

Abstract
Amyloid formation plays an important role in a broad range of diseases and the search for amyloid inhibitors is an active area of research. Amyloid formation takes places in a heterogeneous environment in vivo with the potential for interactions with membranes and with components of the extracellular matrix. Naturally occurring amyloid deposits are associated with sulfated proteoglycans and other factors. However, the vast majority of in vitro assays of amyloid formation and amyloid inhibition are conducted in homogeneous solution where the potential for interactions with membranes or sulfated proteoglycans is lacking and it is possible that different results may be obtained in heterogeneous environments. We show that variants of islet amyloid polypeptide, which are non-amyloidgenic in homogeneous solution, can be readily induced to form amyloid in the presence of glycosaminoglycans. Glycosaminoglycans are found to be more effective than anionic lipid vesicles at inducing amyloid formation on a per charge basis. Several known inhibitors of IAPP amyloid formation are shown to be less effective in the presence of glycosaminoglycans.
Keywords: IAPP, amylin, glycosaminoglycan, extracellular matrix, amyloid, inhibitor
Introduction
Amyloid fiber formation, the aggregation of normally soluble proteins into partially ordered β-rich structures, plays a role in a broad range of diseases including type 2 diabetes and Alzhermer’s disease.1,2 Amyloid, or the process of its formation contributes to the pathology of many diseases3–5 and the search for inhibitors of amyloid formation is an active area of research.6–13
Amyloid formation takes places in a heterogeneous environment in vivo with the potential for interactions with membranes and with components of the extracellular matrix. All naturally occurring amyloid deposits are associated with sulfated proteoglycans and other factors.14–19 However, the vast majority of in vitro assays of amyloid formation and amyloid inhibition are conducted in homogeneous solution where the potential for interactions with membranes or sulfated proteoglycans is lacking and it is possible that different results may be obtained in heterogeneous environments. In addition, studies of proteins in buffer have been widely used to develop propensity scales for amyloid formation,20–22 but it is not certain that these scales will have the same quantitive predictive power in heterogeneous environments.
Islet amyloid polypeptide (IAPP, also known as amylin) forms pancreatic islet amyloid deposits in type-2 diabetes. IAPP amyloid formation is toxic to cultured cells, suggesting that it contributes to the disease by leading to β-cell dysfunction and death.23–27 Recent work also implicates amyloid formation as a cause of graft failure in islet cell transplantation.28,29 A wide range of studies have shown that model membranes which contain anionic lipids accelerate amyloid formation by IAPP,30–32 but less is known about the effects of glycosaminoglycans (GAGs) on IAPP amyloid formation. Immunohistochemical studies have shown that heparan sulfate proteoglycans are associated with islet amyloid isolated from patients with type 2 diabetes,19 and GAGs can enhance amyloid formation by human proIAPP processing intermediates and by human IAPP.33,34 IAPP is one of the most amyloidgenic naturally occurring polypeptides, but it is not known if GAGs can induce amyloid formation in apparently non-amyloidgenic variants of IAPP, nor, with a few exceptions, have the effects of GAGs on IAPP inhibitors been generally considered. Here we examine the effects of GAGs on two apparently non-amyloidgenic variants of IAPP and study the impact of GAGs on IAPP inhibitors. The I26P point mutant of human IAPP (I26P-IAPP), renders the protein non-amyloidgenic and converts it into an inhibitor of amyloid formation by wild type IAPP.12 The doubly N-methylated variant of human IAPP, G24-N-Methyl, I26-N-Methyl-IAPP (NMe-G24, NMe-I26-IAPP), is a potent inhibitor of IAPP amyloid formation in homogeneous solution and is itself not amyloidogenic.11 The mode of action of these inhibitors is not known, but the substitutions are located in a region of the chain which has been highlighted as important for amyloid formation.35,36 We show that these two variants of IAPP can be readily induced to form amyloid by GAGs and that GAGs are more effective than model lipid micelle systems in inducing them to form amyloid. We also demonstrate that some potent inhibitors of IAPP amyloid formation are less effective in the presence of GAGs. Polypeptide-GAG interactions have been postulated to play an important role in islet amyloid formation in vivo,15–19,33,37–40 and our results highlight the importance of considering these effects in vitro and in inhibitor design.
Results and Discussions
Heparan sulfate induces amyloid formation by non-amyloidgenic variants of IAPP
The sequences of wild type IAPP, the I26P-IAPP and NMe-G24, NMe-I26-IAPP variants are shown in Figure 1. No data are available on the effects of GAGs on the ability of either of these variants to form amyloid. We first studied amyloid formation by I26P-IAPP in the presence of the model GAG heparan sulfate (HS). High molecular weight (10 to 14kd) HS was used for these studies. Alexandrescu and coworkers examined the length dependence of heparin upon amyloid formation by wild type IAPP and showed that length dependent effects leveled off above 12 monosaccharide units.39 The high molecular weight HS used here was above the threshold observed for heparin. Figure 2a compares the ability of the I26P point mutant to form amyloid in the absence and presence of HS. The peptide was not amyloidogenic in homogeneous solution over the 10 hr time course of the experiment as judged by thioflavin-T binding assays, transmission electron microscopy (TEM) and circular dichroism (CD). In contrast, wild type IAPP formed amyloid with a T50, the time required for the reaction to reach 50% of the final fluorescence intensity, of 21 mins under these conditions in the absence of GAG. The mutant formed amyloid in the presence of HS. A rapid increase in thioflavin-T fluorescence was observed for I26P-IAPP in the presence of HS with a T50 of 20 mins. TEM confirmed the results of the thioflavin-T studies, and dense matts of amyloid fibers were observed in the presence of HS, but not in its absence (Figure 2b, 2c). CD (Supplementary Material) revealed the presence of significant β-sheet structure in the presence of GAGs.
Figure 1.

Sequence of wild type IAPP, I26P-IAPP and G24-N-methyl, I26-N-methyl-IAPP (NMe denotes N-methylation). Each peptide has an amidated C-terminus and a disulfide bond.
Figure 2.

Amyloid formation by I26P-IAPP in the absence and presence of heparan sulfate. (a) Results of fluorescence-monitored thioflavin-T binding assays are displayed. Black, I26P-IAPP in the absence of heparan sulfate; red, I26P-IAPP in the presence of heparan sulfate. The insert shows an expanded plot of the first 50 mins. (b) TEM image of I26P-IAPP in the presence of heparan sulfate. (c) TEM image of I26P-IAPP in the absence of heparan sulfate. Aliquots were removed at the end of each kinetic experiment for TEM analysis. Scale bars represent 100 nm. The kinetic experiments were conducted in 20 mM Tris-HCl (pH 7.4), 2% HFIP (v/v) with continuous stirring at 25 °C. The concentration of I26P-IAPP was 16 μM. Heparan sulfate, when present, was at 1.3 μM.
We analyzed a second non-amyloidgenic variant of IAPP, NMe-G24, NMe-I26-IAPP. This peptide is non-amyloidgenic and is one of the most effective inhibitors of amyloid formation by IAPP and protects against IAPP induced toxicity in cell culture.11 Figure 3a compares kinetic curves collected in the presence and absence of heparan sulfate. No amyloid formation was observed in the absence of heparan sulfate, even for samples which were incubated for more than 20 times longer than the time required for wild type IAPP to form amyloid (Figure 3a). In contrast, rapid amyloid formation was observed when heparan sulfate was present as judged by thioflavin-T assays and TEM (Figure 3b) with a T50 of 30 mins. CD (Supplementary Material) confirmed the presence of β-sheet structure.
Figure 3.

