

Abstract
Purpose of the review
TNF inhibitors are effective for achieving disease control in several inflammatory diseases. Although anti-TNF agents can inhibit bone loss in vitro, their role in the prevention of clinically relevant outcomes such as osteoporosis and fractures has not been clearly established.
Recent findings
There are many studies of the effects of TNF inhibitors on markers of bone turnover; however few have measured bone mineral density (BMD) or fractures. Most of these studies have small sample sizes and a minority had a placebo control group. Overall these studies suggest that the anti-resorptive effects of anti-TNF therapy are related to control of disease activity.
Summary
The antiresorptive effects of TNF inhibitors are likely related to their anti-inflammatory properties. Studies to date have not demonstrated any advantages of TNF inhibitors over traditional non biologic therapies in the prevention of bone loss and fractures.
Keywords: Anti-TNF, bone loss, rheumatoid arthritis, spondyloarthropaties
Introduction
It is established that patients with chronic inflammatory diseases have more bone loss and a higher risk of fractures compared to the general population [1–5]. Although the causes of bone loss in inflammatory disorders are multiple [6,7], several animal models of inflammation, along with clinical evidence, indicate that inflammatory mediators, including tumor necrosis factor alpha (TNFα), play a major role [8–11].
Numerous in vivo and in vitro experiments provide evidence that TNFα promotes bone resorption directly through activation of cells of the osteoclast lineage [12], and indirectly through the expression of osteoclast activators [13]. TNFα also suppresses bone formation via increased osteoblast apoptosis [14], and reduced differentiation [15] and proliferation [16] of osteoblasts and their progenitors. Thus, TNFα blockade holds the potential to inhibit or reverse bone loss [17–19]. In this regard, anti-TNF therapy has been proposed as a potential dual treatment to control inflammation and to prevent osteoporosis and associated fractures in inflammatory diseases. In this review only pathways that can potentially be targeted by anti-TNF agents are described. Extended reviews that include detailed information of the in vitro and in vivo experiments regarding the effect of different inflammatory cytokines on bone loss are available [20–22].
Bone remodeling
Bone remodeling is a physiological process that repairs microdamage, adapts bone strength to changes in mechanical load, and is critically involved in systemic calcium and phosphate homeostasis. The regulation of this process involves a complex network of multiple signaling pathways within and among the skeletal, immune, endocrine systems and the brain [23–25]. In the classical model, bone remodeling is initiated when osteoclasts resorb damaged or excess bone [26]. Under normal conditions the resorption is closely coupled to bone formation by osteoblasts that refill the resorption pit or Haversian canal. More recently, this model has been expanded to include the osteocyte [27]. Osteocytes embedded in the bone matrix sense and respond to changes in fluid flow and/or mechanical stretching arising from stress, strain or pressure created by mechanical loads on the bone [28, 29] and potentially local microdamage by releasing soluble signal molecules that control bone resorption and formation [30]. In this manner, the osteocyte is thought to orchestrate the processes of bone remodeling at the local level. Hence, perturbation of the delicate balance of coupled bone formation and resorption leads to osteopetrosis, or more commonly, osteoporosis.
Bone resorption requires the proliferation and differentiation of multipotent hemapoietic stem cells into macrophages, which fuse into multinucleated preosteoclasts and further differentiated into mature, active osteoclasts. Macrophage colony stimulating factor (M-CSF) and receptor activator of nuclear factor kappa beta ligand (RANKL), a member of the TNF superfamily, are considered the most critical signaling molecules for osteoclastogenesis [31, 32]. Interaction of M–CSF with its membrane receptor c-FMS is necessary for proliferation and survival of osteoclast precursors [33, 34], priming the bone microenvironment for osteoclast differentiation. Subsequently, RANKL must bond to its receptor RANK on osteoclast progenitors to activate the NF-κ and activator protein (AP) -1 pathways, which trigger fusion, differentiation, and activation of the mature osteoclasts [35]. RANK is expressed on the surface of osteoclast lineage cells; while M-CSF, RANKL, and osteoprotegerin (OPG), a soluble decoy receptor specific for RANKL, are all produced by osteoblastic cells [36]; the balance of the RANKL-RANK-OPG axis is a key regulator of bone resorption.
In healthy subjects bone formation is tightly coupled to bone resorption by a poorly understood mechanism that involves signaling directly between osteoblasts and osteoclasts via the Ephrin family of membrane bound ligands and receptors [37]. Regardless, osteoblast recruitment and differentiation are directly influenced by numerous hormones, cytokines, and growth factors including numerous members of the TGFβ-superfamily, the WNT superfamily, and the TNF superfamily. These and other autocrine/paracrine signals influence the activation, commitment, and differentiation of mesenchymal stem cells and osteoblast progenitors into mature osteoblasts by regulating the expression and activity of several key transcription factors including runt-related transcription factor 2 (RUNX2) [38], the interaction of osterix-NFAT2 (nuclear factor for activated T cells), β-catenin [23, 25], and activating transcription factor 4 (ATF4). Though not entirely sufficient for osteoblastogenesis, RUNX2 is generally considered the master regulator of osteoblast differentiation since it is required for the differentiation of mesenchymal cells into osteoblasts [39].
TNFα and osteoclastogenesis
Activation of the NFκβ pathway by RANK/RANKL signaling is a key driver of osteoclastogenesis and bone formation. Activation of NFκB is also a hallmark of TNFα signaling via TNF receptor 1 (TNFR1) [40], which is expressed on macrophages and osteoclast precursors. Hence, it is not surprising that TNFα also promotes osteoclast formation and bone resorption. TNFα enhances osteoclast differentiation in the presence of minimal concentrations of RANKL [41] and induces the differentiation of pro-osteoclasts into mature osteoclasts in the absence of RANK signaling [42]. During inflammation, TNFα increases the expression of M-CSF and RANKL in several target cells including osteoblasts [43] which promotes osteoclast differentiation indirectly. Finally, TNFα has also been shown to inhibit osteoclast apoptosis through mTOR/S6 kinase [44]. Together, these mechanisms increase the number, and potentially, the lifespan of osteoclasts in pro-inflammatory environments resulting in elevated bone resorption (Figure)
Figure. Effects of Tumor Necrosis Factor α in Bone Cells.
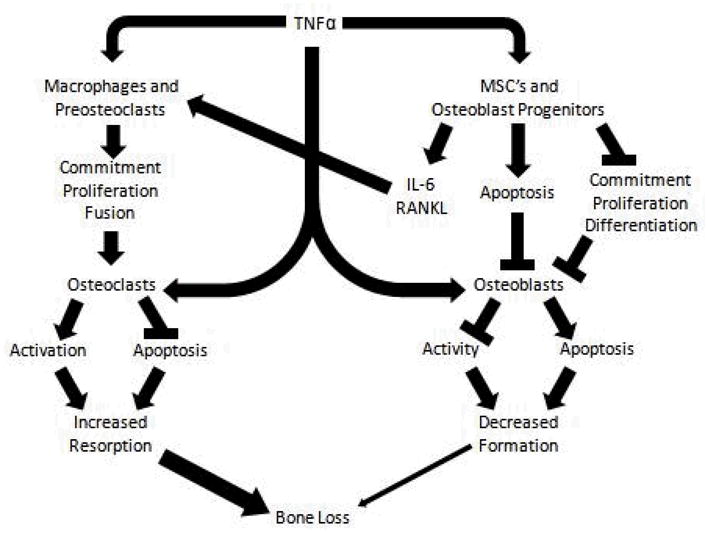
Tumor Necrosis Factor α (TNFα) causes bone resorption by the activation of osteoclastogenesis and the inhibition of osteoblastogenesis. TNFα enhances osteoclastogenesis through the following main pathways: 1) direct effect of TNFα on macrophages and pre-osteoclast cell proliferation or indirectly through the activation of IL6 and RANKL signaling; 2) direct activation of mature osteoclasts and 3) inhibition of osteoclast apoptosis. TNFα suppresses osteoblastogenesis by: 1) inhibition of the proliferation and differentiation of mesenchymal stem cells into osteoblastic cells; 2) inducing apoptosis of osteoblast linage cells; and 3) suppressing osteoblast activity.
