

Abstract
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease of motor neurons. Single-nucleotide polymorphism rs3849942 is associated with ALS, tagging a hexanucleotide repeat mutation in the C9orf72 gene. It is possible that there is more than 1 disease-causing genetic variation at this locus, in which case association might remain after removal of cases carrying the mutation. DNA from patients with ALS was therefore tested for the mutation. Genome-wide association testing was performed first using all samples, and then restricting the analysis to samples not carrying the mutation. rs3849942 and rs903603 were strongly associated with ALS when all samples were included (rs3849942, p = [3 × 2] × 10−6, rank 7/442,057; rs903603, p = [7 × 6] × 10−8, rank 2/442,057). Removal of the mutation-carrying cases resulted in loss of association for rs3849942 (p = [2 × 6] × 10−3, rank 1225/442,068), but had little effect on rs903603 (p = [1 × 9] × 10−5, rank 8/442,068). Those with a risk allele of rs903603 had an excess of apparent homozygosity for wild type repeat alleles, consistent with polymerase chain reaction failure of 1 allele because of massive repeat expansion. These results indicate residual association at the C9orf72 locus suggesting a second disease-causing repeat mutation.
Keywords: Amyotrophic lateral sclerosis, Genetics, C9orf72, Hexanucleotide repeat mutation, GWAS, Residual association
1. Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease of motor neurons that causes relentless paralysis, with death occurring within 2–5 years. The lifetime prevalence of ALS is 1 in 300 (Johnston et al., 2006) with peak onset at approximately age 60 years, and an increased incidence in men (Chio et al., 2009). Genetic studies of ALS have begun to yield insights, with 10 moderate to high penetrance Mendelian risk genes now identified for adult-onset ALS, with or without frontotemporal dementia (FTD): SOD1 (Rosen et al., 1993), TARDBP (Gitcho et al., 2008; Kabashi et al., 2008; Sreedharan et al., 2008), FUS (Kwiatkowski et al., 2009; Vance et al., 2009), ANG (Greenway et al., 2006), OPTN (Maruyama et al., 2010), VCP (Johnson et al., 2010), C9orf72 (DeJesus-Hernandez et al., 2011; Renton et al., 2011), FIG4 (Chow et al., 2009), UBQLN2 (Deng et al., 2011), and PFN1 (Wu et al., 2012). Other variations acting to increase risk might include intermediate expansion of the ATXN2 gene (Elden et al., 2010), ELP3 intron 10 variation, a UNC13A single-nucleotide polymorphism (SNP) (van Es et al., 2009), and indels in the NEFH tail domain (Al-Chalabi et al., 1999; Figlewicz et al., 1994).
In approximately 5% of cases, a family history of ALS is recorded, with the remaining cases labeled sporadic (Byrne and Hardiman, 2010), but this distinction is artificial, with an increased risk of other neurodegenerative diseases including Parkinson's disease, and frontotemporal dementia (Byrne and Hardiman, 2010; Millecamps et al., 2010), and an increased risk of ALS in relatives of those with apparently sporadic disease (Hanby et al., 2011). Furthermore, variants in every Mendelian ALS gene have also been reported in those with apparently sporadic ALS (Al-Chalabi and Lewis, 2011; van Blitterswijk et al., 2012), and the heritability of apparently sporadic ALS has been estimated at 61% (Al-Chalabi et al., 2010).
Genome-wide association studies in ALS and FTD recently identified a locus on chromosome 9 within a known linkage peak for ALS-FTD (Laaksovirta et al., 2010; Shatunov et al., 2010; van Es et al., 2009). The disease-causing mutation has now been identified as a massive expansion mutation of a hexanucleotide repeat between noncoding exons 1A and 1B of C9orf72 (DeJesus-Hernandez et al., 2011; Renton et al., 2011). Unaffected individuals have 2–23 repeats of the (GGGGCC)n hexanucleotide microsatellite, though the pathological repeat mutation might be several hundred repeats. The mutation is interesting in manifesting with at least 4 phenotypes: ALS, ALS-FTD, FTD, and normality.
Although 3 different genome-wide association studies all identified rs3849942 as the most associated SNP at the C9orf72 locus, and it is clear that this is in strong linkage disequilibrium with the repeat mutation (Smith et al., 2013), the UK subpopulation showed stronger genome-wide significant association at a different SNP, rs903603. The most parsimonious explanation is that rs903603 and rs3849942 are in strong linkage disequilibrium and both tag the pathological expansion. If that is the case, one would expect that removal of cases with the repeat mutation from the analysis would obliterate the association at both SNPs. We therefore tested this hypothesis in the UK population.
2. Methods
2.1. Sample collection
Whole blood samples from the Motor Neuron Disease (MND) DNA Bank were used. Selection was based on no family history of ALS, being of white European ancestry, and disease onset January 2002 or after. Control samples were obtained from the Depression Case Control (DeCC) study, the Bipolar Affective Case Control Study (BACCS), from Panos Deloukas of the Wellcome Trust Sanger Institute (Cambridge, UK), and the British 1958 birth cohort DNA collection. This project was ethically approved.
2.2. Sample preparation
DNA was extracted from blood samples by standard methods within 1 week of bleeding and stored at the UK DNA Banking Network in Manchester. Samples were bar-coded in a tracking system to reduce risk of clerical error.
2.3. Genotyping
Genotyping and quality control was carried out as described previously (Shatunov et al., 2010), but in brief, genome-wide association was performed using various Illumina DNA microarrays followed by standard quality control and association testing using PLINK (Purcell et al., 2007).
The mutation was identified using repeat-primed polymerase chain reaction (PCR) as described (DeJesus-Hernandez et al., 2011). Nonmutation repeat length for alleles of the hexanucleotide repeat was quantified using amplified fragment length polymorphism (AFLP), using fluorescently-labeled primers as detailed previously (DeJesus-Hernandez et al., 2011). Both analyses were performed using an automated 3130XL capillary electrophoresis-based Genetic Analyzer and GeneMapper v4.0 software (Applied Biosystems).
