

Abstract
Objective
Overexpression of the sonic hedgehog (SHH) signaling pathway is an essential characteristic of pancreatic cancer stem cells (PCSCs) and arsenic trioxide (ATO) is described as a SHH inhibitor. This study evaluates whether ATO has the potential to inhibit viability of PCSCs via binding to SHH-Gli proteins.
Methods
Cell counting kit-8 and flow cytometry were used for analyzing apoptosis in cells in vitro. The animal model was an athymic nude mouse model bearing subcutaneous xenografts of SW1990 pancreatic cancer cells. The terminal deoxynucleotidyl transferase dUTP nick end labeling assay and immunohistochemistry were used for tumor tissue analysis. The interaction between Gli1 and ATO was examined by a confocal system and an ultraviolet absorption spectrum assay.
Results
ATO induced apoptosis in pancreatic cancer cells, especially CD24+CD44+ cells in vitro. Combination treatment of ATO and low dose gemcitabine inhibited tumor growth by 60.9% (P = 0.004), and decreased the expression of CD24, CD44, and aldehyde dehydrogenase 1 family, member A1 significantly in vivo. ATO changed the structure of the recombinant Gli1 zinc finger peptides in a cell-free condition and the binding action of ATO to recombinant Gli1 was observed in cultured pancreatic cancer cells.
Conclusion
ATO may have the potential to inhibit viability of PCSCs via binding to SHH-Gli proteins in vitro and in vivo.
Keywords: pancreatic cancer, stem cells, gemcitabine, arsenic trioxide, sonic hedgehog, Gli
Introduction
Increasing evidence suggests that the composition of malignant tumors is heterogeneous and that they contain a distinct subpopulation of cells termed cancer stem cells (CSCs). These CSCs, like their normal stem cell counterparts, have the capacity to self-renew and produce differentiated progeny.1,2 These characteristics of CSCs evoke tumor initiation and propagation, while the much larger set of more differentiated cancer cells possesses a limited proliferative potential. Conventional chemotherapy remains the mainstay of pancreatic cancer management. However, even patients whose tumors initially are arrested or regress with therapy eventually experience tumor regrowth while still receiving the primary therapy. This may be attributed to the subpopulation of CSCs originated by a senescent, cytotoxic therapy resistant precursor in pancreatic cancer cells. Pancreatic cancer stem cells (PCSCs) have been isolated and are defined by the expression of the cell surface markers CD24+, CD44+, and ESA+, and these cells account for 0.2%–0.8% of all pancreatic cancer cells in tumors.3,4
The sonic hedgehog (SHH) signaling pathway, which is involved in regulating the growth and survival of tissue stem and progenitor cells, is markedly upregulated in CD24+CD44+ESA+ pancreatic cancer cells when compared with CD24−CD44−ESA− and bulk ones, indicating that aberrant SHH plays a critical role in PCSCs.3 SHH ligands bind to their receptors and activate a signaling cascade that culminates in the nuclear translocation of Gli transcription factors and the regulation of target genes.5 As previously described, inhibition of the SHH signaling pathway might be a strategy for PCSC targeted therapy, and SHH signaling antagonists such as GDC-0449 (vismodegib) and sulforaphane have demonstrated efficacy4,6
Arsenic trioxide (ATO) has been found to block the SHH signaling pathway by inactivating Gli proteins.7–9 Prior work by another group has shown that ATO has significant antitumor effects on several types of solid tumors based on Gli inhibition.9 We hypothesized that ATO has the potential to inhibit viability of PCSCs in culture and in xenografts via binding to Gli proteins of the SHH pathway. We aim to develop a plan that can be applied clinically. For this purpose, the effects of ATO on the human pancreatic cancer cell line, SW1990, and the possible mechanism were evaluated in this study.
Methods
Cell line and reagents
The pancreatic cancer cell line, SW1990, was obtained from the American Type Culture Collection (Manassas, VA, USA). Cells were grown in RPMI (Gibco Invitrogen, Carlsbad, CA, USA) with 10% fetal bovine serum (Gibco Invitrogen). Sterile, pharmaceutical grade ATO powder was obtained from Beijing SL Pharmaceutical Co, Ltd (Beijing, People’s Republic of China) with additives including sodium hydrogen carbonate, mannitol, and glycerin for promoting water solubility. Green fluorescent protein (GFP)-Gli1 expressing adenovirus was provided by Sunbio Medical Biotechnology Co, Ltd (Shanghai, People’s Republic of China).