TNFα and osteoblastogenesis
Although an increase in bone resorption, rather than a decrease in bone formation, is the main mechanism for bone loss during active inflammation [45], TNFα can disrupt the main pathways involved in osteoblast differentiation. A variety of in vitro and animal models have demonstrated that excess TNFα inhibits proliferation and differentiation and increases apoptosis of osteoblasts and their progenitors (Figure). These effects appear to be mediated primarily via TNFR1 activation of multiple downstream signaling pathways [46–50•,51]. The best studied of these is the nuclear factor κB (NF-κB) pathway which suppresses osteoblast differentiation [40] and activity [35]. Traditional downstream targets of NFκβ, including p53 and p21 have also been implicated as important mediators of these effects [52•,53]. Inhibited osteoblast differentiation may be mediated by reduced RUNX2 expression [54] through its ubiquitylation [48], and also through inactivation of pro-osteogenic mitogen-activated protein kinases (MAPK) pathways [25]. TNFα signaling also inhibits osterix expression, a critical regulator of the early stages of osteoblast differentiation, preventing the interaction of NFATc2 with osterix and the resulting activation of osteogenic target genes [55]. Others have demonstrated inhibition of the Wnt- β-catenin pathway through TNFα-induced upregulation of the Wnt inhibtors Dickkopf-related protein 1 (DKK1) [56] and sclerostin [57••].
Clinical studies on anti-TNF agents and
Clinical trials have shown equivalence or superiority of anti-TNF therapy compared to traditional non-biologic regimens in achieving disease control and preventing radiographic joint damage in different inflammatory diseases [58–61]. However, studies that examined the effect of these agents on bone health have primarily measured markers of bone turnover rather than clinically important endpoints. These studies of biochemical markers have not always yielded concordant results in different autoimmune diseases [62–66], but overall have shown modest and transitory increases in bone formation and decreases in bone resorption markers with anti-TNF therapy. These findings support the hypothesis that anti-TNF therapy has little effect on osteoblastogenesis, but primarily affects osteoclastogenesis, improving the bone formation/resorption ratio and thus slowing or arresting bone loss [63,64,67]. However, information about the comparative effects of long term anti-TNF therapy and conventional non biologic regimens on bone distant to sites of inflammatory damage are limited.
In this section we present evidence from clinical studies that examined the effect of anti-TNF therapy on preservation of bone mineral density (BMD) (including local and generalized bone loss) and on the occurrence of fractures compared to non biological treatments for rheumatoid arthritis (RA) and spondyloarthropaties (Table 1 & 2) [67–92].
Table 1.
Anti tumor necrosis factor (anti-TNF) therapy and bone mineral density in rheumatoid arthritis
| Ref | Study | FU time | Treatment groups (n) | BMD Hands | BMD Spine | BMD Hip | Findings |
|---|---|---|---|---|---|---|---|
| [67] | Obs | 1 year | T1:INF (26) | ND | BMD increased (+ 3%, P<0.01) | BMD increased (12%, P<0.001) | Δmarkers of bone turnover did not correlated with ΔBMD, but they changed in the expected direction. |
| [68] | Obs | 1 year | T1:INF (36) | ND | Stable BMD (+1.1%, P>0.05) | Stable BMD (−0.3%, P>0.05) | Disease activity improved but ΔBMD were not associated with disease activity, and use of prednisone or bisphosphonates |
| [69] | Obs | 1 year | T1: INF (102) | BMD loss (−0.8%, P<0.05) Response vs. non-response (−0.6% vs. −1.2%, P>0.05) |
Stable BMD (+0.2% ,P>0.05) Response vs. no-response (+0.7% vs. −0.6%, P>0.05) |
Stable BMD (−0.20% , P>0.05) Response vs. non-response (+0.8% vs. −0.7%, P<0.001) |
Osteocalcin increased at week 14 and then remained stable. β-CTx and RANKL decreased during follow-up; but OPG did not change. PDR use was not associated with ΔBMD |
| [70] | Obs | >2 years | T1:INF (52) | BMD loss (−3.1%, P<0.05) | BMD increased (+2.6%, P<0.05) PDR users vs. no PDR users (+6.2% vs. 0.8%, P=0.002) RA≤1 year vs. RA>10 years (+5.5% vs. −0.2%, P=0.009) |
BMD stable (−0.1%, P>0.05) | ΔBMD in spine was associated with concurrent use of prednisone and RA duration |
| [71•] | Obs | 5285.2 py | T1: anti-TNF T2: MTX T3: other DMARD |
Risk of wrist fractures T1≈T2≈T3 | ND | Risk of hip fractures T1≈T2≈T3 | Risk of non-vertebral fracture was similar between treatment groups. |
| [72] | OL | 6 – 12 months | T1:INF (48) | ND | BMD stable | BMD stable | Markers of bone formation remained stable. Markers of bone resorption decreased the first 6 months of treatment but returned to baseline at 1 year |
| [73] | OL | 1 year | T1:INF/ADA (19) | ND | BMD stable | ND | Inflammatory markers and disease activity improved significantly. |
| [74] | OL | 1 year | T1: ADA(46) | ND | BMD stable +0.3% (P>0.05) | BMD stable +0.3% (P>0.05) | Disease activity significantly improved during follow-up. ΔBMD at hip were associated with concomitant use of PDR. |
| [75] | Cohort | 1 year | T1: INF + MTX (90) T2: MTX (99 historical controls) |
ND | BMD stable in T1 (−0.2% , P>0.05) BMD decrease in T2 (−3.9%, P<0.001) |
BMD stable in T1 (+0.2%, P>0.05) BMD decrease inT2 (−2.5%, P<0.001) |
Markers of bone turnover remained stable in both treatment groups. In the INF treated group, ΔBMD were not associated with treatment response. |
| [76] | RCT | 6 months | T1: INF + MTX + PDR (10) T2:ETA + MTX + PDR (11) T3: MTX + PDR (10) |
BMD stable T1 & T2 (+1.3%, P>0.05) T3 (−4.6%, P>0.05) |
BMD stable T1 & T2 (+0.2%, P>0.05) T3 (− 0.8%, P>0.05) |
BMD stable T1 & T2 (+ 0.1, P>0.05), T3 (− 0.6% , p>0.05) |
In the anti-TNF treated group, markers of bone formation increased while markers of bone resorption decreased. In the MTX+PDR treated group no changes in markers of bone turnover were observed |
| [77] | RCT | 1 year | T1 : INF + MTX (10) T2 : MTX + placebo (10) |
BMD loss (−2.4%, P=0.048) ΔBMD T1≈T2 (−2.1 vs. −2.8%, P=0.82) |
BMD stable (+ 1.3% , P=0.36) ΔBMD T1≈T2 (−0.8% vs. −1.8%, P=0.71) |
BMD femoral neck & hip stable −1.8%, P=0.07 & −1.4%, P=0.07 ΔBMD femoral neck T1 ≪T2 (−0.4% vs. −3.4%, P=0.01) ΔBMD total hip T1≪T2 (−0.2% vs. −2.6%, P=0.03) |
BMD loss was lower INF compared to placebo in femoral neck and hip. Inflammation was associated with bone loss in hands and femoral neck. Radiographic damage was associated with bone loss at spine, femoral neck/hip. |
| [78] | RCT | 26, 52, 104 weeks | T1: ADA+MTX (261) T2: ADA (261) T3: MTX (246) |
BMD loss T3>T2>T1 | ND | ND | Bone loss was associated with age, inflammation and treatment regimen |
