

Summary
Bioactive lipid mediators play a crucial role in the induction and resolution of inflammation. To elucidate their involvement during influenza infection, LC/MS lipidomic profiling of 141 lipid species was performed on a mouse influenza model using two viruses of significantly different pathogenicity. Infection by the low pathogenicity strain, X31/H3N2, induced a pro-inflammatory response followed by a distinct anti-inflammatory response; infection by the high pathogenicity strain, PR8/H1N1, resulted in overlapping pro- and anti-inflammatory states. Integration of the large-scale lipid measurements with targeted gene expression data demonstrated that 5 lipoxygenase metabolites correlated with the pathogenic phase of the infection whereas 12/15-lipoxygenase metabolites were associated with the resolution phase. Hydroxylated linoleic acid, specifically the ratio of 13- to 9-HODE, was identified as a potential biomarker for immune status during an active infection. Importantly, some of the findings from the animal model were recapitulated in studies of human nasopharyngeal lavages obtained during the 2009–2011 influenza seasons.
Introduction
Influenza virus, whether seasonal or pandemic, can cause severe health conditions, often leading to pneumonia. While most infections in humans are self-limiting, highly virulent strains can cause an over-exuberant inflammatory response that is detrimental to the host (Kash et al., 2006). Influenza virus is an enveloped, negativesense, single-stranded RNA virus that contains eight genomic segments that encode up to 13 proteins (Jagger et al., 2012). One of the hallmarks of influenza virus is its high propensity to acquire mutations, which promotes rapid evolution to increase fitness and evade pre-existing immunity in a host population. Antigenic drift, or mutations mainly of the segments encoding the surface proteins, fosters evasion of the host immune system and allows a slightly varied virus to re-infect the population. Antigenic shift, or recombination and exchange of genomic segments between multiple viruses infecting the same cells, allows for the generation of emerging viruses with drastic changes in pathogenicity or host specificity. If a novel virus with increased fitness and pathogenicity emerges, the herd immunity of the human population may be insufficient to prevent sustained transmission, creating devastating epidemic or pandemic events. The most notorious pandemic, caused by the 1918 H1N1 strain, cost the lives of 40–100 million people worldwide (Taubenberger and Kash, 2010). The latest influenza pandemic was caused by a swine-origin H1N1 virus, which is a triple re-assortment of human, avian, and swine strains (Neumann et al., 2009). Although it is well known that the pathogenicity of influenza strains can vary widely, the precise factors and mechanisms that contribute to the clinical outcome of infection have not been defined.
Eicosanoids refer to a family of bioactive lipid mediators that regulate a wide variety of physiological as well as pathophysiological responses and often exhibit potent inflammatory properties (Quehenberger and Dennis, 2011). These mediators are generated from arachidonic acid (AA) and related polyunsaturated fatty acids after their enzymatic release from membrane phospholipids via complex metabolic mechanisms involving over 50 unique enzymes (Buczynski et al., 2009). The three major metabolic pathways for enzymatic eicosanoid biogenesis are the cyclooxygenase pathway (COX-1 and COX-2), producing the prostaglandins and thromboxanes, the lipoxygenase pathway (5-LOX, 12-LOX and 15-LOX), producing the leukotrienes as well as numerous hydroperoxy and hydroxy fatty acids, and the cytochrome P450 pathway producing epoxide and corresponding dihydroxy metabolites of arachidonic acids. Polyunsaturated fatty acids are susceptible to lipid peroxidation, especially under condition of oxidative stress during inflammation producing isoprostanes as well as other non-enzymatic oxidation products including hydroxy fatty acids. In addition to arachidonic acids, the same enzymes can effectively metabolize other polyunsaturated fatty acids, such as linoleic and linolenic acids. Several bioactive lipid mediators have also been appreciated for their anti-inflammatory activity and their participation in the resolution phase of inflammation (Serhan et al., 2008). Arachidonic acid-derived lipoxins via the lipoxygenase pathway, and docosahexaenoic acid- (DHA) and eicosapentaenoic acid-(EPA) derived resolvins, protectins, and maresins have anti-inflammatory and proresolution activities (Serhan et al., 2000). These lipid mediators can prevent further infiltration of immune cells to the site of infection as well as signaling the nonphlogistic phagocytosis of apoptotic immune and epithelial cells, therefore allowing the system to return to homeostasis after microbial infection. While many of these lipid mediators have been studied individually in various model systems, only a few studies have conducted comprehensive quantification of the metabolites in a natural microbial infection (Chiang et al., 2012; Morita et al., 2013; Tobin et al., 2012).
We utilized two influenza viruses, PR8/H1N1 a highly pathogenic strain in mice, and X31/H3N2, a low pathogenicity strain, and profiled 141 lipid metabolites and bioactive mediators during the course of a mouse influenza infection in vivo. The induction kinetics of the protein levels of cytokines/chemokines in the broncho-alveolar lavage and the transcript levels of immune response genes in the host are similar between infections with the two viruses. In contrast, the lipidomic profile provided a more dynamic and comprehensive description of the processes involved in the induction and resolution of inflammation. Metabolites derived from the lipoxygenase and cytochrome P450 pathways, and those derived from linoleic acid, DHA, and EPA had distinct profiles in the two viral infections during the resolution phase of inflammation. Surprisingly, metabolites of the cyclooxygenase pathway showed similar levels and kinetics across different conditions. We have further profiled the lipidome of nasopharyngeal lavages from human influenza clinical samples obtained during the 2009–2011 influenza seasons and validated some of the observations from the animal model. Many of the metabolites and bioactive mediators induced in the high pathogenicity PR8 mouse influenza infection model were also produced at significantly higher levels in individuals with increased clinical symptoms and immune responses.
Results
Mouse Influenza infection
We used X31 and PR8 to determine how the varying pathogenicity of influenza viral strains affects the transcriptional, proteomic, and lipidomic profiles of the host immune response. These two strains differ in their HA and NA segments and contain the identical “internal” genes (PB1, PB2, PA, NP, NS, and M). PR8 is a high-pathogenicity, mouse-adapted virus that is lethal even at a low multiplicity of infection (Francis and Torregrosa, 1945). In contrast, X31 is a low pathogenicity virus that causes a self-limiting infection even at high doses (Allan et al., 1990). Three infection conditions were used to determine the lipidomic profiles of the host during influenza infections: PR8 at a lethal dose (2 × 105 PFU; PR8lethal), PR8 at a sublethal dose (200 PFU; PR8sublethal), and X31 at a sublethal dose (2 × 105 PFU; X31sublethal). We monitored the progress of influenza infection by measuring the percentage of weight loss (Figure 1A). Animals infected with PR8sublethal had a peak weight loss on day 9 post-infection of 22% and recovered to their original weights by day 13 post-infection. Animals infected with X31sublethal differed slightly from animals infected with PR8sublethal; they had a peak weight loss on day 7 postinfection of 16% and recovered to their original weights by day 10 post-infection. Finally, animals infected with PR8lethal experienced rapid weight loss and succumbed 6 days post-infection.
Figure 1.