Amyloid formation by NMe-G24, NMe-I26-IAPP in the absence and presence of heparan sulfate. (a) Results of fluorescence-monitored thioflavin-T binding assays are displayed. Black, NMe-G24, NMe-I26-IAPP in the absence of heparan sulfate; red, NMe-G24, NMe-I26-IAPP in the presence of heparan sulfate. The insert shows an expanded plot of the first 100 mins. (b) TEM image of NMe-G24, NMe-I26-IAPP in the presence of heparan sulfate. (c) TEM image of NMe-G24, NMe-I26-IAPP in the absence of heparan sulfate. Aliquots were removed at the end of each kinetic experiment for TEM analysis. Scale bars represent 100 nm. The kinetic experiments were conducted in 20 mM Tris-HCl (pH 7.4), 2% HFIP (v/v) with continuous stirring at 25 °C. The concentration of NMe-G24, NMe-I26-IAPP was 16 μM. Heparan sulfate, when present, was at 1.3 μM.
The experiments outlined above were carried out using standard protocols for biophysical studies of IAPP. These involved solubilizing the peptide in hexafluoroisopropanol (HFIP) and diluting the stock solution into buffer. This results in 2% residual HFIP by volume. Even this low percentage of organic co-solvent enhances significantly the kinetics of amyloid formation by IAPP.41,42 Thus we wanted to test if our results might be a consequence of the conditions used. We repeated the experiments using a different protocol which avoids the use of HFIP. Buffer was added directly to dried peptide and the time course of amyloid formation was followed. We obtained similar results; both I26P-IAPP and NMe-G24, NMe-I26-IAPP formed amyloid in the presence of heparan sulfate, but not in its absence (Supplementary Material). The T50 of I26P-IAPP in the presence of heparan sulfate under this condition was 465 mins and was 2010 mins for the NMe-G24, NMe-I26-IAPP, wild type IAPP formed amyloid with a T50 of 60 mins in the presence of heparan sulfate under these conditions.
FRET experiments reveal the co-localization of amyloid fibrils and GAGs
To further investigate the mechanism of the amyloid inducing effects of GAGs on I26P-IAPP and NMe-G24, NMe-I26-IAPP, we tested if GAGs are associated with amyloid fibrils. This is important because GAGs might exert their effects by binding to the IAPP variants or through non-specific polyanion effects. We used a recently developed assay based on FRET from thioflavin-T to fluorescein labeled heparin (FLH).39 Labeled heparin with one fluorescein conjugated per heparin was used as a model GAG. The excitation maximum of thioflavin-T when bound to amyloid fibrils is near 450 nm and its emission maximum is near 485 nm. The excitation maximum of fluorescein is 488 nm and its emission maximum is 515 nm. Thus, the two dyes form a convenient FRET pair to monitor the proximity of GAG and amyloid fibrils. Figure 4a shows the kinetic profile of I26P-IAPP amyloid formation induced by FLH, monitored at an excitation wavelength of 440 nm and an emission wavelength of 510 nm. This pair of wavelengths was chosen to detect just the fluorescent signal due to FRET between fluorescein and thioflavin-T bound to fibrils by avoiding signal from direct excitation of bound thioflavin-T. FRET to fluorescein from fibril bound thioflavin-T was detected as amyloid formation by I26P-IAPP proceeds (Figure 4a, red curve). CD (Supplementary Material) and TEM images (Figure 4b) confirmed the existence of fibrils. In a control experiment containing I26P-IAPP and FLH without thioflavin-T, no significant increase in fluorescence intensity was detected (Figure 4a, black curve), although CD (Supplementary Material) and TEM (Figure 4) confirmed the presence of fibrils. We also studied the co-localization of FLH and NMe-G24, NMe-I26-IAPP using FRET. The results were very similar to those observed with I26P-IAPP. (Supplementary Material) The observation of FRET between FLH and thioflavin-T in both cases demonstrates an association of GAG with the peptides. The association may occur during the process of amyloid formation or might result from the IAPP peptides forming amyloid fibers in solution and then binding to GAG. We conducted additional experiments to test these possibilities. We simultaneously monitored direct thioflavin-T fluorescence and thioflavin-T to FLH FRET for the same sample. (Supplementary Material) The two curves showed identical time courses; the simplest explanation is that the peptides bind GAGs before amyloid formation is complete.
Figure 4.

Heparan sulfate is associated with amyloid fibrils. FRET between fluorescein labeled heparin (FLH) and thioflavin-T bound to amyloid fibrils formed by I26P-IAPP. (a) Kinetic profile of I26P-IAPP in the presence of FLH monitored by FRET between FLH and thioflavin-T bound to amyloid fibrils. The fluorescence was measured using an excitation wavelength of 440 nm and an emission wavelegnth of 510 nm. Black, control experiment, I26P-IAPP in the presence of FLH, no thioflavin-T; red, I26P-IAPP in the presence of FLH and thioflavin-T. (b) TEM image of I26P-IAPP in the presence of FLH and thioflavin-T. An aliquot was removed at the end of the reaction for TEM analysis. (c) TEM image of I26P-IAPP in the presence of FLH, no thioflavin-T. An aliquot was removed at the end of the reaction for TEM analysis. Scale bars represent 100 nm. The kinetic experiments were conducted in 20 mM Tris-HCl (pH 7.4), 2% HFIP (v/v) with continuous stirring at 25 °C. The concentration of I26P-IAPP was 16 μM. FLH was at 1.3 μM.
The effect of heparan sulfate on I26P-IAPP amyloid formation can be screened by high ionic strength
IAPP and the two variants studied here all have a net charge between +2 and +4 at physiologically relevant pH, depending upon the exact pKa of the N-terminus and His-18, thus all are capable of making electrostatic interactions with heparan sulfate. In order to study the potential role of electrostatic interactions between I26P-IAPP and heparan sulfate, we examined amyloid formation kinetics in buffers with high concentrations of salt. Amyloid formation by wild type IAPP is very sensitive to salt.43 Thioflavin-T fluorescence assays without HFIP were used to follow amyloid formation as a function of added salt. Figure 5a compares the ability of I26P-IAPP to form amyloid in the presence and absence of heparan sulfate as a function of NaCl in the assay. I26P-IAPP was still non-amyloidgenic in homogeneous solution when 150 mM NaCl was added to the buffer (Figure 5a, black curve). Heparan sulfate still induced amyloid formation under these conditions (Figure 5a, blue curve), but with a lower efficiency as indicated by a 3.3 fold longer lag phase (990 mins), compared to the kinetic profile without NaCl. (Supplementary Material). At higher NaCl, 500 mM, I26P-IAPP did form amyloid in the presence and in the absence of heparan sulfate (Figure 5a). Similar kinetic profiles were obtained with and without heparan sulfate, although slightly higher final fluorescence intensity was observed in the presence of heparan sulfate. TEM images collected at the end of each experiment (Figure 5) are consistent with the thioflavin-T fluorescence measurements. The observation that heparan sulfate no longer accelerates IAPP amyloid formation by I26P-IAPP at high salt relative to control suggests that electrostatic interactions make a significant contribution to GAG-peptide interactions.
Figure 5.

I26P-IAPP amyloid formation in the presence and absence of heparan sulfate at different NaCl concentrations. (a) Results of fluorescence-monitored thioflavin-T binding assays are displayed. The kinetic profiles were collected with 20 mM Tris-HCl and either 150 mM or 500 mM NaCl at pH 7.4, no HFIP, without stirring at 25 °C. Black and blue curves were collected in a buffer with 170 mM total salt concentration (20 mM Tris+150 mM NaCl). Black, I26P-IAPP in the absence of heparan sulfate; blue, I26P-IAPP in the presence of heparan sulfate. Green and pink curves were collected in a buffer with 520 mM salt concentration (20 mM Tris+500 mM NaCl). Green, I26P-IAPP in the absence of heparan sulfate; pink, I26P-IAPP in the presence of heparan sulfate. (b) TEM image of I26P-IAPP in the absence of heparan sulfate with 150 mM NaCl in the buffer. (c) TEM image of I26P-IAPP in the presence of heparan sulfate with 150 mM NaCl in the buffer. (d) TEM image of I26P-IAPP in the absence of heparan sulfate with 500 mM NaCl in the buffer. (e) TEM image of I26P-IAPP in the presence of heparan sulfate with 500 mM NaCl in the buffer. Aliquots were removed at the end of each experiment for TEM analysis. Scale bars represent 100 nm. The concentration of I26P-IAPP was 16 μM. Heparan sulfate, when present, was at 1.3 μM.
