

Abstract
Our study investigated the association of rare allelic variants with extremes of 24-hour urinary calcium excretion because higher urinary calcium excretion is a dominant risk factor for calcium-based kidney stone formation. We resequenced 40 candidate genes potentially related to urinary calcium excretion in individuals from the Nurses' Health Studies I & II and the Health Professionals Follow-up Study. A total of 960 participants were selected based on availability of 24-hour urine collection data and level of urinary calcium excretion (low vs. high). We utilized DNA sample pooling, droplet-based target gene enrichment, multiplexing, and high-throughput sequencing. Approximately 64% of samples (n = 615) showed both successful target enrichment and sequencing data with >20-fold deep coverage. A total of 259 novel allelic variants were identified. None of the rare gene variants (allele frequencies <2%) were found with increased frequency in the low vs. high urinary calcium groups; most of these variants were only observed in single individuals. Unadjusted analysis of variants with allele frequencies ≥2% suggested an association of the Claudin14 SNP rs113831133 with lower urinary calcium excretion (6/520 versus 29/710 haplotypes, P value = 0.003). Our data, together with previous human and animal studies, suggest a possible role for Claudin14 in urinary calcium excretion. Genetic validation studies in larger sample sets will be necessary to confirm our findings for rs113831133. In the tested set of candidate genes, rare allelic variants do not appear to contribute significantly to differences in urinary calcium excretion between individuals.
Introduction
Kidney stone disease is a major cause of morbidity associated with tremendous pain, suffering, and substantial economic impact [1]–[3]. The majority of kidney stones contain calcium, most commonly in the form of calcium (Ca2+) oxalate. Higher urinary Ca2+ excretion is associated with higher risk of calcium-containing kidney stone formation. The etiology of the vast majority of cases of hypercalciuria is unknown and is referred to as idiopathic hypercalciuria [4].
Nephrolithiasis is a multifactorial disease with genetic and environmental factors determining the likelihood of stone formation [5]. We and other investigators have identified multiple environmental risk factors associated with increased risk including lower dietary Ca2+ intake [6]–[8], lower fluid intake [6]–[9], and higher body mass index [8], [10]. A family history of nephrolithiasis is associated with a greater than two-fold increase in the risk of developing a stone [11]. Substantial data demonstrate that calcium-based kidney stones and elevated urinary Ca2+ are linked and likely have a strong genetic component [12]. Genes for rare Mendelian forms of nephrolithiasis and increased urinary Ca2+ excretion have been identified; however, mutations in these genes explain only a very small fraction of kidney stone disease in the general population. For example, mutations in the renal chloride channel CLCN5 cause Dent's disease, a group of X-linked hypercalciuric abnormalities [13]. Inactivating (loss-of-function) mutations in the calcium-sensing receptor (CaSR) cause autosomal-dominant hypocalcemia and hypercalciuria [14]. Another example is familial hypomagnesemia associated with hypercalciuria and nephrocalcinosis (mutations in Claudin16) [15]. Additional genes were identified in various animal models such as the renal epithelial Ca2+ transporter gene (TRPV5), which when mutated causes severe hypercalciuria in the mouse [16].
Rather than testing the “common disease, common variant” hypothesis pursued by genome wide association studies (GWAS), our approach tested the association of the frequencies of rare genetic variants with 24-hour urinary Ca2+ excretion [17]. This approach has succeeded when examining other complex traits such as hypertriglyceridemia [18], hypercholesterolemia [19], and non-alcoholic fatty liver disease [20]. The findings in these studies were consistent with in silico predictions that some sequence variations found in healthy individuals are as deleterious to protein function as mutations that, in other genes, cause monogenic disease. Highly penetrant rare alleles may be an important genetic contributor to common disease seen in the general population as shown for blood pressure variation [21].
The goal of this work was to identify rare, functionally significant genetic variants associated with urinary Ca2+ excretion. Forty candidate genes possibly related to urinary Ca2+ excretion were resequenced at extremes of urinary Ca2+ excretion in 960 individuals from three well-characterized cohorts, the Nurses' Health Studies (NHS) I & II and the Health Professional Follow-Up Study (HPFS).
Methods
A. Study cohorts
The NHS I was established in 1976 with over 120,000 female registered nurses aged 30–55 years. The NHS II was established in 1989 with over 116,000 female nurses aged 25–42 years. The HPFS was established in 1986 with over 51,000 male health care professionals aged 40–75 years. All three cohorts have been followed by biennially mailed questionnaires including questions on lifestyle practices and newly diagnosed diseases such as nephrolithiasis [22]. Additional information was obtained from self-reported cases including symptoms and kidney stone type. In validation studies, permission to obtain medical records was requested from newly diagnosed cases in all three cohorts. The diagnosis of stone disease was confirmed in over 90% of these cases. Twenty-four-hour urine collections were obtained from participants with a history of confirmed nephrolithiasis and from randomly selected controls. Those with a history of kidney stones performed the collections after the diagnosis. All 24-hour urine collections were performed using the Mission Pharmacal system (San Antonio, TX, USA). Urinary Ca2+ was measured by an atomic absorption spectrophotometer [22]. Approximately 10% of individuals with the highest and lowest values of 24-hour urinary Ca2+ excretion from available male and female participants in equal numbers were selected.
This study was approved by the Brigham and Women's Hospital's institutional review board (approval # 2000P001316). The institutional review board (IRB) specifically considered the risks and anticipated benefits, if any, to participants, and the selection, safety and privacy of individuals. Implied consent was considered as appropriate by the IRB for this specific study.
B. DNA samples
DNA samples were collected as part of a general collection of blood samples in the three cohort studies. We limited this study to those who self-reported their race as Caucasian (and this was confirmed as part of a separate GWAS analysis). High quality DNA was extracted (Dana Farber/Harvard Cancer Center) from buffy coats via QIamp 96 spin-protocol (Quiagen Inc., Chatsworth, CA). DNA concentrations were calculated in 96-well format using a Molecular Dynamics spectrophotometer. The 960 DNA samples were ranked by urinary Ca2+ excretion and grouped into pools of 15 or 20 samples prior to target DNA capture. A pilot project testing pooled samples of 15 versus 20 individual samples did not show any differences in DNA capture efficiency and sequence coverage per individual samples between the pools (data not shown). We subjected 52 sample pools (N = 960 individuals; 16 pools of 15 samples and 36 pools of 20 samples) to RainDance target (RDT) DNA capture ( Table 1 ). The amount of genomic DNA provided for RDT capture per pool was 10 µg (0.5–0.66 µg per individual sample).