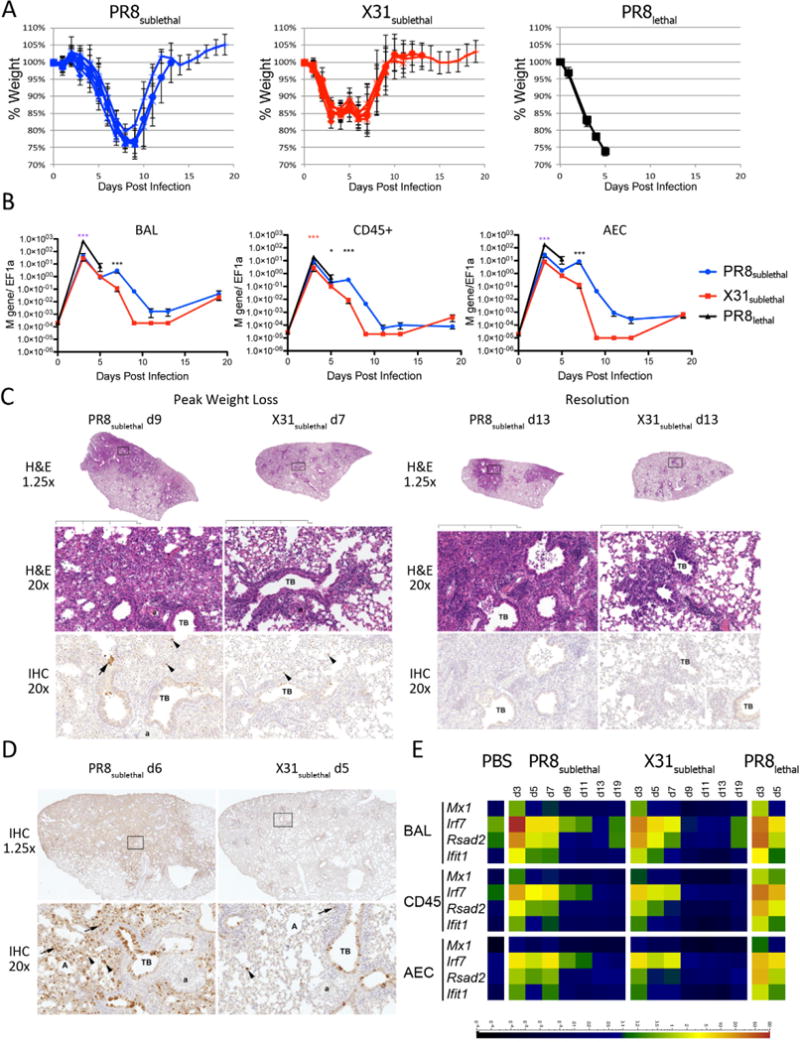
Mouse Influenza Infection Model. (A) Weight loss curves of mice infected with PR8sublethal 200 PFU (Blue), X31sublethal 2× 105 PFU (Red), or PR8lethal 2× 105 PFU (Black) on day 0. Graphs depict mean +/− SEM. (B) Viral loads in BAL (left), CD45+ immune cells (middle), or alveolar epithelium (right) as measured by RT-PCR using Fluidigm. Mann-Whitney tests were performed to determine statistical significance (* 0.01<p <0.05; ** 0.001<p<0.01; *** p<0.001; magenta asterisks denote significance between PR8lethal and both X31sublethal and PR8sublethal infections; blue and red asterisks denote significance between PR8lethal and PR8sublethal or X31sublethal infections, respectively; black asterisks denote significance between PR8sublethal and X31sublethal infections). (C) Representative hematoxylin and eosin (H&E) and anti-influenza antibody –stained sections from PR8 and X31-sublethally infected lungs. Days Post Infection, magnifications, terminal bronchioles (TB), arterioles (a) and alveoli (A) as indicated. Boxed regions on sub-gross images (1.25×) correspond to the regions shown at 20× for both H&E and IHC panels except for PR8 at d9 where the sections are stepped; note that the same terminal bronchiole and arteriole are present in both panels. Boxed inset in lower right X31 IHC panel is an example of the control IgG-stained tissue. Black bar represents 2mm. (D) Representative IHC from PR8 D6 and X31 D5 infected lung sections. Brown staining indicates presence of antigen. Arterioles are indicated (a). (E) Antiviral response represented as heatmap of RT-PCR data detected in BAL, CD45+, or AEC (alveolar epithelial cells). Scale bar represents normalized value to EF1α. See also Figure S1.
We next determined the viral load in the broncho-alveolar lavage (BAL) and in separated cell populations by measuring the levels of the influenza M gene. Cell populations included cells isolated from the BAL, as well as CD45+ immune cells and CD45− epithelial cells isolated from dispase-digested lungs (Figure 1B). Viral loads were measured in cells isolated from sub-lethally infected animals on days 3, 5, 7, 9, 11, 13, and 19 post-infection, whereas viral loads in lethally infected animals were measured on days 3 and 5 post-infection (Figure 1B). The viral load peaked on day 3 post-infection for all three conditions. Not surprisingly, animals infected with PR8lethal had significantly higher viral loads in the alveolar epithelium and BAL on day 3 than animals infected with PR8sublethal or X31subtlethal. Animals infected with X31sublethal had similar viral loads on day 3 and day 5 as animals infected with PR8sublethal, but subsequently cleared the virus after day 7 post-infection. Thereafter, the viral load in animals infected with PR8sublethal was generally higher than that found within animals infected with X31sublethal.
In separate experiments we performed hematoxylin and eosin (H&E) staining and anti-influenza immunohistochemistry (IHC) on lung sections to evaluate the histopathologic characteristics, distribution of changes and localization of influenza antigens (Figure 1C, 1D, S1). In order to compensate for the slightly shifted kinetics in weight loss, we adjusted the collection times for the X31 and PR8 infections. Lung tissues were collected during the induction of inflammation (day 5 for X31; day 6 for PR8), during peak weight loss (day 7 for X31; day 9 for PR8), and during resolution of inflammation (day 13). The IHC staining indicated that while PR8 is capable of spreading into the deeper interstitial lung tissue, X31 is primarily restricted to the major bronchial region with limited positive staining in the lung (Figure 1D). H&E staining of PR8 infected lung tissue indicated polymorphonuclear and monocytic leukocyte infiltration in and around major bronchial and lung interstitial space on day 6 post-infection (Figure 1C). The infiltration spread more extensively in the lung by day 9. On day 13 postinfection, demarcated lymphocytic aggregates were observed in both infections. For X31 infection, the infiltrating cells were generally restricted to the major bronchial areas with lower levels of lymphocyte infiltration at later time points during the resolution phase than for PR8 infection.
We compared the transcript levels of several interferon induced genes (Mx1, Rsad2, Ifit1) and the transcription factor Irf7, all signatures of influenza infection (Sato et al., 2000), to assess the antiviral response. The strongest responses were detected in infiltrating cells isolated from the BAL as well as immune cells from the lung (CD45+) (Figure 1E). Despite the different kinetics of weight loss in animals infected with PR8sublethal and X31sublethal, the abundance of Mx1, Ifit1, Rsad2, and Irf7 transcripts were comparable and reflected the level of viral infection: PR8sublethal induced a slightly stronger and more sustained response than X31sublethal, and PR8lethal induced an even more exuberant response than the sublethal infections.
Cytokine and Chemokine Response
In response to infection of the lung, a concerted signaling cascade is activated to induce secretion of cytokines and chemokines (as well as lipid mediators) that promote vasodilation and a massive infiltration of immune cells to affected tissues. We measured the cytokine and chemokine levels in the BAL by multiplexed immunoassay (Luminex) to establish the levels of protein mediators induced during the course of infection (Figure 2, S2). The shift in the kinetics of weight loss between PR8sublethal and X31sublethal was mirrored by the production of IL-5, MCP-1 and TNFα. By contrast, similar levels of IL-6, IL-1β, and MIP-1a were produced during infection with PR8sublethal and X31sublethal. Finally, infection with PR8sublethal was accompanied by higher levels and prolonged production of KC, IP-10, G-CSF, RANTES, IL-1α, IFNγ, and IL-10 compared to infection with X31sublethal (Figure 2, S2). Interestingly, despite their difference in pathogenicity, PR8lethal and PR8sublethal, produced similar levels of the cytokines and chemokines described above with the exception of MCP-1, RANTES and G-CSF, which were higher during infection with PR8lethal.
Figure 2.
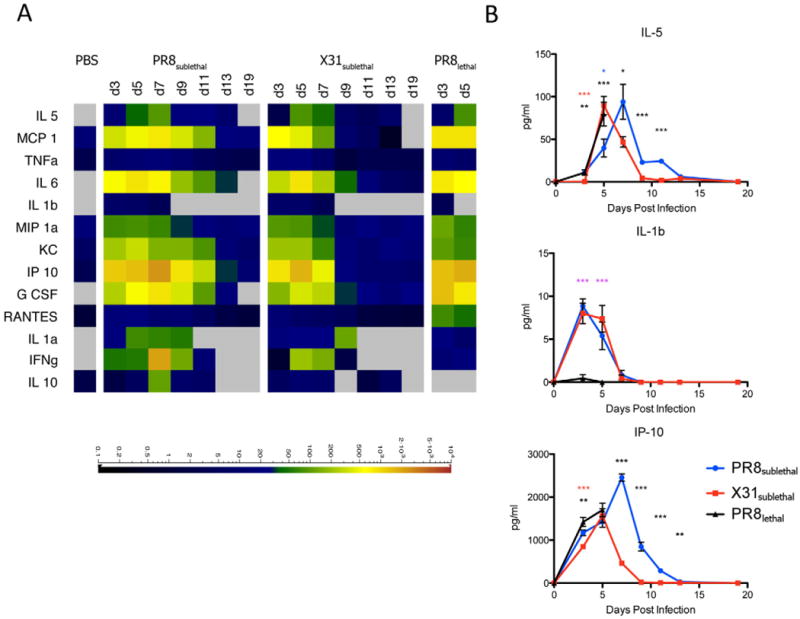
Cytokine and Chemokine Responses in Mouse Influenza Infections. (A) Cytokine and chemokine levels detected in BAL by multiplex Luminex respresented as a heatmap. Scale represents the amount of protein in pg/ml. (B) Line graphs of selected representative cytokines/chemokines depicting the mean +/− SEM. See also Figure S2.
