

Abstract
HSP70 is a member of the family of heat-shock proteins that are known to be up-regulated in neurons following injury and/ or stress. HSP70 over-expression has been linked to neuroprotection in multiple models, including neurodegenerative disorders. In contrast, less is known about the neuroprotective effects of HSP70 in neuronal apoptosis and with regard to modulation of programmed cell death (PCD) mechanisms in neurons. We examined the effects of HSP70 over-expression by transfection with HSP70-expression plasmids in primary cortical neurons and the SH-SY5Y neuronal cell line using four independent models of apoptosis: etoposide, staurosporine, C2-ceramide, and β-Amyloid. In these apoptotic models, neurons transfected with the HSP70 construct showed significantly reduced induction of nuclear apoptotic markers and/or cell death. Furthermore, we demonstrated that HSP70 binds and potentially inactivates Apoptotic protease-activating factor 1, as well as apoptosis-inducing factor, key molecules involved in development of caspase-dependent and caspase-independent PCD, respectively. Markers of caspase-dependent PCD, including active caspase-3, caspase-9, and cleaved PARP were attenuated in neurons over-expressing HSP70. These data indicate that HSP70 protects against neuronal apoptosis and suggest that these effects reflect, at least in part, to inhibition of both caspase-dependent and caspase-independent PCD pathways.
Keywords: AIF, Apaf-1, apoptosis, HSP70, neurons, neuroprotection
The 70-kDa heat-shock proteins (HSP70s) are stress-induced molecules that are expressed in response to various types of central nervous system injuries including stroke, trauma, or neurodegenerative disorders and appear to have neuroprotective actions (Turturici et al. 2011; Gribaldo et al. 1999). HSP70 plays an important role in numerous processes including folding, assembly, and stabilization of newly synthesized proteins, refolding of misfolded proteins, degradation of abnormal proteins, and control of the activity of regulatory proteins (Bukau et al. 2000; Hartl and Hayer-Hartl 2002; Young et al. 2003; Neupert and Brunner 2002; Ryan and Pfanner 2001; Pratt and Toft 2003). Recent findings have suggested that the neuroprotective effects of HSP70 in neurodegenerative diseases such as Parkinson’s disease may be explained not only by its role as a chaperone that attenuates protein aggregation and toxicity, but also through more direct anti-apoptotic effects (Turturici et al. 2011).
Studies using in vitro cell models have shown that heat-shock proteins (HSPs) are synthesized in response to stress and that cells with increased levels of HSPs, either as result of previous stress (pre-conditioning) or after artificial over-expression, display increased resistance to subsequent injury (Li 1983; Mailhos et al. 1993). It has been suggested that HPS70 may protect cells against various kinds of injury/ stress (Li et al. 1992; Mosser et al. 1997; Bellmann et al. 1996) through mechanisms that include stabilization of partially denatured proteins as well as through removal of irreversibly damaged proteins before they can aggregate and disrupt normal cell functions (Kelly et al. 2001). Neurons may also respond to various stressors by inducing the expression of multiple heat-shock proteins, which have been shown to attenuate neuronal death induced by neurotoxic agents such as glutamate (Lowenstein et al. 1991; Rordorf et al. 1991). Mailhos et al. reported that prior heat shock can attenuate neuronal apoptosis (Mailhos et al. 1993). Although these studies have indicated a correlation between the degree of HSPs’ induction and survival for both toxic and apoptotic neuronal programmed cell death (PCD), they did not demonstrate that the neuroprotective effects were dependent on the increased HSPs expression (Amin et al. 1995; Mailhos et al. 1993). Mailhos et al. provided the first direct confirmation of the neuroprotective effect of HSPs when they showed that HSP70 over-expression attenuates thermal stress-induced neuronal death (Mailhos et al. 1994). However, increased levels of HSP70 were unable to protect against stimuli that induced neuronal apoptosis (Mailhos et al. 1994). This difference between the ability of HSP70 to protect neurons against thermal or ischemic stress, and the lack of protection against apoptotic stimuli, was subsequently confirmed (Wyatt et al. 1996; Wagstaff et al. 1999; Zourlidou et al. 2004). Nonetheless, in selected models such as an in vitro model of amyotrophic lateral sclerosis, HSP70 over-expression did attenuate neuronal apoptosis (Patel et al. 2005).
To better clarify the ability of HSP70 to modulate neuronal PCD and delineate the mechanisms involved, we examined a number of well-established inducers of apoptosis using a model of HSP70 over-expression in primary cortical neurons. To ensure that our findings are not restricted to any particular apoptotic model, we produced cell death by four distinct inducers of neuronal apoptosis including etoposide (topo-isomerase II inhibitor) (Nakajima et al. 1994), staurosporine (non-selective protein kinase inhibitor) (Koh et al. 1995), Aβ (25–35) (an in vitro paradigm of β-Amyloid cytotoxicity) (Harada and Sugimoto 1999), and C2-ceramide (an in vitro paradigm of ceramide cytotoxicity) (Movsesyan et al. 2002). Previous studies have shown that etoposide, as well as staurosporine, Aβ (25–35) (Movsesyan et al. 2004), and C2-ceramide (Stoica et al. 2005) activate both caspase-dependent and caspase-independent (AIF-mediated) pathways of neuronal apoptosis. Studies using non-neuronal cells have also demonstrated the ability of HSP70 to independently interact and block Apoptotic protease-activating factor 1 (Apaf-1) (Beere et al. 2000; Saleh et al. 2000) and apoptosis-inducing factor (AIF) (Ravagnan et al. 2001), key components of the caspase-dependent and -independent pathways, respectively. Thus, our work has focused not only on examining the protective effects of HSP70 in neuronal PCD but also to confirm its ability to interfere with these apoptotic pathways in neurons. Our data demonstrate that HSP70 over-expression, achieved by transfecting neurons with HSP70-expression plasmids, significantly protects against etoposide, C2-ceramide, staurosporine, and Aβ (25–35)-induced neuronal apoptosis. Furthermore, our results suggest that HSP70 inhibits apoptosis by at least two mechanisms: (i) attenuation of caspase-independent pathways by interacting with AIF and preventing its translocation to the nucleus, and (ii) inhibition of caspase-dependent pathways by interacting with Apaf-1 and blocking caspase activation.
Methods
Cell cultures
Rat cortical neurons (RCN) were derived from rat embryonic cortices. Cells were seeded onto poly-d-lysine-coated 96-well or 24-well plates or 100-mm Petri dishes (cell density 1 × 106/cm2) and maintained in serum-free conditions using the B27 supplement as described (Yakovlev et al. 2001). Cultures were maintained in Neurobasal medium supplemented with 25 μM Na-glutamate, 0.5 mM L-glutamine, and 2% B27 supplement. Transfection of RCN was performed at 5 days in vitro (DIV). RCN were transfected by using the ProFection® Mammalian Transfection System (Pro-mega, Madison, WI, USA) according the manufacturer’s protocol. The PI assay required analysis of live cells, and to detect transfected cells (Fig. 2) neurons were cotransfected with 2% green fluorescent protein (GFP) and 98% V5-HSP70 plasmids (Hetman et al. 2000). Immunostaining showed that all GFP-cotransfected cells were transfected with V5-HSP70 plasmid (data not shown). Apoptosis inductions were done at 6 DIV by applying Etoposide (Etop) at a final concentration 50 μM, Staurosporine (Staur) 0.5 μM, Aβ (25–35) (Aβ) 50 μM, or C2-ceramide (C2cer) 50 μM. Cell harvesting or immunostaining was performed at 7 DIV.
Fig. 2.