Table 1. DNA pooling strategy for 960 individuals with low (n = 480) vs. high (n = 480) urinary Ca2+ excretion.
| Cohort | No. of pools (sets of 15+20 samples) | No. of individuals |
| NHS I, low urinary Ca2+ | 7 (4×15+3×20) | 120 |
| NHS II, low urinary Ca2+ | 6 (6×20) | 120 |
| HPFS, low urinary Ca2+ | 13 (4×15+9×20) | 240 |
| NHS I, high urinary Ca2+ | 7 (4×15+3×20) | 120 |
| NHS II, high urinary Ca2+ | 6 (6×20) | 120 |
| HPFS, high urinary Ca2+ | 13 (4×15+9×20) | 240 |
NHS = Nurses' Health Study; HPFS = Health Professional Follow-up Study.
C. Candidate genes
Candidate genes were selected by searching public databases (PubMed and Online Mendelian Inheritance in Man (OMIM)). We limited the number of genes to 40 due to cost and technical limitations at the time of study design. Our priority was to achieve sufficient sequence coverage (at least 20×) to detect rare allelic variation. Selection of candidate genes was based on in vivo and in vitro evidence of regulating Ca2+ homeostasis in bone, kidney or intestine. We also selected some genes, such as oxalate and citrate exchangers, which may affect calcium-based stone formation by changing supersaturation rather than urinary Ca2+ excretion per se. The genes resequenced in this study are listed in Table 2 , including gene name, gene/protein function, RefSeq ID and exon number. References underlining the rationale of gene selection are provided. As an additional gene, we included PIK3C2G (phosphoinositide-3-kinase, class 3, gamma polypeptide) based on our unpublished GWAS data. PIK3C2G regulates diverse cellular responses, such as cell proliferation, oncogenic transformation, cell migration, intracellular protein trafficking, and cell survival.
Table 2. Candidate genes included in the resequencing study (n = 40).
| Gene | Gene name/protein function | RefSeq ID | Exons |
| Aconitase [32] | Catalyzes the isomerization of citrate to isocitrate | NM_001098 | 18 |
| CaSR [33] | Calcium-sensing receptor | NM_000388 | 7 |
| Citrate lyase [34] | Catalyzes the formation of acetyl-CoA from citrate | NM_001096 | 29 |
| Claudin 2 [35] | Tight junction protein, proximal tubule (PT) | NM_001171092 | 2 |
| Claudin 8 [35] | Tight junction protein, primarily distal nephron (DCT) | NM_99328 | 1 |
| Claudin 10 [36] | Tight junction protein, thick ascending limb (TAL) and intestine | NM_001160100 | 5 |
| Claudin 14 [37] | Tight junction protein, TAL | NM_001146077 | 3 |
| Claudin 16 [15] | Tight junction protein, TAL | NM_006580 | 4 |
| Claudin 19 [30] | Tight junction protein, TAL and DCT | NM_001123395 | 5 |
| CLCN5 [38] | Chloride channel 5, mutations cause Dent's disease | NM_000084 | 15 |
| CLCNKA [39] | Basolateral chloride channel expressed in TAL | NM_001042704 | 20 |
| CLCNKB [40] | Basolateral chloride channel expressed in TAL and DCT; mutation cause type III Bartter's syndrome | NM_000085 | 20 |
| FGF23 [41] | Fibroblast growth factor 23, phosphatonin | NM_020638 | 3 |
| GCMB [42] | Glial cell missing B, mutations cause familial isolated hypoparathyroidism | NM_004752 | 5 |
| Klotho [43] | Regulator of TRPV5 and FGF23 | NM_004795 | 5 |
| NHERF1 [44] | Hydrogen exchanger regulatory factor 1, mutations cause hypophosphatemia and nephrolithiasis | NM_004252 | 6 |
| NHERF2 [45] | Hydrogen exchanger regulatory factor 2, expressed like NHERF1 in PT | NM_001130012 | 7 |
| NKCC2 [46] | Na+-K−-2Cl−-cotransporter, mutations cause type 1 (neonatal) Bartter's syndrome | NM_000220 | 27 |
| PDZK1 [47] | Hydrogen exchanger regulatory factor 3, PT | NM_002614 | 10 |
| PIK3C2G | Phosphoinositide-3-kinase, class 3, γ polypeptide | NM_004570 | 32 |
| PTH [48] | Parathyroid hormone | NM_000315 | 3 |
| PTHR [49] | Parathyroid hormone receptor 1 | NM_000316 | 16 |
| ROMK [50] | Renal outer medullary K+ channel, mutations cause type 2 Bartter's syndrome | NM_000338 | 4 |
| SLC12A3 [51] | Thiazide-sensitive Na+-Cl− cotransporter, mutation cause Gitelman's syndrome | NM_000339 | 26 |
| SLC13A2 [52] | Na+ citrate transporter NaC1 | NM_001145975 | 12 |
| SLC13A3 [53] | Na+ citrate transporter NaC2 | NM_001193340 | 14 |
| SLC25A1 [54] | Mitochondrial citrate transporter | NM_005984 | 9 |
| SLC26A1 [55] | Oxalate and sulfate anion transporter | NM34425 | 4 |
| SLC26A2 [56] | Oxalate and citrate exchanger | NM_000112 | 2 |
| SLC26A6 [57] | Oxalate and citrate exchanger | NM_001040454 | 21 |
| SLC34A1 [58] | Na+ phosphate co-transporter NaPi2A | NM_003052 | 13 |
| SLC34A3 [59] | Na+ phosphate co-transporter NaPi2C | NM_001177316 | 13 |
| SLC4A1 [60] | AE1 oxalate, mutations cause distal RTA | NM_000342 | 20 |
| SLC4A2 [61] | AE2 oxalate | NM_003040 | 23 |
| SLC4A3 [61] | AE3 oxalate | NM_005070 | 23 |
| TRPV5 [62] | Epithelial Ca2+ channel ECaC1 | NM_019841 | 14 |
| TRPV6 [63] | Epithelial Ca2+ channel ECaC2 | NM_018646 | 15 |
| UMOD [64] | Uromodulin | NM_001008389 | 11 |
| VDR [65] | Vitamin D receptor | NM_000376 | 11 |
| WNK4 [66] | Protein kinase, lysine deficient 4, mutations cause pseudohypoaldosteronism type 2 | NM_032387 | 19 |
D. Primer design
Our list of the 40 genes was provided to RainDance Technologies (RDT) (Lexington, MA) for custom primer design based on the Primer3 algorithm (http://frodo.wi.mit.edu/primer3). The custom panel was prepared and primers were designed to target all 497 exons of the 40 candidate genes, including ∼50 bp of intronic sequence flanking each exon. The 795 amplicons in the panel ranged in size from 200 to 600 bases, with a GC content of 25% to 87%, and represented a total coding sequence of ∼182 kb. All single nucleotide polymorphisms (SNPs) and repeat regions were filtered from the primer selection region. The RDT design was quality checked to ensure that none of the primers were designed over known SNPs and primer sequences were verified to avoid repetitive regions of the genome using the program RepeatMasker (http://www.repeatmasker.org). The primers for the 795 amplicons varied in annealing temperature from 57°C to 60°C, with a primer length range of 15 to 22 bases. Other rules for primer design included BLASTing the primers to the chromosome that contained the gene of interest and in silico PCR to match the designed primers to PCR product and target sequence.