Lipidomic and Transcriptomic Profiling
A variety of bioactive lipid mediators play a critical role in the regulation of inflammation, and we sought to examine their role in the pathogenesis of influenza. Given the complexity of the pathways and multiple routes for degradation and other modifications of these mediators, we examined the dynamic lipidome during influenza infection using LC/MS. Over the course of infection, 141 lipid species (Extended Experimental Procedures) were profiled and 52 lipid metabolites were detected and quantified. The relative percentages of metabolites generated through the COX, LOX, and CYP450 pathways derived from arachidonic acid, as well as metabolites derived from linoleic acid, linolenic acid, DHA, and EPA were determined to assess the composition of bioactive lipids in the BAL (Figure 3A, S3A). For mediators in the COX pathway, levels of prostaglandins increased relative to other metabolites during the early inflammatory response (day 3) but decreased thereafter until returning to the basal level, regardless of whether the mice were infected with PR8sublethal or X31sublethal. In contrast, the percentage of prostaglandins relative to other metabolites was significantly lower during PR8lethal infection. LOX derived metabolites also decreased during the early phase of PR8sublethal and X31sublethal infection while animals infected with X31sublethal had higher levels of LOX metabolites throughout the resolution phase of inflammation. The percentage of metabolites of the cytochrome P450 pathway increased during the induction of inflammation and the kinetics were slightly shifted between PR8 and X31, mirroring the kinetics of weight loss (Figure 1A, S3). In contrast to the COX and CYP450 pathways, percentages of metabolites derived from linoleic acid, linolenic acid, DHA and EPA increased during the later phase of infection. The concentrations of linoleic acid and EPA initially decreased during the early phase of inflammation and subsequently increased throughout the resolution phase. Moreover, the percentage of linoleic acid metabolites was significantly higher in PR8sublethal than X31sublethal. In contrast the low pathogenicity virus, X31sublethal, induced a higher percentage of both DHA- and EPA-derived metabolites than the high pathogenicity virus, PR8sublethal (Figure 3, S3A).
Figure 3.
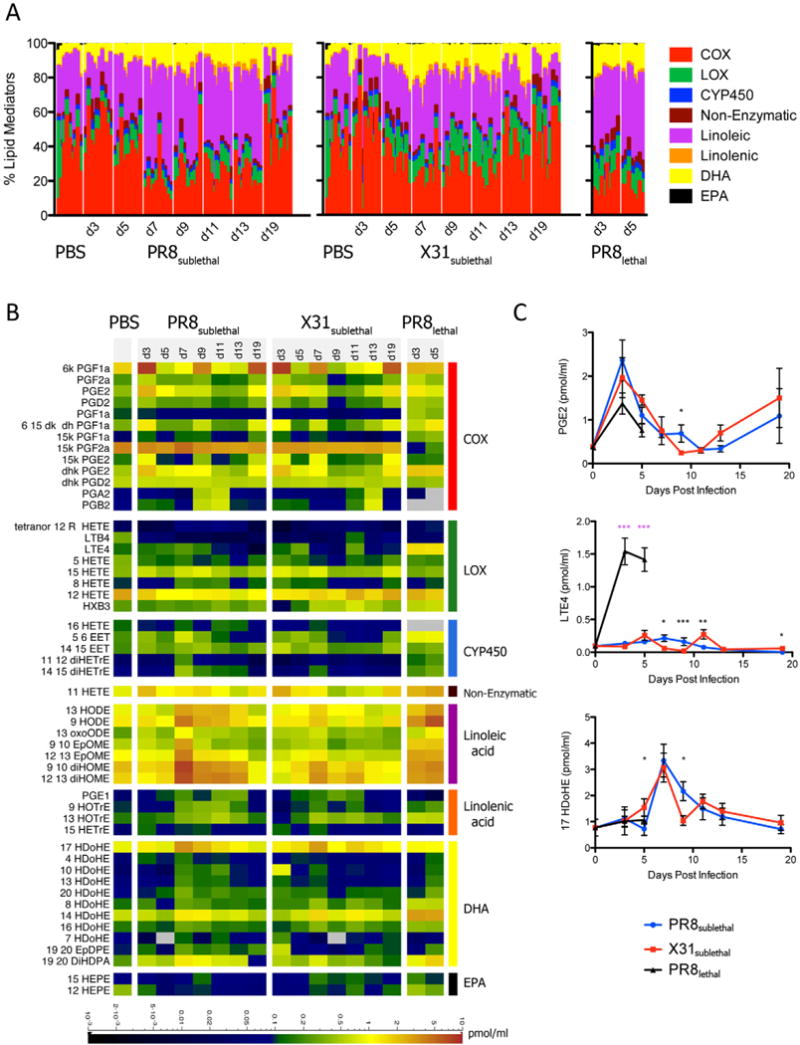
Lipidomic Profiling of Mouse Influenza Infection. (A) Stacked bar graph representing the percentages of arachidonic acid derived cyclooxygenase (COX), lipoxygenase (LOX), and cytochrome P450 (CYP450) metabolites, as well as linoleic, linolenic acid, DHA, and EPA derived metabolites. Each vertical line represents data from a single animal/sample (n=8–10). (B) Quantified lipid mediators represented as a heatmap. Scale bar represents levels measured in pmol/ml. (C) Line graphs of selected representative lipid mediators depicting the mean +/− SEM. See also Figure S3.
While the lipid class composition during infection offered a view of the activation kinetics of specific lipid metabolic pathways, it did not shed light on individual mediators, which have diverse physiological consequences for induction and resolution of inflammation. The COX pathway generates prostaglandins, and their levels were surprisingly similar under all three conditions of infection (X31sublethal, PR8sublethal, and PR8lethal) (Figure 3B, 3C). Most of the prostaglandins peaked early and decreased thereafter (Figure 3B). In general, the mRNA transcripts encoding the enzymes within the cyclooxygenase pathway displayed similar expression kinetics to the specific metabolites that they generate (Figure 3B and Figure S3B). One striking difference between the PR8sublethal and X31sublethal infections was the significantly higher levels of cytochrome P450 and linoleic acid-derived metabolites in animals infected with the high pathogenicity virus (Figure 3B).
DHA and EPA metabolites have recently been shown to promote the resolution of inflammation (Ariel and Serhan, 2007; Bannenberg et al., 2005). While 17-HDoHE, a precursor of resolvins and protectins (D series), was found at similar levels in PR8sublethal and X31sublethal (Figure 3B), two distinct groups of hydroxylated DHA emerged: 4-, 10-, 13-, and 20-HDoHE were detected at a higher levels in PR8sublethal infected animals, while 8-, 14-, and 16-HDoHE were found at higher levels in X31sublethal infected animals. Moreover, for lethally infected animals (PR8lethal), 4-, 10-, 13-, and 20-HDoHE had increasing levels from day 3 to day 5 post infection reaching a level similar to the maximum level found in PR8 sublethal infected mice (Figure 3B). In contrast, 8-, 14-, and 16-HDoHE peaked at day 3 and remained at levels significantly greater than in sublethal infections until day 5 (Figure 3B). Of note, several of these metabolites can be formed either enzymatically or through autoxidation processes. For example, 17-HDoHE is a putative product of the mammalian 12/15 LOX activity, which that can be further metabolized to protectin D1, a potential suppressor of viral replication (Morita et al., 2013; O’Flaherty et al., 2012). Similarly, other positional isoforms of hydroxylated DHA can be formed via 12/15 LOX (Morgan et al., 2010). Even though the source of these hydroxylated DHA metabolites are not entirely established, the profile between PR8 and X31 infection conditions were strikingly distinct. In general, metabolites of EPA, 12-HEPE and 15-HEPE, were detected at a higher level in X31sublethal infected animals than in PR8sublethal infected animals. The relationship between the level of a metabolite and that of the transcript of the corresponding enzyme that generates it can be discerned by comparing Figures 3B and S3B.
Lipidomic Profiling in Human Nasal Washes
The lipidomic profiling in the mouse influenza infection model allowed us to obtain a comprehensive kinetic view of the lipidome across a well-defined time course. Understanding the relevance of these results to human infection is crucial. We conducted lipidomic profiling of human nasopharyngeal lavages obtained from a surveillance study at the St. Jude Children’s Research Hospital during the 2009–2011 influenza seasons. Study participants were enrolled based on detection of the influenza matrix gene (influenza A) or nonstructural NS1 gene (influenza B) by quantitative real-time PCR. Index cases and their asymptomatic household contacts were followed for approximately one month following enrollment, with contacts sampled as cases following a positive detection of influenza by qRT-PCR. Based on hierarchical clustering using clinical scores and multiplexed immunoassay of growth factors, cytokines and chemokines, we identified three groups within qRT-PCR-confirmed influenza-infected subjects: (1) patients with low clinical scores and minimal amounts of cytokines and chemokines, (2) patients with medium clinical scores and positive for a subset of growth factors and cytokines (EGF, GCSF, GRO, IL1Ra2, IL8, and IP10), and (3) patients with high clinical scores and high levels of a wide panel of cytokines (Fig. S4A). A subset of samples was selected from each group for lipidomic profiling (n=9–11/group, total=30). While the human nasal cavity is likely to be a very different physiological environment than the mouse lung, we detected induced levels of many lipid mediators that were identified in the mouse influenza infection model. We found one dramatic difference in the lipid profile between the human and mouse controls; linoleic acid comprises 68% of the lipidome in the nasopharyngeal lavage of human low responders (asymptomatic contacts of the index cases) whereas it makes up 33.7% of the lipidome in the BAL of naïve mice (Figure 3A and 4A). High levels of linoleic acid have been detected in humans in the skin, mucosal surfaces, and nasal fluid, and contribute to antimicrobial activity at these sites (Drake et al., 2008). The samples from patients with high clinical score and increased levels of cytokines and chemokines also contained significantly higher percentages of lipid mediators from the lipoxygenase, CYP450, DHA, and EPA pathways (Figure 4A, S4B). Amounts of individual lipid mediators (including PGE2, LTE4, and 17 HDoHE) were also significantly increased within the high response group compared to the low and medium response groups (Figure 4B, 4C).