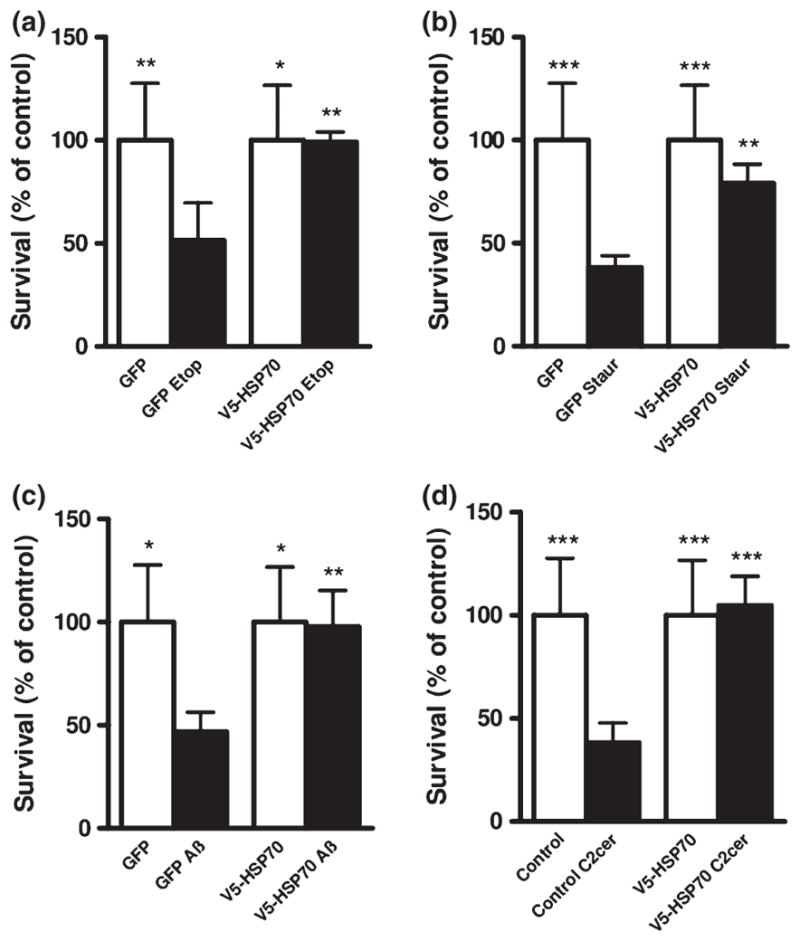
HSP70 over-expression improves neuronal survival. Rat cortical neurons (RCN) cells were transfected with GFP alone or V5-HSP70/GFP and treated for 24 h with (a) etoposide 50 μM (Etop); (b) Staurosporine 0.5 μM (Staur); (c) Aβ (25–35) 50 μM (Aβ); (d) C2-ceramide 50 μM (C2cer). Cell survival of transfected neurons (detected by GFP fluorescence) was quantified by propidium iodide uptake as previously described. The per cent surviving neurons (Propidium iodide negative) was normalized to untreated (control) GFP-transfected RCN. Data represent the mean ± S.D; n = 3 samples. *p < 0.05, **p < 0.01, ***p < 0.001 versus GFP transfected-RCN treated with apoptotic inducers. Analysis by Kruskal–Wallis one-way ANOVA on ranks, all pairwise multiple comparison procedures, followed by post hoc adjustment using Student–Newman–Keul’s Method.
The human neuroblastoma SH-SY5Y cells were seeded in 96-well or 24-well plates or 100-mm Petri dishes and maintained in Dulbecco’s modified Eagle’s medium (Life Technologies, Grand Island, NY, USA) supplemented with 10% fetal calf serum, 100 U/mL penicillin, and 100 U/mL streptomycin in a humid atmosphere of 5% CO2 and 95% air at 37°C. Cells at 70% confluence were transfected with plasmids. Transfection of cells was performed by using Lipofectamine 2000 Reagent (Invitrogen, Carlsbad, CA, USA) according to manufacturer’s protocol. Apoptosis inductions were done 24 h after transfection as described above. Cell viability was measured by retention and de-esterification of calcein AM (Invitrogen). Briefly, culture medium in 96-well plates was replaced with 5 μM calcein AM in Locke’s buffer containing 154 mM NaCl, 5.6 mM glucose, and 5 mM HEPES (pH 7.4). After incubation at 37°C for 30 min, fluorescence was measured using a Biotek Synergy HT multidetection microplate reader at 485 nm excitation and 560 nm emission wavelengths (BioTek, Winooski, VT, USA).
Plasmids
Plasmids encoding human HSP70 (Hspa1a) – pcDNA5/FRT/ TO HSPA1A, HSP70 with V5 tag on N-terminus (V5-HSP70) – pcDNA5/FRT/TO V5 HSPA1A, and HSP70 with His tag on N-terminus (His-HSP70) – pcDNA5/FRT/TO His HSPA1A, and control plasmids encoding GFP – pcDNA5/FRT/TO GFP and V5 –pcDNA5/FRT/TO V5 (Hageman and Kampinga 2009) were obtained from Addgene (Cambridge, MA, USA). pmaxGFP plasmid encoding CopGFP (GFP) was used as a control plasmid. Plasmids were purified using EndoFree Plasmid Maxi Kit (Qiagen, Valencia, CA, USA).
Antibodies
Antibodies from different vendors were used in this study. Abcam: V5 (ab27674), Histone H2A.X (ab11175); Evrogen: CopGFP (GFP) (ab501); Santa Cruz Biotechnology, Santa Cruz, CA, USA: HSP70 (sc-24), AIF (sc-13116), Apaf-1 (sc-65890), cytochrome c (sc-13560); Cell Signaling Technology, Inc., Danvers, MA, USA: Caspase-9 (#9506), Cleaved Caspase-3 (#9661), Cleaved PARP (#9545); Enzo Life Sciences, Inc., Farmingdale, NY, USA: GAPDH (ADI-CSA-335); and Sigma, St Louis, MO, USA: β-actin (A1978).
Cell lysates preparation and western blot
Whole-cell extracts were prepared as described previously (Stoica et al. 2005). A portion of the lysate (20 μg of protein) was then fractionated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE). Western blot was performed as described previously (Stoica et al. 2005). Membranes were washed and protein complexes were visualized using SuperSignal West Dura Extended Duration Substrate (Pierce, Rockford, IL, USA).
Subcellular fractionation
Subcellular fractionation was performed as described (Stoica et al. 2005) with some modifications. RCN were harvested and washed in ice-cold phosphate-buffered saline (PBS). Cell suspension was centrifuged at 500 g for 15 min at 4°C. Cell pellet was resuspended for 10 min on ice in the digitonin lysis buffer (20 mM HEPES, pH 7.4, 80 mM KCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 250 mM Sucrose, 200 μg/mL Digitonin and Protease Inhibitor and Phosphatase Inhibitor (2, 3) cocktails (P8340; P5726 and P0044 Sigma-Aldrich). The lysate was centrifuged at 1000 g for 5 min at 4°C to pellet the nuclei. The supernatant was transferred to a new tube and centrifuged again at 12 000 g for 10 min at 4°C to pellet the mitochondria. The resulting supernatant, representing the cytosolic fraction, was recovered. Nuclear and mitochondrial lysates were prepared in RIPA buffer (Teknova) with Protease Inhibitor Cocktail (P8340 Sigma-Aldrich).
Immunoprecipitation
The following immunoprecipitation (IP) procedure was used: 50 μL of Protein A - Sepharose® 4B beads (Invitrogen, cat10-1041) were coated with 1 μg antibody, by incubating for 2 h at 4°C in 1 mL of non-denaturing lysis buffer (20 mM Tris HCl pH 8, 137 mM NaCl, 1% Nonidet P-40 (NP-40), 2 mM EDTA) containing 0.1% bovine serum albumin. Then, antibody-coated beads were washed three times in non-denaturing lysis buffer and incubated overnight at 4°C with 500 μg of lysate, in a final volume of 800 μL non-denaturing lysis with Protease Inhibitor and Phosphatase Inhibitor (2, 3) cocktails (P8340; P5726 and P0044; Sigma-Aldrich). Beads were finally washed four times with non-denaturing lysis buffer and coimmunoprecipitated proteins were released by boiling in SB (2% SDS, 10% glycerol, 62.5mM Tris-HCl pH 6.8, 100mM dithiothreitol), resolved by SDS–PAGE, and then subjected to western blot analysis. All apoptosis IP experiments were repeated three times. To maximize the number of outcomes per experiment, membranes were stripped and repeatedly subjected to western blot analysis with different primary antibodies. Quantification of data shown in Figs 3e and f and 4c represent pooled averages from three independent experiments.
Fig. 3.

HSP70 over-expression inhibits caspase-3 activation in etoposide-treated neurons. (a) Rat cortical neuronal were transfected with GFP or V5-HSP70 and treated with etoposide 50 μM (Etop) as described above. Transfected cells were visualized by immunostaining with GFP or V-5 antibodies. GFP- and V5-HSP70-transfected neurons are shown in red. Cleaved (active) caspase-3 was visualized by immunostaining with cleaved caspase-3 antibody (green). 4′,6-diamidino-2-phenylindole (DAPI) chromatin staining identified nuclear markers of apoptosis as previously described. Apoptotic nuclei were indicated by red arrows whereas intact non-apoptotic nuclei were indicated by white arrows. (b) The numbers of transfected neurons with cleaved caspase-3 in each group were expressed as per cent control GFP transfected. Scale bar, 25 μm. Data represent the mean ± SEM. *p < 0.05; versus GFP-transfected Etop-treated RCN. Analysis by Kruskal–Wallis one-way ANOVA on ranks, all pairwise multiple comparison procedures, followed by post hoc adjustment using Student–Newman–Keul’s Method.