E. Enrichment of target DNA for sequencing
The capture was performed at two laboratories, RDT in Lexington, MA, and Ambry Genetics in Aliso Viejo, CA. DNA samples were fragmented to 3 to 4 kb by shearing the genomic DNA with the Covaris S2 instrument (Covaris, Woburn, MA) following the manufacturer's instructions. To prepare the input DNA template mixture for targeted amplification, 3 µg of the purified genomic DNA fragments were added to 4.7 µL of high-fidelity buffer (Invitrogen, Carlsbad, CA), 1.26 µL of magnesium sulfate (Invitrogen), 1.6 µL of 10 mmol/L dNTP (Invitrogen), 3.6 µL of 4 mol/L betaine (Sigma-Aldrich, St. Louis, MO), 3.6 µL of Droplet Stabilizer (RDT, Lexington, MA), 1.8 µL of dimethyl sulfoxide (Sigma-Aldrich), and 0.7 µL of 5 units/µL of Platinum High-Fidelity Taq (Invitrogen). The samples were brought to a final volume of 25 µL with nuclease-free water. PCR droplets were generated on the RDT1000 instrument. The enrichment panel consisted of an emulsion that contained a collection of unique primer droplets in which each primer droplet contained a single matched forward and reverse primer for each amplicon in the panel. Each panel contained multiple replicates of each unique primer droplet with consistent volume. The RDT1000 generated a PCR droplet by pairing a single genomic DNA template droplet with a single primer droplet. The paired droplets flowed past an electrode in the RDT chip and were instantly merged to create a single PCR droplet. All of the resulting PCR droplets were dispensed as an emulsion into a PCR tube and then transferred to a standard thermal PCR cycler for amplification (Gene-Amp 9700 thermocycler, Applied Biosystems, Foster City, CA). After PCR amplification, the emulsion was broken to release each individual amplicon from the PCR droplets. For each sample, an equal volume of Droplet Destabilizer (RDT) was added to the emulsion of PCR droplets, the sample was vortexed for 15 sec, and spun in a microcentrifuge at 13,000× g for 5 min. The oil below the aqueous phase was carefully removed from the sample and the remaining sample was purified using a MinElute column (Qiagen, Valencia, CA) following the manufacturer's recommended protocol. The purified amplicon DNA was tested on an Agilent Bioanalyzer (Agilent Technologies, Santa Clara, CA) to confirm that it matches the expected amplicon profile (mixture of amplicons ranging from 200 to 600 bp in size).
F. Targeted deep DNA sequencing
A simplified schematic illustration of our emulsion-based droplet PCR approach and the off-chip work flow before high-throughput sequencing is presented in Figure 1 . Successfully enriched sample pools were barcoded and sequenced on the Hiseq2000 Illumina platform (7 barcoded sample pools per lane). After PCR purification, amplified fragments for each individual were repaired to blunt ends using NEB Quick blunting kit (NEB, catalog # E1201L, 15 min RT). The PCR fragments were then linked using NEB Quick ligation kit (NEB, catalog # M2200L). Ligation was done overnight at 25°C. The ligated products were made into 100 µL volume by adding elution buffer and were then sheared using Covaris E210 (Covaris, Woburn, MA). The sheared fragments were purified using Qiagen QIAquick PCR purification column and eluted in 32 µL of elution buffer. The samples then entered the standard Illumina Genome Analyzer multiplex library introduced preparation protocol. The enrichment was confirmed by running an Agilent BioAnalyzer 7500 DNA chip. A quantitative PCR was done to quantitate the library using KAPA Library quantification kit (KAPA Biosystems, Woburn, MA, USA, catalog # KK4824). Enriched DNA was denatured and diluted to a concentration of 8 pM. Seventy bp single end sequencing was performed using standard IGAII manuals and version 4 kits. Seven sample pools per lane of Illumina sequencing were multiplexed. After sequencing, the reads consisting of 795 fragments covering 262,545 bases were mapped and variants sites identified against the reference sequence using the BWA software [23]. This region is larger than the actual targeted bases (182,345) as RDT included some intronic and intergenic regions to facilitate primer picking. All sequence data aligned for the analysis had sequence coverage exceeding 20-fold. Sequences were compared to those reported by HapMap using a custom perl script to assess the rates of data completion and accuracy. The HapMap data was assumed to be without error when estimating data accuracy.
Figure 1. Simplified schematic illustration of the study design utilizing emulsion-based droplet PCR technology and next-generation sequencing.