Cellular proliferation assay
Cellular proliferation was assessed by plating cells at a density of 5,000 cells per well, in triplicate, in a 96 well plate. Fresh standard growth media containing ATO or vehicle was added at the designated concentrations the morning after plating, once the cells had attached. Viable cells were quantified using cell counting kit-8 (cck-8) reagent (Dojindo Laboratories, Kumamoto, Japan) according to the manufacturer’s protocol on days 1, 2, and 3. Cck-8 solution (10 μL) was added to each well, followed by incubation for 2 hours at 37°C. The absorbance at 450 nm was determined using a multiplate reader (ELX-800; BioTek, Winooski, VT, USA). Cell viability was expressed as the percentage of viable cells relative to the control (untreated) cells. For each concentration of ATO, mean values were calculated from the mean absorbance values of three wells. IC50 values were calculated by sigmoidal dose response curve fit using GraphPad Prism 5 Demo (GraphPad Software, San Diego, CA, USA).
Flow cytometry
Dissociated cells were counted, transferred to 5 mL tubes, and washed twice with phosphate buffered saline (PBS) at a concentration of 1 × 106 cells per 100 μL. An Alexa fluor® Annexin V/Dead Cell Apoptosis kit (Invitrogen, Carlsbad, CA, USA) was used to distinguish and quantitatively determine the percentage of dead, viable, apoptotic, and necrotic cells after treatment with drugs according to the manufacturer’s instructions. Antibodies against human including anti-CD24-phycoerythrin (Biolegend Inc, San Diego, CA, USA) and anti-CD44-APC (allophycocyanin) (Biolegend Inc) were added to the samples, and then the samples were incubated and protected from light for 20 minutes. Samples were washed twice with PBS and resuspended in 100 μL binding buffer. Propidium iodide (PI) and annexin-V reagents were added and incubated for 15 minutes, followed by the addition of 400 μL binding buffer. Flow cytometry was performed using a Beckman Coulter FC500-MPL flow cytometer (Beckman Coulter Inc, Brea, CA, USA) in conjunction with CXP analysis software (Beckman Coulter Inc).
Xenograft experiment
Mice were purchased from the Shanghai Laboratory Animal Center (Chinese Academy of Sciences, Shanghai, People’s Republic of China). The experimental animal protocols were reviewed and approved by the animal care committee of the Shanghai Institutes for Biological Sciences of the Chinese Academy of Sciences as well as the Fudan University Shanghai Cancer Center.
Five-week-old female athymic nude mice were maintained in laminar flow rooms under constant temperature and humidity. First, SW1990 cells (5 × 106 cells) were injected subcutaneously into one flank of five mice. After 3 weeks, mice were sacrificed and tumors were harvested. Two of the medium sized tumors were dissected to 1 mm3 volumes and transplanted into 32 mice. After another 2 weeks, mice showing tumors were randomly assigned to control, Gem, ATO, or the combination group, and administered the following: (1) control group (n = 8): injection of normal saline (vehicle) at the same volume as that used for the combination group; (2) Gem group (n = 8): injection of 15 mg/kg gemcitabine hydrochloride (Lilly, France), intraperitoneal (ip), once weekly; (3) ATO group (n = 8): injection of 5 mg/kg ATO, ip once every other day; and (4) combination group (ATO + Gem; n = 8): 5 mg/kg ATO ip injection once every other day plus 15 mg/kg gemcitabine hydrochloride, ip injection once weekly. Tumor sizes were measured every 3 days and volumes (V) were determined by the equation: V = (L × W2) × 0.5, where L was the length and W was the width of the tumor.10 The mice were treated for 3 weeks and sacrificed on day 22. The tumors were removed and photographed. Tumor tissues were fixed in formalin for hematoxylin and eosin (H&E) staining, terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay, and immunohistochemical (IHC) staining.
IHC and TUNEL assays
Briefy 5 μm sections of formalin fixed paraffin embedded tissues were deparaffinized with xylene and rehydrated through a graded alcohol series. Heat-induced epitope retrieval was performed by immersing the tissue sections in 10 mM citrate buffer (pH 6.0) with 0.05% Tween at 98°C for 20 minutes. Slides were pretreated with 3% hydrogen peroxide at room temperature, 10 mM sodium citrate at 65°C, and equilibration buffer. Slides were exposed to terminal transferase and digoxigenin-labeled dUTP in reaction buffer for 2 hours at 37°C, stopped with wash buffer, and blocked with 10% normal goat serum. Slides were exposed to horseradish peroxidase-conjugated anti-digoxigenin secondary antibodies (Roche Applied Science, Mannheim, Germany) and 3,3′-diaminobenzidine chromagen (Dako, Glostrup, Denmark). The TUNEL assay was performed using the Apotag kit (Roche Applied Science), and the anti-human CD24 antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-human CD44 antibody (Cell Signaling Technology, Danvers, MA, USA), or anti-human aldehyde dehydrogenase 1 family, member A1 (ALDH1 A1) antibody (Santa Cruz Biotechnology) were used for tissue IHC staining according to the guidelines of the providers.10 The results were analyzed by a pathologist in a blinded fashion with Image-Pro Plus 6.0 software (Media Cybernetics, Inc, Maryland USA).11
Peptide recombination and ultraviolet absorbance spectrometry assay
Primers were designed as follows based on the published amino acid sequence of the Gli1 zinc finger domain:12,13 forward primer, CCGGAATTCTGCCGTTGGGATGGCTGCAG, and reverse primer, CCCAAGCTTAATGCACTGTCTTGA CATGTTTTCGC, with an EcoRI site and a HindIII site at the 5′ end of each primer (underlined). The gene fragment coding the zinc finger domain of Gli1 was amplified from a HeLa cell cDNA library and linked into the expression plasmid pMAL-C2X New England Biolabs (NEB) to construct the expression plasmid pMAL-C2X-Gli1-zinc-finger-motif. After confirmation by restriction enzyme digests and DNA sequencing, the expression plasmid was transformed into the BL21 E. coli strain. A high expression strain was selected and induced and the expressed recombinant protein was purified using MBP (maltose-binding proteins) affinity14,15 More information is shown in Figure S1.
