studies question the validity of that interpretation. Like Palazuelos et al. (2009), many of these studies used immunohistochemical techniques to show that levels of the CB2 receptor are increased in disease states in the CNS. These results are not easily interpretable because commercially available antibodies do not selectively recognize CB2 (i.e., all antibodies described to date stain brain sections in CB2−/− mice), and when they were used, the proper controls were not incorporated into their experimental design (e.g., CB2−/− tissue). Unfortunately, the majority of previous studies in which CB2 receptor agonists showed beneficial effects in animal models of neurodegeneration did not include pharmacokinetic measurements to establish the site of action for these compounds, and in general did not include an antagonist to demonstrate specificity for CB2. In the absence of these controls, it is premature to conclude that the protective effects of CB2 agonists derive from CB2 signaling in microglia, since compelling evidence for CB2 receptor expression in these cells is absent. Moreover, CB2 compounds may also bind to CB1 and/or may exert nonspecific effects if inappropriate concentrations are used. Based on these caveats, our results suggest that the interpretation that CB2 signaling in microglia mediates neuroprotection in mouse models of neurodegeneration needs to be re-evaluated. Importantly, CB2 agonists restricted to the periphery would avoid potential side effects due to CB1 agonism in the CNS.
The molecular mechanisms by which CB2 receptor signaling in peripheral immune cells modulates neurodegeneration are likely to be complex and will need to be further explored. Several laboratories reported that CB2 receptor agonists decrease the production of pro-inflammatory cytokines (Klegeris et al., 2003; Rajesh et al., 2008; Su et al., 2012), including those that are elevated in HD (Björkqvist et al., 2008). Consistent with these studies, we found that CB2 receptors regulate IL-6 levels in HD mouse models, and that a neutralizing antibody to IL-6 partially ameliorates behavioral deficits in R6/2 mice. IL-6 crosses the BBB (Banks et al., 1994) and its functional signaling is important for brain development, learning and memory, and CNS responses to disease and injury (Bauer et al., 2007; Jankord et al., 2010). Increased CNS levels of IL-6 have been linked to impairments in memory performance and sickness behavior during inflammation (Bluthé et al., 2000; Sparkman et al., 2006), and transgenic mice bearing additional copies of the IL-6 gene under the control of a brain-specific promoter develop a marked cortical pathology, including severe alterations of the dendritic arborization of cortical neurons (Campbell et al., 1993). While these studies indicate that increased IL-6 levels are sufficient to promote behavioral changes and neurodegeneration in the CNS, the detailed molecular mechanisms that mediate these events remain to be elucidated. For instance, IL-6 produced in the peripheral immune system may be crossing the BBB and exacerbating neurodegeneration through direct effects on neurons or indirect effects on microglia. However, given that treatment with an IL-6 antibody only showed a partial rescue of motor deficits in R6/2 mice, these results suggest that the beneficial effects elicited by CB2 receptor signaling in HD mice are likely to be mediated by signals in addition to IL-6.
More broadly, our data are consistent with other recent studies suggesting that soluble signals derived from peripheral immune cells transduce signals into the CNS that contribute to cognitive dysfunction and/or neurodegeneration. For example, plasma from old mice, that has elevated levels of chemokines, impairs learning and memory in young mice (Villeda et al., 2011). Blocking peripheral immune molecules (e.g., CD40 and TGF-β-Smad2/3) attenuates pathology in a mouse model of Alzheimer's disease (Tan et al., 2002; Town et al., 2008). Given that CB2 receptor signaling also regulates immune cell migration (Miller and Stella, 2008), which is impaired in HD (Kwan et al., 2012), the protective effects of CB2 receptor agonists might be mediated by effects on immune cell migration coincident with dampening of pro-inflammatory cytokine production.
The development of peripherally restricted CB2 receptor agonists holds promise for treating HD and other neurodegenerative diseases. The beneficial effects of enhancing CB2 receptor signaling in HD mice occur rapidly after initial dosing, and this therapeutic effect is still present in severely impaired mice. Elucidating the molecular mechanisms downstream from CB2 receptors that mediate neuroprotection may have important implications for HD research. For example, transgenic expression of IL-6 causes muscle degeneration, which is prevented with an IL-6 antibody (Tsujinaka et al., 1996), and IL-6 might contribute to muscle degeneration and cachexia in HD. Given that an IL-6 antibody is an effective and well tolerated treatment for arthritis (Maini et al., 2006), additional studies to investigate the therapeutic benefits of modulating levels of cytokines, such as IL-6, in HD are clearly warranted.