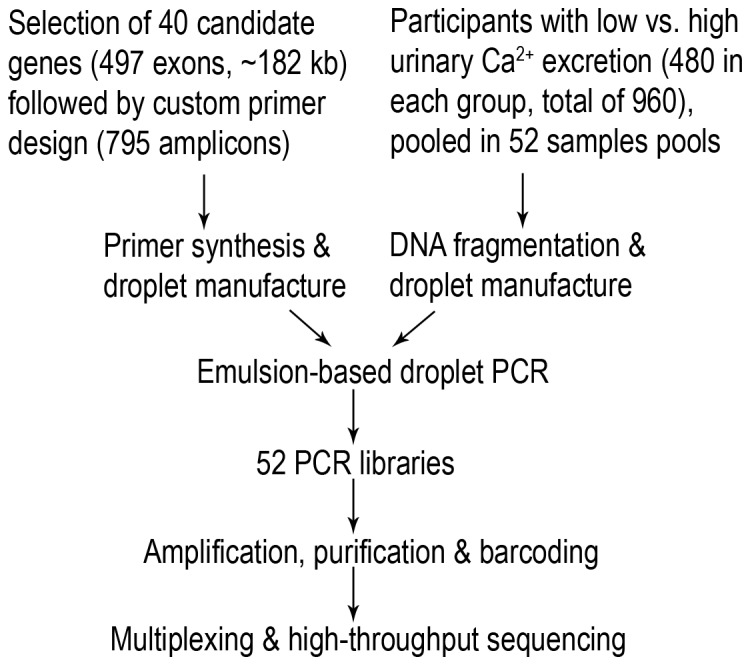
G. Variant identification
Utilizing the Syzygy software [24], we compared all identified single nucleotide variants (SNVs) with three publically available SNP databases including dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP/), 1000 Genome Project (1000G) [25], and Exome Sequencing Project (ESP) (http://evs.gs.washington.edu/EVS/) databases. After filtering known SNPs, we used the transition to transversion ratio (Ti/Tv) to filter out false positive novel variants, which occur frequently on high-throughput sequencing platforms. Ti/Tv can be used to estimate true-positive (TP) to false-positive (FP) SNP data [26]. A transition is the mutation of a purine nucleotide to another purine (A<->G) or a pyrimidine to another pyrimidine (C<->T). In contrast, a transversion (Tv) substitutes a purine for a pyrimidine or vice versa. The initially observed Ti/Tv ratio in our SNV data was a mixture of true-positive and false-positive SNVs. This was based on the assumption that if all detected SNVs are true, Ti/Tv equals ∼3.3. If all SNVs are false Ti/Tv equals 0.5. We assumed that common novel variants were most likely artifacts of sequencing. We therefore considered novel variants only if they were observed less than or equal to 3 times in the whole dataset. In nature, Tis are more common than Tvs. We used the following equation to estimate the number of true positive SNVs: %TP = (Ti/Tvobs-Ti/TvFP)/(Ti/TvTP-Ti/TvFP). Considering that Ti/TvTP is around 2.8–3.3, the %TP should be ∼9–11%.
Results
In order to query the potential relevance of rare allelic variation in genes associated with urinary Ca2+ excretion, we decided to amplify and sequence the exons of 40 genes in 960 well-characterized individuals from the NHS and HPFS populations as outlined in the methods section (see Tables 1 and 2 , Figure 1 )
Efficacy of DNA enrichment and sequencing
Forty-two out of 52 samples pools were successfully captured (N = 730 individual samples). Ten sample pools (N = 230) failed enrichment due to technical issues. The 42 samples pools were barcoded and further pooled (in sets of 7 sample pools) for sequencing on the HiSeq2000 Illumina platform. Eight samples pools had insufficient sequence coverage (<2-fold). Thirty four sample pools (N = 615) showed sequencing data with >20-fold sequence coverage. These raw sequence data were deposited into the NIH short read archive (SRA) database (http://www.ncbi.nlm.nih.gov/sra) under the accession number PRJNA209216. The target DNA showed uniform amplification across all target amplicons.
The distribution of study participants with >20-fold sequence coverage from the three cohorts in low (N = 355) and high (n = 260) urinary calcium excretion groups are shown in Table 3 . The phenotype data provided include urinary solute excretion, age and body mass index (BMI). The number of kidney stone formers was significantly higher in the high urinary Ca2+ group (n = 164 vs. n = 96; Chi Square P value = 0.004).
Table 3. Study participants with successful DNA target amplification and sequencing.
| Lower urinary Ca2+ excretion n = 355 (range 18–165 mg/day) | Higher urinary Ca2+ excretion n = 260 (range 210–465 mg/day) | |||
| Stone formers n = 182 | Non-stone formers n = 173 | Stone formers n = 164* | Non-stone formers n = 96 | |
| Ca2+ (mg/day) | 100±37 | 101±35 | 320±57 | 309±56 |
| Age (years) | 64±11 | 61±8 | 59±10 | 60±8 |
| BMI (kg/m2) | 28±6 | 26±5 | 28±5 | 27±5 |
The frequency of kidney stone formers was significantly higher (*) in the high urinary Ca2+ group (Chi Square P value = 0.004).
Allelic variation in targeted DNA sequence
Samples pools that passed our quality matrix, by enriching for target DNA and showing over 20-fold sequence coverage, were included in our analysis (N = 615 individuals). The total number of identified sequence nucleotide variants (SNVs) with Szygy software was 1,572 ( Figure 2A ). Of these, 429 were known SNPs based on comparison to the three widely used databases (dbSNP, 1000G and ESP databases) ( Figure 2B ). The Ti/Tv of these known SNPs (2.88) differed from the Ti/Tv of the 1,572 SNVs discovered in our study population (0.75). This finding suggested a significant portion of false-positive SNVs. The number of novel variants was reduced to 259 after exclusion of novel SNVs, which were seen 4 or more times and were not present in the tested databases (N = 884, Ti/Tv 0.5). The Ti/Tv of novel allelic variants improved from 0.75 to 2.8, which is both within the expected range of Ti/Tv in naturally occurring mutations (∼2.8–3.3) and similar to the Ti/Tv of the 429 known SNPs (2.88) identified in this study. Table S1 lists all identified novel SNPs (n = 259) including information on chromosomal location, nucleotide substitution, and effect on protein sequence. Novel silent, nonsense and missense SNPs with existing RefSeq accession numbers were deposited into the NCBI ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/). ClinVar accession IDs are provided in Table S1.
Figure 2. The flowchart data analysis of identified sequence nucleotide variants (SNVs).
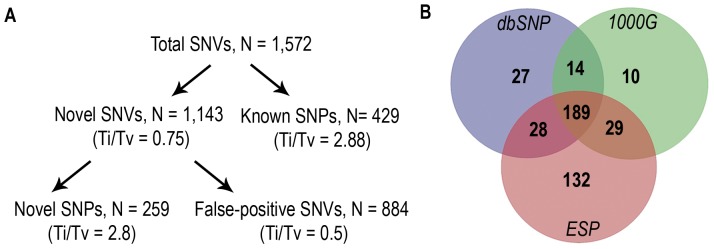
(A). After identifying known SNPs utilizing publically available databases (SNP distribution shown in B), false-positive SNVs were filtered based on frequency in our dataset (present four or more times). The transition to transversion ratio (Ti/Tv) of naturally occurring SNPs supported this analytical approach. dbSNP = Single Nucleotide Polymorphism Database; 1000G = 1000 Genome Project; ESP = Exome Sequencing Project.