2.4. Statistical quality control
Statistical quality control excluded individuals with incongruent sex, those marked for exclusion by the genotyping institution, those with low genotyping per individual <0.02, those with phenotype missingness <0.02, those with low heterozygosity (p < 0.05), those exhibiting identity by descent sharing >5% of alleles with another participant, and those not of white European ancestry.
Excluded SNP markers were those with a minor allele frequency less than 0.015, SNP missingness <0.02, significant departure from Hardy-Weinberg equilibrium (p < 0.001), those marked for exclusion by the genotyping institution, and those on chromosome X.
2.5. Phasing and imputation analyses
Phasing and imputation was completed using 1000 Genomes v2.20101123 autosomal release (http://www.1000genomes.org/), performed using MaCH 1.0.18 (http://www.sph.umich.edu/csg/abecasis/MACH/index.html) and minimac (http://genome.sph.umich.edu/wiki/Minimac). We performed association analysis on imputed data using ProbABEL (http://www.genabel.org/packages/ProbABEL), mach2dat (http://genome.sph.umich.edu/wiki/Mach2dat:_Association_with_MACH_output), and PLINK v1.07 (http://pngu.mgh.harvard.edu/~purcell/plink/index.shtml), after creating input files using the GenGen (http://www.openbioinformatics.org/gengen/) conversion tool. For GenGen, we set the r2 threshold as 0.3 and, based on genotype posterior probabilities derived in MaCH, a quality control threshold of 0.9.
2.6. Association and haplotype analysis
To test for residual association and identify SNPs not related to the mutation we used logistic regression, modeling case-control comparisons with sex as a covariate, stratified by the presence or absence of the repeat mutation.
We set the Bonferroni-corrected threshold of statistical significance at 1.13 × 10−7, and the genome-wide threshold as 5 × 10−8. We used the R statistical package to construct Manhattan and Q-Q Plots, LocusZoom (http://csg.sph.umich.edu/locuszoom/) for regional Manhattan plots, and Haploview for haplotype block maps (http://www.broadinstitute.org/scientific-community/science/programs/medical-and-population-genetics/haploview/haploview). To correct for genomic inflation, p values were adjusted by the median χ² statistic (genomic control factor).
For linear regression analysis of the nonmutated microsatellite repeat lengths as a quantitative variable we used PLINK v1.07 and IBM SPSS 20. For haplotype analyses we used PLINK v1.07 sliding windows between 2 and 100 to explore significantly associated haplotypes using the phased imputed dataset. The prevalence of the most associated haplotypes were refined in Haploview and tested for their association with ALS using an omnibus haplotype association analysis.
We ran all genetic tests under additive, dominant, and recessive models, with little difference in results between dominant and additive models. We have reported the additive model for association of SNP alleles with the repeat mutation, and a dominant model (using the larger allele) for the analysis of repeat lengths.
2.7. Sequencing C9orf72
Four hundred eighty DNA samples of patients with sporadic ALS were pooled in sets of 8. Each pool was tagged with 1 of 12 identifying DNA sequences and then combined into a further pool containing 1 representative of each identifying sequence. Thus each final sample for sequencing contained the DNA of 96 different individuals which could be resolved down to a group of 8. Samples were sequenced using Illumina GAIIx platform generating 38-base pair paired-end, indexed reads. Preliminary analysis and quality control of reads was carried out using Illumina's CASAVA v1.7 software and output files contained all filtered, but unaligned, sequences for each sample stored in a simple exportable FASTQ format. Alignment of short reads against reference messengerRNA sequence (uc003zqq.2, C9orf72, length = 3233) and variations calling was done using Maq 0.7.1 mapping short DNA sequencing reads and calling variants using mapping quality scores with default parameters (Li et al., 2008).
2.8. Sequencing the repeat
Forward (GGTTTAGGAGGTGTGTGTTTTTGT) and reverse primers (CCAGCTTCGGTCAGAGAAAT) were designed to create a 424-base pair amplicon (64% GC-rich) covering the repeat sequence. A touchdown PCR protocol (see Supplementary Section S2) was used to amplify the region of interest followed by gel electrophoresis to inspect the amplicon length.
Samples were purified and analyzed using an ABI 3130xl Genetic Analyzer (Applied Biosystems) with the BigDye Terminator v1.1 Cycle Sequencing Kit. ABI's Sequencing Analysis 5.3.1 software was used to analyze base calls and identify repeat patterns.
3. Results
3.1. C9orf72 genotyping
There were 632 case and 4519 control samples. After quality control there were 599 case and 4136 control samples. There were 585,919 SNPs; after quality control 442,057. Thirty-nine cases tested positive for the mutation; the remaining samples had repeat lengths of less than 23 (Supplementary Section 1; Supplementary Table 1).
3.2. Residual association
Genomic control factor λGC was 1.00–1.03 for all analyses. Genome-wide association of all UK case versus control samples confirmed the findings of the original study (Shatunov et al., 2010), with no new associations (Supplementary Fig. 1 and Supplementary Table 2). The top 3 SNPs were rs10967976 (p = 6.164 × 10−8; odds ratio [OR], 1.41), rs903603 (p = 7.548 × 10−8; OR, 0.71), and rs10812611 (p = 9.819 × 10−8; OR, 1.4), all at 9p21.2. Rs903603 was the most associated SNP in the previous independent analysis (Shatunov et al., 2010). In that study rs10967976 and rs10812611 had been removed for compatibility with the other platforms used in the joint analysis. To confirm that the observed association signal was a result of the mutation and that there were no new associations, we performed association testing excluding the 39 mutation positive cases (Supplementary Figs. 2 and 3, and Supplementary Table 3). There was a residual association at 9p21 with the same top 3 SNPs; rs903603 (p = 1.87e-05; OR, 0.76), rs10967976 (p = 3.63e-05; OR, 1.31), and rs10812611 (p = 5.02e-05; OR, 1.30), suggesting that despite being at the disease locus, they are not tagging the pathological repeat mutation. This is reflected in the change in p value ranks with the most associated SNP from our original study, rs903603, remaining 1 of the 10 most associated SNPs after removal of cases with the mutation, compared with the SNP tagging the repeat mutation, rs3849942, dropping from 7th to 1225th place, as expected (Table 1).