Immunostaining
Rat cortical neuronal were plated on glass coverslips in 24-well plates. After each experiment, the cells were fixed by incubating with 4% paraformaldehyde in PBS, pH 7.5 for 10 min at room temperature. The coverslips were washed three times for 10 min each time with PBS followed by incubation for 1 h at 21°C in blocking buffer (10% normal goat serum (S-1000; Vector Laboratories, Burlingame CA, USA), 0.1% Triton X-100 in PBS). The coverslips were incubated overnight at 21°C with the primary antibodies diluted in blocking buffer. The coverslips were washed three times for 10 min, each time with PBS, and incubated for 1 h at 21°C with the secondary antibodies Alexa Fluor® 546 Goat Anti-Mouse IgG and Alexa Fluor® 633 Goat Anti-Rabbit IgG (A-11018, A-21070; Molecular Probes, Eugene, OR, USA) diluted in blocking buffer solution. The samples were washed three times for 10 min, each time with PBS, incubated for 0.5 h at 21°C with 5 μg/mL of 4″,6-diamidino-2-phenylindole (DAPI) in saline solution, then washed again three times with PBS for 10 min each. Cover slips were mounted on microscope slides and stored in the dark at 4°C until examined. Microscopy imaging was performed using Zeiss Axiovert 135 microscope (Carl Zeiss, Peabody, MA, USA). All immunostaining experiments were repeated three times. Each experiment included triplicate samples (wells containing coverslips with RCN) and images were acquired from 10 random regions of each sample. Data analysis was based on these images. Quantification of nuclear markers shown in Fig. 1b–e, caspase-3 activation shown in Fig. 5b, and PARP cleavage shown in Fig. 6b represent pooled averages from three independent experiments.
Fig. 1.

HSP70 over-expression attenuates nuclear markers of apoptosis. (a) The effect of V5-HSP70 over-expression on nuclear markers of apoptosis following treatment with etoposide was evaluated by 4′,6-diamidino-2-phenylindole (DAPI) staining and fluorescent microscopy. Rat cortical neurons (RCN) transfected with GFP or HSP70-V5 plasmids, and 24 h later were treated with etoposide (Etop). After an additional 24-h time interval, cells were fixed and stained with DAPI. For the analysis of nuclear markers of apoptosis, transfected cells were visualized by GFP fluorescence (green) or by immunostaining with V-5 antibody (red). Transfected cells with intensely stained, condensed, or fragmented chromatin were scored as apoptotic (red arrow). Transfected cells with regular shape, larger and moderately stained chromatin were counted as intact non-apoptotic (white arrow). (b–e) - RCN were transfected, treated with (b) etoposide 50 μM (Etop); (c) Staurosporine 0.5 μM (Staur); (d) Aβ (25–35) 50 μM (Aβ); (e) C2-ceramide 50 μM (C2cer), and nuclear markers of apoptosis in transfected cells were visualized and quantified as described above and in the Methods. The per cent neurons with intact nuclei were normalized to untreated (control) GFP-transfected RCN. Scale bar, 25 μm. Data represent the mean ± SD. *p < 0.05, **p < 0.01 ***p < 0.001 versus GFP-transfected RCN treated with apoptotic inducers. Analysis by Kruskal–Wallis one-way ANOVA on ranks, all pairwise multiple comparison procedures, followed by post hoc adjustment using Student–Newman–Keul’s Method.
Fig. 5.
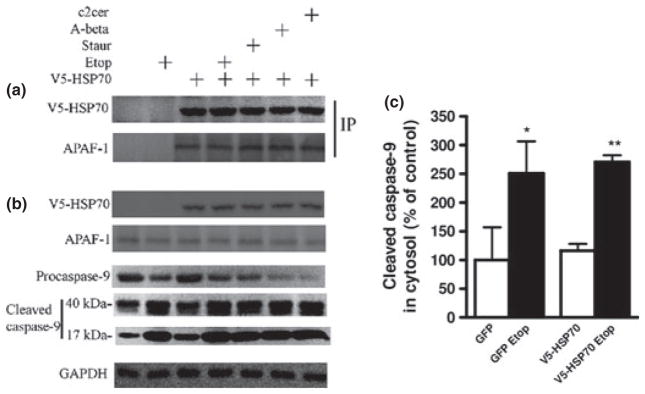
HSP70 constitutively interacts with Apoptotic protease-activating factor 1 (Apaf-1) in neuronal cytosol. Rat cortical neuronal transfection with V5-HSP70, apoptosis induction and subcellular fractionation to generate cytosolic extracts were performed as described. (a) Cytosolic fractions were subjected to immunoprecipitation (IP) with V-5 antibody and immunoblotted with antibodies against V5 and Apaf-1 to demonstrate the HSP70/Apaf-1 interaction. (b) The expression of V5-HSP70 and Apaf-1, as well as the cleavage (activation) of caspase-9, was examined in cytosolic extracts by immunoblot. (c) Levels of cleaved caspase-9 in etoposide 50 μM (Etop)-treated neurons were quantified as fold change to control levels after measurement of band intensity by densitometry in cytosolic fractions and normalization to levels of GAPDH. Data represent the mean ± SD. *p < 0.05, **p < 0.01, versus control neurons (n = 3). Analysis by Kruskal–Wallis one-way ANOVA on ranks, all pairwise multiple comparison procedures, followed by post hoc adjustment using Student–Newman–Keul’s Method.
Fig. 6.

HSP70 interacts with apoptosis-inducing factor (AIF) in the cytosol during neuronal apoptosis rat cortical neuronal transfection with V5-HSP70, apoptosis induction and subcellular fractionation to generate cytosolic, nuclear, and mitochondrial extracts were performed as described. Cytosolic fractions were subjected to immunoprecipitation with V-5 antibody and immunoblotted with antibodies against V5 and AIF (a). Cytosolic (b) and nuclear fractions (c) were fractioned on sodium dodecyl sulfate–polyacrylamide gel and immunoblotted with antibodies against V5 and AIF. Equal loading was demonstrated using GAPDH (cytosolic marker) and Histone H2A.X (nuclear marker). Cytosolic fractions showed no Histone H2A.X contamination (d). V5-HSP70 signal was not present in the nuclear fraction (e). Levels of AIF protein in etoposide 50 μM (Etop)-treated neurons were quantified as fold change to control levels after measurement of band intensity by densitometry in cytosolic (f) and nuclear (g) fractions and normalization to levels of GAPDH for cytosolic and Histone H2A.X for nuclear fraction. Data represent the mean ± SD. *p < 0.05, **p < 0.01, versus control neurons (n = 3). Analysis by Kruskal–Wallis one-way ANOVA on ranks, all pairwise multiple comparison procedures, followed by post hoc adjustment using Student–Newman–Keul’s Method.
Assessment of nuclear markers of apoptosis
4′,6-diamidino-2-phenylindole staining was employed to determine chromatin condensation and fragmentation, a specific nuclear marker of apoptosis (Nardi et al. 1997). Intact cells displayed relatively large nuclei, uniform in size with oval shape, and chromatin homogenously stained at low to moderate intensity. In contrast, the nuclei of apoptotic cells appeared smaller, irregular, with more intense chromatin staining and ultimately showed fragmentation in multiple bright, round spots (Nardi et al. 1997). As a measure of apoptosis for each GFP- or HSP70-transfected/ treatment group, we normalized the percentage of intact nuclei (from the total number of transfected cells in each group) to the percentage of intact nuclei in control GFP-transfected cells.
Propidium iodide uptake assay
Propidium iodide (PI) is a fluorescent exclusion dye widely used as a vital stain in tissue culture systems; PI penetrates only in cells that lack an intact membrane and labels the nucleus (Brana et al. 2002). At the end of each experiment and without fixation, RCN were incubated in conditioned media containing 25 μg/mL of PI for 20 min at 37°C, washed with conditioned media, and then analyzed immediately using a fluorescent microscope using the rhodamine filter (PI) and FITC filter (GFP). PI-negative cells were considered intact, whereas PI-positive cells were considered dying cells. As a measure of survival for each GFP- or HSP70-transfected/treatment group, we normalized the percentage of intact cells (from the total number of transfected cells counted in each group) to the percentage of intact cells in control GFP-transfected cells. The assay was performed as described above in triplicate samples.
Statistical analysis
Data were analyzed by Kruskal–Wallis One-Way ANOVA on Ranks followed by post hoc adjustments using the Student–Newman–Keul’s test. A p < 0.05 was considered statistically significant. Statistical analyses were performed using Sigma Stat 3.5 (Aspire Software International).