| [79••] | RCT | 52, 104 weeks | T1: ADA+MTX (214) T2: MTX (188) |
T2: bone loss higher in those with high/moderate disease activity vs. low/remission T1: bone loss similar in high/moderate disease activity and low/remission. |
ND | ND | In MTX groups bone loss was higher non responders compared to responders, but no differences in the combination group In combination group BMD loss was comparable to MTX group on remission. |
| [80] [81] | RCT | 1–2 years | T1: Sequential monotherapy (81) T2: Step-up combination (84) T3 : Combination + PDR (89) T4 :Combination + INF (88) |
BMD loss at year 1 & 2 T3 & T4≪T1 &T2 (all P=0.05) BMD loss associated with disease severity |
BMD loss T1≈T2≈T3≈T4 |
BMD loss T1≈T2≈T3≈T4 | BMD loss higher in hand than hip & spine BMD loss associated with progression of radiographic destruction Biphosphonates associated with reduce BMD loss in spine and hip. |
| [82••] | RCT | 1 year | T1: Continuous remission T2:Low disease T3: High disease |
BMD gain in T1 but not in T2 and T3 | ND | ND | BMD was not associated with previous or current use of anti-TNF or prednisone. |
Abbreviations: Ref, references; FU, follow-up; BMD, bone mineral density; ND, not done; Δ, changes; ≈, similar; Obs, observational; OL, open label; RCT, randomized clinical trial; INF, infliximab; ADA, adalimumab; MTX, methotrexate; PDR, prednisone; OPG, osteoprogetegerin; β-CTx, beta carboxy-terminal cross-linking telopeptide of type I collagen; RANKL, receptor activator of nuclear factor kappa beta ligand; DMARD: disease modifying anti rheumatic drug; vs., versus.
Table 2.
Anti-tumor necrosis factor (anti-TNF) therapy and bone mineral density in Spondyloarthropaties
| Ref | Study | FU time | Treatment groups (n) | Spine BMD | Hip BMD | Findings |
|---|---|---|---|---|---|---|
| [83] | Obs SpA | 6 months | T1: INF (29) | BMD increased (+3.6%,P=0.001) | BMD total hip increased (+2.2%, p=0.0012) BMD trochanter increased (+2.3%, P=0.00012) BMD total neck stable (+1.1%, P=0.19) |
No changes in BMD with concomitant use of glucocorticoids. Osteocalcin increased at 6 weeks but no differences at 6 months. |
| [84] | Obs CD | Mean 23±11months | T1: INF (15) T2: conventional treatment (30) |
BMD increased T1≫T2 (+8.1% vs.+ 1.0%, P<0.01) |
BMD increased Left hip T1≈T2 (+2.7% vs. +1.3%, P>0.05) Right hip T1≫T2 (+5.6% vs. −0.2%, P<0.05) |
In the INF treated group, ΔBMD in spine was not associated with glucocorticoid use |
| [85] | Obs CD | Mean 2.2 ± 0.99 y | T1: INF (23) T2: conventional treatment (38) |
No biphosphonate, BMD loss T1≈T2 (−4.1% vs. −3.3%, P>0.05) Biphosphonate , BMD loss T1≈T2 (+4.4% vs. +2.0%, P>0.068) Biphosphonate users vs. non-users (+4.0% vs. −3.7%, P<0.001) |
ND | Biphosphonates was associated with spine BMD gain, but effect was partially inhibited by concomitant use of glucocorticoids. INF had a marginal effect in BMD only in those patients that used bisphosphonates |
| [86] | Obs AS | Mean range 13.5 to 15.6 months | T1: conventional treatment (40) T2: biphosphonate (20) T3: anti-TNF (19) T4: anti-TNF+biphosphonate (11) |
BMD increased in all treatment, but marginally significant in T2. | BMD remained stable in all treatment groups, but in T4 BMD increased at trochanter | ΔBMD at trochanter correlate with Δinflammatory markers. In patients without syndesmophytes, ΔBMD at spine and total hip was different between treatments, ΔBMD at femoral neck correlate with Δinflammatory markers. |
| [87] | OL CD | 1 year | T1: INF (46) | ΔBMD similar in all groups BMD increased (2.4%, P=0.002) | BMD increased at trochanter and femoral neck (+2.8%, P=0.03 and +2.6%, P=0.001 respectively) | ΔBMD at spine and femoral neck was independent of glucocorticoids use. |
| [88] | OL SpA | 6 months | T1: ETA (10) T2: SSZ/NSAIDs (10) |
ΔBMD T1≈T2 (+1.1% vs. −1.4%, P=0.19) | ΔBMD femoral neck T1≈T2 (+0.2% vs. −1.5%, P=0.34) ΔBMD total hip T1>T2 (+1.6% vs. −1.3%, P=0.027) |
Measures of disease activity improved in ETA treated group but not in controls. |
| [89] [90] | OL SpA | 1 -2 years | T1:INF/ETA ( n=19) T1:INF/ETA ( n=106) |
BMD increased | BMD hip increased | Markers of bone resorption decreased early and remained low during treatment, but markers of bone formation only increase temporarily (3 months) but return to baseline thereafter. |
| [91] | RCT AS | 24 weeks | T1: INF (201) T2: placebo (78) |
BMD increased T1≫T2 (+2.5% vs. +0.5%, P<0.001) | BMD increased T1≫T2 (0.5% vs. +0.2%, P=0.033) | In the INF treated group ΔBMD in spine was not associated with Δmarkers of bone turnover; but ΔBMD in hip was associated with baseline osteocalcin and bone alkaline phosphatase, and inversely correlated with changes in carboxy –terminal collagen crosslinks |
| [92] | RCT | 30 weeks | T1 : MTX +INF (28) T2 : MTX+ Placebo (14) |
ΔBMD T1≈T2 (+3.6% vs. −1.3%, P=0.06) | ΔBMD total hip T1≈T2 (+1.9% vs. +0.1%, P=0.14) ΔBMD femoral neck T1≈T2 (+2.5% vs. −1.3%, P=0.09) |
BMD increased significantly in INF but not in the MTX monotherapy treated group, ΔBMD were similar in spine, hip and femoral neck |
Abbreviations: Ref, references; FU, follow-up; BMD, bone mineral density; ND, not done; Δ, changes; ≈, similar; Obs, observational; OL, open label; RCT, randomized clinical trial; SpA, spondyloarthropathies; CD, Crohn’s disease;AS, ankylosing spondylitis; INF, infliximab; ETA, etanercept; SSZ, sulfasalazine; NSAIDs, non steroidal anti-inflammatory drugs; MTX, methotrexate; Rx, treatment; vs., versus;
Anti-TNF agents and bone loss in patients with rheumatoid arthritis (RA)
Most of the inflammatory diseases treated with anti-TNF therapy are associated with systemic bone loss. In RA, and to a lesser extent in psoriatic arthritis, periarticular bone loss (which occurs independently of direct contact with inflamed synovium) is often present in the small joints of the hands [93]. It appears that periarticular and generalized bone loss share similar mechanisms. Cytokines are released from the inflammatory pannus and delivered locally to the metacarpal bones, and systemically to the skeleton, activating osteoclastogenesis and bone resorption. Bone loss in the hands has been used as an outcome in some studies because it has been associated with generalized bone loss [94•] and osteoporotic fractures [95]. Progression of hand bone loss during anti-TNF therapy has been observed in several studies [69,77–80]. Dissociation in the anti-resorptive effect of these agents in the hands and in the spine or hip has been described, suggesting that metacarpal bone is more sensitive to inflammatory cytokines that are released from adjacent inflammatory tissue than bone in the hip and spine [69,70,77].