Table 1.
Change in rank by p value for residual and mutation-specific SNPs
| SNP | With hexanucleotide repeat mutation |
Without hexanucleotide repeat mutation |
||||
|---|---|---|---|---|---|---|
| Rank | p | Odds ratio (allele) | Rank | p | Odds ratio (allele) | |
| rs10967976 | First | 6.16E-08 | 1.41 (G) | 23rd | 3.63E-05 | 1.31 (G) |
| rs903603 | Second | 7.55E-08 | 0.71 (A) | Eighth | 1.87E-05 | 0.76 (A) |
| rs10812611 | Third | 9.82E-08 | 1.4 (G) | 34th | 5.02E-05 | 1.3 (G) |
| rs3849942 | Seventh | 3.28E-06 | 1.39 (A) | 1225th | 2.57E-03 | 1.25 (A) |
| rs2814707 | Ninth | 5.00E-06 | 1.38 (A) | 1562nd | 3.30E-03 | 1.24 (A) |
Key: SNP, single-nucleotide polymorphism.
We confirmed that the mutation tagging SNP rs3849942 is strongly associated with ALS when only samples positive for the mutation were tested against controls (p = 9.77 × 10−12; OR, 5.225; rank = 1), and rs903603 was not as strongly associated as rs3849942 (p = 7.73 × 10−7; OR, 0.24; rank = 22) (Supplementary Fig. 4). A Q-Q plot (Supplementary Fig. 5) did not show evidence of inflation of the test statistic.
3.3. Residual association and mutation-specific haplotypes
To explore linkage disequilibrium in the region, we phased and imputed SNPs at C9orf72 and the surrounding region (chr9:27303051–27750375) using the 1000 Genomes Project reference panel. Haplotype association analyses and refinement of haplotype blocks in Haploview identified a 79-SNP haplotype in 100% of the cases with the mutation (n = 39), of which 72 SNPs were from an 82-SNP haplotype published previously (Smith et al., 2013). Using the same method we also identified a distinct 11-SNP haplotype present in 76% of the samples without the mutation (Supplementary Fig. 6).
3.4. Repeat length and risk allelic distribution
We examined the relationship between the hexanucleotide repeat length of phased cases without the mutation (n = 448) and the allelic frequency and distribution of ALS-associated SNPs to explore the linkage disequilibrium pattern of the locus. The mutation-tagging SNP rs3849942 risk allele was associated with increased hexanucleotide repeat numbers for the larger allele in linear regression (β = 5.711; p = 3.02 × 10−84) (Supplementary Table 4). Allele frequency distributions of the repeat length stratified by rs3849942 alleles showed a marked difference between those with 6 or less against 7 or more (Fig. 1). Nonmutation samples with 7 or more repeats (n = 208) showed the strongest genome-wide association with ALS when compared with control samples (n = 4142; OR, 4.99; p = 1.35 × 10−43) (Supplementary Table 5).
Fig. 1.
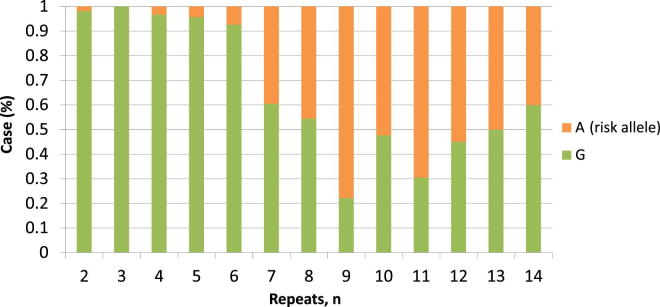
Relationship between rs3849942 alleles and repeat length in nonmutation cases. The risk allele is strongly associated with longer repeat lengths, suggesting it might lie on a haplotype promoting repeat length instability.
A similar relationship between risk alleles and hexanucleotide repeat length was observed for the SNPs showing residual association. Repeat numbers greater than 2 (the minimum length) for the larger allele were significantly associated with risk alleles at all of the 3 SNPs showing residual association (Fig. 2). Modeling repeat length as a quantitative variable in a linear regression confirmed that these SNPs were significant predictors of having greater than 2 repeats (example SNP rs10812611; β = 5.835; p = 1.50 × 10−52) (Supplementary Table 4). Again, association testing of samples with repeat length greater than 2 (n = 385) against all controls (n = 4142) showed that SNPs showing residual association were most significantly associated with ALS; rs903603: OR, 0.39; p = 2.21 × 10−28; rs10812611: OR, 2.78; p = 6.96 × 10−33; and rs10967976: OR, 2.82; p = 1.19 × 10−33 (Supplementary Table 6).
Fig. 2.

The relationship between hexanucleotide allele repeat length and single-nucleotide polymorphisms (SNPs) showing residual association at the C9orf72 locus SNP rs903603 (A), rs10967976 (B), and rs10812611 (C). Just as for the relationship for rs3849942 shown in Fig. 1, the risk allele for SNPs on the alternative risk haplotype is overwhelmingly likely to be associated with repeat sizes greater than 2.
The identified 11-SNP haplotype for the residual association SNPs was found in 100% of the samples with greater than 2 repeats for larger allele (71% in control samples). Extension of the haplotype to include an additional SNP, rs2492816, was very specific for longer repeat lengths. This 12-SNP haplotype was strongly associated with ALS in the sample set excluding cases with the mutation, or with 2 or less repeats (χ² = 114; p = 1.56 × 10−24; Fig. 3). This haplotype was not significantly associated with ALS in cases with 2 hexanucleotide repeats and was not found at all in cases with the pathological repeat mutation.
Fig. 3.

Twelve single-nucleotide polymorphism haplotypes for case samples with repeat length greater than size 2.
An omnibus association analysis comparing mutation-specific and residual association haplotypes in those with the mutation and those with greater than 2 repeats on their larger allele supported the hypothesis that the alleles showing residual association at C9orf72 are significantly associated with ALS, but not by association with the pathological repeat mutation (Table 2).