Results
HSP70 over-expression is neuroprotective in multiple models of apoptosis in primary cortical neurons
To test the ability of HSP70 to inhibit neuronal apoptosis, we transfected primary cortical neurons with HSP70-expressing plasmids, induced apoptosis using four different agents [etoposide, C2-ceramide, staurosporine, or Aβ (25–35)], and examined markers of neuronal apoptosis and cell death. By testing multiple independent apoptotic models, we ensure that our findings are not limited to any one particular apoptosis inducer. All these apoptosis models involve, to varying degrees, both caspase-dependent and AIF-dependent pathways. HSP70 was previously shown to interact and inhibit these apoptosis pathways in non-neuronal cells, but little is known about HSP70 effects on these mechanisms in neurons. The HSP70-expressing plasmid included a V5 tag that allowed us to use V5 tag-specific antibodies to detect the neurons expressing the transfected HSP70 construct; transfection control was performed with GFP-expressing plasmids. In preliminary experiments, we confirmed that V5 tag on N-terminus of HSP70 does not effect on its neuroprotective activity; V5-HSP70, His-HSP70, and non-tagged HSP70 were equally neuroprotective (data not shown). We also verified that GFP-expressing plasmid is an appropriate control plasmid for the study of neuronal cell death and showed that apoptotic neurons maintain GFP expression in a similar manner with Lac-Z (data not shown). DAPI staining was employed to determine chromatin condensation, a specific marker of apoptosis (Majdzadeh et al. 2008; Pfister et al. 2008). As described in the Methods, cells with intensely stained, condensed and/or fragmented nuclei were scored as apoptotic. Cells with moderately stained and larger nuclei were counted as intact. Our results show that in all four different in vitro models of neuronal apoptosis, the proportion of intact non-apoptotic (normal chromatin staining) cells is significantly increased in neurons that express V5-HSP70 as compared to control neurons expressing GFP (Fig. 1).
Although chromatin condensation is a good indicator of apoptosis, it does not directly demonstrate cell death. In contrast, it is generally accepted that loss of integrity of the cellular membrane is an accurate indicator of irreversible cell death. To show that HSP70 over-expression not only attenuates apoptotic markers but also unequivocally inhibits cell death and improves neuronal survival, we used PI staining of the ‘live’ unfixed cultures (Brana et al. 2002). In these conditions, PI is only able to penetrate cells that have lost membrane integrity, a marker of irreversible cell death (Cordeiro et al. 2010). For the PI assay, the neurons were cotransfected with 2% pmaxGFP vector encoding GFP protein as a marker for transfected cells (Hetman et al. 2000).
Using the PI assay, we showed that following all four apoptotic stimuli the proportion of live cells (PI negative) is significantly increased in V5-HSP70-transfected neurons compared with GFP control (Fig. 2). Thus, our data demonstrate that HSP70 over-expression is anti-apoptotic and neuroprotective in multiple neuronal apoptosis models.
HSP70 over-expression inhibits caspase-3 activation and PARP-1 cleavage in etoposide-treated primary cortical neurons
We used fluorescent microscopy to characterize key apoptotic markers in transfected cells. Primary cortical neurons were transfected with GFP-expression plasmids (control) or V5-HSP70 and then either left untreated or treated with etoposide followed by immunofluorescence with V5-HSP70, GFP, and cleaved (active) caspase-3 or cleaved poly (ADP-ribose) polymerase (PARP1)-specific antibodies. We observed that in GFP-transfected neurons, etoposide treatment greatly increased the number of double-positive GFP/cleaved caspase-3 cells; these cells also showed signs of chromatin condensation. In contrast, we detect significantly fewer V5-HSP70/cleaved caspase-3 cells following etoposide treatment (Fig. 3a and b). PARP-1 is a well-described substrate of active caspase-3, and the presence of cleaved PARP-1 fragments is a useful indicator of caspase activation in apoptosis. Our data demonstrate that etoposide treatment significantly increases the number of GPF/cleaved PARP1 neurons (these cells also showed signs of chromatin condensation), whereas the increase in V5-HSP70/cleaved PARP1 neurons is significantly attenuated (Fig. 4a and b).
Fig. 4.

HSP70 over-expression inhibits PARP-1 cleavage in etoposide-treated neurons. (a) Rat cortical neuronal were transfected with GFP or V5-HSP70 and treated with etoposide 50 μM (Etop) as described above. Transfected cells were visualized by immunostaining with GFP or V-5 antibodies. GFP- and V5-HSP70-transfected neurons are shown in red. Cleaved PARP was visualized by immunostaining with cleaved PARP antibody (green). 4′,6-diamidino-2-phenylindole (DAPI) chromatin staining identified nuclear markers of apoptosis as previously described. Red arrows indicate apoptotic nuclei whereas white arrows indicate intact non-apoptotic nuclei. (b) The numbers of transfected neurons with cleaved PARP-1 in each group were expressed as per cent control GFP transfected. Scale bar, 25 μm. Data represent the mean ± SEM. *p < 0.05, versus GFP transfected Etop-treated RCN. Analysis by Kruskal-Wallis one-way ANOVA on ranks, all pairwise multiple comparison procedures, followed by post hoc adjustment using Student-Newman-Keul’s Method.
HSP70 constitutively binds Apaf-1 in the cytosol of primary cortical neurons
Apoptotic protease-activating factor 1 is a cytosolic protein required for the formation of the apoptosome (Apaf-1, cytochrome c, and caspase-9 complex) and initiation of the intrinsic caspase activation pathway (cleavage and activation of caspase-9) (Li et al. 1997). Based on data from non-neuronal cells, it has been suggested that HSP70 inhibits cleavage of caspase-9 by interacting with cytosolic Apaf-1. We investigated the HSP70-mediated modulation of this pathway in various models of neuronal apoptosis. Primary cortical neurons were transfected with V5-HSP70 and apoptosis was induced by etoposide, staurosporine, ceramide, and beta-amyloid. We prepared cytosolic and nuclear extracts for IP and immunoblot.
Our results demonstrate the presence of Apaf-1 in V5-HSP70 IP (Fig. 5a). The levels of V5-HSP70 and Apaf-1 are unchanged in the IP from untreated or apoptotic neurons, suggesting the constitutive nature of their interaction in the cytosol. The total cytosolic expression of both proteins is also unchanged in the treated versus untreated neurons (Fig. 5b). The initiation of the Apaf-1-dependent, intrinsic caspase activation pathways in our apoptotic models is suggested by caspase-9 cleavage: decreased levels of procaspase-9 and increased levels of 40-kDa fragment (active) and 17-kDa fragment (Fig. 5b). We quantified cleaved caspase-9 fragment levels in cytosolic fractions of untreated and etoposide-treated cells (normalization by GAPDH expression) to demonstrate that apoptosis is associated with a significant caspase-9 activation (Fig. 5b and c). Caspase-9 was not coimmunoprecipitated with HSP70 (data not shown). These data are consistent with the hypothesis that HSP70 sequesters Apaf-1 and thus blocks the formation of the apoptosome and activation of the intrinsic caspase pathway in neurons.
HSP70 binds AIF in the cytosol during apoptosis in primary cortical neurons
Apoptosis-inducing factor 1 is a mitochondrial protein, which initiates one of the key mechanisms of caspase-independent apoptosis after its translocation, initially to the cytosol and ultimately to the nucleus where it causes chromatinolysis (Artus et al. 2010). We investigated the AIF apoptotic pathways and its regulation by HSP70 in multiple models of neuronal apoptosis. Primary cortical neurons were transfected with V5-HSP70 and apoptosis was induced by etoposide, staurosporine, ceramide, and Aβ (25–35). We prepared cytosolic and nuclear extracts to be used for IP and immunoblot. Our data show that AIF coimmunoprecipitates with V5-HSP70 in cytosolic fractions; the V5-HSP70–AIF interaction is increased following induction of apoptosis (Fig. 6a). Immunoblot shows that the cytosolic levels of V5-HSP70 were similar in untreated and apoptotic cells, whereas cytosolic AIF is increased by all apoptotic stimuli reflecting the release of AIF from the mitochondria, a key step of the AIF apoptotic pathway (Fig. 6b). This may explain the increased AIF–V5-HSP70 complex in neurons undergoing apoptosis. We quantified AIF levels in untreated and etoposide-treated cells (normalization by GAPDH expression) to demonstrate that apoptosis is associated with a significant increase in cytosolic AIF (Fig. 6f). One of the key final steps in the AIF pathway is translocation of AIF from the cytosol to the nucleus; our data show that apoptotic stimuli greatly increase the levels of AIF in nuclear extracts (Fig. 6c). We quantified AIF levels in untreated and etoposide-treated cells (normalization by H2AX expression) to demonstrate that apoptosis is associated with a significant increase in nuclear AIF (Fig. 6g). Nuclear extracts from treated and untreated cells show steady levels of H2AX, a nuclear marker and AIF partner (Artus et al. 2010) (Fig. 6c and d); the quality of subfractionation is demonstrated by absence of H2AX signal in cytosolic fractions (Fig. 6d). Our observation that V5-HSP70 is detected only in the cytosolic and not nuclear fractions suggests that HSP70 may block nuclear translocation of cointeracting AIF molecules by retaining them in the cytosol (Fig. 6e).