We found only two large randomized clinical trials (RCT) that examined the differences between anti-TNF therapy and other traditional disease modifying anti-rheumatic drugs (DMARDs) regimens on bone loss, the BeSt and the PREMIER trial. The BeSt trial examined the effect of four treatment regimens on bone loss using an intention to treat analysis: sequential monotherapy, step-up therapy, initial combination of synthetic DMARD + high dose of prednisone, and initial combination of synthetic DMARD + infliximab [81]. In this study, patients who received conventional therapy had higher bone loss in the hands than those who receive initial combination therapy with infliximab. This difference remained significant after adjusting for the use of antiresorptive treatments, but disappeared when change in disease activity was included in the analysis. Only radiographic disease progression, post-menopausal status, and inflammation were associated with hand bone loss after adjustment for various covariates [81]. A post-hoc analysis of the data grouping patients by therapeutic response instead of treatment regimen, found that gain in metacarpal bone occurred in association with clinical remission, regardless of the previous or current use of anti-TNF therapy [82••], supporting the hypothesis that anti-TNF therapy decreases bone loss through tight control of disease activity.
However a second well powered RCT, the PREMIER trial, reported in an exploratory analysis that combination of MTX with adalimumab resulted in less hand bone loss than either adalimumab or MTX monotherapies. In this trial, hand bone loss was associated with no use of adalimumab, increasing age and inflammation [78]. A sub-analysis of the data showed that patients in the MTX monotherapy group with high/moderate disease activity (DAS28>3.2) or high levels of C reactive protein (CRP≥10 ng/ml) had greater bone loss in the hands compared to those in remission/low disease activity (DAS≤3.2) or low levels of CRP (<10 ng/ml) [79]. In the group receiving both drugs, bone loss was similar regardless of the disease activity and inflammatory status, and was comparable to patients who had low disease activity with MTX only. These last findings suggest that the benefits of anti-TNF therapy may not be limited to control of inflammation, but also to the ability to block the direct effect of TNFα on osteoclast activation [79••].
Anti-TNF therapy also has been reported to arrest bone loss in the spine and hip in longitudinal studies [67–70,72–74]. However, comparative studies have shown conflicting results [76,80]. For example, Marotte et al reported that hip and spine BMD declined in historical controls that received MTX only, but were preserved in patients treated with the combination of infliximab +MTX. In this study, changes in BMD were not associated with treatment response among patients treated with both drugs [75]. Haugeberg et al also observed that BMD loss at the femoral neck and hip was higher in patients who received MTX+ placebo compared to those who receive MTX+infliximab; but after adjustment for covariates, only measures of inflammation and radiographic damage were associated with bone loss [77]. Whereas in the BeSt trial, declines in hip and spine BMD were associated with progression of radiologic damage and less decline was observed with concomitant use of bisphosponates but not with treatment regimen [81]. In many single arm studies with anti-TNF agents, arrest of bone loss was accompanied by improvement in disease activity [69,74] or a reduction in inflammation [73,74], which suggests that effective control of inflammation, rather than any specific drug therapy, is likely the mechanism that prevents further bone loss. A recent observational study performed in a commercially insured population in the US and Canada found that the risk of non vertebral fractures did not differ in patients receiving synthetic DMARDs (excluding MTX) compared to those receiving MTX (RR= 1.1, 95%CI [0.6–2.0]) or anti-TNF agents (RR=1.2, 95%CI [0.6–2.3]) [71•].
Anti-TNF agents and bone loss in patients with spondyloarthropathies
In addition to RA, spondyloarthropathies (SpA) constitute another group of chronic inflammatory diseases that is associated with systemic bone loss and is treated with anti-TNF agents. Table 2 summarizes the results of studies that have examined the effect of anti-TNF therapy on BMD in SpA. In most of the longitudinal single arm studies anti-TNF therapy has been associated with increased BMD at spine and hip, improved inflammation and disease activity [83,87,89], but results from comparative studies are inconsistent [84–86,88,91,92].
A small observational study [84], and a post-hoc analysis of a large randomized trial [92] reported that treatment with infliximab was associated with a gain in BMD at the spine and hip when compared to conventional treatment. Marzo-Ortega et al observed significant changes in hip, but not in the spine and femoral neck BMD, in patients treated with etanercept compared to those treated with conventional treatment [88]; however the same investigator did not replicate these findings in a small trial where infliximab+MTX was compared with MTX+placebo [91].
On the contrary, two studies suggested that biphosphonates but not anti-TNF therapy may improve BMD in SpA patients. Pazianas et al observed that use of biphosphonates was associated with gain in BMD at spine, and infliximab provided a marginal benefit only in those patients that were taking biphosphonates [85]. In agreement with these results, Kang et al found that BMD in spine was marginally higher in patients that received conventional therapy+biphosponates compared to the other three regimens (conventional therapy, conventional therapy+anti-TNF and conventional therapy+ biphosphonates+anti-TNF); and BMD in the hip was higher in those patients receiving conventional therapy+biphosphonates+anti-TNF therapy compared to the other three regimens [86]. Studies that examined the effect of anti-TNF therapy on the occurrence of fractures in SpA are not available.
Conclusions and Future directions
It is not surprising that anti-TNF therapy has not shown a major benefit in bone loss and fracture prevention in inflammatory disease. There are multiple pathways independent of TNFα that activate osteoclasts, and inflammation generates many mediators that activate these pathways. Therefore studies that target these pathways are needed to determine their effects on clinically important outcomes.
In conclusion, most of the evidence suggests that anti-TNF therapy has indirect anti-resorptive effects on bone through control of inflammation. Currently, the evidence does not suggest that treatment with anti-TNF has any specific beneficial effect on prevention of osteoporosis or fractures beyond control of inflammation compared to conventional non biologic regimens. Many of the published studies have relatively short follow-up periods, limited statistical power, or lack proper controls. TNFα blockade may need longer and sustained disease control to show beneficial effects on bone. However, with the increased use of biphosphonates as concomitant therapy to prevent bone loss and fractures in patients with chronic inflammatory disease, it would be difficult to identify these effects.
Key points.
There can be a dissociation in periarticular and systemic bone loss in rheumatoid arthritis
The anti-resorptive effects of anti-TNF therapy are likely to be result of its anti-inflammatory properties.
In Spondyloarthropathies, treatment with biphosphonates are more likely to prevent bone loss at spine compared to anti-TNF therapy.
Acknowledgments
Sources of Funding: Supported by NIH grant P60 AR056116, The Vanderbilt Orthopaedic Institute Pilot Grant and the Vanderbilt Physician Scientist Award.