Table 2.
Association analyses of haplotypes with ALS in mutation and case samples with hexanucleotide repeat length >2
| Haplotype | Haplotype p-value for association with ALS |
|
|---|---|---|
| Mutation-specific | Residual | |
| Mutation cases (n = 39) | 7.53 × 10−14 | 1.30 × 10−8 |
| Cases with hexanucleotide repeat length >2 (n = 385) | 9.87 × 10−9 | 5.91 × 10−23 |
Key: ALS, amyotrophic lateral sclerosis.
Values in bold highlight association between mutation groups and haplotypes.
3.5. Apparent homozygosity for larger hexanucleotide repeats associates with the risk allele of rs903603
The association of risk alleles with greater than 2 hexanucleotide repeats suggests that residual association SNPs might predispose to instability, and therefore a repeat mutation, as has been postulated previously for the rs3849942 haplotypic background. Thus, 1 possible explanation of the residual association is an interrupted or alternative repeat sequence, also pathologically expanded, but not detected by repeat-primed PCR targeted at (GGGGCC)n (Fig. 4). In that situation one would expect a similar problem to that reported by DeJesus-Hernandez et al. (2011): that cases appear homozygous because their larger repeat allele is too expanded to amplify. Most cases (93.8%; 15/16 cases) with greater than 2 repeats and the rs903603 risk allele were apparently homozygous for the hexanucleotide repeat allele, breaking Hardy–Weinberg equilibrium. In comparison, 66% (150/266) of cases with the same risk allele were heterozygous for the repeat and we found only 4% (3/83) of cases with the risk allele homozygous for 2 repeats (Table 3). We did not observe this >2 repeat homozygosity bias for any other SNPs. For example, 37% of cases with the mutation-specific risk alleles had homozygosity for repeat alleles of length greater than 2, which satisfied Hardy–Weinberg equilibrium.
Fig. 4.

Repeat-primed polymerase chain reaction results. Repeat-primed polymerase chain reaction of example case samples showing a small expansion in the nonpathological size range (A), and a sample known to have a pathological (GGGGCC)n expansion mutation (B).
Table 3.
Frequency of cases by residual rs10967976 alleles stratified by repeat length and zygosity
| Repeat zygosity | Nonrisk allele A (%) | Risk allele G (%) |
|---|---|---|
| Heterozygous | 76 (34) | 150 (66) |
| Homozygous 2 repeats | 80 (96) | 3 (4) |
| Homozygous (repeats >2) | 1 (5) | 18 (95) |
3.6. Trimodal pattern of repeats
We found a trimodal pattern of repeat allele sizes, where the occurrence of 2, 5, or 8 repeats was significantly more frequent than other repeat numbers (Supplementary Figs. 7 and 8). These findings suggest some repeat sizes are more stable than others.
3.7. Sequencing analysis of C9orf72
We identified 2 point mutations, a ketone G/T and an amine A/C substitution, across 31 (n = 272) and 2 (n = 16) pools, respectively. No group analyzed together or separately showed association with the residual or mutation-specific risk haplotypes. There was also no sample showing homozygosity for repeat sizes larger than 2. Thus it is unlikely that the residual association identified is a result of the observed point mutations.
3.8. Sequencing analysis of the C9orf72 repeat
We sequenced the repeat region in 56 samples: 18 cases homozygous for greater than 2 repeats with the risk haplotype showing residual association, 9 homozygous for repeats of size 2, 10 heterozygous for repeat length, and a mixture of non-ALS control samples homozygous for repeat sizes greater than 2 (n = 6), homozygous for repeats of size 2 (n = 7), and heterozygous for repeat length (n = 6). Our previous length and zygosity estimates based on AFLP (Supplementary Fig. 9) and repeat-primed PCR were correct for all these samples.
4. Discussion
We have shown that after removing cases with expansion mutation of the (GGGGCC)n repeat at C9orf72 there remains residual association of SNPs at this locus with ALS. As expected, the SNPs associated with the mutation are no longer associated with ALS in this sample, but rs903603 and SNPs in strong linkage disequilibrium with it are associated, before and after the removal of cases with the mutation. These SNPs showing residual association reside on a distinct haplotype, different from that of the mutation, and like the mutation-tagging SNPs, are also associated with nonpathological expansion of the hexanucleotide repeat, suggesting that they too promote instability of a repeat sequence.
Because the SNPs showing residual association are not associated with the mutation but are associated with ALS, repeat instability, and apparent homozygosity for the repeat, 1 possible explanation is an alternative pathological expansion that is not detected by repeat-primed PCR targeting (GGGGCC)n. Such an alternative repeat sequence might be the result of an interrupted repeat. Interrupted repeat sequences are found in ALS resulting from intermediate expansion of a CAG trinucleotide repeat in the ATXN2 gene, with CAA interrupting the sequence and resulting in an alternative (Corrado et al., 2011). Interrupted sequences are also found in Fragile X syndrome, a condition with a massive pathological repeat expansion (Renčiuk et al., 2009).
Others have reported evidence of pathological C9orf72 hexanucleotide expansions using Southern blot analysis without corresponding detection of repeat-primed PCR in FTD cases, consistent with an alternative repeat sequence (Pickering-Brown et al., 2012).
Current evidence supports the idea that the sheer size of the disease-associated C9orf72 hexanucleotide repeat expansion causes pathology, with no evidence for repeat lengths below 23 being associated with ALS. One would expect that an alternative repeat that is also pathological should also be very large and not readily amplified by PCR. In that case, samples carrying the alternative repeat should appear homozygous for the nonexpanded allele because the pathological allele would appear null using PCR. Consistent with this, we find cases with more than 2 repeats are overwhelmingly likely to appear homozygous.
One factor increasing the likelihood of pathological expansion in transmission to the next generation is the current size of the repeat, and it is particularly interesting that the SNPs showing residual association also strongly associated with alleles showing larger numbers of nonpathological repeats. The C9orf72 region is very GC-rich, known to increase vulnerability toward repeat mutations. Additionally, a familial study examining inheritance of the repeat mutation suggests that larger repeat lengths are indeed more unstable and prone to expansion (Beck et al., 2013).