Over-expression of HSP70 attenuates apoptosis in SH-SY5Y neuronal cell line
The low transfection efficiency of primary cortical neurons did not permit detection of changes at the level of the entire cell population, such as cell viability assays or western blot. To address this limitation, we used the SH-SY5Y neuroblastoma cell line to examine the protective effect of HSP70 over-expression. SH-SY5Y cell line is common model to study neuronal cell death and neurodegeneration (Bhat et al. 2000) and has high efficiency of transfection (more than 50% in our hands).
To test the ability of HSP70 to inhibit apoptosis, SH-SY5Y cells were transfected with GFP-, V5-, or V5-HSP70-expressing plasmids. Apoptosis was induced by using etoposide, C2-ceramide, staurosporine, or Aβ as described above. Cell viability of SH-SY5Y cells was examined using the Calcein AM assay. Our results show that in all four apoptotic models, the cell viability of V5-HSP70-transfected cells was significantly increased (Fig. 7a). We observed no differences in the percentage of survival between control cells and cells transfected by GFP- or V5-expressing plasmids (data not shown).
Fig. 7.
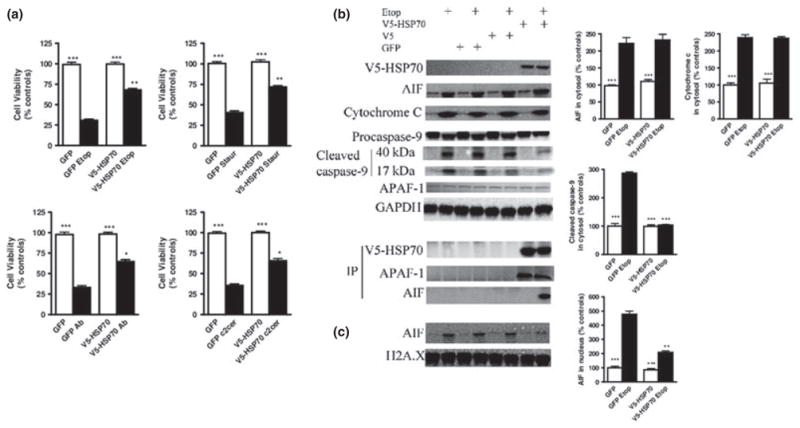
Over-expression of HSP70 attenuates apoptosis in SH-SY5Y cells. (a) V5-HSP70 over-expression improves viability of SH-SY5Y cells following exposure to multiple inducers of apoptosis. Cells were transfected with GFP, V5, or HSP70-V5 plasmids and 24 h later treated with etoposide 50 μM (Etop); Staurosporine 0.5 μM (Staur); Aβ (25–35) 50 μM (Aβ); C2-ceramide 50 μM (C2cer). After an additional 24-h time interval, cell viability was evaluated by Calcein AM assay. (b) V5-HSP70 over-expression does not change cytochrome c and apoptosis-inducing factor (AIF) release from mitochondria to the cytosol, but does attenuate caspase-9 cleavage/activation. HSP70 interacts with Apoptotic protease-activating factor 1 (Apaf-1) and AIF in the cytosol of SHSY-5Y cells. SH-SY5Y transfection with GFP, V5, and V5-HSP70, apoptosis induction, subcellular fractionation, immunoprecipitation, and immunoblot were performed as described. (c) V5-HSP70 over-expression attenuates AIF translocation to the nucleus. Cytosolic and nuclear fractions were fractioned on sodium dodecyl sulfate–polyacrylamide gel and immunoblotted with antibodies against cytochrome c, AIF, V5, caspase-9, GAPDH, and Histone H2A.X. Equal loading was demonstrated using GAPDH (cytosolic marker) and Histone H2A.X (nuclear marker). Levels of AIF, Cytochrome c, cleaved caspase-9 were quantified as fold change to control GFP-transfected cells levels after measurement of band intensity by densitometry in cytosolic (b) and nuclear (c) fractions and normalization to levels of GAPDH for cytosolic and Histone H2A.X for nuclear fraction. Data represent the mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001 versus control GFP-transfected cells. Analysis by Kruskal–Wallis one-way ANOVA on ranks, all pairwise multiple comparison procedures, followed by post hoc adjustment using Student–Newman–Keul’s method.
We also used the SH-SY5Y cells to investigate the mechanisms of HSP70 neuroprotection. SH-SY5Y cells were transfected with V5-HSP70 and apoptosis was induced by etoposide. Cellular subfractionation into cytosolic and nuclear extracts was performed and these extracts were used for IP and immunoblot analysis.
We observed the same level of V5-HSP70 in cytosol in untreated and apoptotic samples. Our data show that the levels of cytosolic AIF and Cytochrome c were increased by the same extent following etoposide treatment in non-transfected or GFP- and V5-transfected cells versus V5-HSP70-transfected samples suggesting that HSP70 over-expression does not regulate the release of these pro-apoptotic molecules from mitochondria. However, just like in primary cortical neurons, AIF coimmunoprecipitates with V5-HSP70 in the cytosolic fractions of SHSY-5Y following etoposide induction of apoptosis (Fig. 7b). In contrast, when we evaluated level of cleaved caspase-9 fragments in cytosolic fractions (a marker of cytochrome c-dependent apoptosis), we demonstrate that over-expression of HSP70 inhibits caspase-9 activation. Our results also demonstrate the presence of Apaf-1 in V5-HSP70 immunoprecipitates from SHSY-5Y cytosolic fractions. Similar to the primary cortical neurons, there are no differences in the levels of Apaf-1 in V5-HSP70 immunoprecipitates from apoptotic and untreated SH-SY5Y cells, suggesting the constitutive nature of the HSP70–Apaf-1 interaction in the cytosol of SHSY-5Y cells (Fig. 7b). These data support the suggestion that HSP70 sequesters Apaf-1 and thus blocks the formation of the apoptosome and activation of the cytochrome c-dependent intrinsic caspase pathway in neurons.
We also examined and quantified AIF translocation to the nucleus in etoposide-treated and untreated cells. Etoposide treatment causes elevation of AIF level in nuclear fraction of non-transfected or GFP- and/or V5-transfected cells. Importantly, over-expression of HSP70 significantly attenuated the etoposide-dependent increase in nuclear AIF compared with etoposide-treated controls (Fig. 7c). These data support the suggestion that HSP70 binds AIF and prevents its translocation to the nucleus, a key step in AIF-dependent apoptosis.
Discussion
This work demonstrates the neuroprotective effects of HSP70 and its modulation of caspase-dependent and caspase-independent pathways in four well-established models of apoptosis in primary cortical neurons. HSPs are molecular chaperones that play a vital physiological role enabling correct folding of freshly synthesized proteins. HSPs are up-regulated in many acute and chronic neurodegeneration models including cerebral ischemia/stroke, trauma, and Parkinson’s disease. It is believed that they have various cytoprotective functions through mechanisms that include refolding of proteins impacted by stress-induced denaturation and inhibition of apoptotic molecules (Turturici et al. 2011). It has been speculated that HSP70-mediated inhibition of cell death may involve two distinct mechanisms: in the case of stimuli that cause stress-induced necrosis associated with protein damage, the key role appears to reflect a chaperone function in repairing the protein machinery, whereas separate and distinct mechanisms appear to be responsible for inhibition of apoptosis when protein damage is not a main feature (Steel et al. 2004). The protective effects of HSP70 in the CNS have been shown in multiple in vivo (Matsumori et al. 2005; van der Weerd et al. 2005; Sinn et al. 2007) and in vitro models (Papadopoulos et al. 1996; Xu and Giffard 1997). Although various in vitro studies have analyzed HSP70 neuronal functions, the interpretation and generality of the results has often been obscured by the multitude of the neuronal cell types and injury conditions employed. In both primary neuronal cell cultures and neuronal cell lines, prior exposure to mild heat or ischemic stress results in protection against later exposure to either toxic conditions (heat, ischemia, or excitotoxins) (Amin et al. 1995; Lowenstein et al. 1991; Rordorf et al. 1991) or apoptotic stimuli (trophic factor withdrawal) (Mailhos et al. 1993, 1994; Wagstaff et al. 1999). The neuroprotective effects appear to be correlated with elevated expression of HSPs, including HSP70, but studies designed to directly link single HSPs over-expression to neuroprotection have provided conflicting results. HSP70 over-expression was neuroprotective following thermal or ischemic stress in primary cultures, such as dorsal root ganglion (Wyatt et al. 1996) or hippocampal (Fink et al. 1997) neurons, as well as in neuronal cell lines (ND7) (Mailhos et al. 1994). HSP70 was also shown to protect primary hippocampal neurons from glutamate and oxidative stress (Kelly et al. 2001). In contrast, HSP70 over-expression in the same cell types did not provide protection against apoptotic stimuli such as trophic factor deprivation (Fink et al. 1997; Mailhos et al. 1994; Wyatt et al. 1996). Other authors also confirmed that HSP70 over-expression in ND7 cells fails to attenuate serum withdrawal or stauro-sporine-induced apoptosis despite effective inhibition of necrotic cell death (Zourlidou et al. 2004). One possible explanation is that additional HSPs, which are induced by pre-conditioning treatments, are essential HSP70 cofactors. However, this possibility is contradicted in at least one study that shows that HSP70 performance was not improved by coexpression of its partner chaperone HSP40 (Zourlidou et al. 2004). Alternatively, HSP70 may be unable to regulate neuronal apoptotic machinery or HSP70 activity may be cell and/or apoptotic-inducer specific. This latter hypothesis is supported by data showing that in HSP70-over-expressing transgenic mice, hippocampal, but not cortical neurons, are protected against oxygen–glucose deprivation and glutamate toxicity (Lee et al. 2001).