SNP data for Claudin14
Table 4 lists all CLDN14 variations identified in our sample set. SNP rs113831133 was more common among individuals in the lower (29 out of 710 haplotypes; 4.1%) compared with the higher (6 out of 520 haplotypes; 1.1%) urinary Ca2+ excretion group (Fisher's exact test: P value = 0.003). When adjusted for multiple comparisons with Bonferroni's correction (for n = 429 known SNPs), our finding for rs113831133 did not reach statistical significance (required P value <0.0001). SNP rs11381133 did not vary by sex (17/650 in females, 2.6%, vs. 18/580 in males, 3.1%, P value = 0.61). The common, synonymous CLDN14 SNP rs219780, previously shown to be associated with kidney stone disease in a large GWAS study [27], did not differ between the two urinary Ca2+ groups (unadjusted P value = 0.82).
Table 4. Distribution of all identified Claudin14 SNPs in the low and high urinary Ca2+ excretion groups with unadjusted Chi square P values.
| Chr. Position | SNP ID (rs…) | SNP class | Nucleotide | Low urine Ca2+ | High urine Ca2+ | P |
| 37833330 | . | missense | c.664G>T | 2.2% (16/710) | 1.5% (8/520) | 0.41 |
| 37833331 | . | silent | c.663G>A | 0.0% (0/710) | 0.2% (1/520) | - |
| 37833934 | . | silent | c.60C>T | 0.1% (1/710) | 0.2% (1/520) | - |
| 37833931 | 117560775 | silent | c.63G>A | 1.4% (10/710) | 3.2% (17/520) | 0.03 |
| 37833694 | 113350364 | silent | c.300C>T | 0.4% (3/710) | 0.0% (0/520) | - |
| 37833699 | . | missense | c.295G>A | 0.0% (0/710) | 0.3% (1/520) | - |
| 37833751 | 219779 | silent | c.243C>T | 24.2% (172/710) | 23.8% (124/520) | 0.89 |
| 37833979 | . | silent | c.15C>T | 0.0% (0/710) | 0.2% (1/520) | - |
| 37833809 | . | missense | c.185A>G | 0.4% (3/710) | 0.0% (0/520) | - |
| 37833892 | . | silent | c.102G>A | 0.1% (1/710) | 0.0% (0/520) | - |
| 37833865 | . | silent | c.129C>T | 0.3% (2/710) | 0.0% (0/520) | - |
| 37833506 | . | missense | c.488C>T | 0.3% (2/710) | 0.2% (1/520) | - |
| 37833983 | 113831133 | missense | c.11C>T | 4.1% (29/710) | 1.1% (6/520) | 0.003 |
| 37833976 | . | silent | c.18G>A | 0.3% (2/710) | 0.0% (0/520) | - |
| 37833661 | 74934405 | silent | c.333A>C | 4.1% (29/710) | 4.4% (23/520) | 0.77 |
| 37833304 | . | silent | c.690C>T | 0.1% (1/710) | 0.1% (1/520) | - |
| 37833307 | 219780 | silent | c.687G>A | 18.4% (131/710) | 18.9% (98/520) | 0.82 |
The missense variant rs113831133, highlighted in bold, was more frequent among individuals with lower urinary Ca2+ excretion. This association did not reach statistical significance when adjusted for multiple comparisons.
Discussion
The goal of this work was to quantify the frequency of rare, presumably functional genetic variants in individuals with lower and higher urinary Ca2+ excretion because urinary Ca2+ is a major risk factor for calcium-based kidney stone disease [4]. A different approach has been pursued previously in a large GWAS for kidney stone disease and has shown only limited success possibly due to the complex mechanistic nature of nephrolithiasis. That GWAS in 3,773 kidney stone cases and 42,510 controls from Iceland and the Netherlands reported an association of CLDN14 with nephrolithiasis. The synonymous CLDN14 variant rs219780(C) (minor allele frequency ∼20%) showed a significant association with kidney stone disease (P value = 4×10−12) and low bone mineral density (for the hip P value = 0.00039) [27].
In this study, we tested the association of the frequencies of rare allelic variants with possible effect on urinary Ca2+ excretion by resequencing 40 known genes that could potentially affect or correlate with urinary Ca2+ excretion either in human disease or animal models. Our main hypothesis was that differing number of rare variants between individuals with lower and higher urinary Ca2+ excretion would be identified in at least some of these candidate genes. Rare, non-synonymous variants in low and high urinary Ca2+ excretion groups could contribute to the level of urinary Ca2+ excretion (lowering or increasing excretion and thereby protecting from or predisposing to calcium-based stone formation). This approach has succeeded for other phenotypes such as hyperlipidemia [18],[19] and non-alcoholic fatty liver disease [20], where the frequency of rare gene variants was significantly different in the extremes of the studied phenotype. The results in those studies were consistent with in silico predictions that rare amino acid changing sequence variations found in healthy individuals are as deleterious to protein function as gene mutations causing Mendelian disease. Such sequence variations may explain a significant fraction of phenotypic variation in human as suggested for blood pressure variation in the Framingham Heart Study population [21].
We identified 1,572 single nucleotide variants (SNVs), most of which appeared common in our subjects. Initially, we filtered out known gene variants based on three large SNP databases and then applied a simple but very efficient method the Ti/Tv of naturally occurring mutations [26]. We filtered potentially false-positive SNVs, significantly reducing the initially seen number of SNVs and improving the Ti/Tv from 0.75 to a “normal” range of 2.88. Of the 259 novel SNPs in 40 different genes, none showed a significantly increased frequency in the low versus the high urinary Ca2+ excretion groups. These extremely rare candidate gene variants identified mostly in single individuals could be functionally contributing to the level of urinary Ca2+ excretion, because they are mostly amino acid changing and occur frequently only in one of the two groups with extreme urinary Ca2+ excretion. Identification of the individuals carrying these very rare variants and testing their relatives (for degree of urinary Ca2+ excretion and presence of variant) would help to answer if these variants contribute to the phenotype. Examining the conservation of the affected residue across species as well as the in vitro and in vivo effects on gene function would be necessary to postulate a cause-effect relation for these very rare variants.