Figure 4.
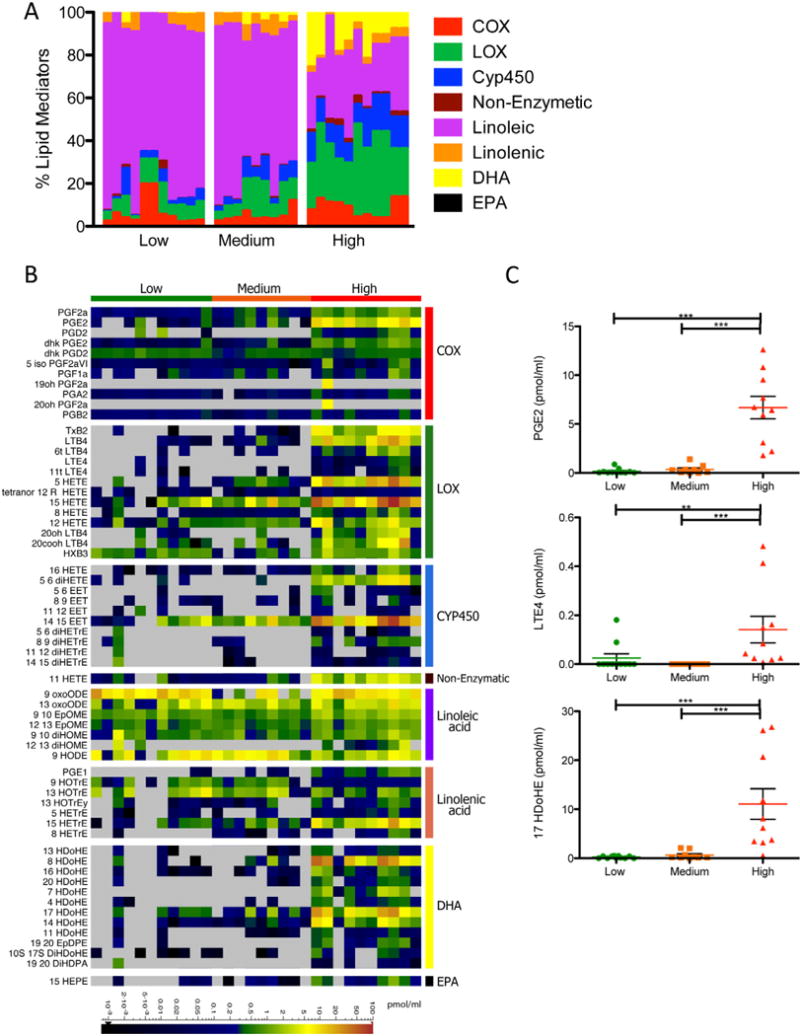
Lipidomic Profiling of Human Nasal Washes. (A) Stacked bar graph represents the percentages of arachidonic acid derived cyclooxygenase (COX), lipoxygenase (LOX), and cytochrome P450 (CYP450) metabolites, as well as linoleic, linolenic acid, DHA, and EPA derived metabolites. Each vertical line represents data from a single patient/sample, which is categorized into either the low, medium or high clinical score/immune response group (n=9–11). (B) Quantified lipid mediators represented as a heatmap. Scaled bar represents levels measured in pmol/ml. (C) Column vertical scatter plot of selected representative lipid mediators depicting the mean +/− SEM. See also Figure S4.
Pro-inflammatory and Anti-inflammatory States During Influenza Infection
In order to look at global changes in the mouse and human data, we grouped the bioactive lipids based on their pro-inflammatory or anti-inflammatory/pro-resolution properties and created quantitative indices to characterize the pro-inflammatory and anti-inflammatory contributions of the host response at each stage of the infection (see Extended Experimental Procedures). These indices were calculated for each infected mouse or patient at every time point/condition. To create the index for the mouse infection model, we first calculated the fold change of individual lipid mediators in infected animals over naïve animals and only included values greater than or equal to two. The fold changes for each mediator were then normalized to the maximum value across all samples, setting the maximum value to one. We separated lipid mediators into groups with pro- or anti-inflammatory activity and then summed the normalized values of each mediator within the pro- or anti-inflammatory group to define the total activity. The maximum activity in each group is then equal to the total number of mediators in the group. To generate the index for each time point, the percentage of the pro- or anti-inflammatory mediator activity was calculated by dividing total activity by the maximum activity in the pro- or anti-inflammatory groups.
As discussed above, the early pro-inflammatory response was similar between PR8sublethal and X31sublethal infections as indicated by levels of mediators such as prostaglandins on day 3 post-infection (Figure 5A). This was reflected in a comparable increase of pro-inflammatory indices on day 3 for both sublethal infections (Figure 5B, S5A). For PR8sublethal infection, a secondary wave of pro-inflammatory mediators occurred on day 7 post-infection, including linoleic acid-derived epoxide and dihydroxy linoleic acid. This contributed to a significant increase in the pro-inflammatory indices for PR8sublethal, which began to subside on day 9 post-infection (Figure 5B, S5A). The mediators 9, 10 EpOME, 12, 13 EpOME, 9, 10 diHOME and 12, 13 diHOME, which are potent chemoattractants for neutrophils, were present at much lower levels in X31sublehtal infected animals on day 7 post-infection. For the anti-inflammatory index, the peak of the response began on day 7 post-infection, and was not significantly different between PR8sublethal and X31sublethal. The anti-inflammatory indices decreased slightly on day 9 post-infection and sustained these levels until day 13 post-infection. In contrast to the sublethal infections, PR8lethal induced significant increases of both pro- and anti-inflammatory indices consecutively on day 3 and day 5 post-infection (Figure 5B, S5A).
Figure 5.
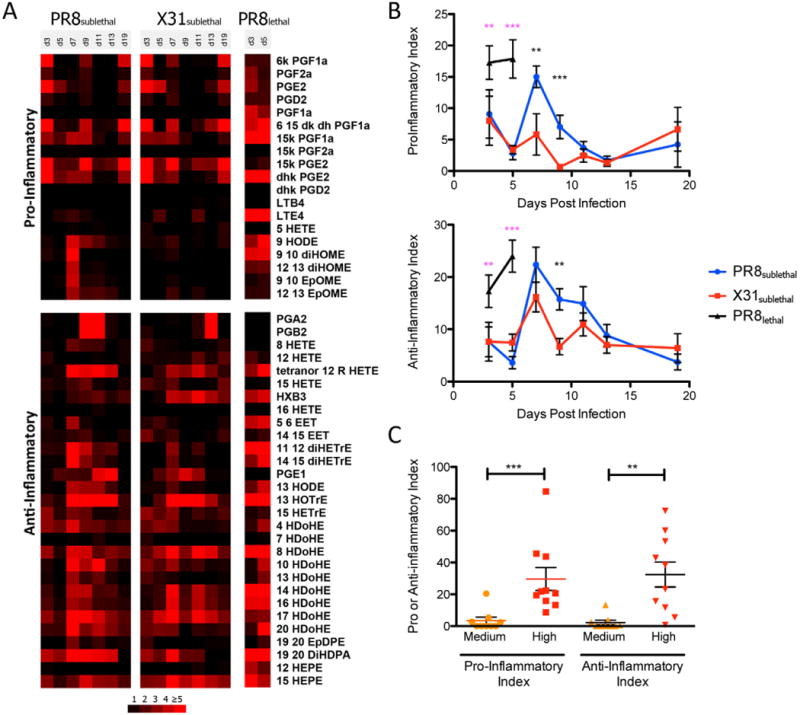
Pro- and Anti-inflammatory Indices of Mouse and Human Lipidomic Profiles. (A) Heatmap represents the mean fold change in each lipid mediator (separated by having either pro- or anti-inflammatory activity). (B) Line graphs of pro- and anti-inflammatory indices depicting the mean +/− SEM. (C) Column vertical scatter plot of pro- and anti-inflammatory indices for individual patient samples. Horizontal lines indicate the mean +/− SEM. See also Figure S5.
Using the same approach to determine the pro-inflammatory and anti-inflammatory indices of patient samples (using low clinical symptoms and immune response as the control), the high response group had significant increases in the pro- and anti-inflammatory indices compared to the medium response group (Figure 5C, S5B, S5C).
Lipoxygenase Pathway
We next dissected the lipoxygenase pathway in more detail since it contains well-defined pro- and anti-inflammatory mediators. 5-lipoxygenase-derived leukotrienes have long been known as pro-inflammatory mediators: LTB4 is a potent chemotactic factor for neutrophil recruitment, and LTC4 and LTE4 are potent mediators for immediate hypersensitivity, bronchoconstriction, smooth muscle contraction, and increased vascular permeability. Recently, 12-lipoxygenase- and 15-lipoxygenase-derived metabolites such as 12-HETE, hepoxilin, 15-HETE, and lipoxins have been increasingly appreciated as anti-inflammatory mediators (Kühn and O’Donnell, 2006). Specifically, 12-HETE and 15-HETE are potent activator of PPARγ (Kronke et al., 2009), which can dampen inflammation via inhibition of NF-kB and modulate macrophage function (Ricote et al., 1998). Lipoxins can block further recruitment of neutrophils (Serhan et al., 1995) and stimulate nonpholgistic clearance of apoptotic cells (Godson et al., 2000). While many of these molecules have been studied individually in various settings, a comprehensive but focused analysis of the metabolites and mediators within this pathway would yield a better understanding of the local inflammatory milieu during an active microbial infection.