Footnotes
Conflict of interest: None
Reference List
Papers of particular interest, published within the annual period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Van Staa TP, Geusens P, Bijlsma JW, et al. Clinical assessment of the long-term risk of fracture in patients with rheumatoid arthritis. Arthritis Rheum. 2006;54(10):3104–3112. doi: 10.1002/art.22117. [DOI] [PubMed] [Google Scholar]
- 2.Kim SY, Schneeweiss S, Liu J, et al. Risk of osteoporotic fracture in a large population-based cohort of patients with rheumatoid arthritis. Arthritis Res Ther. 2010;12(4):R154. doi: 10.1186/ar3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haugeberg G, Uhlig T, Falch JA, et al. Bone mineral density and frequency of osteoporosis in female patients with rheumatoid arthritis: results from 394 patients in the Oslo County Rheumatoid Arthritis register. Arthritis Rheum. 2000;43(3):522–530. doi: 10.1002/1529-0131(200003)43:3<522::AID-ANR7>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 4.Weiss RJ, Wick MC, Ackermann PW, Montgomery SM. Increased fracture risk in patients with rheumatic disorders and other inflammatory diseases -- a case-control study with 53,108 patients with fracture. J Rheumatol. 2010;37(11):2247–2250. doi: 10.3899/jrheum.100363. [DOI] [PubMed] [Google Scholar]
- 5.Bernstein CN, Blanchard JF, Leslie W, et al. The incidence of fracture among patients with inflammatory bowel disease. A population-based cohort study. AnnIntern Med. 2000;133(10):795–799. doi: 10.7326/0003-4819-133-10-200011210-00012. [DOI] [PubMed] [Google Scholar]
- 6.Cooper C, Coupland C, Mitchell M. Rheumatoid arthritis, corticosteroid therapy and hip fracture. AnnRheum Dis. 1995;54(1):49–52. doi: 10.1136/ard.54.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Card T, West J, Hubbard R, Logan RF. Hip fractures in patients with inflammatory bowel disease and their relationship to corticosteroid use: a population based cohort study. Gut. 2004;53(2):251–255. doi: 10.1136/gut.2003.026799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keffer J, Probert L, Cazlaris H, et al. Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO J. 1991;10(13):4025–4031. doi: 10.1002/j.1460-2075.1991.tb04978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin CL, Moniz C, Chambers TJ, Chow JW. Colitis causes bone loss in rats through suppression of bone formation. Gastroenterology. 1996;111(5):1263–1271. doi: 10.1053/gast.1996.v111.pm8898640. [DOI] [PubMed] [Google Scholar]
- 10.Gough AK, Lilley J, Eyre S, et al. Generalised bone loss in patients with early rheumatoid arthritis. Lancet. 1994;344(8914):23–27. doi: 10.1016/s0140-6736(94)91049-9. [DOI] [PubMed] [Google Scholar]
- 11.Ding C, Parameswaran V, Udayan R, et al. Circulating levels of inflammatory markers predict change in bone mineral density and resorption in older adults: a longitudinal study. J Clin Endocrinol Metab. 2008;93(5):1952–1958. doi: 10.1210/jc.2007-2325. [DOI] [PubMed] [Google Scholar]
- 12.Kobayashi K, Takahashi N, Jimi E, et al. Tumor necrosis factor alpha stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL-RANK interaction. J Exp Med. 2000;191(2):275–286. doi: 10.1084/jem.191.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boyce BF, Schwarz EM, Xing L. Osteoclast precursors: cytokine-stimulated immunomodulators of inflammatory bone disease. Curr Opin Rheumatol. 2006;18(4):427–432. doi: 10.1097/01.bor.0000231913.32364.32. [DOI] [PubMed] [Google Scholar]
- 14.Pascher E, Perniok A, Becker A, Feldkamp J. Effect of 1alpha,25(OH)2-vitamin D3 on TNF alpha-mediated apoptosis of human primary osteoblast-like cells in vitro. Horm Metab Res. 1999;31(12):653–656. doi: 10.1055/s-2007-978815. [DOI] [PubMed] [Google Scholar]
- 15.Gilbert L, He X, Farmer P, et al. Inhibition of osteoblast differentiation by tumor necrosis factor-alpha. Endocrinology. 2000;141(11):3956–3964. doi: 10.1210/endo.141.11.7739. [DOI] [PubMed] [Google Scholar]
- 16.Yoshihara R, Shiozawa S, Imai Y, Fujita T. Tumor necrosis factor alpha and interferon gamma inhibit proliferation and alkaline phosphatase activity of human osteoblastic SaOS-2 cell line. Lymphokine Res. 1990;9(1):59–66. [PubMed] [Google Scholar]
- 17.Lee CK, Lee EY, Chung SM, et al. Effects of disease-modifying antirheumatic drugs and antiinflammatory cytokines on human osteoclastogenesis through interaction with receptor activator of nuclear factor kappaB, osteoprotegerin, and receptor activator of nuclear factor kappaB ligand. Arthritis Rheum. 2004;50(12):3831–3843. doi: 10.1002/art.20637. [DOI] [PubMed] [Google Scholar]
- 18.Redlich K, Gortz B, Hayer S, et al. Repair of local bone erosions and reversal of systemic bone loss upon therapy with anti-tumor necrosis factor in combination with osteoprotegerin or parathyroid hormone in tumor necrosis factor-mediated arthritis. Am J Pathol. 2004;164(2):543–555. doi: 10.1016/S0002-9440(10)63144-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saito H, Kojima T, Takahashi M, et al. A tumor necrosis factor receptor loop peptide mimic inhibits bone destruction to the same extent as anti-tumor necrosis factor monoclonal antibody in murine collagen-induced arthritis. Arthritis Rheum. 2007;56(4):1164–1174. doi: 10.1002/art.22495. [DOI] [PubMed] [Google Scholar]
- 20.D'Amelio P, Fornelli G, Roato I, Isaia GC. Interactions between the immune system and bone. World J Orthop. 2011;2(3):25–30. doi: 10.5312/wjo.v2.i3.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Agrawal M, Arora S, Li J, et al. Bone, inflammation, and inflammatory bowel disease. Curr Osteoporos Rep. 2011;9(4):251–257. doi: 10.1007/s11914-011-0077-9. [DOI] [PubMed] [Google Scholar]
- 22.Schett G, David JP. The multiple faces of autoimmune-mediated bone loss. Nat Rev Endocrinol. 2010;6(12):698–706. doi: 10.1038/nrendo.2010.190. [DOI] [PubMed] [Google Scholar]
- 23.Zaidi M. Skeletal remodeling in health and disease. Nat Med. 2007;13(7):791–801. doi: 10.1038/nm1593. [DOI] [PubMed] [Google Scholar]
- 24.Teti A. Bone development: overview of bone cells and signaling. Curr Osteoporos Rep. 2011;9(4):264–273. doi: 10.1007/s11914-011-0078-8. [DOI] [PubMed] [Google Scholar]
- 25.Redlich K, Smolen JS. Inflammatory bone loss: pathogenesis and therapeutic intervention. Nat Rev Drug Discov. 2012;11(3):234–250. doi: 10.1038/nrd3669. [DOI] [PubMed] [Google Scholar]
- 26.Ross FP. Osteoclast Biology and Bone Resorption. In: Rosen CJ, editor. Primer on the Metabolic Bone Diseases and Disorders of Mineral metabolism. 7. American Society for Bone and Mineral Research; Washington, D.C: American Society for Bone and Mineral Research; 2009. pp. 16–22. [Google Scholar]
- 27.Bonewald LF. Osteocytes. In: Rosen CJ, editor. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. 7. American Society for Bone and Mineral Research; Washington, D.C: American Society for Bone and Mineral Research; 2009. pp. 22–26. [Google Scholar]