Peptides were solubilized at a concentration of 25 μM in phosphate buffer (pH 7.0), and various molar equivalents of ATO were added. The solutions were placed in a 0.1 mm path length quartz cuvette. Spectra were averaged over eight scans from 240–340 nm on a ultraviolet-visible spectrophotometer (UV2800; UNICO, Shanghai, People’s Republic of China).
Confocal fluorescence microscopy
To detect the interaction between ATO and Gli, SW1990 cells were transfected with GFP-Gli1 expressing adenovirus (AD-GFP-Gli1, provided by Sunbio Medical Biotechnology Co, Ltd) according to the manufacturer’s protocol. Localization was determined by the detection of green fluorescence released from GFP tagged proteins. Twenty-four hours after adenovirus transfection, cells grown on cover glasses were treated with ATO at 2.5 μM for 12 hours. Adenovirus containing GFP (AD-GFP) was used as the negative control. Cells expressing GFP-fused constructs were labeled with 2.5 μM ReAsH-EDT2 (Invitrogen, molecular probes, Inc.) for 30 minutes at 37°C in serum-free Opti-MEM (Invitrogen). After thoroughly washing with 250 μM BAL (Invitrogen, molecular probes, Inc.) buffer, cells were live-imaged using an Olympus Fluo View FV300 confocal laser scanning biological microscope system and software (Olympus, Tokyo, Japan).
Statistics
The quantitative results were represented as the mean ± standard error of at least three independent experiments. Significant differences were determined with 2-tailed Student’s t tests performed using Office Excel 2003 or GraphPad Prism 5 Demo (GraphPad Software, San Diego, CA, USA). P-values < 0.05 were considered statistically significant.
Results
ATO induced apoptosis in CD24+CD44+ SW1990 cells in vitro
The cell proliferation assay with cck-8 demonstrated that ATO inhibited the viability of the SW1990 cell line in a time dependent manner. After 24 hour treatment, only when the ATO concentration was above 10 μM, the proliferation of SW1990 cells was able to be inhibited. However, extending the treatment time to 48 hours decreased the effective concentration to 5 μM, and after a 72 hour treatment, the viability of the cells decreased at a drug concentration of approximately 2.5 μM (Figure 1A).
Figure 1.

Arsenic trioxide induced apoptosis in CD24+CD44+ SW1990 cells.
Notes: (A) Time dependent effects of arsenic trioxide (ATO) on pancreatic cancer cell viability. (B) ATO induced apoptosis in cells expressing CD24+CD44+. The blue points indicate the CD24+CD44+ cells in the whole population. Cells were treated with 2.5 or 5 μm ATO for 72 hours.
Abbreviation: PI, propidium iodide.
Different staining patterns observed in the flow cytometry assay enabled us to identify different cell populations: live cells (both annexin V and PI negative), early apoptotic cells (annexin V positive and PI negative), late apoptotic or necrotic cells (both annexin V and PI positive), and dead cells (annexin V negative and PI positive). When compared with the vehicle group, significant differences were observed in the apoptosis of either the whole group or the CD24+CD44+ subgroup of cells after 2.5 μM or 5 μM ATO treatment. Furthermore, when compared with the whole group, the apoptosis of CD24+CD44+ cells increased approximately 2.9 or 3.4 fold (Figure 1B and Table 1), indicating that ATO may act more effectively on the CD24+CD44+ cells.
Table 1.