The levels of 5-lipoxygenase mediators were similar in the BAL of sublethally infected mice with the exception of LTE4, in which more abundant in PR8sublethal infected animals on day 7 and day 9 post-infection (Figure 3C). This difference resulted in higher percentages of 5-lipoxygenase derived mediators in PR8sublethal infection (Figure 6A, S6A). In concordance with its pathogenicity, PR8lethal infection elicited much greater amounts of LTE4, which contributed to a significant increase in the percentages of 5-lipoxygenase derived metabolites compared to the sublethal infections (Figure 3C, 6A, S6A). Transcription of 5-lipoxygenase (alox5) was down-regulated at the onset of infection (Figure 6B), similar to the regulation observed in macrophages stimulated in vitro with lipopolysaccharide Kdo2-Lipid A (Dennis et al., 2010). 5-lipoxygenase activating protein, alox5ap, on the other hand, was significantly increased in the lethal infection, which correlated with the level of the lipid LTE4 (Figure 6B, 3C).
Figure 6.
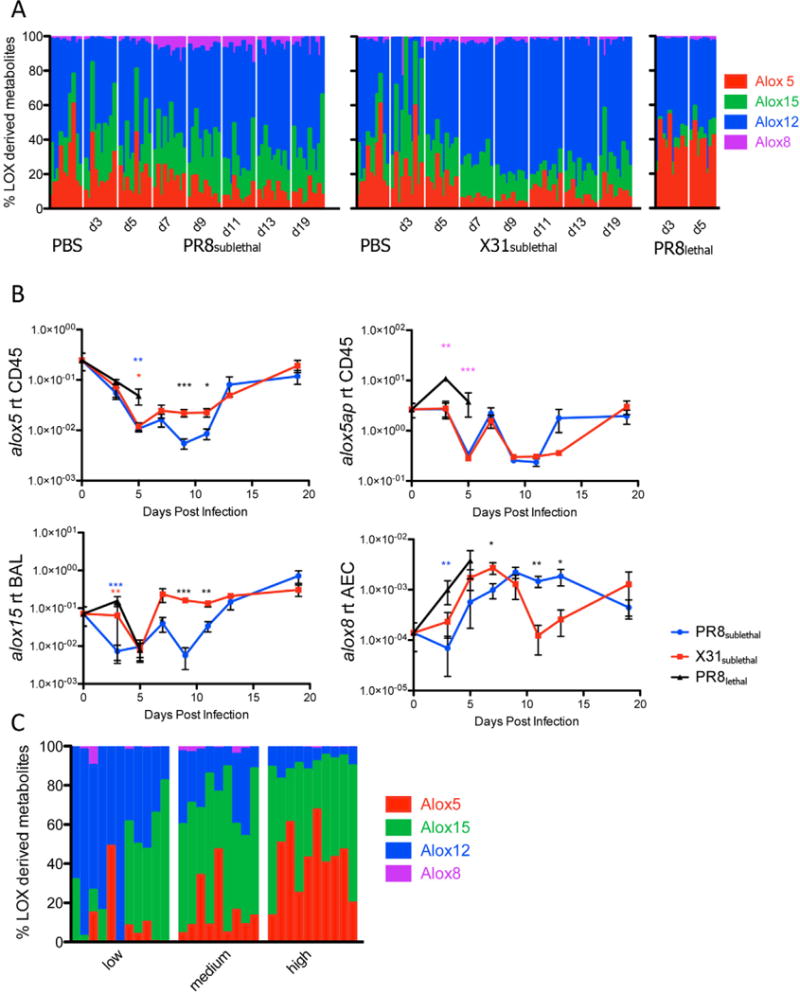
Lipoxygenase Enzymes and Derived Metabolites in Mouse and Human Influenza Infection. Stacked bar graph represents the percentages of lipoxygenase 5, 15, 12, or 8 derived metabolites of all lipoxygenase derived metabolites in mouse (A) or human (C) samples. Each vertical line represents data from a single sample (n=8–11 per time point). (B) Line graphs of 5 lipoxygenase (alox5), 5-lipoxygenase activating protein (alox5ap) in CD45+ cells, 15 lipoxygenase (alox15) in BAL, and 8 lipoxygenase (alox8) in alveolar epithelium depicting the mean +/− SEM. See also Figure S6.
Mediators of the 12/15-lipoxygenase pathway, 12-HETE and hepoxilin B3 (HXB3), had increased levels in the late phase of X31sublethal infection when compared to PR8sublethal (Figure S6A). These differences contributed to significantly higher percentages of 12-lipoxygenase derived mediators for X31sublethal when compared to PR8sublethal from day 7 to day 13 post-infection (Figure 6A, S6A). The level of 15-HETE was similar between the sublethal infections. The transcriptional level of 15-lipoxygenase (alox15) is significantly increased in X31sublethal infection at 9 and 11 days post-infection in comparison to PR8sublethal (Figure 6B, S6B). The discrepancy suggests that the increased quantity of Alox15 protein may be acting on other substrates such as linoleic acid or linolenic acid. 8-lipoxygenase (alox8) exhibited a greater level of expression in PR8sublethal infected animals than in X31sublethal infected animals at the late phase of infection. This transcriptional induction in the epithelium correlated with increased detection of 8-HETE in the BAL fluid (Figure 6B, S6A). In general, a high percentage of pro-inflammatory 5-lipoxygenase mediators was tightly correlated with the highly pathogenic phase of the PR8 virus, whereas an elevated percentage of anti-inflammatory 12/15-lipoxygenase mediators was associated with the resolution phase of the X31 infection (Figure 6A, S6A).
This trend was recapitulated in studies of human nasopharyngeal lavage fluid obtained during the 2009–2011 influenza seasons. Here, the percentages of 5-lipoxygenase derived mediators were strongly correlated with disease symptom from low to medium to high responders (Figure 6C, Figure S6B). The high response group exhibited significantly elevated levels of 5-HETE, LTB4, and LTE4 compared to the low and medium responders. In contrast, percentages of 12-lipoxygenase derived metabolites were anti-correlated with the level of disease response (Figure 6C, S6B).
Hydroxylated Linoleic acid Metabolites as Biomarkers for the Status of Influenza Infection
While the comprehensive study of bioactive lipid profiles offered a complete picture of the host response to influenza, the methodology also allowed for identification of useful biomarkers that indicate the immunological status of an active infection. Linoleic acid is further metabolized into 9 or 13 hydroxylated linoleic acid (9 HODE and 13 HODE). While 13 HODE is generated by 15-lipoxygenase, 9 HODE production can be catalyzed by multiple enzymes and non-enzymatic reactions (Obinata and Izumi, 2009) (Figure 7A). 9 HODE and 13 HODE have previously been proposed as markers for lipid peroxidation in various chronic diseases (Spiteller and Spiteller, 1997). More recently, 9 HODE has been shown to have a pro-inflammatory role (Hattori et al., 2007; Obinata and Izumi, 2009) while 13 HODE is anti-inflammatory (Altmann et al., 2007; Belvisi and Mitchell, 2009). We calculated the ratio of 13 to 9 HODE to determine whether it accurately describes the pro- versus anti-inflammatory state of an infection. An advantage of using ratios of mediators is that while the values of individual lipid mediators may vary greatly between different animals and different patients, the ratio between two mediators is normalized as they arise from a common precursor. Another reason for choosing 9 and 13 HODE is that both mediators were detected robustly across all samples (29/30 clinical samples for 9 HODE and 30/30 clinical samples for 13 HODE). In contrast, many of the other mediators were undetectable in either the low or medium response groups making them less suitable as biomarkers.
Figure 7.