- 28.Han Y, Cowin SC, Schaffler MB, Weinbaum S. Mechanotransduction and strain amplification in osteocyte cell processes. Proc Natl Acad Sci USA. 2004;101(47):16689–16694. doi: 10.1073/pnas.0407429101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li J, Rose E, Frances D, et al. Effect of oscillating fluid flow stimulation on osteocyte mRNA expression. J Biomech. 2012;45(2):247–251. doi: 10.1016/j.jbiomech.2011.10.037. [DOI] [PubMed] [Google Scholar]
- 30.Rochefort GY, Pallu S, Benhamou CL. Osteocyte: the unrecognized side of bone tissue. Osteoporos Int. 2010;21(9):1457–1469. doi: 10.1007/s00198-010-1194-5. [DOI] [PubMed] [Google Scholar]
- 31.Abe E, Ishimi Y, Takahashi N, et al. A differentiation-inducing factor produced by the osteoblastic cell line MC3T3-E1 stimulates bone resorption by promoting osteoclast formation. J Bone Miner Res. 1988;3(6):635–645. doi: 10.1002/jbmr.5650030609. [DOI] [PubMed] [Google Scholar]
- 32.Kodama H, Nose M, Niida S, Yamasaki A. Essential role of macrophage colony-stimulating factor in the osteoclast differentiation supported by stromal cells. J Exp Med. 1991;173(5):1291–1294. doi: 10.1084/jem.173.5.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suda T, Tanaka S, Takahashi N. Macrophage colon-stimulating factor (M-CSF) is essential for differentiation rather than proliferation of osteoclast progenitors. Osteoporos Int. 1993;3 (Suppl 1):111–113. doi: 10.1007/BF01621881. [DOI] [PubMed] [Google Scholar]
- 34.Tanaka S, Takahashi N, Udagawa N, et al. Macrophage colony-stimulating factor is indispensable for both proliferation and differentiation of osteoclast progenitors. J Clin Invest. 1993;91(1):257–263. doi: 10.1172/JCI116179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krum SA, Chang J, Miranda-Carboni G, Wang CY. Novel functions for NFkappaB: inhibition of bone formation. Nat Rev Rheumatol. 2010;6(10):607–611. doi: 10.1038/nrrheum.2010.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stein GS, Lian JB, van Wijnen AJ, et al. Runx2 control of organization, assembly and activity of the regulatory machinery for skeletal gene expression. Oncogene. 2004;23(24):4315–4329. doi: 10.1038/sj.onc.1207676. [DOI] [PubMed] [Google Scholar]
- 37.Zhao C, Irie N, Takada Y, et al. Bidirectional ephrinB2-EphB4 signaling controls bone homeostasis. Cell Metab. 2006;4(2):111–121. doi: 10.1016/j.cmet.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 38.Franceschi RT, Xiao G. Regulation of the osteoblast-specific transcription factor, Runx2: responsiveness to multiple signal transduction pathways. J Cell Biochem. 2003;88(3):446–454. doi: 10.1002/jcb.10369. [DOI] [PubMed] [Google Scholar]
- 39.Ducy P, Zhang R, Geoffroy V, et al. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell. 1997;89(5):747–754. doi: 10.1016/s0092-8674(00)80257-3. [DOI] [PubMed] [Google Scholar]
- 40.Kruppa G, Thoma B, Machleidt T, et al. Inhibition of tumor necrosis factor (TNF)-mediated NF-kappa B activation by selective blockade of the human 55-kDa TNF receptor. J Immunol. 1992;148(10):3152–3157. [PubMed] [Google Scholar]
- 41.Lam J, Takeshita S, Barker JE, et al. TNF-alpha induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J Clin Invest. 2000;106(12):1481–1488. doi: 10.1172/JCI11176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fuller K, Murphy C, Kirstein B, et al. TNFalpha potently activates osteoclasts, through a direct action independent of and strongly synergistic with RANKL. Endocrinology. 2002;143(3):1108–1118. doi: 10.1210/endo.143.3.8701. [DOI] [PubMed] [Google Scholar]
- 43.Komine M, Kukita A, Kukita T, et al. Tumor necrosis factor-alpha cooperates with receptor activator of nuclear factor kappaB ligand in generation of osteoclasts in stromal cell-depleted rat bone marrow cell culture. Bone. 2001;28(5):474–483. doi: 10.1016/s8756-3282(01)00420-3. [DOI] [PubMed] [Google Scholar]
- 44.Glantschnig H, Fisher JE, Wesolowski G, et al. M-CSF, TNFalpha and RANK ligand promote osteoclast survival by signaling through mTOR/S6 kinase. Cell Death Differ. 2003;10(10):1165–1177. doi: 10.1038/sj.cdd.4401285. [DOI] [PubMed] [Google Scholar]
- 45.Redlich K, Hayer S, Ricci R, et al. Osteoclasts are essential for TNF-alpha-mediated joint destruction. J Clin Invest. 2002;110(10):1419–1427. doi: 10.1172/JCI15582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abbas S, Zhang YH, Clohisy JC, Abu-Amer Y. Tumor necrosis factor-alpha inhibits pre-osteoblast differentiation through its type-1 receptor. Cytokine. 2003;22(1–2):33–41. doi: 10.1016/s1043-4666(03)00106-6. [DOI] [PubMed] [Google Scholar]
- 47.Gilbert LC, Rubin J, Nanes MS. The p55 TNF receptor mediates TNF inhibition of osteoblast differentiation independently of apoptosis. Am J Physiol Endocrinol Metab. 2005;288(5):E1011–E1018. doi: 10.1152/ajpendo.00534.2004. [DOI] [PubMed] [Google Scholar]
- 48.Kaneki H, Guo R, Chen D, et al. Tumor necrosis factor promotes Runx2 degradation through up-regulation of Smurf1 and Smurf2 in osteoblasts. J Biol Chem. 2006;281(7):4326–4333. doi: 10.1074/jbc.M509430200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mukai T, Otsuka F, Otani H, et al. TNF-alpha inhibits BMP-induced osteoblast differentiation through activating SAPK/JNK signaling. Biochem Biophys Res Commun. 2007;356(4):1004–1010. doi: 10.1016/j.bbrc.2007.03.099. [DOI] [PubMed] [Google Scholar]
- 50•.Lee HL, Yi T, Woo KM, et al. Msx2 mediates the inhibitory action of TNF-alpha on osteoblast differentiation. Exp Mol Med. 2010;42(6):437–445. doi: 10.3858/emm.2010.42.6.045. MSX2 is a known regulator of the early stages of osteoblast commitment and differentiation. The regulation of MSX2 by TNFα presents one of the first described mechanism of TNFα action that may be specific to the early stages of osteoblastogenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ochi H, Hara Y, Tagawa M, et al. The roles of TNFR1 in lipopolysaccharide-induced bone loss: dual effects of TNFR1 on bone metabolism via osteoclastogenesis and osteoblast survival. J Orthop Res. 2010;28(5):657–663. doi: 10.1002/jor.21028. [DOI] [PubMed] [Google Scholar]
- 52••.Ghali O, Chauveau C, Hardouin P, et al. TNF-alpha's effects on proliferation and apoptosis in human mesenchymal stem cells depend on RUNX2 expression. J Bone Miner Res. 2010;25(7):1616–1626. doi: 10.1002/jbmr.52. The dependence of TNFα effects upon RUNX2 expression suggests that RUNX2 may determine the differential effects of TNFα on osteoblastic cells compared to other cells in the mesenchymal linage. This finding suggests that RUNX2 dependent mechanisms may present opportunities for osteoblast-restricted blockade of the TNFα signaling. [DOI] [PubMed] [Google Scholar]