We also analyzed our data for rare variants with allele frequency of 2–5%. Of these, the non-synonymous CLDN14 SNP rs113831133 (minor allele frequency for ∼3.3% for c.11C>T, p.Thr4Met, dbSNP) showed a lower allele frequency in individuals with high urinary Ca2+ excretion (∼1.1% vs. 4.1% in the lower urinary Ca2+ group), suggesting that if present it may lower urinary Ca2+ excretion, probably by decrease in CLDN14 function. The functional significance of the CLDN14 SNP rs113831133 in the thick ascending limb is unknown. Since heterozygous CLDN14-deficient mice have no renal phenotype, a dominant negative effect of rs113831133 appears more likely than haploinsufficiency [28]. The rs113831133 finding is in particular interesting since the synonymous CLDN14 SNP rs219780 has been implicated previously in kidney stone disease [27]. The frequency of this SNP was not significantly different in our study groups, though our study included a smaller sample size and our focus was on urine Ca2+ excretion rather than kidney stone formation. There are several different studies implicating an important role for CLDN14 in urinary Ca2+ excretion. CLDN14 has been shown to be a negative regulator of the CLDN16/19 complex in the TAL, which has an important role in the paracellular Ca2+ reabsorption in the TAL [28]. Mutations in both CLDN16 and 19 have been shown to cause familial hypercalciuria in human. In addition, recent animal studies showed a significant role for CLDN14 in urinary Ca2+ reabsorption [29], [30]. Homozygous CLDN14-deficient mice display lower urinary Ca2+ excretion than wild-type controls when challenged with a high calcium diet [28]. Renal tubule-specific Casr-deficient mice display decreased urinary Ca2+ excretion compared to control animals. CLDN14 is significantly downregulated (∼80%) in this mouse model [31].
Our study had several limitations. The selection of candidate genes was biased and several other candidate genes potentially related to urinary Ca2+ excretion were not included in our investigation. Therefore, an unbiased approach including all genes would be most favorable. This could be accomplished by exome capture followed by next-generation sequencing including all coding regions of the genome. Another limitation is the relatively low sample number reducing the power to test the main hypothesis of this study. In order to further examine the role of rare, potentially functional variants, e.g. with minor allele frequency of 1% or lower, a larger sample group is needed. Our study was designed to include sequencing data of 960 individuals, however, due to technical difficulties we were only able to include 615 samples.
Conclusions
Our study does not support the hypothesis that rare, presumably functional allelic variants in the tested genes influence urinary Ca2+ excretion. Although an association of CLDN14 with urinary Ca2+ excretion was observed, this finding didn't reach statistical significance. Yet, our data combined with the previous GWAS, recent human and animal data suggest a potential role for CLDN14 in urinary Ca2+ excretion. Further studies of CLDN14 are required to study its contribution to urinary Ca2+ excretion and calcium-based kidney stone disease.
Supporting Information
Novel SNPs identified in low and high urinary Ca2+ excretion cohorts.
(DOCX)
Funding Statement
This study was funded by the National Institute for Health, PO1 grant DK070756 (to D.B.M, M.R.P. and G.C.C.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Pearle MS, Calhoun EA, Curhan GC (2005) Urologic diseases in America project: urolithiasis. J Urol 173: 848–857. [DOI] [PubMed] [Google Scholar]
- 2. Yoshida O, Okada Y (1990) Epidemiology of urolithiasis in Japan: a chronological and geographical study. Urol Int 45: 104–111. [DOI] [PubMed] [Google Scholar]
- 3. Stamatelou KK, Francis ME, Jones CA, Nyberg LM, Curhan GC (2003) Time trends in reported prevalence of kidney stones in the United States: 1976–1994. Kidney Int 63: 1817–1823. [DOI] [PubMed] [Google Scholar]
- 4. Worcester EM, Coe FL (2010) Clinical practice. Calcium kidney stones. N Engl J Med 363: 954–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Attanasio M (2011) The genetic components of idiopathic nephrolithiasis. Pediatr Nephrol 26: 337–346. [DOI] [PubMed] [Google Scholar]
- 6. Curhan GC, Willett WC, Rimm EB, Stampfer MJ (1993) A prospective study of dietary calcium and other nutrients and the risk of symptomatic kidney stones. N Engl J Med 328: 833–838. [DOI] [PubMed] [Google Scholar]
- 7. Curhan GC, Willett WC, Speizer FE, Spiegelman D, Stampfer MJ (1997) Comparison of dietary calcium with supplemental calcium and other nutrients as factors affecting the risk for kidney stones in women. Ann Intern Med 126: 497–504. [DOI] [PubMed] [Google Scholar]
- 8. Curhan GC, Willett WC, Knight EL, Stampfer MJ (2004) Dietary factors and the risk of incident kidney stones in younger women: Nurses' Health Study II. Arch Intern Med 164: 885–891. [DOI] [PubMed] [Google Scholar]
- 9. Borghi L, Meschi T, Amato F, Briganti A, Novarini A, et al. (1996) Urinary volume, water and recurrences in idiopathic calcium nephrolithiasis: a 5-year randomized prospective study. J Urol 155: 839–843. [PubMed] [Google Scholar]
- 10. Taylor EN, Stampfer MJ, Curhan GC (2005) Obesity, weight gain, and the risk of kidney stones. JAMA 293: 455–462. [DOI] [PubMed] [Google Scholar]
- 11. Curhan GC, Willett WC, Rimm EB, Stampfer MJ (1997) Family history and risk of kidney stones. J Am Soc Nephrol 8: 1568–1573. [DOI] [PubMed] [Google Scholar]
- 12. Moe OW, Bonny O (2005) Genetic hypercalciuria. J Am Soc Nephrol 16: 729–745. [DOI] [PubMed] [Google Scholar]
- 13. Wrong OM, Norden AG, Feest TG (1994) Dent's disease; a familial proximal renal tubular syndrome with low-molecular-weight proteinuria, hypercalciuria, nephrocalcinosis, metabolic bone disease, progressive renal failure and a marked male predominance. QJM 87: 473–493. [PubMed] [Google Scholar]