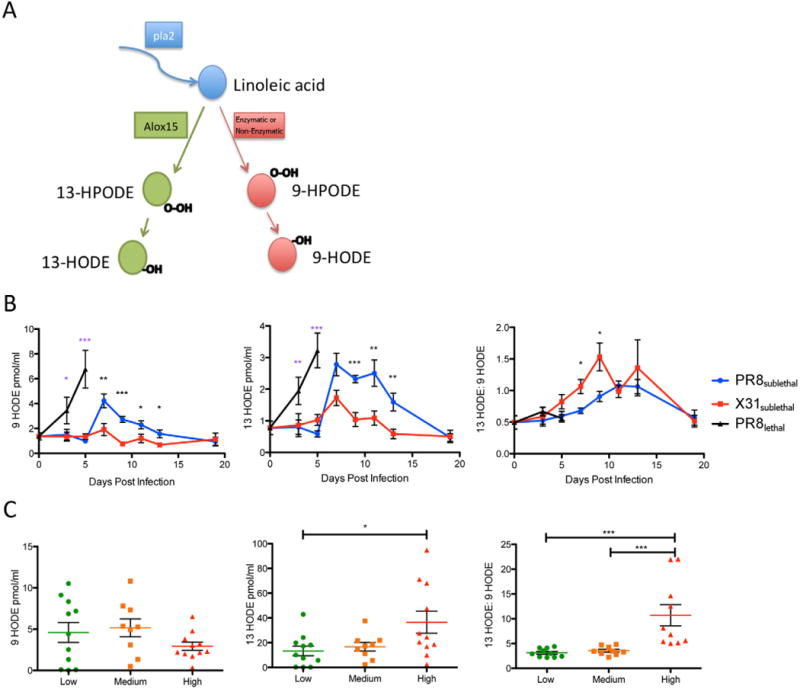
Hydroxylated Linoleic acid Metabolites as a Biomarker for Immune Status During Influenza Infection. (A) Pathway for linoleic acid to generate 9-hydroxylated or 13-hydroxylated linoleic acid. (B) Line graphs of 9 HODE, 13 HODE, and 13:9 HODE in the mouse lung during influenza infection (red: X31sublethal; blue: PR8sublethal; black: PR8lethal). (C) Column vertical scatter plot of 9 HODE, 13 HODE, and 13:9 HODE in human nasal wash samples. Horizontal lines indicate mean +/− SEM. See also Figure S7.
When we compared the levels of 9 HODE, 13 HODE and the ratio of 13 HODE/9 HODE during influenza infection in mice, PR8sublethal infection induced a significantly increased level of 9 HODE on day 7 post-infection compared to X31sublethal infection. In contrast, both PR8sublethal and X31sublethal induced similar production of 13 HODE. Interestingly, the ratios of 13:9 HODE are increased for both PR8sublethal and X31sublethal during the resolution phase of the infection, indicating an increasingly anti-inflammatory state. Moreover, the ratio is significantly higher in X31sublethal even at early time points (day 7 and 9 post-infection), which supports the notion that the low pathogenicity virus induced a response skewed toward an anti-inflammatory profile (Figure 7B).
We tested the robustness of these parameters by combining three additional experiments performed on separate days and animals from different vendors and harvested at different time points. While the raw values of 9 and 13 HODE varied between animals and experiments, the ratio of 13:9 HODE remained consistent (Figure S7A, B).
In the human nasopharyngeal lavage, levels of 9 HODE were slightly lower in the high response group although the difference was not significant (Figure 7C) while levels of the anti-inflammatory mediator, 13 HODE, were elevated with moderate significance (p<0.05). When we compared the 13:9 HODE ratios between the groups, however, the difference became more dramatic and highly significant (p<0.001) (Figure 7C). Further measurements on additional samples and clinical data will be required to determine whether the 13:9 HODE ratio can be used to further stratified the severity and/or course of infection.
Discussion
Lipidomic profiling of influenza infection provided a comprehensive map of eicosanoids and other related bioactive lipids during an active infection of the lung. We detected and quantified metabolites and mediators within the cyclooxygenase, lipoxygenase, and cytochrome P450 pathways derived from arachidonic acid, as well as metabolites and mediators derived from linoleic acid, linolenic acid, DHA, and EPA. The protein levels of cytokines and chemokines and the transcript levels of numerous antiviral genes suggested a straightforward, positive correlation between the level of immune response and the pathogenicity of the infection, with the response increasing from infection with X31, the low pathogenicity virus, to PR8, the high pathogenicity virus (PR8sublethal to PR8lethal). In contrast, the lipidomic profiling uncovered a more complex response, with the levels of mediators involved in both pro- and anti-inflammatory processes exhibiting more complex dynamics. Surprisingly, the cyclooxygenase pathway mediators, consisting of the classical signaling prostaglandins, were produced with similar kinetics during infections by the different influenza strains. Concordantly, enzymes within the COX pathway were activated to similar levels transcriptionally. In contrast to the COX-derived lipid mediators, metabolites derived from other pathways (LOX, CYP450) and precursors (Linoleic acid, DHA, EPA) diverged strongly between the different infections.
Hepoxilin and 12-HETE, mediators derived from the 12/15 lipoxygenase pathway, were detected at a higher level in X31sublethal than PR8sublethal infected animals. Hepoxilin (HX) has been suggested to be involved in anti-inflammatory mechanisms and stable hepoxilin analogs inhibit macrophage influx as well as fibrosis in the lung (Jankov et al., 2002). 12-HETE also has anti-inflammatory activities: for example, it is capable of blocking TNFα-induced IL-6 secretion from macrophages (Kronke et al., 2009). In contrast, 8-HETE, which is derived from 8 lipoxygenase, was detected at a higher level in PR8sublethal than X31sublethal infected animals. While the biological activity of 8-HETE within the respiratory system is poorly characterized, the metabolite has been detected in human lung vasculature (Kiss et al., 2000). The increased production of 8-HETE late in infection suggests that it plays a role in epithelial repair after influenza infection. It is important to note that mice do not have separate 12- and 15-lipoxygenases but rather a combined mouse 12/15-lipoxygenase which is related in sequence to the human 12-lipoxygenase as well as human 15-lipoxygenase type I (reticulocyte-type) and type II (epidermis-type) enzymes (Kühn et al., 2002).
Overall, while metabolites of the LOX pathways displayed a heterogeneous profile in the mouse infection model, a clear difference between PR8 and X31 infection was found primarily in metabolites contributing to anti-inflammatory and pro-resolution activities. In human samples, however, a general increase was observed in both the pro-inflammatory (5-lipoxygenase derived) and anti-inflammatory (12, 15, 8-lipoxygenase derived) metabolites of the LOX pathway in high responders. Interestingly, when comparing the percentages of mediators derived from different lipoxygenase enzymes, a consistent pattern emerged in the mouse and human data. In the mouse model, the percentage of 5-lipoxygenase metabolites was elevated during the pathogenic phase of the PR8 infection, whereas the percentage of 12/15-lipoxygenase metabolites was elevated during the resolution phase of the X31 infection. In the human samples, increasing percentages of 5-lipoxygenase derived metabolites and decreasing percentages of 12-lipoxygenase derived metabolites correlated with increasing clinical symptoms and immune response. However, a direct comparison between the mouse and human data is difficult due to the lack of information on the time course and pathogenicity of the viral strains in the human infection samples as well as the unknown contribution of host genetic diversity to susceptibility to viral infection. When infected with influenza, 12/15-lipoxygenase gene-deficient mice recovered more slowly than wild-type mice, similar to the observations of Morita et al. (data not shown; Morita et al., 2013). However, 12/15 gene-deficient mice are of limited utility in modeling human infections as the function of this enzyme is performed in humans by at least four different enzymes that are expressed in different cell types. Detailed pharmacological studies using specific inhibitors would be required to determine the effect of these enzymes at different phases of infection.
Cytochrome P450 enzymes can metabolize arachidonic acid to produce 5,6- or 14,15-EET with subsequent metabolic transformation to diHETrE by the soluble epoxide hydrolase (sEH). EETs have potent vasodilator activities and they have been determined to have anti-inflammatory activities by activating the peroxisome proliferator-activated receptor alpha (PPAR□) pathway (Thomson et al., 2012; Wray et al., 2009). CypP450 metabolites were detected at significantly higher levels in PR8sublethal infections than in X31sublethal infected animals. Similar to arachidonic acid-derived EET and diHETrE, cytochrome P450 enzymes can metabolize linoleic acid to produce 9,10 or 12,13 EpOME and sEH further converts them to 9,10- or 12, 13-diHOME. The epoxides of linoleic acid are leukotoxins produced by activated neutrophils and macrophages. Elevated levels are associated with ARDS (acute respiratory distress syndrome) and cause pulmonary edema, vasodilation, and cardiac failure in animal models (Ishizaki et al., 1999; 1995). These EpOME mediators and their sEH-derived mediators, diHOME, are chemotactic to neutrophils and exert their toxicity by disrupting mitochondrial functions (Sisemore, 2001; Totani et al., 2000). The elevated levels of these mediators in animals infected with the highly pathogenic influenza strain may explain the severe damage occurring in the lung during infection.
Recently, a new class of endogenous lipid mediators (derived from DHA and EPA) that promote resolution of inflammation provides great promise to treat inflammatory diseases (Ji et al., 2011). Although assays for resolvins and protectins were included in the panel, these anti-inflammatory/ pro-resolution molecules were not detected in the animal model or human clinical samples. This is interesting because the production of PD1 is suppressed in lung tissues of influenza-infected mice at early time points (Morita et al., 2013). Additionally, PD1 and 17-HDoHE were detected at higher levels in exhaled breath condensate from healthy individuals compared to asthma patients (Levy et al., 2007). Further studies will determine whether these differences arise from the different sampling methods employed (BAL versus lung tissue; nasopharyngeal lavage versus exhaled breath condensate) generated different results. Aside from different tissue, the infection model and the time frame of sampling is quite different between our study and previous reports. Even though our profiling approach was comprehensive, the biological activities of several of the metabolites detected in our screens are unknown and further systematic studies will be necessary to determine their involvement in the inflammatory process and return to homeostasis within the respiratory system.