GAGs are more effective than anionic vesicles at inducing amyloid formation by I26P-IAPP
A wide range of studies have shown that lipid membranes containing negatively charged lipids promote the formation of IAPP amyloid.30,32,44,45 Although the mechanism of the effect is not completely clear, it is believed that electrostatic interactions between positively charged peptides and negatively charged lipids play an important role. If the effects of GAGs and lipid vesicles depended only on the concentration of negative charges rather than on their spatial distribution, then we would expect similar effects when the total number of negatively charged sites were matched. To test if this is the case, we compared the effects of lipid vesicles and heparan sulfate on amyloid formation by I26P-IAPP. Anionic dioleoylphosphatidylglycerol (DOPG) which contains a single negative charge was chosen as a model lipid. DOPG has been widely used in model studies of IAPP membrane interactions.31,46,47
DOPG induces amyloid formation by I26P-IAPP, but much less efficiently on a per negative charge basis than heparan sulfate. The concentration of heparan sulfate (0.97 μM) and DOPG (48 μM) used here correspond to a concentration of 48 μM negative charges. The lag phase of I26P-IAPP amyloid formation in the presence of DOPG (675 mins) was almost 10 fold longer than observed with heparan sulfate (68 mins). (Figure 6) Peptide-lipid binding and lipid induced fiber formation are highly dependent on the concentration of lipids and on the lipid to peptide ratio. A 5 fold increase in the concentration of DOPG increased T50 two fold. (Supplementary Material) This dependence was weaker for HS. A 10 fold increase in HS concentration led to only a 1.5 fold increase in rate. (Supplementary Material) These results demonstrate that heparan sulfate is more efficient than DOPG in inducing amyloid formation by I26P-IAPP at the same net total charge under the conditions used. Moreover, the effects of HS are less dependent on its concentration.
Figure 6.

Comparison of amyloid formation by I26P-IAPP in the presence of GAG and lipids. (a) Results of fluorescence-monitored thioflavin-T binding assays are displayed. Black, I26P-IAPP in the presence of heparan sulafte; red, I26P-IAPP in the presence of 100 nm DOPG vesicles. (b) TEM image of I26P-IAPP in the presence of heparan sulfate. (c) TEM image of I26P-IAPP in the presence of 100 nm DOPG vesicles. Aliquots were removed at the end of each reaction for TEM analysis. Scale bars represent 100 nm. The kinetic profiles were collected with 20 mM Tris at pH 7.4, no HFIP, no stirring at 25°C. The concentration of IAPP is 16 μM. Heparan sulfate, when present was at 0.97 μM. DOPG, when present, was at 48 μM. The concentration of heparan sulfate and DOPG were chosen so that the samples would contain the same number of negatively charged sites.
I26P-IAPP and NMe-G24, I26-IAPP can be induced to form amyloid by other GAGs
It is natural to inquire if the effect of heparan sulfate is specific to the structure of the GAG used. We studied amyloid formation by the IAPP variants in the presence of two other GAGs, chondroitin sulfate and dermatan sulfate. All three of these GAGs are composed of repeating disaccharide units. The most common unit of heparan sulfate is glucuronic acid (GlcA) linked to N-acetylglucosamine (GlcNAc). Chondrointin sulfate is composed mainly of alternating D-glucuronic acid (GlcA) and N-acetyl-D-galactosamine (GalNAc). When the GlcA is epimerized into L-iduronic acid (IdoA), the resulting GAG is denoted dermatan sulfate.
Figure 7a compares I26P-IAPP amyloid formation induced by the three GAGs. Chondroitin sulfate and dermatan sulfate also promoted amyloid formation by I26P-IAPP. A slightly shorter lag phase and lower final fluorescence intensity was observed in the presence of chondroitin sulfate (Figure 7a, red curve) than with heparan sulfate (Figure 7a, black curve), while the lag phase for dermatan sulfate induced amyloid formation was between that observed for chondroitin sulfate and heparan sulfate. TEM (Figure 7) and CD (Supplementary Material) confirmed that fibrils had formed at the end of each experiment. Similar results were observed with NMe-G24, NMe-I26-IAPP. The peptide was amyloidgenic in the presence of each of the three GAGs and the trend in the efficiency in inducing amyloid formation was the same as observed for I26P-IAPP as judged by the lag times (Supplementary Material). The results show that the amyloid inducing effect of GAGs on non-amyloidgenic IAPP variants is not specific to heparan sulfate. The difference in GAGs’ structures and the arrangement of the charges are likely responsible for the slight variations in the kinetic parameters.
Figure 7.

Comparison of I26P-IAPP amyloid formation in the presence of different GAGs. (a) Kinetic profiles. Black, I26P-IAPP in the presence of heparan sulfate; red, I26P-IAPP in the presence of chondroitin sulfate; green, I26P-IAPP in the presence of dermatan sulfate. (b) TEM image of I26P-IAPP in the presence of chondroitin sulfate. (c) TEM image of I26P-IAPP in the presence of dermatan sulfate. Aliquots were removed at the end of each reaction for TEM analysis. Scale bars represent 100 nm. The kinetic experiments were conducted in 20 mM Tris-HCl (pH 7.4), 2% HFIP (v/v) with continuous stirring at 25 °C. The concentration of I26P-IAPP was 16 μM, and the concentration of GAG was 1.3 μM.
IAPP amyloid inhibitors are less effective in the presence of GAGs
I26P-IAPP and NMe-G24, NMe-I26-IAPP are both inhibitors of IAPP amyloid formation in homogeneous solution. We tested their efficiency in the presence of heparan sulfate. Figure 8a displays the results of an inhibition experiment using I26P-IAPP. In the absence of GAG, the lag phase of amyloid formation by IAPP was a factor of 3 times longer when the inhibitor was present, consistent with previous reports.8 Quite different results were obtained when GAG was present in the mixture; an initial, rapid increase in the thioflavin fluorescence was observed followed by an intermediate plateau. Similar biphasic behavior has been observed during GAG catalyzed amyloid formation by proIAPP processing intermediates and is thought to be due to the rapid formation of a GAG bound intermediate.33 This was followed by a second growth phase leading to a final plateau. TEM analysis of aliquots removed during the first plateau revealed the presence of thin fibers as well as shorter fibril like objects (Figure 8). Numerous fibers were observed in the final plateau (Figure 8). The results of the thioflavin-T assays are difficult to unambiguously interpret since both polypeptides can form amyloid in the presence of GAGs. Thus the final thioflavin-T signal could arise from amyloid formation by one or both of the polypeptides. None the less, it is clear that mixtures of wild type IAPP with I26P-IAPP or with NMe-G24, NMe-I26-IAPP behave very differently in the presence of GAGs than in their absence.
Figure 8.