Apoptosis in SW1990 cells treated with arsenic trioxide (percentage, mean ± standard deviation)
| SW1990 cells | ATO concentrations
|
||
|---|---|---|---|
| 0 μm | 2.5 μm | 5 μm | |
| Total | 1.4 ± 0.3 | 3.7 ± 0.8* | 4.6 ± 1.7* |
| CD24+CD44+ | 0.7 ± 0.2 | 10.7 ± 2.4* | 15.5 ± 2.8* |
Notes: Data are from flow cytometry analysis of apoptotic cells. SW1990 cells were treated with vehicle, or 2.5, or 5 μm arsenic trioxide for 72 hours. Percentage values indicate the proportion of apoptotic cells.
Denotes P < 0.01 versus 0 μm.
Abbreviation: ATO, arsenic trioxide.
ATO plus gemcitabine synergistically inhibited tumorigenesis of pancreatic cancer cells. Based on our in vitro results and the current theories regarding CSC, we proposed that the inhibition of CSC viability by ATO could improve effects of conventional chemotherapies to retard tumor growth significantly. To investigate this idea, four management arms were delivered to mouse models established by transplantation of SW1990 xenograft tumors. In contrast to the studies performed by other investigators,16–18 the dosage of gemcitabine applied in this study (15 mg/kg weekly) was much lower. Meanwhile, the dosage of ATO (5 mg/kg every other day) was similar to other studies.19,20 When compared with the control, the combination treatment with ATO and gemcitabine, as well as the treatment with ATO or gemcitabine alone, inhibited tumor growth by 60.9% (P = 0.004), 22.8% (P = 0.205), and 23.6%(P = 0.189), respectively (Figure 2A and B). As we designed for this experiment, neither low dose gemcitabine nor ATO could inhibit the growth of tumors, but their combination could.
Figure 2.
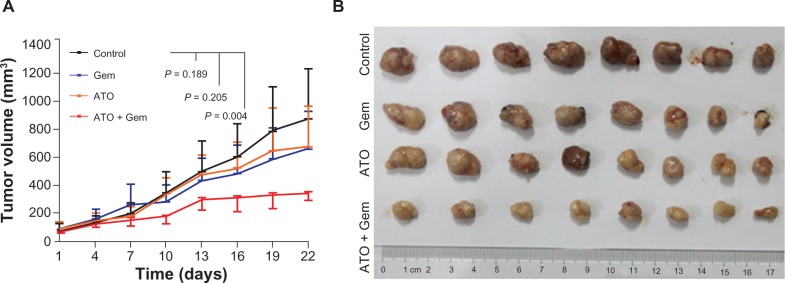
Arsenic trioxide inhibited tumorigenesis of SW1990 cells in vivo.
Notes: (A) Tumor growth chart showing the effects of each management arm. Points represent the mean values of tumor volumes in each group. The bars represent standard deviation. (B) Pictures of tumors.
Abbreviations: ATO, arsenic trioxide; gem, gemcitabine hydrochloride.
ATO inhibited viability of PCSCs in vivo
Using the TUNEL assay, apoptosis induced by the treatments could be observed in tumor tissues. According to the results, the apoptosis induced by the ATO and gemcitabine combined treatment increased by approximately 3.4, 3.6, or 5.5 fold when compared to ATO, gemcitabine alone, or the control, respectively (Figure 3A, B and Table 2). As shown by the IHC assay, although low dose gemcitabine treatment could increase CD24, CD44, and ALDH1A1 staining intensities, for the ATO and gemcitabine combination group, the staining intensities of CD24, CD44, and ALDH1A1 were stable when compared to the control, and decreased significantly when compared to the Gem group (Figure 3A, B and Table 3), indicating that ATO may act as a drug to suppress expression of the markers and inhibit viability of PCSCs.
Figure 3.

Alterations in apoptosis signaling in xenograft tumor samples and illustration of the potential mechanism.
Notes: Tumors were collected after a 3-week treatment and the prepared tissue slides were analyzed by immunohistochemistry (IHC) using anti-CD24, anti-CD44, and anti-aldehyde dehydrogenase 1 family, member A1 (AlDH1A1) antibodies, accompanied by hematoxylin and eosin and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assays. (A) representative figures of hematoxylin and eosin, The black bars are scale bars, arrows indicate positive cells. TUNEL, and IHC assays using treated tumor samples. Scale bars are 20 μm. (B) Apoptosis and relative expression of CD24, CD44, and AlDH1A1 in tumors. Apoptosis in tumor tissues were detected by TUNEL assay. Relative expression of CD24, CD44, and AlDH1A1 was detected by IHC, and analyzed with image-Pro Plus software (Media Cybernetics, Inc, Maryland, USA). in the three lower panels, MOD, mean optical density, the columns are mean values of MOD of each group, the bars represent standard deviation. *denotes P < 0.05 and **denotes P < 0.01.
Abbreviations: AlDH1A1, aldehyde dehydrogenase 1 family, member A1; ATO, arsenic trioxide; Gem, gemcitabine hydrochloride; H&E, hematoxylin and eosin; MOD, mean optical density; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling.