The aim of this study was to directly examine the anti-apoptotic effects of HSP70 in a well-established in vitro model of primary cortical neurons using four independent apoptosis stimuli. Our data demonstrate that primary cortical neurons showing selective over-expression of HSP70 are not only significantly protected against late apoptotic features such as chromatin condensation, but also ultimately show improved survival following etoposide, ceramide, staurosporine, or beta-amyloid treatment compared with control. Independently, we showed that HSP70 over-expression increased survival following apoptosis induction in the SH-SY5Y neuroblastoma cell line. It was previously shown that in the ND7 cell line, based on peripheral nervous system neurons, HSP70 did not protect against staurosporine-induced cell death (Zourlidou et al. 2004). One possible explanation for this discrepancy may be the intrinsic differences between their in vitro model and the CNS neuronal model (primary cortical neurons) or the SH-SY5Y cells we employed. Nonetheless, it is significant that we validated the HSP70 anti-apoptotic effects in four independent models of neuronal cell death using both primary cortical neurons and the SH-SY5Y cells. We conclude that, at least within the parameters of our models, HSP70 can modulate neuronal apoptotic machinery resulting in significant inhibition of neuronal cell death.
Most data regarding the mechanisms of HSP70-mediated inhibition of apoptosis originate from studies performed in non-neuronal cells. HSP70 modulates caspase-dependent apoptotic pathways through binding apoptotic protease activating factor-1 (Apaf-1) and preventing the formation of a functional apoptosome, the Apaf-1/cytochrome c/caspase-9 activation complex (Saleh et al. 2000; Beere et al. 2000). The interaction between HSP70 and Apaf-1 prevents the recruitment of caspase-9 to the apoptosome, thereby inhibiting procaspase-9 cleavage and activation (Saleh et al. 2000; Beere et al. 2000). The binding of HSP70 to Apaf-1 requires the N-terminal ATP-binding domain (ABD) of HSP70, and over-expression of a HSP70 construct without the ABD fails to interact with Apaf-1 and to attenuate cell death following stimuli that induce caspase-dependent apoptosis, such as etoposide (Ravagnan et al. 2001). Not all studies support this mechanism for HSP70-dependent inhibition of caspase pathways. Steel et al. 2004 found no HSP70/apoptosome interaction and instead suggested that HSP70 acts upstream of the mitochondria and inhibits several models of caspase-dependent apoptosis by blocking cytochrome c release from the mitochondria (Steel et al. 2004). Interestingly, in their system, HSP70 did not attenuate etoposide-induced cytochrome c release/apoptosis. Clemons et al. 2005 also demonstrated the ability of HSP70 to attenuate apoptosis by acting upstream of the mitochondria through inhibition of the release of cytochrome c and potentially of other mitochondrial pro-apoptotic proteins (Clemons et al. 2005). Stankiewicz et al. 2005 proposed that HSP70 acts upstream of the mitochondria through inhibition of Bax activation and prevention of release of pro-apoptotic factors from the mitochondria intermembrane space, and that HSP70 does not influence apoptosis once the mitochondrial events have occurred (Stankiewicz et al. 2005). Our data provide the first evidence that HSP70 constitutively interacts with Apaf-1 in primary cortical neurons and SHSY-5Y neuroblastoma cell line. Active caspase-9 amplifies the caspase cascade by caspase-3 cleavage and activation (Li et al. 1997; Srinivasula et al. 1998) followed by cleavage of caspase substrates, such as PARP, and ultimately cell death (Le Rhun et al. 1998). Although we detect the translocation of cytochrome c from the mitochondria to the cytosol and the cleavage of caspase-9 in all tested apoptotic primary cortical neuronal models, the low transfection efficiency in primary neurons does not permit observation of HSP70-mediated effects in homogenous cell extracts. To address this issue, we used SH-SY5Y cells where higher transfection efficiencies can be achieved. Our data show that although HSP70 over-expression does not attenuate release of cytochrome c, it significantly attenuates caspase-9 activation following treatment with etoposide. This suggests that the HSP70-dependent neuroprotective mechanisms are downstream of cytochrome c release and may instead involve the HSP70-Apaf-1 interaction, which prevents the formation of the apoptosome. Examination of individual primary cortical neurons by immunostaining reveals that over-expression of HSP70 inhibits cleavage of both caspase-3 and PARP in etoposide-induced apoptosis. On the basis of these data, we propose that HSP70-dependent attenuation of etoposide-induced neuronal cell death is explained at least in part through binding and blocking of Apaf-1 and inhibition of the caspase-dependent apoptosis pathway.
Apoptosis-inducing factor is an inducer of caspase-independent PCD and is released from mitochondria early in the apoptotic process (Susin et al. 1999). Ravagnan et al. 2001 showed, using non-neuronal cells, that HSP70 specifically interacts with AIF and that HSP70 over-expression protects cells from AIF-dependent cell death (Ravagnan et al. 2001). In contrast to the HSP70–Apaf-1 interaction, which is dependent on the ABD, the HSP70–AIF interaction required the HSP70 C-terminal peptide-binding domain; an HSP70 construct lacking ABD preserves interaction with AIF and is protective in AIF-dependent models of apoptosis such as induced by staurosporine (Ravagnan et al. 2001b). Significantly, it was shown that HSP70 constructs without ABD and/ or impaired chaperone function are able to attenuate apoptosis in response to both proteotoxic (heat) and/or non-proteotoxic (TNF alpha) provided they have a functional peptide-binding domain, suggesting the importance of this domain for HSP70 cell-protective role (Chow et al. 2009). We demonstrate AIF release and translocation to the nucleus in cortical neurons and SHSY-5Y neuroblastoma cells following all apoptotic stimuli examined. We also show that in all tested apoptotic models, HSP70 binds AIF in the cytoplasm. Therefore, our data suggest that the neuroprotective effects of HSP70 reflect intervention downstream of mitochondria permeabilization and the release of apoptotic factors. Importantly, we never detected HSP70 signal in the nucleus, suggesting that in neurons, as in other cell types (Ravagnan et al. 2001), the AIF –HSP70 complex cannot be translocated to the nucleus, a key step in the AIF-mediated PCD pathway. Because the low transfection efficiency in primary cortical neurons, we were not able to detect the HSP70-dependent inhibition of AIF translocation to the nucleus in homogenous extracts by western blot. Again, the SH-SY5Y cells allowed us to perform a more direct biochemical analysis of HSP70 effects on the AIF-dependent neuronal cell death. Our data show that similarly with cytochrome c, HSP70 over-expression does not attenuate the release of AIF from mitochondria into the cytosol. We also demonstrate that AIF coimmunoprecipitates with V5-HSP70 in the cytosolic fractions of SH-SY5Y cells following induction of apoptosis. Importantly, HSP70-transfected cells show a significant inhibition of AIF translocation from the cytosol to the nucleus, a key step in AIF-dependent apoptosis. Taken together, our results suggest that HSP70 modulates caspase-independent pathway apoptosis in primary cortical neurons and SH-SY5Y cells through interaction with AIF and by preventing its translocation to the nucleus.
Our data show that in primary cortical neurons, all tested apoptosis inducers lead to activation of both caspase-dependent and -independent pathways. Previous studies have suggested that selective blockage of a specific cell death pathway, such as caspases, may only temporarily improve survival (Volbracht et al. 2001). Therefore, it is possible that the robust neuroprotective effects of HSP70 over-expression are at least in partly because of its capacity to inhibit multiple PCD pathways.