Inhibition of IAPP amyloid formation by I26P-IAPP in the absence and presence of heparan sulfate. (a) The results of thioflavin-T binding assays are plotted. Black, wild type IAPP in the absence of heparan sulfate; red, a 1:1 mixture of wild type IAPP and I26P-IAPP in the absence of heparan sulfate; green, a 1:1 mixture of wild type IAPP and I26P-IAPP in the presence of heparan sulfate. (b) TEM image of wild type IAPP in the absence of heparan sulfate. An aliquot was removed at the end of the reaction for TEM analysis as indicated by the black rectangle in panel A. (c) TEM image of a 1:1 mixture of wild type IAPP and I26P-IAPP in the absence of heparan sulfate. An aliquot was removed at the end of the reaction for TEM analysis as indicated by the red triangle in panel A. (d) TEM image of a 1:1 mixture of wild type IAPP and I26P-IAPP in the presence of heparan sulfate in the first plateau. An aliquot was removed in the middle of the first plateau (10 mins) for TEM analysis as indicated by the green circle in panel A. (e) TEM image of a 1:1 mixture of wild type IAPP and I26P-IAPP in the presence of heparan sulfate. An aliquot was removed at the end of the reaction for TEM analysis as indicated by the green star in panel A. Scale bars represent 100 nm. The kinetic experiments were conducted in 20 mM Tris-HCl (pH 7.4), 2% HFIP (v/v) with continuous stirring at 25 °C. The concentration of wild type IAPP was 16 μM. Heparan sulfate, when present, was at 2.6 μM.
This experiment was conducted by mixing IAPP, inhibitor and HS at the beginning of the experiment. The effect of the GAG could arise from sequestration of the inhibitor by HS. We conducted an additional experiment to test whether GAGs accelerated amyloid formation if they were added in the lag phase of the I26P-IAPP: IAPP mixture. In this experiment, IAPP and inhibitor were allowed to form a complex before addition of HS. HS still accelerated amyloid formation (Figure 9), suggesting that simple sequestration of the inhibitor is not the only cause of the HS induced effects. This is reasonable since both IAPP and I26P-IAPP can bind HS and there is no reason that HS should selectively remove I26P-IAPP from solution.
Figure 9.
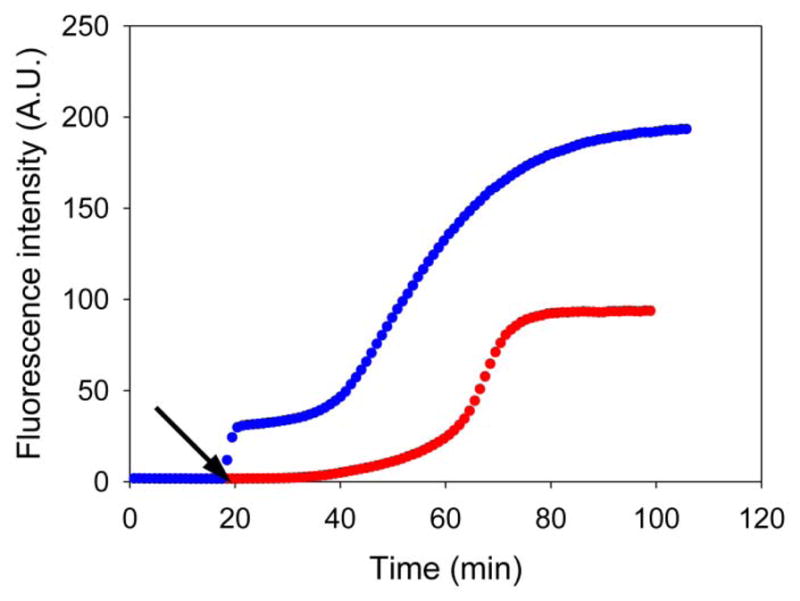
Heparan sulfate can induce amyloid formation by a mixture of IAPP and I26P-IAPP. The results of thioflavin-T binding assays are plotted. Red, a 1:1 mixture of wild type IAPP and I26P-IAPP in the absence of heparan sulfate; blue, a 1:1 mixture of wild type IAPP and I26P-IAPP in the presence of heparan sulfate, heparan sulfate was added at the time point as indicated by the black arrow.
We next examined the ability of NMe-G24, NMe-I26-IAPP to inhibit amyloid formation. Figure 10a compares thioflavin-T kinetic curves for wild type IAPP in the absence of heparan sulfate, a 1:1 mixture of wild type IAPP and inhibitor without heparan sulfate and a 1:1 mixture of wild type IAPP and inhibitor in the presence of heparan sulfate. NMe-G24, NMe-I26-IAPP is, as expected, an effective inhibitor in the absence of heparan sulfate. Under our conditions wild type IAPP forms amyloid with a lag time of 17 mins, in contrast, no significant increase in thioflavin-T fluorescence is observed for 150 mins in the presence of inhibitor although eventually a significant increase is detected around 400 mins. The inhibitor is less effective when heparan sulfate is present. A rapid increase in thioflavin-T fluorescence is observed, which is followed, quickly, by a second transition to a final plateau. TEM reveals some fibril like aggregates in the first plateau (Figure 10d). Dense matts of fibers were observed at the end of the reaction (Figure 10e).
Figure 10.

Inhibition of IAPP amyloid formation by NMe-G24, NMe-I26-IAPP in the absence and presence of heparan sulfate. (a) The results of thioflavin-T binding assays are plotted. Black, wild type IAPP; red, a 1:1 mixture of wild type IAPP and NMe-G24, NMe-I26-IAPP in the absence of heparan sulfate; green, a 1:1 mixture of wild type IAPP and NMe-G24, NMe-I26-IAPP in the presence of heparan sulfate. (b) An expansion of the first 200 mins of panel A. The same color coding is used. (c) TEM image of a 1:1 mixture of wild type IAPP and NMe-G24, NMe-I26-IAPP in the absence of heparan sulfate. An aliquot was removed at the end of the reaction (563 min) for TEM analysis. (d) TEM image of a 1:1 mixture of wild type IAPP and NMe-G24, NMe-I26-IAPP in the presence of heparan sulfate in the first plateau. An aliquot was removed in the middle of the first plateau (17 min) for TEM analysis. (e) TEM image of a 1:1 mixture of wild type IAPP and NMe-G24, NMe-I26-IAPP in the presence of heparan sulfate. An aliquot was removed at the end of the reaction (333 min) for TEM analysis. Scale bars represent 100 nm. The kinetic experiments were conducted in 20 mM Tris-HCl (pH 7.4), 2% HFIP (v/v) with continuous stirring at 25 °C. The concentration of wild type IAPP was 16 μM. Heparan sulfate, when present, was at 2.6 μM.
We also analyzed the ability of the small molecule (−)-epigallocatechin 3-gallate (EGCG) to inhibit amyloid formation in the presence of heparan sulfate in order to test if GAGs can affect small molecule inhibitors as well as peptide-based inhibitors. EGCG is a green tea-derived flavanol that has been reported to inhibit amyloid formation by a wide range of natively unfolded polypeptides including IAPP.9,48–51 Previous work from our laboratory has shown that EGCG effectively inhibits in vitro IAPP amyloid formation and dissociates pre-formed fibrils into small aggregates.9,51 In the presence of heparan sulfate without EGCG, IAPP amyloid formation is greatly accelerated compared to the same reaction in homogeneous solution (Figure 11). No increase of thioflavin-T fluorescence is observed for the 1:1 mixture of IAPP with EGCG without heparan sulfate (Figure 11). However, when heparan sulfate and EGCG are present, an initial rapid increase of thioflavin-T fluorescence was observed followed by a slow decrease, which may indicate that aggregates formed immediately and then gradually dissociated into small aggregates or were restructured (Figure 11, red curve). The initial increase of fluorescence was lower for higher concentrations of added EGCG (Supplementary Material), but the effect was still observable even when EGCG was in 20 fold excess. TEM images confirmed the existence of small aggregates at the end of each experiment. (Figure 11, Supplementary Material) The aggregates did not show any obvious change in morphology as the concentration of EGCG is increased (Supplementary Material).
Figure 11.