- 53.Wahl EC, Aronson J, Liu L, et al. Restoration of regenerative osteoblastogenesis in aged mice: modulation of TNF. J Bone Miner Res. 2010;25(1):114–123. doi: 10.1359/jbmr.090708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gilbert L, He X, Farmer P, et al. Expression of the osteoblast differentiation factor RUNX2 (Cbfa1/AML3/Pebp2alpha A) is inhibited by tumor necrosis factor-alpha. J Biol Chem. 2002;277(4):2695–2701. doi: 10.1074/jbc.M106339200. [DOI] [PubMed] [Google Scholar]
- 55.Lu X, Gilbert L, He X, et al. Transcriptional regulation of the osterix (Osx, Sp7) promoter by tumor necrosis factor identifies disparate effects of mitogen-activated protein kinase and NF kappa B pathways. J Biol Chem. 2006;281(10):6297–6306. doi: 10.1074/jbc.M507804200. [DOI] [PubMed] [Google Scholar]
- 56.Diarra D, Stolina M, Polzer K, et al. Dickkopf-1 is a master regulator of joint remodeling. Nat Med. 2007;13(2):156–163. doi: 10.1038/nm1538. [DOI] [PubMed] [Google Scholar]
- 57••.Findlay DM, Atkins GJ. TWEAK and TNF regulation of sclerostin: a novel pathway for the regulation of bone remodelling. Adv Exp Med Biol. 2011;691:337–348. doi: 10.1007/978-1-4419-6612-4_34. Sclerostin is a key regulator of Wnt signaling produced by osteocytes. Consequenlty, osteocyte secretion of sclerostin is a major regulator of bone remodeling and BMD. This paper describes a novel mechanism by which TNFα may influence bone via direc effects on osteocytes, suggesting that osteocytes trageted therapeutics hold potential to mitigate the effects of TNFα on bone formation and resorption. [DOI] [PubMed] [Google Scholar]
- 58.van der Heijde D, Kavanaugh A, Gladman DD, et al. Infliximab inhibits progression of radiographic damage in patients with active psoriatic arthritis through one year of treatment: Results from the induction and maintenance psoriatic arthritis clinical trial 2. Arthritis Rheum. 2007;56(8):2698–2707. doi: 10.1002/art.22805. [DOI] [PubMed] [Google Scholar]
- 59.Lipsky PE, van der Heijde DM, St Clair EW, et al. Infliximab and methotrexate in the treatment of rheumatoid arthritis. Anti-Tumor Necrosis Factor Trial in Rheumatoid Arthritis with Concomitant Therapy Study Group. N Engl J Med. 2000;343(22):1594–1602. doi: 10.1056/NEJM200011303432202. [DOI] [PubMed] [Google Scholar]
- 60.St Clair EW, van der Heijde DM, Smolen JS, et al. Combination of infliximab and methotrexate therapy for early rheumatoid arthritis: a randomized, controlled trial. Arthritis Rheum. 2004;50(11):3432–3443. doi: 10.1002/art.20568. [DOI] [PubMed] [Google Scholar]
- 61.van der Heijde D, Kivitz A, Schiff MH, et al. Efficacy and safety of adalimumab in patients with ankylosing spondylitis: results of a multicenter, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2006;54(7):2136–2146. doi: 10.1002/art.21913. [DOI] [PubMed] [Google Scholar]
- 62.Yasunori K, Masaaki T, Tetsuyuki N, et al. Reduction of urinary levels of pyridinoline and deoxypyridinoline and serum levels of soluble receptor activator of NF-kappaB ligand by etanercept in patients with rheumatoid arthritis. Clin Rheumatol. 2008;27(9):1093–1101. doi: 10.1007/s10067-008-0870-8. [DOI] [PubMed] [Google Scholar]
- 63.Miheller P, Muzes G, Zagoni T, et al. Infliximab therapy improves the bone metabolism in fistulizing Crohn's disease. Dig Dis. 2006;24(1–2):201–206. doi: 10.1159/000091299. [DOI] [PubMed] [Google Scholar]
- 64.Abreu MT, Geller JL, Vasiliauskas EA, et al. Treatment with infliximab is associated with increased markers of bone formation in patients with Crohn's disease. J Clin Gastroenterol. 2006;40(1):55–63. doi: 10.1097/01.mcg.0000190762.80615.d4. [DOI] [PubMed] [Google Scholar]
- 65.Barnabe C, Hanley DA. Effect of tumor necrosis factor alpha inhibition on bone density and turnover markers in patients with rheumatoid arthritis and spondyloarthropathy. Semin Arthritis Rheum. 2009;39(2):116–122. doi: 10.1016/j.semarthrit.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 66.Veerappan SG, O'Morain CA, Daly JS, Ryan BM. Review article: the effects of antitumour necrosis factor-alpha on bone metabolism in inflammatory bowel disease. Aliment Pharmacol Ther. 2011;33(12):1261–1272. doi: 10.1111/j.1365-2036.2011.04667.x. [DOI] [PubMed] [Google Scholar]
- 67.Lange U, Teichmann J, Muller-Ladner U, Strunk J. Increase in bone mineral density of patients with rheumatoid arthritis treated with anti-TNF-alpha antibody: a prospective open-label pilot study. Rheumatology (Oxford) 2005;44(12):1546–1548. doi: 10.1093/rheumatology/kei082. [DOI] [PubMed] [Google Scholar]
- 68.Vis M, Voskuyl AE, Wolbink GJ, et al. Bone mineral density in patients with rheumatoid arthritis treated with infliximab. Ann Rheum Dis. 2005;64(2):336–337. doi: 10.1136/ard.2003.017780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vis M, Havaardsholm EA, Haugeberg G, et al. Evaluation of bone mineral density, bone metabolism, osteoprotegerin and receptor activator of the NFkappaB ligand serum levels during treatment with infliximab in patients with rheumatoid arthritis. Ann Rheum Dis. 2006;65(11):1495–1499. doi: 10.1136/ard.2005.044198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Eekman DA, Vis M, Bultink IE, et al. Stable bone mineral density in lumbar spine and hip in contrast to bone loss in the hands during long-term treatment with infliximab in patients with rheumatoid arthritis. Ann Rheum Dis. 2011;70(2):389–390. doi: 10.1136/ard.2009.127787. [DOI] [PubMed] [Google Scholar]
- 71••.Kim SY, Schneeweiss S, Liu J, Solomon DH. Effects of disease-modifying antirheumatic drugs on nonvertebral fracture risk in rheumatoid arthritis: a population-based cohort study. J Bone Miner Res. 2012;27(4):789–796. doi: 10.1002/jbmr.1489. Although anti-TNF therapy has shown the potential to reduce bone loss in experimental studies, the comparative effectiveness of the agents in the risk of fractures has never been examined. This is the first observational study that examined the risk of non-vertebral fractures between three treatment regimens for RA using administrative data. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chopin F, Garnero P, le Henanff A, et al. Long-term effects of infliximab on bone and cartilage turnover markers in patients with rheumatoid arthritis. Ann Rheum Dis. 2008;67(3):353–357. doi: 10.1136/ard.2007.076604. [DOI] [PubMed] [Google Scholar]
- 73.Serelis J, Kontogianni MD, Katsiougiannis S, et al. Effect of anti-TNF treatment on body composition and serum adiponectin levels of women with rheumatoid arthritis. Clin Rheumatol. 2008;27(6):795–797. doi: 10.1007/s10067-008-0855-7. [DOI] [PubMed] [Google Scholar]
