

Abstract
Double-strand breaks (DSB) occur in chromatin following replication fork collapse and chemical or physical damage [Symington and Gautier (Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 2011;45:247–271.)] and may be repaired by homologous recombination (HR) and non-homologous end-joining. Nucleosomes are the fundamental units of chromatin and must be remodeled during DSB repair by HR [Andrews and Luger (Nucleosome structure(s) and stability: variations on a theme. Annu. Rev. Biophys. 2011;40:99–117.)]. Physical initiation of HR requires RAD51, which forms a nucleoprotein filament (NPF) that catalyzes homologous pairing and strand exchange (recombinase) between DNAs that ultimately bridges the DSB gap [San Filippo, Sung and Klein. (Mechanism of eukaryotic HR. Annu. Rev. Biochem. 2008;77:229–257.)]. RAD51 forms an NPF on single-stranded DNA and double-stranded DNA (dsDNA). Although the single-stranded DNA NPF is essential for recombinase initiation, the role of the dsDNA NPF is less clear. Here, we demonstrate that the human RAD51 (HsRAD51) dsDNA NPF disassembles nucleosomes by unwrapping the DNA from the core histones. HsRAD51 that has been constitutively or biochemically activated for recombinase functions displays significantly reduced nucleosome disassembly activity. These results suggest that HsRAD51 can perform ATP hydrolysis-dependent nucleosome disassembly in addition to its recombinase functions.
INTRODUCTION
Genome instability resulting from defects in recombinational repair of DNA double-strand breaks (DSBs) has been linked to hereditary breast cancer (BRCA1/2) as well as hematopoietic and other solid tumors (Ataxia telangiectasia mutated, Nijmegen breakage syndrome, Fanconi anemia, Bloom’s syndrome, among others) (1–5). Recombinational repair engenders a complex cascade of responses that include cellular signaling integrated with the physical processes of DSB repair (6,7) and can result in high fidelity homologous recombination (HR) as well as lower fidelity non-homologous end-joining (8–11). HR uses homologous chromatid or chromosomal sequences to bridge the DSB where the biochemical reactions must manage the disrupted chromatin composite on the broken donor DNA to search and pair with the assembled chromatin of a bridging homologous acceptor DNA (12).
RAD51 plays a central role in eukaryotic and archael HR and is homologous to the prototypical bacterial RecA (10,13,14). RecA/RAD51 forms and maintains a stable nucleoprotein filament (NPF) that catalyzes the initial homologous pairing and strand exchange (recombinase) of HR (13,14,15). RAD51 may nucleate an NPF on both single-stranded DNA (ssDNA) and double-stranded DNA (dsDNA) (16,17). However, unusual cationic salts appear to bias a stable ATP-bound RAD51 NPF to ssDNA that then promotes recombinase initiation functions (18–20). Substitution of a conserved Asp (D) residue in the ATP cap domain with a Lys (K) residue that is generally conserved in the RecA family [RAD51(D316K)] relieves the unusual salt requirements, enhances discrimination between ssDNA and dsDNA and induces the formation of a stable and active NPF (21). The conservation of D316 appears to suggest that the RAD51 family may be poised to regulate the ssDNA and dsDNA NPF via this residue.
RAD51 recombinase activity has been observed with chromatin substrates reconstituted from heterogeneous Hela and Drosophila cellular histone octamers (HO) as well as recombinant Xenopus HOs (22–24). Three consistent observations have emerged: (i) RAD51 may catalyze the homology search and pairing reaction even if the homology site is within the nucleosome, (ii) RAD54 appears to convert a paranemic RAD51 pairing structure to a plectonemic strand exchange product and (iii) RAD51 recombinase may stimulate a RAD54 chromatin remodeling activity. Even though RAD51 appears capable of catalyzing recombination initiation within a nucleosome, the process presents a topological problem at locations where the wrapped DNA makes multiple contacts with the HO. These observations suggest that recombination is likely to require synchronized and/or regulated nucleosome processing. The mechanics of nucleosome processing is largely unknown.
The overexpression of ScRad51 induces spontaneous foci on yeast chromatin and appears toxic in the absence of SWI/SNF translocases that include ScRad54, ScRdh54 and Uls1 (25). These results suggest that cellular dsDNA binding by RAD51 may be significant and tightly regulated. ScRad51 was recently reported to remodel nucleosomes (26). Interestingly, it appeared that ScRad51 nucleosome remodeling largely involved a ‘pushing’ mechanism along dsDNA, with ∼30% of the nucleosomes being dissociated during the process (26). These observations were surprising, as the multiple contacts between the core histones within the nucleosome DNA would seem to engender a substantial energetic barrier to a remodeling process that involved pushing and concentrating nucleosomes in discrete regions (27).