We found a trimodal pattern of repeat allele sizes with significant departure from a uniform distribution for repeat lengths 2, 5, and 8. This pattern is also apparent in previous publications (DeJesus-Hernandez et al., 2011; Smith et al., 2013), although not explicitly described. The tendency to increase the allele size by 18 nucleotides might be related to the underlying pathological mechanism of expansion, either because the mechanism results in preferential expansion in units of 18 base pairs, or because expansion in other unit sizes is unstable and leads to massive pathological expansion.
A weakness of this study is that we relied on repeat-primed PCR to identify cases with the mutation. It is possible that some cases with the mutation were missed and this explains the residual association. Several factors make this unlikely. First, the mutation is tagged by rs3849942, and as expected, this SNP lost association with ALS after the removal of mutation-positive cases, and rs903603 remained in the top 10 associated SNPs. Second, the frequency of identified mutation cases in our sample is among the highest in the world (Smith et al., 2013), suggesting we are not missing a large proportion of these. Third, we performed a standard PCR of the hexanucleotide repeat and assayed the length polymorphism of the alleles (AFLP); in all cases, there were no heterozygous cases with the expansion, suggesting that the detection method worked. Fourth, in all cases in which sequence data were available, the repeat size on sequencing matched our estimate of size based on AFLP and repeat-primed PCR.
Another explanation for our findings is a substitution or similar mutation, rather than an alternative repeat mutation. This seems unlikely because for several years many groups, including ours, performed sequencing through the region finding no segregating point mutations or indels. Furthermore, the point mutations we identified on sequencing do not occur in individuals carrying the alternative risk haplotype. It does however remain possible that for a small subset of individuals with sporadic ALS, point mutations of C9orf72 are pathogenic, but it is then difficult to explain the association between rs903603 risk alleles and hexanucleotide repeats greater than a length of 2. Nevertheless, we cannot exclude this possibility.
A third possibility is that SNPs showing residual association tag repeat alleles greater than size 2 because such repeats are themselves pathogenic. Case and control samples share the same frequency and distribution of repeat lengths (Rutherford et al., 2012) making this unlikely, but recent evidence suggests that moderate expansion in the number of repeats can actually be pathogenic (Gomez-Tortosa et al., 2013).
We present evidence of residual association at the C9orf72 locus in ALS, not accounted for by the known pathological GGGGCC hexanucleotide repeat expansion but consistent with the hypothesis of an interrupted GGGGCC or alternative sequence repeat. We present a new pathological haplotype associated with ALS at this locus that is associated with having greater than 2 repeats for the larger allele, suggesting this haplotype predisposes to repeat instability. Further investigation including Southern blot analysis of individuals carrying the risk haplotype and sequence analysis of the region will help to identify the cause of the residual association signal we have identified.
Disclosure statement
Ammar Al-Chalabi discloses the following. Personal payments: royalties from the books, The Brain (Oneworld Publications) and Complex Disease Genetics, a Laboratory Manual (Cold Spring Harbor Laboratory Press); institutional payments: consultancy to Biogen Idec and Cytokinetics Inc; grant income: from various charities and governmental organizations. The remaining authors have no conflicts of interest to disclose.
Patients provided ethically approved consent for withdrawal of blood used for genotyping.
Acknowledgements
This work was funded by a Medical Research Council PhD studentship awarded through the MRC Centre for Neurodegeneration Research, by a grant from the Motor Neuron Disease Association of Great Britain and Northern Ireland, and by the European Community's Health Seventh Framework Programme (FP7/2007–2013) 259867 to A.A.C. and P.J.S., and a grant (075615/Z/04/z) from the Welcome Trust to V.L.B.
The authors thank the Motor Neurone Disease Association of Great Britain and Northern Ireland, the Wellcome Trust, the ALS Association, the Angel Fund, and the ALS Therapy Alliance for support. A.A.C. receives salary support from the National Institute for Health Research (NIHR) Dementia Biomedical Research Unit, and C.M.L. from the NIHR Biomedical Research Centre in Mental Health, both at South London and Maudsley NHS Foundation Trust and King's College London. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR, or the Department of Health.
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.neurobiolaging.2013.03.003.
Appendix A. Supplementary data
Supplementary Fig. 1.

Genome-wide association study of all case (599) versus control (4136) samples. The red line is the genome correction threshold (5 × 10−8) and blue line is the Bonferroni threshold (1.13 × 10−7).
Supplementary Fig. 2.

Genome-wide association study of mutation cases removed (560) versus control (4136) samples. The red line is the genome correction threshold (5 × 10−8) and the blue line is the Bonferroni threshold (1.13 × 10−7).
Supplementary Fig. 3.

Q-Q plot for genome-wide association study comparing nonrepeat mutation case (560) versus control (4136) samples.
Supplementary Fig. 4.

Manhattan plot of genome-wide association study comparing repeat mutation case (39) versus control (4136) samples. The red line is the genome correction threshold (5 × 10−8) and the blue line is the Bonferroni threshold (1.13 × 10−7).
Supplementary Fig. 5.

Q-Q plot for genome-wide association study comparing repeat mutation case (39) versus control (4136) samples (λGCof 1.02): 79 single-nucleotide polymorphism mutation-specific haplotype association analysis of mutation case (39) versus control (1182) samples: χ² = 64.62; p = 9.08 × 10−14 and 11 single-nucleotide polymorphism residual haplotype association analysis of nonmutation case (560) versus control (1182) samples: χ² = 15.58; p = 7.90 × 10−5.
Supplementary Fig. 6a.

A seventy-nine single-nucleotide polymorphism haplotype for cases with the pathological expansion (A) and an 11 single-nucleotide polymorphism haplotype for those without (B).
Supplementary Fig. 6b.

Relative frequency of nonmutation case and control samples by the largest repeat length.
Relative frequency of nonrepeat mutation case (n = 239) and control (n = 357) samples by repeat length as an additive model.
Supplementary Fig. 9.