Table 2.
Apoptosis in tumors of each group analyzed by terminal deoxynucleotidyl transferase dUTP nick end labeling assay (percentage, mean ± standard deviation)
Notes: Tumor tissues were from mice in each group and apoptosis analysis was performed manually.
Denotes P < 0.05
denotes P < 0.01 versus the control.
Abbreviations: ATO, arsenic trioxide; gem, gemcitabine hydrochloride ; dUTP, 2’-deoxyuridine-5’-triphosphate.
Table 3.
Mean optical density analysis on the immunohistochemistry slides (×10−2, mean ± standard deviation)
| Markers | Groups
|
|||
|---|---|---|---|---|
| Control | Gem | ATO | ATO + Gem | |
| CD24 | 1.07 ± 0.33 | 1.79 ± 0.54* | 1.11 ± 0.28 | 1.04 ± 0.37 |
| CD44 | 1.05 ± 0.36 | 2.08 ± 0.48** | 0.73 ± 0.22 | 0.56 ± 0.25 |
| ALDH1A1 | 0.06 ± 0.05 | 0.37 ± 0.11** | 0.06 ± 0.04 | 0.07 ± 0.04 |
Notes: Immunohistochemistry slides were prepared with the tumors from mice in each treatment group and the mean optical density of label color (brown) in immunohistochemistry were analyzed with image-Pro Plus software (Media Cybernetics, Inc, Maryland, USA).
Denotes P < 0.05
denotes P < 0.01 versus the control. No significant difference was observed among other groups for the same marker.
Abbreviations: AlDH1A1, aldehyde dehydrogenase 1 family, member A1; ATO, arsenic trioxide; Gem, gemcitabine hydrochloride.
ATO was bound to Gli at the zinc finger domain
Using the recombinant peptide consisting of the amino acid sequence of the five zinc finger motifs of the Gli1 protein, the ultraviolet absorbance spectrometry assay showed that ATO was capable of changing the optical absorbance of the peptide. ATO alone was not able to generate absorbance in water, while the peptides generated an absorbance peak at approximately 260 nm. When various molar equivalents of ATO were added to the peptide solutions as indicated in Figure 4A, the optical absorbance of the peptides decreased at wavelengths ranging from 240 to 340 nm, suggesting that ATO changed the structure of the peptides and that changes in the binding action of ATO may occur.
Figure 4.

Arsenic trioxide binds to Gli1.
Notes: (A) Near-ultraviolet absorbance spectrometry assay of the Gli1 zinc finger peptides titrated with arsenic trioxide (ATO). The absorbance decreased with various molar equivalents of ATO added, indicating a binding action of ATO to the peptide of the Gli1 zinc finger domain. (B) Colocalization of GFP-labeled Gli1 with Reash could be weakened by ATO treatment in cultured SW1990 cells. Scale bars are 10 μm.
Abbreviations: equ, molar equivalents versus the concentration of recombinant Gli1 zinc finger peptides in the solution examined; GFP, green fluorescent protein.
Furthermore, employing the ReAsH-EDT2 reagent, the colocalization of Gli1 and ATO within cultured SW1990 cells was examined by confocal fluorescence microscopy. The results demonstrated that ReAsH-EDT2 did not bind to AD-GFP proteins but did bind to AD-GFP-Gli1, releasing a red emission that became yellow when merged with the green color of GFP (Figure 4B). Within the cells treated by ATO for 12 hours, the interaction between AD-GFP-Gli1 and ReAsH-EDT2 was weakened (Figure 4B), indicating that the binding of ReAsH-EDT2 to Gli1 might be replaced by ATO.
Discussion
ATO has been used in Chinese folk remedies for more than 2,000 years and has been approved by the US Food and Drug Administration as a second line therapy for acute promyelocytic leukemia patients who have had no response to, or who have relapsed on, all transretinoic acid therapy.21,22 Previous reports regarding ATO treatment of solid tumors have provided the evidence for several possible mechanisms, including nuclear factor-κB, the mitochondrial pathway, Bcl-2, c-Jun N-terminal kinase, Sox2, and the Notch pathway.23,24 The effect of ATO in acute promyelocytic leukemia is to induce the degradation of the promyelocytic leukemia-retinoic acid receptor α (PML-RAR) fusion protein via binding to the cysteine residue in the zinc finger domain of the PML-RAR.21 Similar to the PML-RAR protein, Gli proteins also have zinc finger domains, and there is evidence to suggest that ATO is capable of inhibiting the SHH pathway through a similar binding action as with PML-RAR. Furthermore, ATO was proposed to block SHH signaling through preventing ciliary accumulation of Gli.8,9 Data from our study indicated that ATO may replace the zinc ions in the zinc fingers of the Gli proteins, leading to protein inactivity similar to that observed for PML-RAR. In other words, the replacement of the zinc ions chelated in the zinc finger motifs of Gli proteins by arsenic may be the key factor that allows ATO to act as a SHH inhibitor.