Heparan sulfate interferes with the ability of EGCG to inhibit IAPP amyloid formation. (a) Results of fluorescence-monitored thioflavin-T binding assays are displayed. Black, IAPP in the presence of heparan sulfate; grey, a 1:1 mixture of IAPP and EGCG in the absence of heparan sulfate; red, a 1:1 mixture of IAPP and EGCG in the presence of heparan sulfate. (b) TEM image of an aliquot removed at the end of the experiment represented by the black curve in panel A, at the time point corresponding to the black diamond. (c) TEM image of an aliquot removed at the end of the experiment represented by the grey curve in panel A, at the time point corresponding to the grey circle. (d) TEM image of an aliquot removed from the sample represented by the red curve in panel A, at the time point corresponding to the red star. (e) TEM image of an aliquot removed at the end of the experiment represented by the red curve in panel A, at the time point corresponding to the red triangle. Scale bar represent 100 nm. Kinetic experiments were conducted under experimental conditions with 20 mM Tris-HCl, no HFIP and no stirring at 25 °C. The concentration of IAPP was 16 μM. Heparan sulfate, when present, was at 1.3 μM.
EGCG was still able to dissociate amyloid fibrils formed by IAPP in the presence of heparan sulfate (Figure 12a). The structures of the aggregates formed were similar to those formed when EGCG is added at the beginning of the reaction as judged by TEM images (Figure 12, Supplemental Material). CD confirmed that all of the aggregates contain β-sheet structure (Supplementary Material). These results indicate that EGCG is still able to inhibit IAPP amyloid formation and dissociate mature amyloid fibrils into small aggregates in the presence of heparan sulfate, but is less effective than in a homogeneous environment.
Figure 12.

EGCG can dissociate amyloid fibrils formed by IAPP in the presence of heparan sulfate. (a) Results of fluorescence-monitored thioflavin-T binding assays are displayed. Black, IAPP in the presence of heparan sulfate; red, a 1:1 mixture of IAPP and EGCG in the presence of heparan sulfate, EGCG was added at the time point indicated by the red arrow. (b) TEM image of an aliquot taken at the end of the experiment represented by the red curve in panel A, at the time point corresponding to the red triangle. Scale bar represent 100 nm. Kinetic experiments were conducted under experimental conditions with 20 mM Tris-HCl, no HFIP and no stirring at 25 °C. The concentration of IAPP was 16 μM. Heparan sulfate was at 1.3 μM.
Conclusions
The analysis presented here reveals that IAPP amyloid formation in heterogeneous environment is very different than in homogeneous solutions: Apparently non-amyloidgenic variants can be readily induced to form amyloid in the presence of GAGs and some peptide based and small molecule amyloid inhibitors are less effective. The GAGs tested here are more effective at inducing amyloid formation than are anionic lipid vesicles on a per net charge basis even when the total number of anionic sites is the same, suggesting that the spatial arrangement of negatively charged sites is important. This conjecture is supported by a recent study of the effects of varying the number of sulfated saccharide monomers in heparin on amyloid formation by wild type human IAPP.39 Small fragments were less effective than large ones, but the length dependence leveled off beyond 12 sulfated saccharide units. Control experiments showed that the dependence was due to a length effect rather than an increase in the number of monomer units. The length dependent effects are believed to arise from the ability of the longer heparin fragments to adopt specific structures. Solution NMR studies have shown that heparin forms a left-handed helix structure with four saccharides per turn52 and the length dependence studies imply that heparin exerts its most potent effects on IAPP amyloid formation when it can form helix structures.39 IAPP contains a positively charged N-terminus, a Lys at position 1, an Arg at position-11 and a His at position 18. All of these groups have been shown to be important for IAPP GAG interactions and the charge arrangement in helical heparin fragments is complimentary to the arrangement of positive charges in models of IAPP amyloid fibers.33,39,53 Modeling of Aβ GAG interactions also suggests a role for complimentary charged interfaces in the catalytic effects of GAGs on Aβ amyloid formation.54
HSPGs are associated with in vivo islet amyloid and the interaction of proIAPP processing intermediates with HSPGs of the extracellular has been postulated to play a role in inducing amyloid formation.34,38 Studies with inhibitors of GAG synthesis show that decreasing GAG synthesis and protein glycosylation in islets results in reduced islet amyloid formation in vitro.55 This result is consistent with earlier in vivo findings that these inhibitors reduce amyloid formation in mouse models of Amyloid A amyloidosis.56 These in vitro and in vivo studies suggest that limiting IAPP GAG interaction might be a therapeutic method for amyloid related diseases.
Relatively little attention has been paid to the role of IAPP GAG interactions. The results presented here show they can strongly impact amyloid formation and the efficiency of amyloid inhibitors. Thus potential interactions with GAGs should be considered in inhibitor design and in the analysis of amyloidgenicity. GAGs have also been shown to induce amyloid formation by other polypeptides which appear to be non-amyloidgenic in homogeneous solution.57 Thus the effects observed with IAPP may be one example of a broader role for GAGs in amyloid formation.
Materials and Methods
Peptide Synthesis and Purification
Peptides were synthesized on a 0.25 mmol scale using a CEM Liberty Microwave Peptide Synthesizer utilizing 9-fluornylmethoxycarbonyl (Fmoc) chemistry. 5-(4′-fmoc-aminomethyl-3′,5-dimethoxyphenol) valeric acid (PAL-PEG) resin was used to form an amidated C-terminus. Fmoc protected pseudoproline (oxazolidine) dipeptide derivatives were incorporated to facilitate the synthesis as previously described.58,59 Standard Fmoc reaction cycles were used. The first residue attached to the resin, all β-branched residues and all pseudoproline dipeptide derivatives were double-coupled. N-methylated isoleucine was triple-coupled. The alanine that directly followed the N-methylated isoleucine was coupled 5 times. N-methylated glycine, the phenylalanine and the asparagines directly following the N-methylated glycine were double coupled. The peptides were cleaved from the resin through the use of standard trifluoroacetic acid (TFA) methods. Crude peptides were oxidized by dimethyl sulfoxide (DMSO) at room temperature. The peptides were purified via reverse-phase high-performance liquid chromatography (RP-HPLC) using a Vydac C18 preparative column.60 Analytical HPLC were used to check the purity of the peptides before each experiment. The identity of the pure peptides was confirmed by MALDI-TOF MS. Wild type IAPP expected 3902.9, observed 3902.7; I26P-IAPP expected 3888.3, observed 3888.2; NMe-G24, NMe-I26-IAPP expected, 3930.9, observed, 3930.8.
Sample preparation
Peptide stock solutions were prepared in 100% hexafluoroisopropanol (HFIP) at 1.6 mM. Low molecular weight heparan sulfate (10,000–14,000 molecular weight), chondrointin sulfate and dermatan sulfate were purchased from Sigma. Fluorescein-labeled heparin (FLH) with an average molecular weight of 18,000 was purchased from Invitrogen. All GAG stock solutions were prepared by dissolving GAG in 20 mM Tris-HCl (pH 7.4) at 2.2 mg/ml. EGCG was purchased from Sigma and dissolved in 20 mM Tris-HCl (pH 7.4) at 1.6 mM immediately before use.
Preparation of large unilamellar vesicles (LUVs)
The LUVs were composed of 100% 1,2-dioleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (DOPG), from Avanti Polar Lipids. Stock solutions of DOPG in chloroform at a concentration of 10 mg/mL were first evaporated with a stream of nitrogen gas and then dried under a vacuum overnight to completely remove the residual organic solvent. The resulting lipid film was hydrated in 20 mM Tris-HCl, pH 7.4 buffer for 1 hour, at a lipid concentration of 8 mM. The multilamellar vesicles were subjected to 10 freeze-thaw cycles and extruded 11 times through 100 nm pore size filters (Whatman, GE). The phospholipid concentration was determined by the method of Stewart.61 A fresh vesicle solution was used for each experiment.