By creating quantitative pro- and anti-inflammatory indices, we defined lipid profiles that readily distinguished infections by the low pathogenicity strain, X31, and the highly pathogenic strain, PR8. X31sublethal induced a normal sequence of events, with induction of a pro-inflammatory state on day 3 after infection, followed by induction of an anti-inflammatory state on day 7 which restored homeostasis to the system. Interestingly, while PR8sublethal also induced a pro-inflammatory state on day 3, there was a second, more robust induction of pro-inflammatory mediators on day 7 occurring simultaneously with the induction of anti-inflammatory mediators. This overlap of pro-and anti-inflammatory states was also quite apparent in the PR8lethal infection, in which both indices were significantly higher than the sublethal infections on day 3 and 5 postinfection. The clinical data was similar to the mouse data, suggesting that pro- and anti-inflammatory states overlap within infected humans as well. Further studies are warranted to determine whether this simultaneous pro- and anti-inflammatory lipid response is beneficial or detrimental to the host during an infection with a highly pathogenic influenza strain.
From the mouse lipidome, we have identified 9 HODE, 13 HODE, and their ratio as potential biomarkers for immune status during an infection. While there are numerous parameters to assess the pro-inflammatory status of an infection, parameters characterizing the resolution of inflammation are lacking. Because these metabolites can be robustly detected by ELISA and are derived from a common precursor, making their abundance ratio self-normalizing, they offer great potential as a biomarker of immune status during influenza infection. A larger cohort of patients and samples is required to confirm the correlation between the ratio of 13:9 HODE and the status of disease and recovery.
Lipidomic profiling provides an additional parameter for understanding the host response during a microbial infection. Although transcriptomic analysis of the response to infection has been conducted routinely, our data suggest that while some transcript levels of the enzymes correlate with the lipid levels, the majority of the changes seen in the lipid response were regulated post-transcriptionally. It is likely that the response to infection is accelerated by the uncoupling of metabolite production from the transcriptional regulation of the specific upstream enzymes.
Experimental Procedures
Mouse Influenza Infection
Female C57Bl/6 animals (8–12 weeks) (Jackson Laboratory and Charles River) were kept in specific pathogen-free conditions. Experiments conducted were approved by the Institute for Systems Biology IACUC. Animals were anesthetized with a ketamine/xylazine mixture and infected intranasally with 200 or 2× 105 PFU of influenza virus strain PR8 or 2× 105 PFU of X31 in 30μl sterile PBS. Mock-infected animals were inoculated with 30μl sterile PBS. Mice were monitored and weighed daily.
Human Clinical Samples
This study was carried out in accordance with Good Clinical Practice (GCP) as required by the U.S. Code of Federal Regulations applicable to clinical studies (45 CFR 46), the International Commission on Harmonization (ICH) guidelines for Good Clinical Practice (GCP) E6, and the NIH Clinical Terms of Award. Children and adults with influenza-like illness receiving medical counsel were approached by study personnel to participate. Study participants were enrolled based on detection of the influenza matrix gene (influenza A) or nonstructural NS1 gene (influenza B) by quantitative real-time PCR. Individuals with acute respiratory illness meeting the case definition for influenza (fever or feverishness accompanied by cough or sore throat) who were symptomatic for 96 hours or less, and their household or family contacts (symptomatic or asymptomatic) were eligible to participate in the study, and followed for approximately one month post-enrollment. Contacts that became influenza-positive were sampled cases after conversion. Nasopharyngeal swabs and lavages were collected upon enrollment and at each subsequent study visit.
Lipidomic profiling by LC Mass Spectrometry
Lipid mediators were analyzed by liquid chromatography (LC) and mass spectrometry (MS) essentially as described previously (Dumlao et al., 2011; Quehenberger et al., 2010). Briefly, 0.9 ml of BAL or nasal washes were supplemented with a mix consisting of 25 deuterated internal standards (Cayman Chemical) and lipid metabolites isolated by solid phase extraction on a Strata X column (Phenomenex). The extracted samples were evaporated, reconstituted in a small volume and the eicosanoids were separated by reverse phase LC using a Synergy C18 column (Phenomenex). The eicosanoids were analyzed by tandem quadrupole MS (MDS SCIEX 4000 QTRAP, Applied Biosystems) operated in the negative-ionization mode via multiple-reaction monitoring (MRM) using transitions that were optimized for selectivity and sensitivity. Authentic standards were analyzed under identical conditions and eicosanoid quantitation was achieved by the stable isotope dilution method and comparison with 141 quantitation standards. Data analysis was performed using the MultiQuant 2.1 software (Applied Biosystems). A complete list of all metabolites included in the MRM profile is listed in Table S2.
Statistical analysis
The two-tailed Mann-Whitney test was used to determine statistical significance between population means (for mouse infections, red: X31sublethal; blue: PR8sublethal; black: PR8lethal. Magenta asterisks denote significance between PR8lethal and both X31sublethal and PR8sublethal infections; blue and red asterisks denote significance between PR8lethal and PR8sublethal or X31sublethal infections, respectively; black asterisks denote significance between PR8sublethal and X31sublethal infections). Statistical significance: * 0.01<p <0.05; ** 0.001<p<0.01; *** p<0.001. For correlation studies, the two-tailed Spearman test was used to calculate correlation significance. Heatmaps and clustering analysis were generated using Genedata, and Java TreeView.
Data Dissemination
Raw files and processed data for lipidomics profilings are available at the Systems Influenza Website (www.systemsinfluenza.org) and at Metabolights (www.ebi.ac.uk/metabolights/; MTBLS31).
Supplementary Material
Acknowledgments
We also thank Alan Diercks and Peter Askovich for helpful advice and critical reading of the manuscript. This work was supported by National Institute of Allergy and Infectious Diseases Contract #HHSN272200800058C, “A Systems Biology Approach to Infectious Disease Research”, by the American Lebanese Syrian Associated Charities (ALSAC), and NIH/NIAID Center of Excellence for Influenza Research and Surveillance (HHSN266200700005C), and NIGMS U54 GM069338(EAD).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allan W, Tabi Z, Cleary A, Doherty PC. Cellular events in the lymph node and lung of mice with influenza. Consequences of depleting CD4+ T cells. J Immunol. 1990;144:3980–3986. [PubMed] [Google Scholar]
- Altmann R, Hausmann M, Spöttl T, Gruber M, Bull AW, Menzel K, Vogl D, Herfarth H, Schölmerich J, Falk W, et al. 13-Oxo-ODE is an endogenous ligand for PPARgamma in human colonic epithelial cells. Biochem Pharmacol. 2007;74:612–622. doi: 10.1016/j.bcp.2007.05.027. [DOI] [PubMed] [Google Scholar]
- Ariel A, Serhan CN. Resolvins and protectins in the termination program of acute inflammation. Trends Immunol. 2007;28:176–183. doi: 10.1016/j.it.2007.02.007. [DOI] [PubMed] [Google Scholar]
- Bannenberg GL, Chiang N, Ariel A, Arita M, Tjonahen E, Gotlinger KH, Hong S, Serhan CN. Molecular circuits of resolution: formation and actions of resolvins and protectins. J Immunol. 2005;174:4345–4355. doi: 10.4049/jimmunol.174.7.4345. [DOI] [PubMed] [Google Scholar]
- Belvisi MG, Mitchell JA. Targeting PPAR receptors in the airway for the treatment of inflammatory lung disease. Br J Pharmacol. 2009;158:994–1003. doi: 10.1111/j.1476-5381.2009.00373.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buczynski M, Dumlao D, Dennis E. An integrated omics analysis of eicosanoid biology. J Lipid Res. 2009 doi: 10.1194/jlr.R900004-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang N, Fredman G, Bäckhed F, Oh SF, Vickery T, Schmidt BA, Serhan CN. Infection regulates pro-resolving mediators that lower antibiotic requirements. Nature. 2012;484:524–528. doi: 10.1038/nature11042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis EA, Deems RA, Harkewicz R, Quehenberger O, Brown HA, Milne SB, Myers DS, Glass CK, Hardiman G, Reichart D, et al. A mouse macrophage lipidome. Journal of Biological Chemistry. 2010;285:39976–39985. doi: 10.1074/jbc.M110.182915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake DR, Brogden KA, Dawson DV, Wertz PW. Thematic review series: skin lipids. Antimicrobial lipids at the skin surface. J Lipid Res. 2008;49:4–11. doi: 10.1194/jlr.R700016-JLR200. [DOI] [PubMed] [Google Scholar]