Pancreatic cancer is a highly lethal disease. The 5-year survival rate for pancreatic cancer is 4% compared with 86% for a US patient diagnosed with breast cancer and 97% for a US patient diagnosed with prostate cancer.25 Preclinical and clinical evidence exists for the important role SHH signaling has in pancreatic tumorigenesis with aberrant expression of SHH ligands occurring in 70% of cases.26 It was described that SHH secreted signaling proteins are overexpressed in CSC pools.3 For this reason, the SHH pathway could be an interesting target for trials of PCSC eradication strategies. ATO demonstrated potent antiproliferative and proapoptotic effects in cultured pancreatic cancer cell lines, however, the effects of ATO for treating pancreatic cancer patients are still unclear despite the promising in vitro data.27
Gemcitabine is a deoxycytidine analog and is widely used as the first line treatment for unresectable pancreatic cancer. However, the median overall survival of gemcitabine treated patients with advanced pancreatic cancer is still only 5–6 months.28 To improve the prognosis of patients, low dose gemcitabine therapy combined with radiotherapy or other chemotherapeutic agents has been used, but with few exceptions, no improvements in the overall survival rates of patients with advanced pancreatic cancer have been reported.29,30 In this study, we used ATO as a SHH inhibitor together with low dose gemcitabine for in vivo management SW1990 xenografts. The results showed that although ATO or low dose gemcitabine alone could not inhibit tumor growth, the combination of ATO and gemcitabine did. Also, ATO treatment stabilized the expression of CSC markers including CD24, CD44, and ALDH1A1 either when used alone or when used together with gemcitabine, while gemcitabine alone provoked, indicating that ATO may have the potential to inhibit the viability of PCSCs in vivo.
Different chemical structures generate different ultraviolet absorbance spectra, and we used an ultraviolet absorbance spectrometry assay for detecting the structural change of peptides.31 To discover whether the effects of ATO were related to SHH-Gli proteins, we applied the recombinant peptides of Gli1 zinc finger motifs to perform an ultraviolet absorbance spectrometry assay. The results suggested that the Gli1 zinc finger peptides could generate ultraviolet absorbance in water at wavelengths ranging from 240 to 340 nm, but ATO did not. However, when ATO was added to the peptide solution, the ultraviolet absorbance decreased, which means that ATO changed the original structure of the peptides via binding to them. To verify whether the binding action of ATO could occur in cells, we commissioned a biological company (Sunbio Medical Biotechnology Co Ltd, Shanghai, People’s Republic of China) to construct a GFP-Gli1 expressing adenovirus and transfect cultured SW1990 cells. The cells were then treated with ReAsh which is an arsenic dye. Under the confocal fluorescence microscope, not only was ReAsh discovered to bind the recombinant protein to weaken ReAsh’s action, but also ATO, which suggested that Gli proteins may be the target of ATO treatment.
In summary, the results provide the evidence that ATO has the potential to inhibit viability of PCSCs via binding to SHH-Gli proteins in vitro and in vivo, although only one pancreatic cell line was investigated in this study. Our data also suggested further investigation into the use of ATO as a SHH inhibitor in combination with low dose gemcitabine for pancreatic cancer cases is needed.
Supplementary figure
Gli1 zinc finger peptide synthesis.
Notes: (A) The amino acid sequence of the five zinc finger motifs of the Gli1 protein. The “–” symbol indicates a gap. (B) Restriction enzyme analysis of the recombinant plasmid pMAL-C2X-Gli1-Zf by EcoRI and HindIII. Lane M1, Dl-2000 DNA marker; lane 1, products of the recombinant pMAL-C2X-Gli1-Zf plasmid digested with EcoRI and HindIII. (C) Polymerase chain reaction analysis of the cloned strains. Lane M2, DL-2000 DNA marker, lanes 1–6, products of the six clones. (D) SDS-PAGE: Lane M3, TaKaRa protein marker (broad); lane 1, uninduced cell pellet; lane 2, induced cell pellet containing the Gli1 zinc finger motifs as indicated by the arrow.
Abbreviations: Zfs, zinc finger motifs; SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis; Bp, base pair; KD, kilo-Dalton.
Acknowledgments
This work was supported by grants from the China Postdoctoral Science Foundation (numbers 2011M500735 and 2012T50398) and the National Natural Science Foundation of China (number 81072942).
Footnotes
Disclosure
The authors report no conflict of interest in this work.