- 74.Wijbrandts CA, Klaasen R, Dijkgraaf MG, et al. Bone mineral density in rheumatoid arthritis patients 1 year after adalimumab therapy: arrest of bone loss. Ann Rheum Dis. 2009;68(3):373–376. doi: 10.1136/ard.2008.091611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Marotte H, Pallot-Prades B, Grange L, et al. A 1-year case-control study in patients with rheumatoid arthritis indicates prevention of loss of bone mineral density in both responders and nonresponders to infliximab. Arthritis Res Ther. 2007;9(3):R61. doi: 10.1186/ar2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Seriolo B, Paolino S, Sulli A, et al. Bone metabolism changes during anti-TNF-alpha therapy in patients with active rheumatoid arthritis. Ann NY Acad Sci. 2006;1069:420–427. doi: 10.1196/annals.1351.040. [DOI] [PubMed] [Google Scholar]
- 77.Haugeberg G, Conaghan PG, Quinn M, Emery P. Bone loss in patients with active early rheumatoid arthritis: infliximab and methotrexate compared with methotrexate treatment alone. Explorative analysis from a 12-month randomised, double-blind, placebo-controlled study. Ann Rheum Dis. 2009;68(12):1898–1901. doi: 10.1136/ard.2008.106484. [DOI] [PubMed] [Google Scholar]
- 78.Hoff M, Kvien TK, Kalvesten J, et al. Adalimumab therapy reduces hand bone loss in early rheumatoid arthritis: explorative analyses from the PREMIER study. Ann Rheum Dis. 2009;68(7):1171–1176. doi: 10.1136/ard.2008.091264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79••.Hoff M, Kvien TK, Kalvesten J, et al. Adalimumab reduces hand bone loss in rheumatoid arthritis independent of clinical response: subanalysis of the PREMIER study. BMC Musculoskelet Disord. 2011;12:54. doi: 10.1186/1471-2474-12-54. Hand bone loss is considered the first sign of bone involvement in RA, therefore it is used in various clinical studies as an important outcome to monitor treatment efficacy. In this paper, treatment with adalimumab decreased bone loss regardless of clinical response suggesting a direct effect of TNFα in osteoclast activation that is blocked by Adalimumab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Guler-Yuksel M, Bijsterbosch J, Goekoop-Ruiterman YP, et al. Changes in bone mineral density in patients with recent onset, active rheumatoid arthritis. Ann Rheum Dis. 2008;67(6):823–828. doi: 10.1136/ard.2007.073817. [DOI] [PubMed] [Google Scholar]
- 81.Guler-Yuksel M, Allaart CF, Goekoop-Ruiterman YP, et al. Changes in hand and generalised bone mineral density in patients with recent-onset rheumatoid arthritis. Ann Rheum Dis. 2009;68(3):330–336. doi: 10.1136/ard.2007.086348. [DOI] [PubMed] [Google Scholar]
- 82••.Dirven L, Guler-Yuksel M, de Beus WM, et al. Changes in hand bone mineral density and the association with the level of disease activity in patients with rheumatoid arthritis: bone mineral density measurements in a multicenter randomized clinical trial. Arthritis Care Res(Hoboken) 2011;63(12):1691–1699. doi: 10.1002/acr.20612. This is the first clinical study that reported bone gain in the hands during RA treatment. In this study, gain in hand bone was associated with disease control, which suggest that adequates disease control should stop and reverse periarticular bone loss. [DOI] [PubMed] [Google Scholar]
- 83.Allali F, Breban M, Porcher R, et al. Increase in bone mineral density of patients with spondyloarthropathy treated with anti-tumour necrosis factor alpha. Ann Rheum Dis. 2003;62(4):347–349. doi: 10.1136/ard.62.4.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mauro M, Radovic V, Armstrong D. Improvement of lumbar bone mass after infliximab therapy in Crohn's disease patients. Can J Gastroenterol. 2007;21(10):637–642. doi: 10.1155/2007/216162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pazianas M, Rhim AD, Weinberg AM, et al. The effect of anti-TNF-alpha therapy on spinal bone mineral density in patients with Crohn's disease. Ann NY Acad Sci. 2006;1068:543–556. doi: 10.1196/annals.1346.055. [DOI] [PubMed] [Google Scholar]
- 86.Kang KY, Lee KY, Kwok SK, et al. The change of bone mineral density according to treatment agents in patients with ankylosing spondylitis. Joint Bone Spine. 2011;78(2):188–193. doi: 10.1016/j.jbspin.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 87.Bernstein M, Irwin S, Greenberg GR. Maintenance infliximab treatment is associated with improved bone mineral density in Crohn's disease. Am J Gastroenterol. 2005;100(9):2031–2035. doi: 10.1111/j.1572-0241.2005.50219.x. [DOI] [PubMed] [Google Scholar]
- 88.Marzo-Ortega H, McGonagle D, Haugeberg G, et al. Bone mineral density improvement in spondyloarthropathy after treatment with etanercept. Ann Rheum Dis. 2003;62(10):1020–1021. doi: 10.1136/ard.62.10.1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Briot K, Garnero P, le HA, et al. Body weight, body composition, and bone turnover changes in patients with spondyloarthropathy receiving anti-tumour necrosis factor {alpha} treatment. Ann Rheum Dis. 2005;64(8):1137–1140. doi: 10.1136/ard.2004.028670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Briot K, Gossec L, Kolta S, et al. Prospective assessment of body weight, body composition, and bone density changes in patients with spondyloarthropathy receiving anti-tumor necrosis factor-alpha treatment. J Rheumatol. 2008;35(5):855–861. [PubMed] [Google Scholar]
- 91.Marzo-Ortega H, McGonagle D, Jarrett S, et al. Infliximab in combination with methotrexate in active ankylosing spondylitis: a clinical and imaging study. Ann Rheum Dis. 2005;64(11):1568–1575. doi: 10.1136/ard.2004.022582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Visvanathan S, van der HD, Deodhar A, et al. Effects of infliximab on markers of inflammation and bone turnover and associations with bone mineral density in patients with ankylosing spondylitis. Ann Rheum Dis. 2009;68(2):175–182. doi: 10.1136/ard.2007.084426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Harrison BJ, Hutchinson CE, Adams J, et al. Assessing periarticular bone mineral density in patients with early psoriatic arthritis or rheumatoid arthritis. Ann Rheum Dis. 2002;61(11):1007–1011. doi: 10.1136/ard.61.11.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94••.Desai SP, Gravallese EM, Shadick NA, et al. Hand bone mineral density is associated with both total hip and lumbar spine bone mineral density in post-menopausal women with RA. Rheumatology(Oxford) 2010;49(3):513–519. doi: 10.1093/rheumatology/kep385. Hand bone loss has been associated with radiographic disease progression. In this paper, hand BMD was an important predictor of spine and hip BMD in postmenopausal women with established RA, suggesting that hand BMD could potentially be used to asses systemic bone loss in RA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Haugeberg G, Lodder MC, Lems WF, et al. Hand cortical bone mass and its associations with radiographic joint damage and fractures in 50–70 year old female patients with rheumatoid arthritis: cross sectional Oslo-Truro-Amsterdam (OSTRA) collaborative study. Ann Rheum Dis. 2004;63(10):1331–1334. doi: 10.1136/ard.2003.015065. [DOI] [PMC free article] [PubMed] [Google Scholar]