Fluorescence Assays
Two types of ThioflavinT binding assays were utilized, one for samples with hexafluoroisopropanol (HFIP) and the other for samples which did not contain HFIP. Amyloid formations by I26P-IAPP and NMe-G24, NMe-I26-IAPP and their ability to inhibit amyloid formation by wild type human IAPP in the presence and absence of GAGs were first monitored by thioflavin-T binding assays conducted in the presence of 2% hexafluoroisopropanol (HFIP) and with continuous stirring at 25 °C. Fluorescence measurements were performed using an Applied Photon Technology fluorescence spectrophotometer with 450 nm excitation and 485 nm emission. The slit widths for excitation and emission were set at 4 nm and a 1.0 cm cuvette was used. Each point was averaged for 1 minute. Solutions were prepared by diluting filtered stock solution (0.45 μM Acrodisc syringe filter with GHP membrane) into Tris-HCl buffer and thioflavin-T solution immediately before the measurement. The final concentration was 16 μM peptide and 32 μM thioflavinT in 20 mM Tris-HCl (pH 7.4). In the I26P-IAPP and NMe-G24, NMe-I26-IAPP amyloid formation experiments, GAGs (heparan sulfate, chondroitin sulfate and dermatan sulfate), when present, were at 1.3 μM. In the inhibition experiments, heparan sulfate, when present, was at 2.6 μM. FRET experiments involving fluorescein-labeled heparin (FLH) were also monitored using this assay in solutions which contained 2% HFIP. Fluorescence measurements were performed on the same instrument using 440 nm excitation and 510 nm emission. The final solution composition was 16 μM peptide, 1.3 μM FLH and 32 μM thioflavinT, when present.
Amyloid formation by I26P-IAPP and NMe-G24, NMe-I26-IAPP in the presence and absence of heparan sulfate were also monitored by thioflavin-T binding assays in the absence of HFIP. These assays were conducted without stirring using a Beckman Coulter DTX 880 plate reader with multimode detector using 430 nm excitation and 485 nm emission at 25°C. Fluorescence solutions were prepared by lyophilizing filtered stock solution (0.45 μM Acrodisc syringe filter with GHP membrane) for 22 hrs and then dissolving dry peptides into Tris-HCl buffer and thioflavin-T solution immediately before the measurement. The final concentration was 16 μM peptide and 32 μM thioflavin-T in 20 mM Tris-HCl (pH 7.4). Heparan sulfate, when present, was at 1.3 μM. Experiments involving high ionic strength, lipid micelle or EGCG were also monitored using this assay.
Circular Dichroism (CD)
Far-UV CD experiments were performed at 25°C on an Applied Photophysics Chirascan CD spectrophotometer. Aliquots from the kinetic experiments were removed at relevant time points during the experiments and the spectra were recorded. Spectra were the average of three repeats recorded over a range of 190–260 nm, at 1 nm intervals. A 0.1 cm quartz cuvette was used and a background spectrum was subtracted from the collected data.
Transmission Electron Microscopy (TEM)
TEM images were collected at the Life Science Microscopy Center at the State University of New York at Stony Brook. 15 μL aliquots of the samples used for the kinetic studies were removed at relevant time points, placed on a carbon-coated 300-mesh copper grid for 1 min and then negatively stained with saturated uranyl acetate for 1 min.
Supplementary Material
Acknowledgments
We thank the National Institute of Health (GM078114) for financial support. We also thank Dr. Fanling Meng and Dr. Andishen Abedini for helpful discussions.
Abbreviations
- IAPP
islet amyloid polypeptide
- I26P-IAPP
I26P point mutant of human IAPP
- NMe-G24
NMe-I26-IAPP, G24-N-Methyl, I26-N-Methyl-IAPP
- GAG
glycosaminoglycan
- HS
heparan sulfate
- FLH
fluorescein labeled heparin
- HFIP
hexafluoroisopropanol
- DMSO
dimethyl sulfoxide
- LUV
large unilamellar vesicles
- DOPG
1,2-dioleoyl-sn-glycero-3-phospho-(1′-rac-glycerol)
- GlcA
glucuronic acid
- GlcNAc
N-acetylglucosamine
- GalNAc
N-acetyl--galactosamine
- IdoA
iduronic acid
- CD
circular dichroism
- TEM
transmission electron microscopy
- EGCG
(−)-epigallocatechin 3-gallate
References
- 1.Vendruscolo M, Zurdo J, MacPhee CE, Dobson CM. Protein folding and misfolding: a paradigm of self-assembly and regulation in complex biological systems. Philos Trans R Soc Lond Ser A-Math Phys Eng Sci. 2003;361:1205–1222. doi: 10.1098/rsta.2003.1194. [DOI] [PubMed] [Google Scholar]
- 2.Sipe JD. Amyloidosis. Crit Rev Clin Lab Sci. 1994;31:325–354. doi: 10.3109/10408369409084679. [DOI] [PubMed] [Google Scholar]
- 3.Kirkitadze MD, Bitan G, Teplow DB. Paradigm shifts in Alzheimer’s disease and other neuro degenerative disorders: The emerging role of oligomeric assemblies. J Neurosci Res. 2002;69:567–577. doi: 10.1002/jnr.10328. [DOI] [PubMed] [Google Scholar]
- 4.Lorenzo A, Razzaboni B, Weir GC, Yankner BA. Pancreatic islet cell toxicity of amylin associated with type-2 diabetes mellitus. Nature. 1994;368:756–760. doi: 10.1038/368756a0. [DOI] [PubMed] [Google Scholar]
- 5.Silveira JR, Raymond GJ, Hughson AG, Race RE, Sim VL, Hayes SF, Caughey B. The most infectious prion protein particles. Nature. 2005;437:257–261. doi: 10.1038/nature03989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blazer LL, Neubig RR. Small molecule protein-protein interaction inhibitors as CNS therapeutic agents: Current progress and future hurdles. Neuropsychopharmacology. 2009;34:126–141. doi: 10.1038/npp.2008.151. [DOI] [PubMed] [Google Scholar]
- 7.Takahashi T, Mihara H. Peptide and protein mimetics inhibiting amyloid beta-peptide aggregation. Acc Chem Res. 2008;41:1309–1318. doi: 10.1021/ar8000475. [DOI] [PubMed] [Google Scholar]
- 8.Meng FL, Raleigh DP, Abedini A. Combination of kinetically selected inhibitors in trans leads to highly effective inhibition of amyloid formation. J Am Chem Soc. 2010;132:14340–14342. doi: 10.1021/ja1046186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meng F, Abedini A, Plesner A, Verchere CB, Raleigh DP. The flavanol (−)-epigallocatechin 3-gallate inhibits amyloid formation by islet amyloid polypeptide, disaggregates amyloid fibrils, and protects cultured cells against IAPP-induced toxicity. Biochemistry. 2010;49:8127–8133. doi: 10.1021/bi100939a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meng F, Abedini A, Plesner A, Middleton CT, Potter KJ, Zanni MT, Verchere CB, Raleigh DP. The sulfated triphenyl methane derivative acid fuchsin is a potent inhibitor of amyloid formation by human islet amyloid polypeptide and protects against the toxic effects of amyloid formation. J Mol Biol. 2010;400:555–566. doi: 10.1016/j.jmb.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yan LM, Tatarek-Nossol M, Velkova A, Kazantzis A, Kapurniotu A. Design of a mimic of nonamyloidogenic and bioactive human islet amyloid polypeptide (IAPP) as nanomolar affinity inhibitor of IAPP cytotoxic fibrillogenesis. Proc Natl Acad Sci USA. 2006;103:2046–2051. doi: 10.1073/pnas.0507471103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abedini A, Meng FL, Raleigh DP. A single-point mutation converts the highly amyloidogenic human islet amyloid polypeptide into a potent fibrillization inhibitor. J Am Chem Soc. 2007;129:11300–11301. doi: 10.1021/ja072157y. [DOI] [PubMed] [Google Scholar]