In summary, our results suggest CB2 receptor signaling in peripheral immune cells has an important role in HD and other neurodegeneration disorders. Further elucidation of the molecular mechanisms that underlie these effects may lead to novel therapeutic strategies to treat these disorders.
Footnotes
This study was supported by the J. David Gladstone Institutes (P.J.M.), the CHDI foundation (P.J.M. and N.S.), the Taube-Koret Center for Huntington's Disease Research (P.J.M.), and Huntington's Disease Society of America (D.D.). The J. David Gladstone Institutes received support from National Center for Research Resources Grant RR18928-01. Behavioral data were obtained with the help of the Gladstone Institutes' Behavioral Core (supported by National Institutes of Health Grant P30NS065780). We thank G. Howard for editorial assistance, the Nikon Imaging Center at University of California San Francisco for image acquisition, and Eliezer Masliah and Anthony Adame for synaptophysin quantification training.
The authors declare declare no competing financial interests.
References
- Arévalo-Martin A, Vela JM, Molina-Holgado E, Borrell J, Guaza C. Therapeutic action of cannabinoids in a murine model of multiple sclerosis. J Neurosci. 2003;23:2511–2516. doi: 10.1523/JNEUROSCI.23-07-02511.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashton JC, Glass M. The cannabinoid CB2 receptor as a target for inflammation-dependent neurodegeneration. Curr Neuropharmacol. 2007;5:73–80. doi: 10.2174/157015907780866884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks WA, Kastin AJ, Gutierrez EG. Penetration of interleukin-6 across the murine blood-brain barrier. Neurosci Lett. 1994;179:53–56. doi: 10.1016/0304-3940(94)90933-4. [DOI] [PubMed] [Google Scholar]
- Bauer S, Kerr BJ, Patterson PH. The neuropoietic cytokine family in development, plasticity, disease and injury. Nat Rev Neurosci. 2007;8:221–232. doi: 10.1038/nrn2054. [DOI] [PubMed] [Google Scholar]
- Björkqvist M, Wild EJ, Thiele J, Silvestroni A, Andre R, Lahiri N, Raibon E, Lee RV, Benn CL, Soulet D, Magnusson A, Woodman B, Landles C, Pouladi MA, Hayden MR, Khalili-Shirazi A, Lowdell MW, Brundin P, Bates GP, Leavitt BR, et al. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington's disease. J Exp Med. 2008;205:1869–1877. doi: 10.1084/jem.20080178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluthé RM, Michaud B, Poli V, Dantzer R. Role of IL-6 in cytokine-induced sickness behavior: a study with IL-6 deficient mice. Physiol Behav. 2000;70:367–373. doi: 10.1016/s0031-9384(00)00269-9. [DOI] [PubMed] [Google Scholar]
- Buckley NE, McCoy KL, Mezey E, Bonner T, Zimmer A, Felder CC, Glass M, Zimmer A. Immunomodulation by cannabinoids is absent in mice deficient for the cannabinoid CB(2) receptor. Eur J Pharmacol. 2000;396:141–149. doi: 10.1016/s0014-2999(00)00211-9. [DOI] [PubMed] [Google Scholar]
- Campbell IL, Abraham CR, Masliah E, Kemper P, Inglis JD, Oldstone MB, Mucke L. Neurologic disease induced in transgenic mice by cerebral overexpression of interleukin 6. Proc Natl Acad Sci U S A. 1993;90:10061–10065. doi: 10.1073/pnas.90.21.10061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalrymple A, Wild EJ, Joubert R, Sathasivam K, Björkqvist M, Petersén A, Jackson GS, Isaacs JD, Kristiansen M, Bates GP, Leavitt BR, Keir G, Ward M, Tabrizi SJ. Proteomic profiling of plasma in Huntington's disease reveals neuroinflammatory activation and biomarker candidates. J Proteome Res. 2007;6:2833–2840. doi: 10.1021/pr0700753. [DOI] [PubMed] [Google Scholar]