Here, we examine nucleosome remodeling catalyzed by human RAD51 (HsRAD51). Our studies are consistent with the majority of remodeling occurring by an unwrapping mechanism. We found conditions that enhance stable ATP-bound HsRAD51 and recombinase activity were refractory to nucleosome unwrapping, whereas conditions that promote ATP-hydrolysis enhanced nucleosome unwrapping. These results suggest that HsRAD51 may toggle between recombinase and nucleosome unwrapping activities.
MATERIALS AND METHODS
HsRAD51 and ScRad51 expression and purification
HsRAD51 and ScRAD51 were expressed and purified as previously described (28,29). Escherichia coli RecA was purchased from New England Biolabs. All RecA/RAD51 proteins were active for strand exchange under their respective published optimal conditions.
DNA synthesis
The 601E, 601D, 5SD, 5SE, MP2 and components for the 601D-linker and 601E-linker molecules for fluorescence studies were prepared by PCR from plasmid containing the 601 or Xenopus laevis 5S rDNA nucleosome positioning sequence (NPS) and a TspRI restriction site 10 bp downstream of the NPS. Oligonucleotides (see Supplementary Table S1, Sigma) were labeled with a Cy3 NHS ester (GE healthcare) at an amino group attached at the 5′ end or to a modified internal thymine and then purified by RP-High pressure (or high performance) liquid chromatography (HPLC) on a 218TP C18 column (Grace/Vydac). Following PCR amplification (30), each DNA molecule was purified by HPLC with a Gen-Pak Fax column (Waters). The 601D-linker and 601E-linker were synthesized by digesting with TspRI in NEB buffer #4 (New England Biolabs) PCR product containing the 601 NPS and TspRI site and PCR product containing the TspRI site and a 90 bp linker DNA. Digested NPS and linker fragments were purified by Gen-Pak Fax column, ligated with T4 ligase in supplied buffer plus 2 mM ATP (New England Biolabs) and purified by Gen-Pak Fax column to remove unligated fragments.
Histone expression and labeling
Xenopus laevis recombinant histones were expressed and purified as previously described (31). Plasmids encoding histones H2A(K119C), H2B, H3 and H4 were generous gifts from Dr Karolin Luger (Colorado State University) and Dr Jonathan Widom (Northwestern University). Mutation H3(C110A) was introduced by site-directed mutagenesis (Stratagene). Histones: H2A(K119C), H2B, H3(C110A), H4 or H2A, H2B, H3, H4 were combined at equal molar ratios, refolded and purified as previously described (31). H2A(K119C) was labeled after octamer refolding with Cy5-maleamide (GE Healthcare) as previously described (32). The purity of each octamer was confirmed bySDS–PAGE and mass spectrometry. The labeling efficiency was 70–80% as determined by ultraviolet/visible spectroscopy (UV-VIS) and mass spectroscopy.
Nucleosome reconstitution
Nucleosomes for fluorescence measurements were reconstituted from 2 µg of Cy3-labeled DNA, 6 µg of lambda DNA and 2 µg of Cy5-labeled HO by salt double dialysis and purified by sucrose gradient as previously described (32). Non-labeled nucleosomes were reconstituted from 12 µg of MP2-147 DNA with 10 µg of unlabeled HO and purified by sucrose gradient. Purity of each nucleosome was checked by 5% native PAGE with 0.3× 45 mM Tris-borate, 1 mM EDTA (TBE) at 300V (Supplementary Figure S1A). Localization studies of nucleosomes reconstituted with these and similar substrates has been published previously (33).