Amplified fragment length polymorphism of an example case showing homozygosity for 5 repeats.
References
- Al-Chalabi A., Andersen P.M., Nilsson P., Chioza B., Andersson J.L., Russ C., Shaw C.E., Powell J.F., Nigel Leigh P. Deletions of the heavy neurofilament subunit tail in amyotrophic lateral sclerosis. Hum. Mol. Genet. 1999;8:157–164. doi: 10.1093/hmg/8.2.157. [DOI] [PubMed] [Google Scholar]
- Al-Chalabi A., Fang F., Hanby M.F., Leigh P.N., Shaw C.E., Ye W., Rijsdijk F. An estimate of amyotrophic lateral sclerosis heritability using twin data. J. Neurol. Neurosurg. Psychiatry. 2010;81:1324–1326. doi: 10.1136/jnnp.2010.207464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Chalabi A., Lewis C.M. Modelling the effects of penetrance and family size on rates of sporadic and familial disease. Hum. Hered. 2011;71:281–288. doi: 10.1159/000330167. [DOI] [PubMed] [Google Scholar]
- Beck J., Poulter M., Hensman D., Rohrer J.D., Mahoney C.J., Adamson G., Campbell T., Uphill J., Borg A., Fratta P., Orrell R.W., Malaspina A., Rowe J., Brown J., Hodges J., Sidle K., Polke J.M., Houlden H., Schott J.M., Fox N.C., Rossor M.N., Tabrizi S.J., Isaacs A.M., Hardy J., Warren J.D., Collinge J., Mead S. Large C9orf72 hexanucleotide repeat expansions are seen in multiple neurodegenerative syndromes and are more frequent than expected in the UK population. Am. J. Hum. Genet. 2013;92:345–353. doi: 10.1016/j.ajhg.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne S., Hardiman O. Familial aggregation in amyotrophic lateral sclerosis. Ann. Neurol. 2010;67:554. doi: 10.1002/ana.21883. [DOI] [PubMed] [Google Scholar]
- Chio A., Mora G., Calvo A., Mazzini L., Bottacchi E., Mutani R. Epidemiology of ALS in Italy: a 10-year prospective population-based study. Neurology. 2009;72:725–731. doi: 10.1212/01.wnl.0000343008.26874.d1. [DOI] [PubMed] [Google Scholar]
- Chow C.Y., Landers J.E., Bergren S.K., Sapp P.C., Grant A.E., Jones J.M., Everett L., Lenk G.M., McKenna-Yasek D.M., Weisman L.S., Figlewicz D., Brown R.H., Meisler M.H. Deleterious variants of FIG4, a phosphoinositide phosphatase, in patients with ALS. Am. J. Hum. Genet. 2009;84:85–88. doi: 10.1016/j.ajhg.2008.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrado L., Mazzini L., Oggioni G., Luciano B., Godi M., Brusco A., D'Alfonso S. ATXN-2 CAG repeat expansions are interrupted in ALS patients. Hum. Genet. 2011;130:575–580. doi: 10.1007/s00439-011-1000-2. [DOI] [PubMed] [Google Scholar]
- DeJesus-Hernandez M., Mackenzie Ian R., Boeve B.F., Boxer A.L., Baker M., Rutherford N.J., Nicholson A.M., Finch N.A., Flynn H., Adamson J., Kouri N., Wojtas A., Sengdy P., Hsiung G.Y., Karydas A., Seeley W.W., Josephs K.A., Coppola G., Geschwind D.H., Wszolek Z.K., Feldman H., Knopman D.S., Petersen R.C., Miller B.L., Dickson D.W., Boylan K.B., Graff-Radford N.R., Rademakers R. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H.X., Chen W., Hong S.T., Boycott K.M., Gorrie G.H., Siddique N., Yang Y., Fecto F., Shi Y., Zhai H., Jiang H., Hirano M., Rampersaud E., Jansen G.H., Donkervoort S., Bigio E.H., Brooks B.R., Ajroud K., Sufit R.L., Haines J.L., Mugnaini E., Pericak-Vance M.A., Siddique T. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 2011;477:211–215. doi: 10.1038/nature10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elden A.C., Kim H.J., Hart M.P., Chen-Plotkin A.S., Johnson B.S., Fang X., Armakola M., Geser F., Greene R., Lu M.M., Padmanabhan A., Clay-Falcone D., McCluskey L., Elman L., Juhr D., Gruber P.J., Rub U., Auburger G., Trojanowski J.Q., Lee V.M., Van Deerlin V.M., Bonini N.M., Gitler A.D. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature. 2010;466:1069–1075. doi: 10.1038/nature09320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figlewicz D.A., Krizus A., Martinoli M.G., Meininger V., Dib M., Rouleau G.A., Julien J.P. Variants of the heavy neurofilament subunit are associated with the development of amyotrophic lateral sclerosis. Hum. Mol. Genet. 1994;3:1757–1761. doi: 10.1093/hmg/3.10.1757. [DOI] [PubMed] [Google Scholar]
- Gitcho M.A., Baloh R.H., Chakraverty S., Mayo K., Norton J.B., Levitch D., Hatanpaa K.J., White C.L., Bigio E.H., Caselli R., Baker M., Al-Lozi M.T., Morris J.C., Pestronk A., Rademakers R., Goate A.M., Cairns N.J. TDP-43 A315T mutation in familial motor neuron disease. Ann. Neurol. 2008;63:535–538. doi: 10.1002/ana.21344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Tortosa E., Gallego J., Guerrero-Lopez R., Marcos A., Gil-Neciga E., Sainz M.J., Diaz A., Franco-Macias E., Trujillo-Tiebas M.J., Ayuso C., Perez-Perez J. C9ORF72 hexanucleotide expansions of 20-22 repeats are associated with frontotemporal deterioration. Neurology. 2013;80:366–370. doi: 10.1212/WNL.0b013e31827f08ea. [DOI] [PubMed] [Google Scholar]