- Galiègue S, Mary S, Marchand J, Dussossoy D, Carrière D, Carayon P, Bouaboula M, Shire D, Le Fur G, Casellas P. Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte subpopulations. Eur J Biochem. 1995;232:54–61. doi: 10.1111/j.1432-1033.1995.tb20780.x. [DOI] [PubMed] [Google Scholar]
- García C, Palomo-Garo C, García-Arencibia M, Ramos J, Pertwee R, Fernández-Ruiz J. Symptom-relieving and neuroprotective effects of the phytocannabinoid Delta-THCV in animal models of Parkinson's disease. Br J Pharmacol. 2011;163:1495–1506. doi: 10.1111/j.1476-5381.2011.01278.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray M, Shirasaki DI, Cepeda C, Andr é VM, Wilburn B, Lu XH, Tao J, Yamazaki I, Li SH, Sun YE, Li XJ, Levine MS, Yang XW. Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J Neurosci. 2008;28:6182–6195. doi: 10.1523/JNEUROSCI.0857-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin G, Wray EJ, Tao Q, McAllister SD, Rorrer WK, Aung MM, Martin BR, Abood ME. Evaluation of the cannabinoid CB2 receptor-selective antagonist, SR144528: further evidence for cannabinoid CB2 receptor absence in the rat central nervous system. Eur J Pharmacol. 1999;377:117–125. doi: 10.1016/s0014-2999(99)00402-1. [DOI] [PubMed] [Google Scholar]
- Gu X, Li C, Wei W, Lo V, Gong S, Li SH, Iwasato T, Itohara S, Li XJ, Mody I, Heintz N, Yang XW. Pathological cell-cell interactions elicited by a neuropathogenic form of mutant Huntingtin contribute to cortical pathogenesis in HD mice. Neuron. 2005;46:433–444. doi: 10.1016/j.neuron.2005.03.025. [DOI] [PubMed] [Google Scholar]
- Gu X, André VM, Cepeda C, Li SH, Li XJ, Levine MS, Yang XW. Pathological cell-cell interactions are necessary for striatal pathogenesis in a conditional mouse model of Huntington's disease. Mol Neurodegener. 2007;2:8. doi: 10.1186/1750-1326-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockly E, Richon VM, Woodman B, Smith DL, Zhou X, Rosa E, Sathasivam K, Ghazi-Noori S, Mahal A, Lowden PA, Steffan JS, Marsh JL, Thompson LM, Lewis CM, Marks PA, Bates GP. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington's disease. Proc Natl Acad Sci U S A. 2003;100:2041–2046. doi: 10.1073/pnas.0437870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogeveen AT, Willemsen R, Meyer N, de Rooij KE, Roos RA, van Ommen GJ, Galjaard H. Characterization and localization of the Huntington disease gene product. Hum Mol Genet. 1993;2:2069–2073. doi: 10.1093/hmg/2.12.2069. [DOI] [PubMed] [Google Scholar]
- Jankord R, Zhang R, Flak JN, Solomon MB, Albertz J, Herman JP. Stress activation of IL-6 neurons in the hypothalamus. Am J Physiol Regul Integr Comp Physiol. 2010;299:R343–351. doi: 10.1152/ajpregu.00131.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Moore DH, Makriyannis A, Abood ME. AM1241, a cannabinoid CB2 receptor selective compound, delays disease progression in a mouse model of amyotrophic lateral sclerosis. Eur J Pharmacol. 2006;542:100–105. doi: 10.1016/j.ejphar.2006.05.025. [DOI] [PubMed] [Google Scholar]
- Klegeris A, Bissonnette CJ, McGeer PL. Reduction of human monocytic cell neurotoxicity and cytokine secretion by ligands of the cannabinoid-type CB2 receptor. Br J Pharmacol. 2003;139:775–786. doi: 10.1038/sj.bjp.0705304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan W, Magnusson A, Chou A, Adame A, Carson MJ, Kohsaka S, Masliah E, Möller T, Ransohoff R, Tabrizi SJ, Björkqvist M, Muchowski PJ. Bone marrow transplantation confers modest benefits in mouse models of Huntington's disease. J Neurosci. 2012;32:133–142. doi: 10.1523/JNEUROSCI.4846-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leblhuber F, Walli J, Jellinger K, Tilz GP, Widner B, Laccone F, Fuchs D. Activated immune system in patients with Huntington's disease. Clin Chem Lab Med. 1998;36:747–750. doi: 10.1515/CCLM.1998.132. [DOI] [PubMed] [Google Scholar]