- Dumlao DS, Buczynski MW, Norris PC, Harkewicz R, Dennis EA. High-throughput lipidomic analysis of fatty acid derived eicosanoids and N-acylethanolamines. BBA – Molecular and Cell Biology of Lipids. 2011;1811:724–736. doi: 10.1016/j.bbalip.2011.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis T, Torregrosa D. Combined Infection of Mice with H. influenzae and Influenza Virus by the Intranasal Route. J Infect Dis. 1945;76:70–77. [Google Scholar]
- Godson C, Mitchell S, Harvey K, Petasis NA, Hogg N, Brady HR. Cutting edge: lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. J Immunol. 2000;164:1663–1667. doi: 10.4049/jimmunol.164.4.1663. [DOI] [PubMed] [Google Scholar]
- Hattori T, Obinata H, Ogawa A, Kishi M, Tatei K, Ishikawa O, Izumi T. G2A Plays Proinflammatory Roles in Human Keratinocytes under Oxidative Stress as a Receptor for 9-Hydroxyoctadecadienoic Acid. J Investig Dermatol. 2007;128:1123–1133. doi: 10.1038/sj.jid.5701172. [DOI] [PubMed] [Google Scholar]
- Ishizaki T, Ozawa T, Voelkel NF. Leukotoxins and the lung. Pulmonary Pharmacology & Therapeutics. 1999;12:145–155. doi: 10.1006/pupt.1999.0179. [DOI] [PubMed] [Google Scholar]
- Ishizaki T, Shigemori K, Nakai T, Miyabo S, Ozawa T, Chang SW, Voelkel NF. Leukotoxin, 9,10-epoxy-12-octadecenoate causes edematous lung injury via activation of vascular nitric oxide synthase. Am J Physiol. 1995;269:L65–L70. doi: 10.1152/ajplung.1995.269.1.L65. [DOI] [PubMed] [Google Scholar]
- Jagger BW, Wise HM, Kash JC, Walters KA, Wills NM, Xiao YL, Dunfee RL, Schwartzman LM, Ozinsky A, Bell GL, et al. An Overlapping Protein-Coding Region in Influenza A Virus Segment 3 Modulates the Host Response. Science. 2012;337:199–204. doi: 10.1126/science.1222213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankov RP, Luo X, Demin P, Aslam R, Hannam V, Tanswell AK, Pace-Asciak CR. Hepoxilin analogs inhibit bleomycin-induced pulmonary fibrosis in the mouse. J Pharmacol Exp Ther. 2002;301:435–440. doi: 10.1124/jpet.301.2.435. [DOI] [PubMed] [Google Scholar]
- Ji R-R, Xu Z-Z, Strichartz G, Serhan CN. Emerging roles of resolvins in the resolution of inflammation and pain. Trends in Neurosciences. 2011;34:599–609. doi: 10.1016/j.tins.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kash JC, Tumpey TM, Proll SC, Carter V, Perwitasari O, Thomas MJ, Basler CF, Palese P, Taubenberger JK, García-Sastre A, et al. Genomic analysis of increased host immune and cell death responses induced by 1918 influenza virus. Nature. 2006;443:578–581. doi: 10.1038/nature05181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss L, Schutte H, Mayer K, Grimm H, Padberg W, Seeger W, Grimminger F. Synthesis of arachidonic acid-derived lipoxygenase and cytochrome P450 products in the intact human lung vasculature. Am J Respir Crit Care Med. 2000;161:1917–1923. doi: 10.1164/ajrccm.161.6.9906058. [DOI] [PubMed] [Google Scholar]
- Kronke G, Katzenbeisser J, Uderhardt S, Zaiss MM, Scholtysek C, Schabbauer G, Zarbock A, Koenders MI, Axmann R, Zwerina J, et al. 12/15-Lipoxygenase Counteracts Inflammation and Tissue Damage in Arthritis. The Journal of Immunology. 2009;183:3383–3389. doi: 10.4049/jimmunol.0900327. [DOI] [PubMed] [Google Scholar]
- Kühn H, O’Donnell VB. Inflammation and immune regulation by 12/15-lipoxygenases. Prog Lipid Res. 2006;45:334–356. doi: 10.1016/j.plipres.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Kühn H, Walther M, Kuban RJ. Mammalian arachidonate 15-lipoxygenases structure, function, and biological implications. Prostaglandins Other Lipid Mediat. 2002:68–69. 263–290. doi: 10.1016/s0090-6980(02)00035-7. [DOI] [PubMed] [Google Scholar]
- Levy BD, Kohli P, Gotlinger K, Haworth O, Hong S, Kazani S, Israel E, Haley KJ, Serhan CN. Protectin D1 is generated in asthma and dampens airway inflammation and hyperresponsiveness. J Immunol. 2007;178:496–502. doi: 10.4049/jimmunol.178.1.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan LT, Thomas CP, Kühn H, O’Donnell VB. Thrombin-activated human platelets acutely generate oxidized docosahexaenoic-acid-containing phospholipids via 12-lipoxygenase. Biochem J. 2010;431:141–148. doi: 10.1042/BJ20100415. [DOI] [PubMed] [Google Scholar]
- Morita M, Kuba K, Ichikawa A, Nakayama M, Katahira J, Iwamoto R, Watanebe T, Sakabe S, Daidoji T, Nakamura S, et al. The Lipid Mediator Protectin D1 Inhibits Influenza Virus Replication and Improves Severe Influenza. Cell. 2013;153:112–125. doi: 10.1016/j.cell.2013.02.027. [DOI] [PubMed] [Google Scholar]
- Neumann G, Noda T, Kawaoka Y. Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature. 2009;459:931–939. doi: 10.1038/nature08157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obinata H, Izumi T. G2A as a receptor for oxidized free fatty acids. Prostaglandins Other Lipid Mediat. 2009;89:66–72. doi: 10.1016/j.prostaglandins.2008.11.002. [DOI] [PubMed] [Google Scholar]
- O’Flaherty JT, Hu Y, Wooten RE, Horita DA, Samuel MP, Thomas MJ, Sun H, Edwards IJ. 15-Lipoxygenase Metabolites of Docosahexaenoic Acid Inhibit Prostate Cancer Cell Proliferation and Survival. PLoS ONE. 2012;7:e45480. doi: 10.1371/journal.pone.0045480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quehenberger O, Armando AM, Brown AH, Milne SB, Myers DS, Merrill AH, Bandyopadhyay S, Jones KN, Kelly S, Shaner RL, et al. Lipidomics reveals a remarkable diversity of lipids in human plasma. J Lipid Res. 2010;51:3299–3305. doi: 10.1194/jlr.M009449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quehenberger O, Dennis EA. The human plasma lipidome. N Engl J Med. 2011;365:1812–1823. doi: 10.1056/NEJMra1104901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N, et al. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity. 2000;13:539–548. doi: 10.1016/s1074-7613(00)00053-4. [DOI] [PubMed] [Google Scholar]
- Serhan CN, Clish CB, Brannon J, Colgan SP, Chiang N, Gronert K. Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing. J Exp Med. 2000;192:1197–1204. doi: 10.1084/jem.192.8.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN, Maddox JF, Petasis NA, Akritopoulou-Zanze I, Papayianni A, Brady HR, Colgan SP, Madara JL. Design of lipoxin A4 stable analogs that block transmigration and adhesion of human neutrophils. Biochemistry. 1995;34:14609–14615. doi: 10.1021/bi00044a041. [DOI] [PubMed] [Google Scholar]
- Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. 2008;8:349–361. doi: 10.1038/nri2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisemore M. Cellular Characterization of Leukotoxin Diol-Induced Mitochondrial Dysfunction. Arch Biochem Biophys. 2001;392:32–37. doi: 10.1006/abbi.2001.2434. [DOI] [PubMed] [Google Scholar]
- Spiteller P, Spiteller G. 9-Hydroxy-10,12-octadecadienoic acid (9-HODE) and 13-hydroxy-9,11-octadecadienoic acid (13-HODE): excellent markers for lipid peroxidation. Chem Phys Lipids. 1997;89:131–139. [Google Scholar]
- Taubenberger JK, Kash JC. Influenza virus evolution, host adaptation, and pandemic formation. Cell Host Microbe. 2010;7:440–451. doi: 10.1016/j.chom.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson SJ, Askari A, Bishop-Bailey D. Anti-Inflammatory Effects of Epoxyeicosatrienoic Acids. International Journal of Vascular Medicine. 2012;2012:1–7. doi: 10.1155/2012/605101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobin DM, Roca FJ, Oh SF, McFarland R, Vickery TW, Ray JP, Ko DC, Zou Y, Bang ND, Chau TTH, et al. Host Genotype-Specific Therapies Can Optimize the Inflammatory Response to Mycobacterial Infections. Cell. 2012;148:434–446. doi: 10.1016/j.cell.2011.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totani Y, Saito Y, Ishizaki T, Sasaki F, Ameshima S, Miyamori I. Leukotoxin and its diol induce neutrophil chemotaxis through signal transduction different from that of fMLP. Eur Respir J. 2000;15:75–79. doi: 10.1183/09031936.00.15107500. [DOI] [PubMed] [Google Scholar]
- Wray JA, Sugden MC, Zeldin DC, Greenwood GK, Samsuddin S, Miller-Degraff L, Bradbury JA, Holness MJ, Warner TD, Bishop-Bailey D. The Epoxygenases CYP2J2 Activates the Nuclear Receptor PPARα In Vitro and In Vivo. PLoS ONE. 2009;4:e7421. doi: 10.1371/journal.pone.0007421. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.