In summary, our results show that in primary cortical neurons and SH-SY5Y cells, HSP70 modulates both Apaf-1 caspase-dependent and AIF caspase-independent pathways resulting in attenuation of apoptosis and ultimately inhibition of neuronal cell death. The lack of success of other studies to demonstrate HSP70-dependent inhibition of apoptosis may reflect differences between the cell types and apoptosis models used. The demonstrated ability of HSP70 to block both caspase-dependent and caspase-independent PCD pathways in primary cortical neurons underscores the potential for developing selective HSP70 over-expression methods to provide neuroprotection for conditions associated with cortical neuronal apoptosis.
Acknowledgments
We thank Shruti Kabadi and Giorgi Kharebava for expert technical assistance. This work was supported by grants 5 RC1 HD064758-03, RO1 NS061839-04 to Alan Faden.
Abbreviations used
- ABD
ATP-binding domain
- AIF
apoptosis-inducing factor
- Apaf-1
Apoptotic protease-activating factor 1
- DAPI
4′,6-diamidino-2-phenylindole
- GFP
green fluorescent protein
- HSP70
70-kDa heat-shock proteins
- PBS
phosphate-buffered saline
- PCD
programmed cell death
- PI
Propidium iodide
- RCN
rat cortical neurons
Footnotes
Conflict of interest and Disclosure
The authors declare that they have no competing financial interests.
References
- Amin V, Cumming DV, Coffin RS, Latchman DS. The degree of protection provided to neuronal cells by a preconditioning stress correlates with the amount of heat shock protein 70 it induces and not with the similarity of the subsequent stress. Neurosci Lett. 1995;200:85–88. doi: 10.1016/0304-3940(95)12074-e. [DOI] [PubMed] [Google Scholar]
- Artus C, Boujrad H, Bouharrour A, et al. AIF promotes chromatinolysis and caspase-independent programmed necrosis by interacting with histone H2AX. EMBO J. 2010;29:1585–1599. doi: 10.1038/emboj.2010.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beere HM, Wolf BB, Cain K, et al. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol. 2000;2:469–475. doi: 10.1038/35019501. [DOI] [PubMed] [Google Scholar]
- Bellmann K, Jaattela M, Wissing D, Burkart V, Kolb H. Heat shock protein hsp70 overexpression confers resistance against nitric oxide. FEBS Lett. 1996;391:185–188. doi: 10.1016/0014-5793(96)00730-2. [DOI] [PubMed] [Google Scholar]
- Bhat RV, Shanley J, Correll MP, Fieles WE, Keith RA, Scott CW, Lee CM. Regulation and localization of tyrosine216 phosphorylation of glycogen synthase kinase-3beta in cellular and animal models of neuronal degeneration. Proc Natl Acad Sci USA. 2000;97:11074–11079. doi: 10.1073/pnas.190297597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brana C, Benham C, Sundstrom L. A method for characterising cell death in vitro by combining propidium iodide staining with immunohistochemistry. Brain Res Brain Res Protoc. 2002;10:109–114. doi: 10.1016/s1385-299x(02)00201-5. [DOI] [PubMed] [Google Scholar]
- Bukau B, Deuerling E, Pfund C, Craig EA. Getting newly synthesized proteins into shape. Cell. 2000;101:119–122. doi: 10.1016/S0092-8674(00)80806-5. [DOI] [PubMed] [Google Scholar]
- Chow AM, Steel R, Anderson RL. Hsp72 chaperone function is dispensable for protection against stress-induced apoptosis. Cell Stress Chaperones. 2009;14:253–263. doi: 10.1007/s12192-008-0079-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemons NJ, Buzzard K, Steel R, Anderson RL. Hsp72 inhibits Fas-mediated apoptosis upstream of the mitochondria in type II cells. J Biol Chem. 2005;280:9005–9012. doi: 10.1074/jbc.M414165200. [DOI] [PubMed] [Google Scholar]
- Cordeiro MF, Guo L, Coxon KM, et al. Imaging multiple phases of neurodegeneration: a novel approach to assessing cell death in vivo. Cell Death Dis. 2010;1:e3. doi: 10.1038/cddis.2009.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink SL, Chang LK, Ho DY, Sapolsky RM. Defective herpes simplex virus vectors expressing the rat brain stress-inducible heat shock protein 72 protect cultured neurons from severe heat shock. J Neurochem. 1997;68:961–969. doi: 10.1046/j.1471-4159.1997.68030961.x. [DOI] [PubMed] [Google Scholar]
- Gribaldo S, Lumia V, Creti R, Conway de Macario E, Sanangelantoni A, Cammarano P. Discontinuous occurrence of the hsp70 (dnaK) gene among Archaea and sequence features of HSP70 suggest a novel outlook on phylogenies inferred from this protein. J Bacteriol. 1999;181:434–443. doi: 10.1128/jb.181.2.434-443.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hageman J, Kampinga HH. Computational analysis of the human HSPH/HSPA/DNAJ family and cloning of a human HSPH/ HSPA/DNAJ expression library. Cell Stress Chaperones. 2009;14:1–21. doi: 10.1007/s12192-008-0060-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada J, Sugimoto M. Activation of caspase-3 in beta-amyloid-induced apoptosis of cultured rat cortical neurons. Brain Res. 1999;842:311–323. doi: 10.1016/s0006-8993(99)01808-9. [DOI] [PubMed] [Google Scholar]
- Hartl FU, Hayer-Hartl M. Molecular chaperones in the cytosol: from nascent chain to folded protein. Science. 2002;295:1852–1858. doi: 10.1126/science.1068408. [DOI] [PubMed] [Google Scholar]
- Hetman M, Cavanaugh JE, Kimelman D, Xia Z. Role of glycogen synthase kinase-3beta in neuronal apoptosis induced by trophic withdrawal. J Neurosci. 2000;20:2567–2574. doi: 10.1523/JNEUROSCI.20-07-02567.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly S, Bieneman A, Horsburgh K, Hughes D, Sofroniew MV, McCulloch J, Uney JB. Targeting expression of hsp70i to discrete neuronal populations using the Lmo-1 promoter: assessment of the neuroprotective effects of hsp70i in vivo and in vitro. J Cereb Blood Flow Metab. 2001;21:972–981. doi: 10.1097/00004647-200108000-00010. [DOI] [PubMed] [Google Scholar]
- Koh JY, Wie MB, Gwag BJ, Sensi SL, Canzoniero LM, Demaro J, Csernansky C, Choi DW. Staurosporine-induced neuronal apoptosis. Exp Neurol. 1995;135:153–159. doi: 10.1006/exnr.1995.1074. [DOI] [PubMed] [Google Scholar]
- Lee JE, Yenari MA, Sun GH, Xu L, Emond MR, Cheng D, Steinberg GK, Giffard RG. Differential neuroprotection from human heat shock protein 70 overexpression in in vitro and in vivo models of ischemia and ischemia-like conditions. Exp Neurol. 2001;170:129–139. doi: 10.1006/exnr.2000.7614. [DOI] [PubMed] [Google Scholar]
- Li GC. Induction of thermotolerance and enhanced heat shock protein synthesis in Chinese hamster fibroblasts by sodium arsenite and by ethanol. J Cell Physiol. 1983;115:116–122. doi: 10.1002/jcp.1041150203. [DOI] [PubMed] [Google Scholar]
- Li GC, Li L, Liu RY, Rehman M, Lee WM. Heat shock protein hsp70 protects cells from thermal stress even after deletion of its ATP-binding domain. Proc Natl Acad Sci USA. 1992;89:2036–2040. doi: 10.1073/pnas.89.6.2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- Lowenstein DH, Chan PH, Miles MF. The stress protein response in cultured neurons: characterization and evidence for a protective role in excitotoxicity. Neuron. 1991;7:1053–1060. doi: 10.1016/0896-6273(91)90349-5. [DOI] [PubMed] [Google Scholar]
- Mailhos C, Howard MK, Latchman DS. Heat shock protects neuronal cells from programmed cell death by apoptosis. Neuroscience. 1993;55:621–627. doi: 10.1016/0306-4522(93)90428-i. [DOI] [PubMed] [Google Scholar]
- Mailhos C, Howard MK, Latchman DS. Heat shock proteins hsp90 and hsp70 protect neuronal cells from thermal stress but not from programmed cell death. J Neurochem. 1994;63:1787–1795. doi: 10.1046/j.1471-4159.1994.63051787.x. [DOI] [PubMed] [Google Scholar]