- 14. Pollak MR, Brown EM, Estep HL, McLaine PN, Kifor O, et al. (1994) Autosomal dominant hypocalcaemia caused by a Ca(2+)-sensing receptor gene mutation. Nat Genet 8: 303–307. [DOI] [PubMed] [Google Scholar]
- 15. Simon DB, Lu Y, Choate KA, Velazquez H, Al-Sabban E, et al. (1999) Paracellin-1, a renal tight junction protein required for paracellular Mg2+ resorption. Science 285: 103–106. [DOI] [PubMed] [Google Scholar]
- 16. Hoenderop JG, van Leeuwen JP, van der Eerden BC, Kersten FF, van der Kemp AW, et al. (2003) Renal Ca2+ wasting, hyperabsorption, and reduced bone thickness in mice lacking TRPV5. J Clin Invest 112: 1906–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Maher B (2008) Personal genomes: The case of the missing heritability. Nature 456: 18–21. [DOI] [PubMed] [Google Scholar]
- 18. Romeo S, Pennacchio LA, Fu Y, Boerwinkle E, Tybjaerg-Hansen A, et al. (2007) Population-based resequencing of ANGPTL4 uncovers variations that reduce triglycerides and increase HDL. Nat Genet 39: 513–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fahmi S, Yang C, Esmail S, Hobbs HH, Cohen JC (2008) Functional characterization of genetic variants in NPC1L1 supports the sequencing extremes strategy to identify complex trait genes. Hum Mol Genet 17: 2101–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, et al. (2008) Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 40: 1461–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ji W, Foo JN, O'Roak BJ, Zhao H, Larson MG, et al. (2008) Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat Genet 40: 592–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Curhan GC, Taylor EN (2008) 24-h uric acid excretion and the risk of kidney stones. Kidney Int 73: 489–496. [DOI] [PubMed] [Google Scholar]
- 23. Li H, Durbin R (2010) Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26: 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rivas MA, Beaudoin M, Gardet A, Stevens C, Sharma Y, et al. (2011) Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat Genet 43: 1066–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Consortium GP (2010) A map of human genome variation from population-scale sequencing. Nature 467: 1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, et al. (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43: 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Thorleifsson G, Holm H, Edvardsson V, Walters GB, Styrkarsdottir U, et al. (2009) Sequence variants in the CLDN14 gene associate with kidney stones and bone mineral density. Nat Genet 41: 926–930. [DOI] [PubMed] [Google Scholar]
- 28. Gong Y, Renigunta V, Himmerkus N, Zhang J, Renigunta A, et al. (2012) Claudin-14 regulates renal Ca(+)(+) transport in response to CaSR signalling via a novel microRNA pathway. EMBO J 31: 1999–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Konrad M, Schaller A, Seelow D, Pandey AV, Waldegger S, et al. (2006) Mutations in the tight-junction gene claudin 19 (CLDN19) are associated with renal magnesium wasting, renal failure, and severe ocular involvement. Am J Hum Genet 79: 949–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hou J, Renigunta A, Gomes AS, Hou M, Paul DL, et al. (2009) Claudin-16 and claudin-19 interaction is required for their assembly into tight junctions and for renal reabsorption of magnesium. Proc Natl Acad Sci U S A 106: 15350–15355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Toka HR, Al-Romaih K, Koshy JM, Dibartolo S, Kos CH, et al. (2012) Renal tubule-specific calcium-sensing receptor deficient mice display PTH-independent hypocalciuria. J Am Soc Nephrol [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Melnick JZ, Preisig PA, Moe OW, Srere P, Alpern RJ (1998) Renal cortical mitochondrial aconitase is regulated in hypo- and hypercitraturia. Kidney Int 54: 160–165. [DOI] [PubMed] [Google Scholar]
- 33. Pearce SH, Williamson C, Kifor O, Bai M, Coulthard MG, et al. (1996) A familial syndrome of hypocalcemia with hypercalciuria due to mutations in the calcium-sensing receptor. N Engl J Med 335: 1115–1122. [DOI] [PubMed] [Google Scholar]
- 34. Tosukhowong P, Tungsanga K, Phongudom S, Sriboonlue P (2005) Effects of potassium-magnesium citrate supplementation on cytosolic ATP citrate lyase and mitochondrial aconitase activity in leukocytes: a window on renal citrate metabolism. Int J Urol 12: 140–144. [DOI] [PubMed] [Google Scholar]
- 35. Hou J, Rajagopal M, Yu AS (2013) Claudins and the kidney. Annu Rev Physiol 75: 479–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Breiderhoff T, Himmerkus N, Stuiver M, Mutig K, Will C, et al. (2012) Deletion of claudin-10 (Cldn10) in the thick ascending limb impairs paracellular sodium permeability and leads to hypermagnesemia and nephrocalcinosis. Proc Natl Acad Sci U S A 109: 14241–14246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dimke H, Desai P, Borovac J, Lau A, Pan W, et al. (2013) Activation of the Ca(2+)-sensing receptor increases renal claudin-14 expression and urinary Ca(2+) excretion. Am J Physiol Renal Physiol 304: F761–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lloyd SE, Pearce SH, Fisher SE, Steinmeyer K, Schwappach B, et al. (1996) A common molecular basis for three inherited kidney stone diseases. Nature 379: 445–449. [DOI] [PubMed] [Google Scholar]
- 39. Uchida S (2000) Physiological role of CLC-K1 chloride channel in the kidney. Nephrol Dial Transplant 15 Suppl 6: 14–15. [DOI] [PubMed] [Google Scholar]
- 40. Simon DB, Bindra RS, Mansfield TA, Nelson-Williams C, Mendonca E, et al. (1997) Mutations in the chloride channel gene, CLCNKB, cause Bartter's syndrome type III. Nat Genet 17: 171–178. [DOI] [PubMed] [Google Scholar]
- 41. Rendina D, Mossetti G, De Filippo G, Cioffi M, Strazzullo P (2006) Fibroblast growth factor 23 is increased in calcium nephrolithiasis with hypophosphatemia and renal phosphate leak. J Clin Endocrinol Metab 91: 959–963. [DOI] [PubMed] [Google Scholar]
- 42. Thomee C, Schubert SW, Parma J, Le PQ, Hashemolhosseini S, et al. (2005) GCMB mutation in familial isolated hypoparathyroidism with residual secretion of parathyroid hormone. J Clin Endocrinol Metab 90: 2487–2492. [DOI] [PubMed] [Google Scholar]