- Li SH, Schilling G, Young WS, 3rd, Li XJ, Margolis RL, Stine OC, Wagster MV, Abbott MH, Franz ML, Ranen NG, Folstein SE, Hedreen JC, Ross CA. Huntington's disease gene (IT15) is widely expressed in human and rat tissues. Neuron. 1993;11:985–993. doi: 10.1016/0896-6273(93)90127-d. [DOI] [PubMed] [Google Scholar]
- Li YY, Li YN, Ni JB, Chen CJ, Lv S, Chai SY, Wu RH, Yüce B, Storr M. Involvement of cannabinoid-1 and cannabinoid-2 receptors in septic ileus. Neurogastroenterol Motil. 2010;22:350–e388. doi: 10.1111/j.1365-2982.2009.01419.x. [DOI] [PubMed] [Google Scholar]
- Maini RN, Taylor PC, Szechinski J, Pavelka K, Bröll J, Balint G, Emery P, Raemen F, Petersen J, Smolen J, Thomson D, Kishimoto T. Double-blind randomized controlled clinical trial of the interleukin-6 receptor antagonist, tocilizumab, in European patients with rheumatoid arthritis who had an incomplete response to methotrexate. Arthritis Rheum. 2006;54:2817–2829. doi: 10.1002/art.22033. [DOI] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- Maresz K, Carrier EJ, Ponomarev ED, Hillard CJ, Dittel BN. Modulation of the cannabinoid CB2 receptor in microglial cells in response to inflammatory stimuli. J Neurochem. 2005;95:437–445. doi: 10.1111/j.1471-4159.2005.03380.x. [DOI] [PubMed] [Google Scholar]
- Martín-Moreno AM, Brera B, Spuch C, Carro E, García-García L, Delgado M, Pozo MA, Innamorato NG, Cuadrado A, de Ceballos ML. Prolonged oral cannabinoid administration prevents neuroinflammation, lowers beta-amyloid levels and improves cognitive performance in Tg APP 2576 mice. J Neuroinflammation. 2012;9:8. doi: 10.1186/1742-2094-9-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menalled L, El-Khodor BF, Patry M, Suárez-Fariñas M, Orenstein SJ, Zahasky B, Leahy C, Wheeler V, Yang XW, MacDonald M, Morton AJ, Bates G, Leeds J, Park L, Howland D, Signer E, Tobin A, Brunner D. Systematic behavioral evaluation of Huntington's disease transgenic and knock-in mouse models. Neurobiol Dis. 2009;35:319–336. doi: 10.1016/j.nbd.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AM, Stella N. CB2 receptor-mediated migration of immune cells: it can go either way. Br J Pharmacol. 2008;153:299–308. doi: 10.1038/sj.bjp.0707523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscovitch-Lopatin M, Weiss A, Rosas HD, Ritch J, Doros G, Kegel KB, Difiglia M, Kuhn R, Bilbe G, Paganetti P, Hersch S. Optimization of an HTRF assay for the detection of soluble mutant Huntingtin in human buffy coats: a potential biomarker in blood for Huntington disease. PLoS Curr. 2010;2:RRN1205. doi: 10.1371/currents.RRN1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- Murikinati S, Jüttler E, Keinert T, Ridder DA, Muhammad S, Waibler Z, Ledent C, Zimmer A, Kalinke U, Schwaninger M. Activation of cannabinoid 2 receptors protects against cerebral ischemia by inhibiting neutrophil recruitment. FASEB J. 2010;24:788–798. doi: 10.1096/fj.09-141275. [DOI] [PubMed] [Google Scholar]
- Palazuelos J, Aguado T, Pazos MR, Julien B, Carrasco C, Resel E, Sagredo O, Benito C, Romero J, Azcoitia I, Fernández-Ruiz J, Guzmán M, Galve-Roperh I. Microglial CB2 cannabinoid receptors are neuroprotective in Huntington's disease excitotoxicity. Brain. 2009;132:3152–3164. doi: 10.1093/brain/awp239. [DOI] [PubMed] [Google Scholar]