HsRad51 nucleosome disassembly by gel shift
Fluorescent-labeled 601E or 601E-linker nucleosomes (10 nM) in 20 mM Tris (pH 8.0), 2 mM MgCl2, 500 µM ATP (ADP or ATPγS), 3% glycerol, 1 mM dithiothreitol, 0.2 mg/ml acetylated bovine serum albumin and 130 mM KCl were incubated at 37°C for 30 min with 0–900 nM HsRAD51 (as indicated) in 10 µl total reaction volume. Reactions were quenched with 10 µg/µl oligo-dA ssDNA and 20 mM ethylenediamine-tetraacetic acid (pH 8.0) (EDTA) and incubated at 42°C for 90 min to destabilize the HsRAD51 filament. Reactions were resolved by 5% native PAGE gel in 0.5× TTE [45 mM Tris (pH 8), 14 mM Taurine, 0.5 mM EDTA], were prerun at 7 V/cm for 1 h and run at 7 V/cm for 2 h with continuous buffer circulation. Gels were imaged by the Cy3 fluorophore using a Typhoon Trio variable mode imager (GE Healthcare). Images were quantified using Image Quant software (Invitrogen).
Nucleosome disassembly kinetics by Förster (fluorescence) resonance energy transfer
Fluorescent-labeled nucleosomes (2 nM) and unlabeled MP2-147 nucleosomes (10 nM), 12 nM nucleosome total (33), were preincubated at 37°C for 5 min in 130 mM KCl or 25 mM KCl (E. coli RecA), 2 mM MgCl2 or CaCl2, 250 µM ATP, ADP or ATPγS, 20 mM Tris (pH 8.0), 3% glycerol, 1 mM dithiothreitol, 0.05 mg/ml acetylated bovine serum albumin and 1× COT/NBA (1 mM 1-3-5-7-Cyclooctatetraene + 1 mM Nitrobenzoic acid with 2 mM Trolox when necessary) as a triplet state quencher. Unlabeled nucleosomes were included to enhance the stability of the fluorescent-labeled nucleosomes as previously described (30). The total concentration of nucleosomes was 12 nM (2 µM DNA nt). Indicated concentrations of HsRAD51, HsRad51(D316K), Saccharomyces cerevisae Rad51 (yRad51), or E. coli RecA was then rapidly mixed with the nucleosomes placed into a submicro quartz cuvette (Starna, 16.12F-Q-1.5/Z8.5) in a Flouromax-4 (Horiba) photon counting flourometer with circulating waterbath at 37°C. The Cy3 label was excited at 510 nM, and the changes in FRET between Cy3 and Cy5 monitored by measuring Cy5 fluorescence emission at 670 nm. The Cy5 fluorescence emission as a function of time, F(t), from 0 to 300 s is fit to a single exponential decay, F(t) = Ae(−t/τ) + Fo, where A is the amplitude of fluorescence decay, τ is the characteristic time of fluorescence decay and Fo is the final Cy5 emission decay fluorescence. The equilibrium FRET efficiency between Cy3 and Cy5 was determined by the (ratio)A method as previously described (32,34). The (ratio)A method only depends on the labeling efficiency of the donor molecule, which is purified to homogeneity by HPLC as previously described (30). Thus, the donor labeled fraction is 1.0, and the acceptor labeled fraction is not included in FRET efficiency calculations. Because 50% of the acceptor fluorophore will be located near the DNA end that does not contain the donor flourophore, the (ratio)A derived FRET efficiency was corrected by a factor of 2 as described by Widom and colleagues (35).
RESULTS
HsRAD51 disassembles nucleosomes
To examine the interplay between HsRAD51 and chromatin, we constructed DNA substrates containing either a 601 (36) or a 5S rDNA (5S) (37) NPS as well as a Cy3 fluorophore located near the entry-exit (E) or dyad regions (D; Figure 1A; see ‘Materials and Methods’ section) (38). We examined the possibility that HsRAD51 might require the linker DNA between nucleosomes to form an NPF by constructing DNA substrates that either contained only the 147 bp NPS sequence or the NPS plus a 100 bp 3′-extension (linker, Figure 1A). These DNA substrates were assembled into nucleosomes with HO that was labeled with Cy5 at the H2A(K119C) substitution residue [Supplementary Figure S1A; (39)]. Nucleosome assembly positions the Cy5 acceptor near enough to the Cy3 donor for efficient FRET [Figure 1B and Supplementary Figure S1B; (38)]. For these studies, complete saturation of the NPS dsDNA requires ∼0.5–1 µM HsRAD51 depending on whether the NPS DNA contained a linker region.
Figure 1.