References
- 1.Valent P, Bonnet D, De Maria R, et al. Cancer stem cell definitions and terminology: the devil is in the details. Nat Rev Cancer. 2012;12(11):767–775. doi: 10.1038/nrc3368. [DOI] [PubMed] [Google Scholar]
- 2.Pantic I. Cancer stem cell hypotheses: impact on modern molecular physiology and pharmacology research. J Biosci. 2011;36(5):957–961. doi: 10.1007/s12038-011-9155-5. [DOI] [PubMed] [Google Scholar]
- 3.Li C, Heidt DG, Dalerba P, et al. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67(3):1030–1037. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- 4.Singh BN, Fu J, Srivastava RK, Shankar S. Hedgehog signaling antagonist GDC-0449 (vismodegib) inhibits pancreatic cancer stem cell characteristics: molecular mechanisms. PLoS One. 2011;6(11):e27306. doi: 10.1371/journal.pone.0027306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taipale J, Beachy PA. The Hedgehog and Wnt signalling pathways in cancer. Nature. 2001;411(6835):349–354. doi: 10.1038/35077219. [DOI] [PubMed] [Google Scholar]
- 6.Rodova M, Fu J, Watkins DN, Srivastava RK, Shankar S. Sonic Hedgehog signaling inhibition provides opportunities for targeted therapy by sulforaphane in regulating pancreatic cancer stem cell self-renewal. PLoS One. 2012;7(9):e46083. doi: 10.1371/journal.pone.0046083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim J, Aftab BT, Tang JY, et al. Itraconazole and arsenic trioxide inhibit Hedgehog pathway activation and tumor growth associated with acquired resistance to smoothened antagonists. Cancer Cell. 2013;23(1):23–34. doi: 10.1016/j.ccr.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim J, Lee JJ, Kim J, Gardner D, Beachy PA. Arsenic antagonizes the Hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proc Natl Acad Sci U S A. 2010;107(30):13432–13437. doi: 10.1073/pnas.1006822107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beauchamp EM, Ringer L, Bulut G, et al. Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking Hedgehog/Gli pathway. J Clin Invest. 2011;121(1):148–160. doi: 10.1172/JCI42874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jimeno A, Feldmann G, Suarez-Gauthier A, et al. A direct pancreatic cancer xenograft model as a platform for cancer stem cell therapeutic development. Mol Cancer Ther. 2009;8(2):310–314. doi: 10.1158/1535-7163.MCT-08-0924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang CJ, Zhou ZG, Holmqvist A, et al. Survivin expression quantified by Image Pro-Plus compared with visual assessment. Appl Immunohistochem Mol Morphol. 2009;17(6):530–535. doi: 10.1097/PAI.0b013e3181a13bf2. [DOI] [PubMed] [Google Scholar]
- 12.Nakashima M, Tanese N, Ito M, et al. A novel gene, GliH1, with homology to the Gli zinc finger domain not required for mouse development. Mech Dev. 2002;119(1):21–34. doi: 10.1016/s0925-4773(02)00291-5. [DOI] [PubMed] [Google Scholar]
- 13.Zhang F, Jetten AM. Genomic structure of the gene encoding the human Gli-related, Kruppel-like zinc finger protein GLIS2. Gene. 2001;280(1–2):49–57. doi: 10.1016/s0378-1119(01)00764-8. [DOI] [PubMed] [Google Scholar]
- 14.Foster KW, Ren S, Louro ID, et al. Oncogene expression cloning by retroviral transduction of adenovirus E1A-immortalized rat kidney RK3E cells: transformation of a host with epithelial features by c-MYC and the zinc finger protein GKLF. Cell Growth Differ. 1999;10(6):423–434. [PubMed] [Google Scholar]
- 15.Cui YB, Cai HX, Zhou Y, et al. Cloning, expression, and characterization of Der f 7, an allergen of dermatophagoides farinae from China. J Med Entomol. 2010;47(5):868–876. doi: 10.1603/me09180. [DOI] [PubMed] [Google Scholar]
- 16.Bruns CJ, Shrader M, Harbison MT, et al. Effect of the vascular endothelial growth factor receptor-2 antibody DC101 plus gemcitabine on growth, metastasis and angiogenesis of human pancreatic cancer growing orthotopically in nude mice. Int J Cancer. 2002;102(2):101–108. doi: 10.1002/ijc.10681. [DOI] [PubMed] [Google Scholar]
- 17.Hennig R, Ventura J, Segersvard R, et al. LY293111 improves efficacy of gemcitabine therapy on pancreatic cancer in a fluorescent orthotopic model in athymic mice. Neoplasia. 2005;7(4):417–425. doi: 10.1593/neo.04559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ito D, Fujimoto K, Mori T, et al. In vivo antitumor effect of the mTOR inhibitor CCI-779 and gemcitabine in xenograft models of human pancreatic cancer. Int J Cancer. 2006;118(9):2337–2343. doi: 10.1002/ijc.21532. [DOI] [PubMed] [Google Scholar]