- Pryce G, Ahmed Z, Hankey DJ, Jackson SJ, Croxford JL, Pocock JM, Ledent C, Petzold A, Thompson AJ, Giovannoni G, Cuzner ML, Baker D. Cannabinoids inhibit neurodegeneration in models of multiple sclerosis. Brain. 2003;126:2191–2202. doi: 10.1093/brain/awg224. [DOI] [PubMed] [Google Scholar]
- Rajesh M, Mukhopadhyay P, Hask ó G, Huffman JW, Mackie K, Pacher P. CB2 cannabinoid receptor agonists attenuate TNF-alpha-induced human vascular smooth muscle cell proliferation and migration. Br J Pharmacol. 2008;153:347–357. doi: 10.1038/sj.bjp.0707569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaldi-Carmona M, Barth F, Millan J, Derocq JM, Casellas P, Congy C, Oustric D, Sarran M, Bouaboula M, Calandra B, Portier M, Shire D, Brelière JC, Le Fur GL. SR 144528, the first potent and selective antagonist of the CB2 cannabinoid receptor. J Pharmacol Exp Ther. 1998;284:644–650. [PubMed] [Google Scholar]
- Schatz AR, Lee M, Condie RB, Pulaski JT, Kaminski NE. Cannabinoid receptors CB1 and CB2: a characterization of expression and adenylate cyclase modulation within the immune system. Toxicol Appl Pharmacol. 1997;142:278–287. doi: 10.1006/taap.1996.8034. [DOI] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- Shoemaker JL, Seely KA, Reed RL, Crow JP, Prather PL. The CB2 cannabinoid agonist AM-1241 prolongs survival in a transgenic mouse model of amyotrophic lateral sclerosis when initiated at symptom onset. J Neurochem. 2007;101:87–98. doi: 10.1111/j.1471-4159.2006.04346.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons DA, Casale M, Alcon B, Pham N, Narayan N, Lynch G. Ferritin accumulation in dystrophic microglia is an early event in the development of Huntington's disease. Glia. 2007;55:1074–1084. doi: 10.1002/glia.20526. [DOI] [PubMed] [Google Scholar]
- Singh UP, Singh NP, Singh B, Price RL, Nagarkatti M, Nagarkatti PS. Cannabinoid receptor-2 (CB2) agonist ameliorates colitis in IL-10(−/−) mice by attenuating the activation of T cells and promoting their apoptosis. Toxicol Appl Pharmacol. 2012;258:256–267. doi: 10.1016/j.taap.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparkman NL, Buchanan JB, Heyen JR, Chen J, Beverly JL, Johnson RW. Interleukin-6 facilitates lipopolysaccharide-induced disruption in working memory and expression of other proinflammatory cytokines in hippocampal neuronal cell layers. J Neurosci. 2006;26:10709–10716. doi: 10.1523/JNEUROSCI.3376-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffens S, Veillard NR, Arnaud C, Pelli G, Burger F, Staub C, Karsak M, Zimmer A, Frossard JL, Mach F. Low dose oral cannabinoid therapy reduces progression of atherosclerosis in mice. Nature. 2005;434:782–786. doi: 10.1038/nature03389. [DOI] [PubMed] [Google Scholar]
- Su TF, Zhao YQ, Zhang LH, Peng M, Wu CH, Pei L, Tian B, Zhang J, Shi J, Pan HL, Li M. Electroacupuncture reduces the expression of proinflammatory cytokines in inflamed skin tissues through activation of cannabinoid CB2 receptors. Eur J Pain. 2012;16:624–635. doi: 10.1002/j.1532-2149.2011.00055.x. [DOI] [PubMed] [Google Scholar]
- Tan J, Town T, Crawford F, Mori T, DelleDonne A, Crescentini R, Obregon D, Flavell RA, Mullan MJ. Role of CD40 ligand in amyloidosis in transgenic Alzheimer's mice. Nat Neurosci. 2002;5:1288–1293. doi: 10.1038/nn968. [DOI] [PubMed] [Google Scholar]