- 13.Chafekar SM, Malda H, Merkx M, Meijer EW, Viertl D, Lashuel HA, Baas F, Scheper W. Branched KLVFF tetramers strongly potentiate inhibition of beta-amyloid aggregation. ChemBioChem. 2007;8:1857–1864. doi: 10.1002/cbic.200700338. [DOI] [PubMed] [Google Scholar]
- 14.van Horssen J, Wesseling P, van den Heuvel LP, de Waal RM, Verbeek MM. Heparan sulphate proteoglycans in Alzheimer’s disease and amyloid-related disorders. Lancet Neurol. 2003;2:482–492. doi: 10.1016/s1474-4422(03)00484-8. [DOI] [PubMed] [Google Scholar]
- 15.Snow AD, Wight TN. Proteoglycans in the pathogenesis of Alzheimer’s disease and other amyloidoses. Neurobiol Aging. 1989;10:481–497. doi: 10.1016/0197-4580(89)90108-5. [DOI] [PubMed] [Google Scholar]
- 16.Inoue S. International Review of Cytology - a Survey of Cell Biology. Vol. 210. Academic Press Inc; San Diego: 2001. Basement membrane and beta amyloid fibrillogenesis in Alzheimer’s disease; pp. 121–161. [DOI] [PubMed] [Google Scholar]
- 17.Potter-Perigo S, Hull RL, Tsoi C, Braun KR, Andrikopoulos S, Teague J, Verchere CB, Kahn SE, Wight TN. Proteoglycans synthesized and secreted by pancreatic islet beta-cells bind amylin. Arch Biochem Biophys. 2003;413:182–190. doi: 10.1016/s0003-9861(03)00116-4. [DOI] [PubMed] [Google Scholar]
- 18.Ancsin JB. Amyloidogenesis: historical and modern observations point to heparan sulfate proteoglycans as a major culprit. Amyloid. 2003;10:67–79. doi: 10.3109/13506120309041728. [DOI] [PubMed] [Google Scholar]
- 19.Young ID, Ailles L, Narindrasorasak S, Tan R, Kisilevsky R. Localization of the basement membrane heparan sulfate proteoglycan in islet amyloid deposits in type II diabetes mellitus. Arch Pathol Lab Med. 1992;116:951–954. [PubMed] [Google Scholar]
- 20.Bonifacio MJ, Sakaki Y, Saraiva MJ. ‘In vitro’ amyloid fibril formation from transthyretin: The influence of ions and the amyloidogenicity of TTR variants. BBA-Mol Basis Dis. 1996;1316:35–42. doi: 10.1016/0925-4439(96)00014-2. [DOI] [PubMed] [Google Scholar]
- 21.McParland VJ, Kad NM, Kalverda AP, Brown A, Kirwin-Jones P, Hunter MG, Sunde M, Radford SE. Partially unfolded states of beta(2)-microglobulin and amyloid formation in vitro. Biochemistry. 2000;39:8735–8746. doi: 10.1021/bi000276j. [DOI] [PubMed] [Google Scholar]
- 22.Kayed R, Bernhagen J, Greenfield N, Sweimeh K, Brunner H, Voelter W, Kapurniotu A. Conformational transitions of islet amyloid polypeptide (IAPP) in amyloid formation in vitro. J Mol Biol. 1999;287:781–796. doi: 10.1006/jmbi.1999.2646. [DOI] [PubMed] [Google Scholar]
- 23.Kahn SE, Andrikopoulos S, Verchere CB. Islet amyloid: A long-recognized but underappreciated pathological feature of type 2 diabetes. Diabetes. 1999;48:241–253. doi: 10.2337/diabetes.48.2.241. [DOI] [PubMed] [Google Scholar]
- 24.Cooper GJ, Willis AC, Clark A, Turner RC, Sim RB, Reid KB. Purification and characterization of a peptide from amyloid-rich pancreases of type 2 diabetic patients. Proc Natl Acad Sci USA. 1987;84:8628–8632. doi: 10.1073/pnas.84.23.8628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clark A, Wells CA, Buley ID, Cruickshank JK, Vanhegan RI, Matthews DR, Cooper GJ, Holman RR, Turner RC. Islet amyloid, increased A-cells, reduced B-cells and exocrine fibrosis: quantitative changes in the pancreas in type 2 diabetes. Diabetes Res Clin Pract. 1988;9:151–159. [PubMed] [Google Scholar]
- 26.Westermark P, Wernstedt C, Wilander E, Hayden DW, Obrien TD, Johnson KH. Amyloid fibrils in human insulinoma and islets of Langerhans of the diabetic cat are derived from a neuropeptide-like protein also present in normal islet cells. Proc Natl Acad Sci USA. 1987;84:3881–3885. doi: 10.1073/pnas.84.11.3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clark A, Nilsson MR. Islet amyloid: a complication of islet dysfunction or an aetiological factor in Type 2 diabetes? Diabetologia. 2004;47:157–169. doi: 10.1007/s00125-003-1304-4. [DOI] [PubMed] [Google Scholar]
- 28.Andersson A, Bohman S, Borg LAH, Paulsson JF, Schultz SW, Westermark GT, Westermark P. Amyloid deposition in transplanted human pancreatic islets: a conceivable cause of their long-term failure. Exp Diabetes Res. 2008;2008 doi: 10.1155/2008/562985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Potter KJ, Abedini A, Marek P, Klimek AM, Butterworth S, Driscoll M, Baker R, Nilsson MR, Warnock GL, Oberholzer J, Bertera S, Trucco M, Korbutt GS, Fraser PE, Raleigh DP, Verchere CB. Islet amyloid deposition limits the viability of human islet grafts but not porcine islet grafts. Proc Natl Acad Sci USA. 2010;107:4305–4310. doi: 10.1073/pnas.0909024107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knight JD, Miranker AD. Phospholipid catalysis of diabetic amyloid assembly. J Mol Biol. 2004;341:1175–1187. doi: 10.1016/j.jmb.2004.06.086. [DOI] [PubMed] [Google Scholar]
- 31.Knight JD, Hebda JA, Miranker AD. Conserved and cooperative assembly of membrane-bound alpha-helical states of islet amyloid polypeptide. Biochemistry. 2006;45:9496–9508. doi: 10.1021/bi060579z. [DOI] [PubMed] [Google Scholar]
- 32.Jayasinghe SA, Langen R. Lipid membranes modulate the structure of islet amyloid polypeptide. Biochemistry. 2005;44:12113–12119. doi: 10.1021/bi050840w. [DOI] [PubMed] [Google Scholar]
- 33.Meng F, Abedini A, Song B, Raleigh DP. Amyloid formation by pro-islet amyloid polypeptide processing intermediates: Examination of the role of protein heparan sulfate interactions and implications for islet amyloid formation in type 2 diabetes. Biochemistry. 2007;46:12091–12099. doi: 10.1021/bi7004834. [DOI] [PubMed] [Google Scholar]
- 34.Abedini A, Tracz SM, Cho JH, Raleigh DP. Characterization of the heparin binding site in the N-terminus of human pro-islet amyloid polypeptide: Implications for amyloid formation. Biochemistry. 2006;45:9228–9237. doi: 10.1021/bi0510936. [DOI] [PubMed] [Google Scholar]
- 35.Westermark P, Engstrom U, Johnson KH, Westermark GT, Betsholtz C. Islet amyloid polypeptide: pinpointing amino acid residues linked to amyloid fibril formation. Proc Natl Acad Sci USA. 1990;87:5036–5040. doi: 10.1073/pnas.87.13.5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shim SH, Gupta R, Ling YL, Strasfeld DB, Raleigh DP, Zanni MT. Two-dimensional IR spectroscopy and isotope labeling defines the pathway of amyloid formation with residue-specific resolution. Proc Natl Acad Sci USA. 2009;106:6614–6619. doi: 10.1073/pnas.0805957106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Castillo GM, Cummings JA, Yang WH, Judge ME, Sheardown MJ, Rimvall K, Hansen JB, Snow AD. Sulfate content and specific glycosaminoglycan backbone of perlecan are critical for perlecan’s enhancement of islet amyloid polypeptide (amylin) fibril formation. Diabetes. 1998;47:612–620. doi: 10.2337/diabetes.47.4.612. [DOI] [PubMed] [Google Scholar]