- Greenway M.J., Andersen P.M., Russ C., Ennis S., Cashman S., Donaghy C., Patterson V., Swingler R., Kieran D., Prehn J., Morrison K.E., Green A., Acharya K.R., Brown R.H., Hardiman O. ANG mutations segregate with familial and ‘sporadic’ amyotrophic lateral sclerosis. Nat. Genet. 2006;38:411–413. doi: 10.1038/ng1742. [DOI] [PubMed] [Google Scholar]
- Hanby M.F., Scott K.M., Scotton W., Wijesekera L., Mole T., Ellis C.E., Leigh P.N., Shaw C.E., Al-Chalabi A. The risk to relatives of patients with sporadic amyotrophic lateral sclerosis. Brain. 2011;134:3454–3457. doi: 10.1093/brain/awr248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J.O., Mandrioli J., Benatar M., Abramzon Y., Van Deerlin V.M., Trojanowski J.Q., Gibbs J.R., Brunetti M., Gronka S., Wuu J., Ding J., McCluskey L., Martinez-Lage M., Falcone D., Hernandez D.G., Arepalli S., Chong S., Schymick J.C., Rothstein J., Landi F., Wang Y.D., Calvo A., Mora G., Sabatelli M., Monsurrò M.R., Battistini S., Salvi F., Spataro R., Sola P., Borghero G., Galassi G., Scholz S.W., Taylor J.P., Restagno G., Chiò A., Traynor B.J. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. 2010;68:857–864. doi: 10.1016/j.neuron.2010.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston C., Stanton B., Turner M., Gray R., Blunt A., Butt D., Ampong M.A., Shaw C., Leigh P., Al-Chalabi A. Amyotrophic lateral sclerosis in an urban setting. J. Neurol. 2006;253:1642–1643. doi: 10.1007/s00415-006-0195-y. [DOI] [PubMed] [Google Scholar]
- Kabashi E., Valdmanis P.N., Dion P., Spiegelman D., McConkey B.J., Velde C.V., Bouchard J.-P., Lacomblez L., Pochigaeva K., Salachas F., Pradat P.-F., Camu W., Meininger V., Dupre N., Rouleau G.A. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 2008;40:572–574. doi: 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski T.J., Jr., Bosco D.A., LeClerc A.L., Tamrazian E., Vanderburg C.R., Russ C., Davis A., Gilchrist J., Kasarskis E.J., Munsat T., Valdmanis P., Rouleau G.A., Hosler B.A., Cortelli P., de Jong P.J., Yoshinaga Y., Haines J.L., Pericak-Vance M.A., Yan J., Ticozzi N., Siddique T., McKenna-Yasek D., Sapp P.C., Horvitz H.R., Landers J.E., Brown R.H., Jr. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- Laaksovirta H., Peuralinna T., Schymick J.C., Scholz S.W., Lai S.L., Myllykangas L., Sulkava R., Jansson L., Hernandez D.G., Gibbs J.R., Nalls M.A., Heckerman D., Tienari P.J., Traynor B.J. Chromosome 9p21 in amyotrophic lateral sclerosis in Finland: a genome-wide association study. Lancet Neurol. 2010;9:978–985. doi: 10.1016/S1474-4422(10)70184-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Ruan J., Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama H., Morino H., Ito H., Izumi Y., Kato H., Watanabe Y., Kinoshita Y., Kamada M., Nodera H., Suzuki H., Komure O., Matsuura S., Kobatake K., Morimoto N., Abe K., Suzuki N., Aoki M., Kawata A., Hirai T., Kato T., Ogasawara K., Hirano A., Takumi T., Kusaka H., Hagiwara K., Kaji R., Kawakami H. Mutations of optineurin in amyotrophic lateral sclerosis. Nature. 2010;465:223–226. doi: 10.1038/nature08971. [DOI] [PubMed] [Google Scholar]
- Millecamps S., Salachas F., Cazeneuve C., Gordon P., Bricka B., Camuzat A., Guillot-Noël L., Russaouen O., Bruneteau G., Pradat P.F., Le Forestier N., Vandenberghe N., Danel-Brunaud V., Guy N., Thauvin-Robinet C., Lacomblez L., Couratier P., Hannequin D., Seilhean D., Le Ber I., Corcia P., Camu W., Brice A., Rouleau G., LeGuern E., Meininger V. SOD1, ANG, VAPB, TARDBP, and FUS mutations in familial amyotrophic lateral sclerosis: genotype-phenotype correlations. J. Med. Genet. 2010;47:554–560. doi: 10.1136/jmg.2010.077180. [DOI] [PubMed] [Google Scholar]
- Pickering-Brown S., Rollinson P.S., Snowden J., Gerhard A., Neary D., Mann D. FTLD repeat expansions in C9orf72: evidence for variability in the repeat sequence. Alzheimers Dement. 2012;8:93. [Google Scholar]
- Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renčiuk D., Zemánek M., Kejnovská I., Vorlíčková M. Quadruplex-forming properties of FRAXA (CGG) repeats interrupted by (AGG) triplets. Biochimie. 2009;91:416–422. doi: 10.1016/j.biochi.2008.10.012. [DOI] [PubMed] [Google Scholar]
- Renton A.E., Majounie E., Waite A., Simón-Sánchez J., Rollinson S., Gibbs J.R., Schymick J.C., Laaksovirta H., van Swieten J.C., Myllykangas L., Kalimo H., Paetau A., Abramzon Y., Remes A.M., Kaganovich A., Scholz S.W., Duckworth J., Ding J., Harmer D.W., Hernandez D.G., Johnson J.O., Mok K., Ryten M., Trabzuni D., Guerreiro R.J., Orrell R.W., Neal J., Murray A., Pearson J., Jansen I.E., Sondervan D., Seelaar H., Blake D., Young K., Halliwell N., Callister J.B., Toulson G., Richardson A., Gerhard A., Snowden J., Mann D., Neary D., Nalls M.A., Peuralinna T., Jansson L., Isoviita V.M., Kaivorinne A.L., Hölttä-Vuori M., Ikonen E., Sulkava R., Benatar M., Wuu J., Chiò A., Restagno G., Borghero G., Sabatelli M., Heckerman D., Rogaeva E., Zinman L., Rothstein J.D., Sendtner M., Drepper C., Eichler E.E., Alkan C., Abdullaev Z., Pack S.D., Dutra A., Pak E., Hardy J., Singleton A., Williams N.M., Heutink P., Pickering-Brown S., Morris H.R., Tienari P.J., Traynor B.J. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen D., Siddique T., Patterson D., Figlewicz D., Sapp P., Hentati A., Donaldson D., Goto J., O'Regan J., Deng H. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Rutherford N.J., DeJesus- Hernandez M.B., Baker M.C., Kryston T.B., Brown P.E., Lomen-Hoerth C.M., Boylan K.M., Wszolek Z.K., Rademakers R.P. C9ORF72 hexanucleotide repeat expansions in patients with ALS from the Coriell Cell Repository. Neurology. 2012;79:482–483. doi: 10.1212/WNL.0b013e31826170f1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shatunov A., Mok K., Newhouse S., Weale M.E., Smith B., Vance C., Johnson L., Veldink J.H., van Es M.A., van den Berg L.H., Robberecht W., Van Damme P., Hardiman O., Farmer A.E., Lewis C.M., Butler A.W., Abel O., Andersen P.M., Fogh I., Silani V., Chiò A., Traynor B.J., Melki J., Meininger V., Landers J.E., McGuffin P., Glass J.D., Pall H., Leigh P.N., Hardy J., Brown R.H., Jr., Powell J.F., Orrell R.W., Morrison K.E., Shaw P.J., Shaw C.E., Al-Chalabi A. Chromosome 9p21 in sporadic amyotrophic lateral sclerosis in the UK and seven other countries: a genome-wide association study. Lancet Neurol. 2010;9:986–994. doi: 10.1016/S1474-4422(10)70197-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith B.N., Newhouse S., Shatunov A., Vance C., Topp S., Johnson L., Miller J., Lee Y., Troakes C., Scott K.M., Jones A., Gray I., Wright J., Hortobagyi T., Al-Sarraj S., Rogelj B., Powell J., Lupton M., Lovestone S., Sapp P.C., Weber M., Nestor P.J., Schelhaas H.J., Asbroek A.A., Silani V., Gellera C., Taroni F., Ticozzi N., Van den Berg L., Veldink J., Van Damme P., Robberecht W., Shaw P.J., Kirby J., Pall H., Morrison K.E., Morris A., de Belleroche J., Vianney de Jong J.M., Baas F., Andersen P.M., Landers J., Brown R.H., Weale M.E., Al-Chalabi A., Shaw C.E. The C9ORF72 expansion mutation is a common cause of ALS+/−FTD in Europe and has a single founder. Eur. J. Hum. Genet. 2013;21:102–108. doi: 10.1038/ejhg.2012.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreedharan J., Blair I., Tripathi V., Hu X., Vance C., Rogelj B., Ackerley S., Durnall J., Williams K., Buratti E. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Blitterswijk M., van Es M.A., Hennekam E.A., Dooijes D., van Rheenen W., Medic J., Bourque P.R., Schelhaas H.J., van der Kooi A.J., de Visser M., de Bakker P.I., Veldink J.H., van den Berg L.H. Evidence for an oligogenic basis of amyotrophic lateral sclerosis. Hum. Mol. Genet. 2012;21:3776–3784. doi: 10.1093/hmg/dds199. [DOI] [PubMed] [Google Scholar]
- Vance C., Rogelj B., Hortobagyi T., De Vos K.J., Nishimura A.L., Sreedharan J., Hu X., Smith B., Ruddy D., Wright P., Ganesalingam J., Williams K.L., Tripathi V., Al-Saraj S., Al-Chalabi A., Leigh P.N., Blair I.P., Nicholson G., de Belleroche J., Gallo J.M., Miller C.C., Shaw C.E. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Es M.A., Veldink J.H., Saris C.G., Blauw H.M., van Vught P.W., Birve A., Lemmens R., Schelhaas H.J., Groen E.J., Huisman M.H., van der Kooi A.J., de Visser M., Dahlberg C., Estrada K., Rivadeneira F., Hofman A., Zwarts M.J., van Doormaal P.T., Rujescu D., Strengman E., Giegling I., Muglia P., Tomik B., Slowik A., Uitterlinden A.G., Hendrich C., Waibel S., Meyer T., Ludolph A.C., Glass J.D., Purcell S., Cichon S., Nothen M.M., Wichmann H.E., Schreiber S., Vermeulen S.H., Kiemeney L.A., Wokke J.H., Cronin S., McLaughlin R.L., Hardiman O., Fumoto K., Pasterkamp R.J., Meininger V., Melki J., Leigh P.N., Shaw C.E., Landers J.E., Al-Chalabi A., Brown R.H., Robberecht W., Andersen P.M., Ophoff R.A., van den Berg L.H. Genome-wide association study identifies 19p13.3 (UNC13A) and 9p21.2 as susceptibility loci for sporadic amyotrophic lateral sclerosis. Nat. Genet. 2009;41:1083–1087. doi: 10.1038/ng.442. [DOI] [PubMed] [Google Scholar]
- Wu C.H., Fallini C., Ticozzi N., Keagle P.J., Sapp P.C., Piotrowska K., Lowe P., Koppers M., McKenna-Yasek D., Baron D.M., Kost J.E., Gonzalez-Perez P., Fox A.D., Adams J., Taroni F., Tiloca C., Leclerc A.L., Chafe S.C., Mangroo D., Moore M.J., Zitzewitz J.A., Xu Z.S., van den Berg L.H., Glass J.D., Siciliano G., Cirulli E.T., Goldstein D.B., Salachas F., Meininger V., Rossoll W., Ratti A., Gellera C., Bosco D.A., Bassell G.J., Silani V., Drory V.E., Brown R.H., Jr., Landers J.E. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature. 2012;488:499–503. doi: 10.1038/nature11280. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Relative frequency of nonmutation case and control samples by the largest repeat length.
Relative frequency of nonrepeat mutation case (n = 239) and control (n = 357) samples by repeat length as an additive model.