- 43. Yoshida T, Fujimori T, Nabeshima Y (2002) Mediation of unusually high concentrations of 1,25-dihydroxyvitamin D in homozygous klotho mutant mice by increased expression of renal 1alpha-hydroxylase gene. Endocrinology 143: 683–689. [DOI] [PubMed] [Google Scholar]
- 44. Karim Z, Gerard B, Bakouh N, Alili R, Leroy C, et al. (2008) NHERF1 mutations and responsiveness of renal parathyroid hormone. N Engl J Med 359: 1128–1135. [DOI] [PubMed] [Google Scholar]
- 45. Palmada M, Poppendieck S, Embark HM, van de Graaf SF, Boehmer C, et al. (2005) Requirement of PDZ domains for the stimulation of the epithelial Ca2+ channel TRPV5 by the NHE regulating factor NHERF2 and the serum and glucocorticoid inducible kinase SGK1. Cell Physiol Biochem 15: 175–182. [DOI] [PubMed] [Google Scholar]
- 46. Simon DB, Karet FE, Hamdan JM, DiPietro A, Sanjad SA, et al. (1996) Bartter's syndrome, hypokalaemic alkalosis with hypercalciuria, is caused by mutations in the Na-K-2Cl cotransporter NKCC2. Nat Genet 13: 183–188. [DOI] [PubMed] [Google Scholar]
- 47. Seidler U, Singh A, Chen M, Cinar A, Bachmann O, et al. (2009) Knockout mouse models for intestinal electrolyte transporters and regulatory PDZ adaptors: new insights into cystic fibrosis, secretory diarrhoea and fructose-induced hypertension. Exp Physiol 94: 175–179. [DOI] [PubMed] [Google Scholar]
- 48. Parkinson DB, Thakker RV (1992) A donor splice site mutation in the parathyroid hormone gene is associated with autosomal recessive hypoparathyroidism. Nat Genet 1: 149–152. [DOI] [PubMed] [Google Scholar]
- 49. Ba J, Brown D, Friedman PA (2003) Calcium-sensing receptor regulation of PTH-inhibitable proximal tubule phosphate transport. Am J Physiol Renal Physiol 285: F1233–1243. [DOI] [PubMed] [Google Scholar]
- 50. Simon DB, Karet FE, Rodriguez-Soriano J, Hamdan JH, DiPietro A, et al. (1996) Genetic heterogeneity of Bartter's syndrome revealed by mutations in the K+ channel, ROMK. Nat Genet 14: 152–156. [DOI] [PubMed] [Google Scholar]
- 51. Simon DB, Nelson-Williams C, Bia MJ, Ellison D, Karet FE, et al. (1996) Gitelman's variant of Bartter's syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat Genet 12: 24–30. [DOI] [PubMed] [Google Scholar]
- 52. Okamoto N, Aruga S, Matsuzaki S, Takahashi S, Matsushita K, et al. (2007) Associations between renal sodium-citrate cotransporter (hNaDC-1) gene polymorphism and urinary citrate excretion in recurrent renal calcium stone formers and normal controls. Int J Urol 14: 344–349. [DOI] [PubMed] [Google Scholar]
- 53. Bergeron MJ, Clemencon B, Hediger MA, Markovich D (2013) SLC13 family of Na(+)-coupled di- and tri-carboxylate/sulfate transporters. Mol Aspects Med 34: 299–312. [DOI] [PubMed] [Google Scholar]
- 54. Nota B, Struys EA, Pop A, Jansen EE, Fernandez Ojeda MR, et al. (2013) Deficiency in SLC25A1, encoding the mitochondrial citrate carrier, causes combined D-2- and L-2-hydroxyglutaric aciduria. Am J Hum Genet 92: 627–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dawson PA, Russell CS, Lee S, McLeay SC, van Dongen JM, et al. (2010) Urolithiasis and hepatotoxicity are linked to the anion transporter Sat1 in mice. J Clin Invest 120: 706–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Heneghan JF, Akhavein A, Salas MJ, Shmukler BE, Karniski LP, et al. (2010) Regulated transport of sulfate and oxalate by SLC26A2/DTDST. Am J Physiol Cell Physiol 298: C1363–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wang Z, Wang T, Petrovic S, Tuo B, Riederer B, et al. (2005) Renal and intestinal transport defects in Slc26a6-null mice. Am J Physiol Cell Physiol 288: C957–965. [DOI] [PubMed] [Google Scholar]
- 58. Chau H, El-Maadawy S, McKee MD, Tenenhouse HS (2003) Renal calcification in mice homozygous for the disrupted type IIa Na/Pi cotransporter gene Npt2. J Bone Miner Res 18: 644–657. [DOI] [PubMed] [Google Scholar]
- 59. Ichikawa S, Sorenson AH, Imel EA, Friedman NE, Gertner JM, et al. (2006) Intronic deletions in the SLC34A3 gene cause hereditary hypophosphatemic rickets with hypercalciuria. J Clin Endocrinol Metab 91: 4022–4027. [DOI] [PubMed] [Google Scholar]
- 60. Zhang Z, Liu KX, He JW, Fu WZ, Yue H, et al. (2012) Identification of two novel mutations in the SLC4A1 gene in two unrelated Chinese families with distal renal tubular acidosis. Arch Med Res 43: 298–304. [DOI] [PubMed] [Google Scholar]
- 61. Alper SL, Darman RB, Chernova MN, Dahl NK (2002) The AE gene family of Cl/HCO3− exchangers. J Nephrol 15 Suppl 5: S41–53. [PubMed] [Google Scholar]
- 62. Loh NY, Bentley L, Dimke H, Verkaart S, Tammaro P, et al. (2013) Autosomal dominant hypercalciuria in a mouse model due to a mutation of the epithelial calcium channel, TRPV5. PLoS One 8: e55412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Suzuki Y, Pasch A, Bonny O, Mohaupt MG, Hediger MA, et al. (2008) Gain-of-function haplotype in the epithelial calcium channel TRPV6 is a risk factor for renal calcium stone formation. Hum Mol Genet 17: 1613–1618. [DOI] [PubMed] [Google Scholar]
- 64. Gudbjartsson DF, Holm H, Indridason OS, Thorleifsson G, Edvardsson V, et al. (2010) Association of variants at UMOD with chronic kidney disease and kidney stones-role of age and comorbid diseases. PLoS Genet 6: e1001039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ruggiero M, Pacini S, Amato M, Aterini S, Chiarugi V (1999) Association between vitamin D receptor gene polymorphism and nephrolithiasis. Miner Electrolyte Metab 25: 185–190. [DOI] [PubMed] [Google Scholar]
- 66. Mayan H, Munter G, Shaharabany M, Mouallem M, Pauzner R, et al. (2004) Hypercalciuria in familial hyperkalemia and hypertension accompanies hyperkalemia and precedes hypertension: description of a large family with the Q565E WNK4 mutation. J Clin Endocrinol Metab 89: 4025–4030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Novel SNPs identified in low and high urinary Ca2+ excretion cohorts.
(DOCX)