- 38.Park K, Verchere CB. Identification of a heparin binding domain in the N-terminal cleavage site of pro-islet amyloid polypeptide - Implications for islet amyloid formation. J Biol Chem. 2001;276:16611–16616. doi: 10.1074/jbc.M008423200. [DOI] [PubMed] [Google Scholar]
- 39.Jha S, Patil SM, Gibson J, Nelson CE, Alder NN, Alexandrescu AT. Mechanism of amylin fibrillization enhancement by heparin. J Biol Chem. 2011;286:22894–22904. doi: 10.1074/jbc.M110.215814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Solomon JP, Bourgault S, Powers ET, Kelly JW. Heparin binds 8 kDa gelsolin cross-beta-sheet oligomers and accelerates amyloidogenesis by hastening fibril extension. Biochemistry. 2011;50:2486–2498. doi: 10.1021/bi101905n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abedini A, Raleigh DP. The role of His-18 in amyloid formation by human islet amyloid polypeptide. Biochemistry. 2005;44:16284–16291. doi: 10.1021/bi051432v. [DOI] [PubMed] [Google Scholar]
- 42.Padrick SB, Miranker AD. Islet amyloid: Phase partitioning and secondary nucleation are central to the mechanism of fibrillogenesis. Biochemistry. 2002;41:4694–4703. doi: 10.1021/bi0160462. [DOI] [PubMed] [Google Scholar]
- 43.Marek PJ, Patsalo V, Green DF, Raleigh DP. Ionic strength effects on amyloid formation by amylin are a complicated interplay among Debye screening, ion selectivity, and Hofmeister effects. Biochemistry. 2012 doi: 10.1021/bi300574r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lopes DHJ, Meister A, Gohlke A, Hauser A, Blume A, Winter R. Mechanism of islet amyloid polypeptide fibrillation at lipid interfaces studied by infrared reflection absorption spectroscopy. Biophys J. 2007;93:3132–3141. doi: 10.1529/biophysj.107.110635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brender JR, Lee EL, Cavitt MA, Gafni A, Steel DG, Ramamoorthy A. Amyloid fiber formation and membrane disruption are separate processes localized in two distinct regions of IAPP, the type-2-diabetes-related peptide. J Am Chem Soc. 2008;130:6424–6429. doi: 10.1021/ja710484d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Domanov YA, Kinnunen PKJ. Islet amyloid polypeptide forms rigid lipid-protein amyloid fibrils on supported phospholipid bilayers. J Mol Biol. 2008;376:42–54. doi: 10.1016/j.jmb.2007.11.077. [DOI] [PubMed] [Google Scholar]
- 47.Evers F, Jeworrek C, Tiemeyer S, Weise K, Sellin D, Paulus M, Struth B, Tolan M, Winter R. Elucidating the mechanism of lipid membrane-induced IAPP fibrillogenesis and its inhibition by the red wine compound resveratrol: a synchrotron X-ray reflectivity study. J Am Chem Soc. 2009;131:9516–9521. doi: 10.1021/ja8097417. [DOI] [PubMed] [Google Scholar]
- 48.Bieschke J, Russ J, Friedrich RP, Ehrnhoefer DE, Wobst H, Neugebauer K, Wanker EE. EGCG remodels mature alpha-synuclein and amyloid-beta fibrils and reduces cellular toxicity. Proc Natl Acad Sci USA. 2010;107:7710–7715. doi: 10.1073/pnas.0910723107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ehrnhoefer DE, Bieschke J, Boeddrich A, Herbst M, Masino L, Lurz R, Engemann S, Pastore A, Wanker EE. EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat Struct Mol Biol. 2008;15:558–566. doi: 10.1038/nsmb.1437. [DOI] [PubMed] [Google Scholar]
- 50.Ehrnhoefer DE, Duennwald M, Markovic P, Wacker JL, Engemann S, Roark M, Legleiter J, Marsh JL, Thompson LM, Lindquist S, Muchowski PJ, Wanker EE. Green tea (−)-epigallocatechin-gallate modulates early events in huntingtin misfolding and reduces toxicity in Huntington’s disease models. Hum Mol Genet. 2006;15:2743–2751. doi: 10.1093/hmg/ddl210. [DOI] [PubMed] [Google Scholar]
- 51.Cao P, Raleigh DP. Analysis of the inhibition and remodeling of islet amyloid polypeptide amyloid fibers by flavanols. Biochemistry. 2012;51:2670–2683. doi: 10.1021/bi2015162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang Z, McCallum SA, Xie J, Nieto L, Corzana F, Jimenez-Barbero J, Chen M, Liu J, Linhardt RJ. Solution structures of chemoenzymatically synthesized heparin and its precursors. J Am Chem Soc. 2008;130:12998–13007. doi: 10.1021/ja8026345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Luca S, Yau WM, Leapman R, Tycko R. Peptide conformation and supramolecular organization in amylin fibrils: constraints from solid-state NMR. Biochemistry. 2007;46:13505–13522. doi: 10.1021/bi701427q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Valle-Delgado JJ, Alfonso-Prieto M, de Groot NS, Ventura S, Samitier J, Rovira C, Fernandez-Busquets X. Modulation of A beta(42) fibrillogenesis by glycosaminoglycan structure. Faseb J. 2010;24:4250–4261. doi: 10.1096/fj.09-153551. [DOI] [PubMed] [Google Scholar]
- 55.Hull RL, Zraika S, Udayasankar J, Kisilevsky R, Szarek WA, Wight TN, Kahn SE. Inhibition of glycosaminoglycan synthesis and protein glycosylation with WAS-406 and azaserine result in reduced islet amyloid formation in vitro. Am J Physiol-Cell Physiol. 2007;293:C1586–C1593. doi: 10.1152/ajpcell.00208.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kisilevsky R, Szarek WA, Ancsin JB, Elimova E, Marone S, Bhat S, Berkin A. Inhibition of amyloid A amyloidogenesis in vivo and in tissue culture by 4-deoxy analogues of peracetylated 2-acetamido-2-deoxy-alpha- and beta-d-glucose - Implications for the treatment of various amyloidoses. Am J Pathol. 2004;164:2127–2137. doi: 10.1016/s0002-9440(10)63771-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maji SK, Perrin MH, Sawaya MR, Jessberger S, Vadodaria K, Rissman RA, Singru PS, Nilsson KPR, Simon R, Schubert D, Eisenberg D, Rivier J, Sawchenko P, Vale W, Riek R. Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science. 2009;325:328–332. doi: 10.1126/science.1173155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Marek P, Woys AM, Sutton K, Zanni MT, Raleigh DP. Efficient microwave-assisted synthesis of human islet amyloid polypeptide designed to facilitate the specific incorporation of labeled amino acids. Org Lett. 2010;12:4848–4851. doi: 10.1021/ol101981b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Abedini A, Raleigh DP. Incorporation of pseudoproline derivatives allows the facile synthesis of human IAPP, a highly amyloidogenic and aggregation-prone polypeptide. Org Lett. 2005;7:693–696. doi: 10.1021/ol047480+. [DOI] [PubMed] [Google Scholar]
- 60.Abedini A, Singh G, Raleigh DP. Recovery and purification of highly aggregation-prone disulfide-containing peptides: Application to islet amyloid polypeptide. Anal Biochem. 2006;351:181–186. doi: 10.1016/j.ab.2005.11.029. [DOI] [PubMed] [Google Scholar]
- 61.Stewart JC. Colorimetric determination of phospholipids with ammonium ferrothiocyanate. Anal Biochem. 1980;104:10–14. doi: 10.1016/0003-2697(80)90269-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.