- Majdzadeh N, Wang L, Morrison BE, Bassel-Duby R, Olson EN, D’Mello SR. HDAC4 inhibits cell-cycle progression and protects neurons from cell death. Dev Neurobiol. 2008;68:1076–1092. doi: 10.1002/dneu.20637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumori Y, Hong SM, Aoyama K, et al. Hsp70 overexpression sequesters AIF and reduces neonatal hypoxic/ ischemic brain injury. J Cereb Blood Flow Metab. 2005;25:899–910. doi: 10.1038/sj.jcbfm.9600080. [DOI] [PubMed] [Google Scholar]
- Mosser DD, Caron AW, Bourget L, Denis-Larose C, Massie B. Role of the human heat shock protein hsp70 in protection against stress-induced apoptosis. Mol Cell Biol. 1997;17:5317–5327. doi: 10.1128/mcb.17.9.5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Movsesyan VA, Yakovlev AG, Dabaghyan EA, Stoica BA, Faden AI. Ceramide induces neuronal apoptosis through the caspase-9/caspase-3 pathway. Biochem Biophys Res Commun. 2002;299:201–207. doi: 10.1016/s0006-291x(02)02593-7. [DOI] [PubMed] [Google Scholar]
- Movsesyan VA, Stoica BA, Faden AI. MGLuR5 activation reduces beta-amyloid-induced cell death in primary neuronal cultures and attenuates translocation of cytochrome c and apoptosis-inducing factor. J Neurochem. 2004;89:1528–1536. doi: 10.1111/j.1471-4159.2004.02451.x. [DOI] [PubMed] [Google Scholar]
- Nakajima M, Kashiwagi K, Ohta J, Furukawa S, Hayashi K, Kawashima T, Hayashi Y. Etoposide induces programmed death in neurons cultured from the fetal rat central nervous system. Brain Res. 1994;641:350–352. doi: 10.1016/0006-8993(94)90165-1. [DOI] [PubMed] [Google Scholar]
- Nardi N, Avidan G, Daily D, Zilkha-Falb R, Barzilai A. Biochemical and temporal analysis of events associated with apoptosis induced by lowering the extracellular potassium concentration in mouse cerebellar granule neurons. J Neurochem. 1997;68:750–759. doi: 10.1046/j.1471-4159.1997.68020750.x. [DOI] [PubMed] [Google Scholar]
- Neupert W, Brunner M. The protein import motor of mitochondria. Nat Rev Mol Cell Biol. 2002;3:555–565. doi: 10.1038/nrm878. [DOI] [PubMed] [Google Scholar]
- Papadopoulos MC, Sun XY, Cao J, Mivechi NF, Giffard RG. Over-expression of HSP-70 protects astrocytes from combined oxygen-glucose deprivation. NeuroReport. 1996;7:429–432. doi: 10.1097/00001756-199601310-00013. [DOI] [PubMed] [Google Scholar]
- Patel YJ, Payne Smith MD, de Belleroche J, Latchman DS. Hsp27 and Hsp70 administered in combination have a potent protective effect against FALS-associated SOD1-mutant-induced cell death in mammalian neuronal cells. Brain Res Mol Brain Res. 2005;134:256–274. doi: 10.1016/j.molbrainres.2004.10.028. [DOI] [PubMed] [Google Scholar]
- Pfister JA, Ma C, Morrison BE, D’Mello SR. Opposing effects of sirtuins on neuronal survival: sIRT1-mediated neuroprotection is independent of its deacetylase activity. PLoS ONE. 2008;3:e4090. doi: 10.1371/journal.pone.0004090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt WB, Toft DO. Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp Biol Med (Maywood) 2003;228:111–133. doi: 10.1177/153537020322800201. [DOI] [PubMed] [Google Scholar]
- Ravagnan L, Gurbuxani S, Susin SA, et al. Heat-shock protein 70 antagonizes apoptosis-inducing factor. Nat Cell Biol. 2001;3:839–843. doi: 10.1038/ncb0901-839. [DOI] [PubMed] [Google Scholar]
- Le Rhun Y, Kirkland JB, Shah GM. Cellular responses to DNA damage in the absence of Poly(ADP-ribose) polymerase. Biochem Biophys Res Commun. 1998;245:1–10. doi: 10.1006/bbrc.1998.8257. [DOI] [PubMed] [Google Scholar]
- Rordorf G, Koroshetz WJ, Bonventre JV. Heat shock protects cultured neurons from glutamate toxicity. Neuron. 1991;7:1043–1051. doi: 10.1016/0896-6273(91)90348-4. [DOI] [PubMed] [Google Scholar]
- Ryan MT, Pfanner N. Hsp70 proteins in protein translocation. Adv Protein Chem. 2001;59:223–242. doi: 10.1016/s0065-3233(01)59007-5. [DOI] [PubMed] [Google Scholar]
- Saleh A, Srinivasula SM, Balkir L, Robbins PD, Alnemri ES. Negative regulation of the Apaf-1 apoptosome by Hsp70. Nat Cell Biol. 2000;2:476–483. doi: 10.1038/35019510. [DOI] [PubMed] [Google Scholar]
- Sinn DI, Chu K, Lee ST, et al. Pharmacological induction of heat shock protein exerts neuroprotective effects in experimental intracerebral hemorrhage. Brain Res. 2007;1135:167–176. doi: 10.1016/j.brainres.2006.11.098. [DOI] [PubMed] [Google Scholar]
- Srinivasula SM, Ahmad M, Fernandes-Alnemri T, Alnemri ES. Autoactivation of procaspase-9 by Apaf-1-mediated oligomerization. Mol Cell. 1998;1:949–957. doi: 10.1016/s1097-2765(00)80095-7. [DOI] [PubMed] [Google Scholar]
- Stankiewicz AR, Lachapelle G, Foo CP, Radicioni SM, Mosser DD. Hsp70 inhibits heat-induced apoptosis upstream of mitochondria by preventing Bax translocation. J Biol Chem. 2005;280:38729–38739. doi: 10.1074/jbc.M509497200. [DOI] [PubMed] [Google Scholar]
- Steel R, Doherty JP, Buzzard K, Clemons N, Hawkins CJ, Anderson RL. Hsp72 inhibits apoptosis upstream of the mitochondria and not through interactions with Apaf-1. J Biol Chem. 2004;279:51490–51499. doi: 10.1074/jbc.M401314200. [DOI] [PubMed] [Google Scholar]
- Stoica BA, Movsesyan VA, Knoblach SM, Faden AI. Ceramide induces neuronal apoptosis through mitogen-activated protein kinases and causes release of multiple mitochondrial proteins. Mol Cell Neurosci. 2005;29:355–371. doi: 10.1016/j.mcn.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Zamzami N, et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- Turturici G, Sconzo G, Geraci F. Hsp70 and its molecular role in nervous system diseases. Biochem Res Int. 2011;2011:618127. doi: 10.1155/2011/618127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volbracht C, Leist M, Kolb SA, Nicotera P. Apoptosis in caspase-inhibited neurons. Mol Med. 2001;7:36–48. [PMC free article] [PubMed] [Google Scholar]
- Wagstaff MJ, Collaco-Moraes Y, Smith J, de Belleroche JS, Coffin RS, Latchman DS. Protection of neuronal cells from apoptosis by Hsp27 delivered with a herpes simplex virus-based vector. J Biol Chem. 1999;274:5061–5069. doi: 10.1074/jbc.274.8.5061. [DOI] [PubMed] [Google Scholar]
- van der Weerd L, Lythgoe MF, Badin RA, Valentim LM, Akbar MT, de Belleroche JS, Latchman DS, Gadian DG. Neuroprotective effects of HSP70 overexpression after cerebral ischaemia–an MRI study. Exp Neurol. 2005;195:257–266. doi: 10.1016/j.expneurol.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Wyatt S, Mailhos C, Latchman DS. Trigeminal ganglion neurons are protected by the heat shock proteins hsp70 and hsp90 from thermal stress but not from programmed cell death following nerve growth factor withdrawal. Brain Res Mol Brain Res. 1996;39:52–56. doi: 10.1016/0169-328x(95)00352-s. [DOI] [PubMed] [Google Scholar]
- Xu L, Giffard RG. HSP70 protects murine astrocytes from glucose deprivation injury. Neurosci Lett. 1997;224:9–12. doi: 10.1016/s0304-3940(97)13444-9. [DOI] [PubMed] [Google Scholar]
- Yakovlev AG, Ota K, Wang G, Movsesyan V, Bao WL, Yoshihara K, Faden AI. Differential expression of apoptotic protease-activating factor-1 and caspase-3 genes and susceptibility to apoptosis during brain development and after traumatic brain injury. J Neurosci. 2001;21:7439–7446. doi: 10.1523/JNEUROSCI.21-19-07439.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JC, Barral JM, Ulrich Hartl F. More than folding: localized functions of cytosolic chaperones. Trends Biochem Sci. 2003;28:541–547. doi: 10.1016/j.tibs.2003.08.009. [DOI] [PubMed] [Google Scholar]
- Zourlidou A, Payne Smith MD, Latchman DS. HSP27 but not HSP70 has a potent protective effect against alpha-synuclein-induced cell death in mammalian neuronal cells. J Neurochem. 2004;88:1439–1448. doi: 10.1046/j.1471-4159.2003.02273.x. [DOI] [PubMed] [Google Scholar]