- The Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Town T, Laouar Y, Pittenger C, Mori T, Szekely CA, Tan J, Duman RS, Flavell RA. Blocking TGF-beta-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat Med. 2008;14:681–687. doi: 10.1038/nm1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschöp J, Kasten KR, Nogueiras R, Goetzman HS, Cave CM, England LG, Dattilo J, Lentsch AB, Tschöp MH, Caldwell CC. The cannabinoid receptor 2 is critical for the host response to sepsis. J Immunol. 2009;183:499–505. doi: 10.4049/jimmunol.0900203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujinaka T, Fujita J, Ebisui C, Yano M, Kominami E, Suzuki K, Tanaka K, Katsume A, Ohsugi Y, Shiozaki H, Monden M. Interleukin 6 receptor antibody inhibits muscle atrophy and modulates proteolytic systems in interleukin 6 transgenic mice. J Clin Invest. 1996;97:244–249. doi: 10.1172/JCI118398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzano KJ, Tafesse L, Lee G, Harrison JE, Boulet JM, Gottshall SL, Mark L, Pearson MS, Miller W, Shan S, Rabadi L, Rotshteyn Y, Chaffer SM, Turchin PI, Elsemore DA, Toth M, Koetzner L, Whiteside GT. Pharmacological and pharmacokinetic characterization of the cannabinoid receptor 2 agonist, GW405833, utilizing rodent models of acute and chronic pain, anxiety, ataxia and catalepsy. Neuropharmacology. 2005;48:658–672. doi: 10.1016/j.neuropharm.2004.12.008. [DOI] [PubMed] [Google Scholar]
- van der Burg JM, Björkqvist M, Brundin P. Beyond the brain: widespread pathology in Huntington's disease. Lancet Neurol. 2009;8:765–774. doi: 10.1016/S1474-4422(09)70178-4. [DOI] [PubMed] [Google Scholar]
- Van Sickle MD, Duncan M, Kingsley PJ, Mouihate A, Urbani P, Mackie K, Stella N, Makriyannis A, Piomelli D, Davison JS, Marnett LJ, Di Marzo V, Pittman QJ, Patel KD, Sharkey KA. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science. 2005;310:329–332. doi: 10.1126/science.1115740. [DOI] [PubMed] [Google Scholar]
- Villeda SA, Luo J, Mosher KI, Zou B, Britschgi M, Bieri G, Stan TM, Fainberg N, Ding Z, Eggel A, Lucin KM, Czirr E, Park JS, Couillard-Després S, Aigner L, Li G, Peskind ER, Kaye JA, Quinn JF, Galasko DR, et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature. 2011;477:90–94. doi: 10.1038/nature10357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wexler NS, Lorimer J, Porter J, Gomez F, Moskowitz C, Shackell E, Marder K, Penchaszadeh G, Roberts SA, Gayan J, Brocklebank D, Cherny SS, Cardon LR, Gray J, Dlouhy SR, Wiktorski S, Hodes ME, Conneally PM, Penney JB, Gusella J, et al. Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington's disease age of onset. Proc Natl Acad Sci U S A. 2004;101:3498–3503. doi: 10.1073/pnas.0308679101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild E, Magnusson A, Lahiri N, Krus U, Orth M, Tabrizi SJ, Björkqvist M. Abnormal peripheral chemokine profile in Huntington's disease. PLoS Curr. 2011;3:RRN1231. doi: 10.1371/currents.RRN1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yiangou Y, Facer P, Durrenberger P, Chessell IP, Naylor A, Bountra C, Banati RR, Anand P. COX-2, CB2 and P2X7-immunoreactivities are increased in activated microglial cells/macrophages of multiple sclerosis and amyotrophic lateral sclerosis spinal cord. BMC Neurol. 2006;6:12. doi: 10.1186/1471-2377-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Martin BR, Adler MW, Razdan RK, Jallo JI, Tuma RF. Cannabinoid CB(2) receptor activation decreases cerebral infarction in a mouse focal ischemia/reperfusion model. J Cereb Blood Flow Metab. 2007;27:1387–1396. doi: 10.1038/sj.jcbfm.9600447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwilling D, Huang SY, Sathyasaikumar KV, Notarangelo FM, Guidetti P, Wu HQ, Lee J, Truong J, Andrews-Zwilling Y, Hsieh EW, Louie JY, Wu T, Scearce-Levie K, Patrick C, Adame A, Giorgini F, Moussaoui S, Laue G, Rassoulpour A, Flik G, et al. Kynurenine 3-monooxygenase inhibition in blood ameliorates neurodegeneration. Cell. 2011;145:863–874. doi: 10.1016/j.cell.2011.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]