- 19.Jutooru I, Chadalapaka G, Sreevalsan S, et al. Arsenic trioxide downregulates specificity protein (sp) transcription factors and inhibits bladder cancer cell and tumor growth. Exp Cell Res. 2010;316(13):2174–2188. doi: 10.1016/j.yexcr.2010.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ahn RW, Chen F, Chen H, Stern ST, et al. A novel nanoparticulate formulation of arsenic trioxide with enhanced therapeutic efficacy in a murine model of breast cancer. Clin Cancer Res. 2010;16(14):3607–3617. doi: 10.1158/1078-0432.CCR-10-0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang XW, Yan XJ, Zhou ZR, et al. Arsenic trioxide controls the fate of the PML-RARalpha oncoprotein by directly binding PML. Science. 2010;328(5975):240–243. doi: 10.1126/science.1183424. [DOI] [PubMed] [Google Scholar]
- 22.Beauchamp EM, Uren A. A new era for an ancient drug: arsenic trioxide and Hedgehog signaling. Vitam Horm. 2012;88:333–354. doi: 10.1016/B978-0-12-394622-5.00015-8. [DOI] [PubMed] [Google Scholar]
- 23.Sun H, Zhang S. Arsenic trioxide regulates the apoptosis of glioma cell and glioma stem cell via down-regulation of stem cell marker Sox2. Biochem Biophys Res Commun. 2011;410(3):692–697. doi: 10.1016/j.bbrc.2011.06.060. [DOI] [PubMed] [Google Scholar]
- 24.Zhen Y, Zhao S, Li Q, Li Y, Kawamoto K. Arsenic trioxide-mediated Notch pathway inhibition depletes the cancer stem-like cell population in gliomas. Cancer Lett. 2010;292(1):64–72. doi: 10.1016/j.canlet.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 25.Nakamura K, Sasajima J, Mizukami Y, et al. Hedgehog promotes neovascularization in pancreatic cancers by regulating Ang-1 and IGF-1 expression in bone-marrow derived pro-angiogenic cells. PLoS One. 2010;5(1):e8824. doi: 10.1371/journal.pone.0008824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kelleher FC. Hedgehog signaling and therapeutics in pancreatic cancer. Carcinogenesis. 2011;32(4):445–451. doi: 10.1093/carcin/bgq280. [DOI] [PubMed] [Google Scholar]
- 27.Kindler HL, Aklilu M, Nattam S, Vokes EE. Arsenic trioxide in patients with adenocarcinoma of the pancreas refractory to gemcitabine: a phase II trial of the University of Chicago Phase II consortium. Am J Clin Oncol. 2008;31(6):553–556. doi: 10.1097/COC.0b013e318178e4cd. [DOI] [PubMed] [Google Scholar]
- 28.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2007. CA Cancer J Clin. 2007;57(1):43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 29.Ciliberto D, Botta C, Correale P, et al. Role of gemcitabine-based combination therapy in the management of advanced pancreatic cancer: a meta-analysis of randomised trials. Eur J Cancer. 2013;49(3):593–603. doi: 10.1016/j.ejca.2012.08.019. [DOI] [PubMed] [Google Scholar]
- 30.Ko AH, Dito E, Schillinger B, et al. A phase II study evaluating bevacizumab in combination with fixed-dose rate gemcitabine and low-dose cisplatin for metastatic pancreatic cancer: Is an anti-VEGF strategy still applicable? Invest New Drugs. 2008;26(5):463–471. doi: 10.1007/s10637-008-9127-2. [DOI] [PubMed] [Google Scholar]
- 31.Derrick TS, Kashi RS, Durrani M, Jhingan A, Middaugh CR. Effect of metal cations on the conformation and inactivation of recombinant human factor VIII. J Pharm Sci. 2004;93(10):2549–2557. doi: 10.1002/jps.20167. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Gli1 zinc finger peptide synthesis.
Notes: (A) The amino acid sequence of the five zinc finger motifs of the Gli1 protein. The “–” symbol indicates a gap. (B) Restriction enzyme analysis of the recombinant plasmid pMAL-C2X-Gli1-Zf by EcoRI and HindIII. Lane M1, Dl-2000 DNA marker; lane 1, products of the recombinant pMAL-C2X-Gli1-Zf plasmid digested with EcoRI and HindIII. (C) Polymerase chain reaction analysis of the cloned strains. Lane M2, DL-2000 DNA marker, lanes 1–6, products of the six clones. (D) SDS-PAGE: Lane M3, TaKaRa protein marker (broad); lane 1, uninduced cell pellet; lane 2, induced cell pellet containing the Gli1 zinc finger motifs as indicated by the arrow.
Abbreviations: Zfs, zinc finger motifs; SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis; Bp, base pair; KD, kilo-Dalton.