HsRAD51 alters nucleosome positioning. (A) Diagram of Cy3-labeled DNA’s containing either the 147 bp 601 or Xenopus 5S rDNA NPS with a 100 bp linker that may be used for quantifying HsRAD51 interactions with a nucleosome. (B) Schematic of the FRET system using the Cy3-labeled 601E-linker (left) or 601D-linker (right) DNAs reconstituted into nucleosomes with Cy5-labeled HO. The nucleotide position of the Cy3 fluorophore is shown in Supplementary Table S1, whereas the Cy5 fluorophore was linked to H2A(K119C) by maleimide chemistry to the HO. Spatial positioning of the Cy3 and Cy5 fluorophores on the nucleosome crystal structure is shown in Supplementary Figure S1B. (C) Representative fluorescence emission spectra of 601D-linker nucleosomes (12 nM) excited at 510 nM in the presence 130 mM KCl, ATP (250 µM) and 2 mM MgCl2 (Mg2+) with HsRAD51 at 0 (black), 100 (blue), 200 (green) and 500 nM (orange). (D) Representative fluorescence emission spectra of 601D-linker nucleosomes (12 nM) excited at 510 nM in the presence 130 mM KCl, ATP (250 µM) and 2 mM CaCl2 (Ca2+) with HsRAD51 at 0 (black), 100 (blue), 200 (green) and 500 nM (orange). (E) Fluorescence emission spectra of 601D-linker nucleosomes (12 nM) excited at 610 nM in the presence 130 mM KCl, ATP (250 µM) and 2 mM Mg2+ (left) or 2 mM Ca2+ (right) and incubated with hsRAD51 at 0 (black), 100 (blue), 200 (green) and 500 nM (orange). (F) Normalized nucleosome fraction as a function of HsRAD51 concentration. HsRad51 + 2 mM Mg2+ (black open square), HsRad51 + 2 mM Ca2+ (red open circle) and HsRad51(D316K) + 2 mM Mg2+ (open triangle), normalized nucleosome fraction determined by gel shift analysis for HsRad51 + 2 mM Mg2+ (pink open square). Each point represents the average and standard deviation from at least three independent experiments. (G) Gel analysis of nucleosome disassembly. 601E-linker nucleosomes (10 nM) were incubated with indicated concentration of HsRAD51, ATP (250 µM) and 2 mM Mg2+. Multiple bands are the results of at least two populations of nucleosomes that occur with tailed positioning DNAs; where the histone octomer may deviate from the consensus positioning sequence. The average fraction of nucleosome DNA (Nuc) from at least three independent experiments was determined by densitometry (Typhoon), normalized and plotted in Panel (F).
Representative fluorescence emission spectra of the nucleosome substrates excited at 510 nm resulted in quenched Cy3 emission at 560 nm and stimulated Cy5 FRET emission at 670 nm (see Figure 1C and D, black). We determined the FRET efficiency for increasing concentrations of HsRAD51 by the (ratio)A method from these emission spectra as well as the 670 nm emission from the nucleosome substrates when directly excited at 610 nm (Figure 1E) (34). The emission from direct excitation was not affected by HsRAD51 concentration, suggesting that any FRET changes must result from distance changes between donor and acceptor fluorophores, and not protein quenching. HsRAD51 catalyzes the formation of D-loop’s by homologous pairing and strand exchange in the presence of divalent calcium (Ca2+), but not divalent magnesium (Mg2+) cations (18). This is because Ca2+ appears to maintain an active ATP-bound HsRA51 filament while Mg2+ promotes ATP hydrolysis resulting in either protein turnover or a mixed adenosine nucleotide NPF (21). We observed a significant decrease of the FRET efficiency of the 601D-linker nucleosome in the presence of Mg2+ compared with Ca2+ (Figure 1C and D; plotted as fraction of starting FRET/nucleosomes in Figure 1F). These results are consistent with the hypothesis that HsRAD51 may more effectively reposition nucleosomes under conditions where ATP hydrolysis occurs.
Changes in FRET reflect nanometer distance changes between the donor and acceptor flourophores (34) with a reduction in FRET resulting from HO repositioning along the DNA and/or octamer removal from the DNA. To determine the extent of nucleosome repositioning or disassembly that resulted in reduced FRET, we first performed a gel shift analysis (Figure 1G). In these studies, we may directly examine nucleosome disassembly by incubating 601E-linker nucleosomes with increasing HsRAD51 and determined the fraction DNA bound in nucleosomes compared with nucleosome-free DNA. We observed a concentration-dependent decrease in the nucleosome gel-shift band in the presence of HsRAD51 (Figure 1G). These results suggest that the steady-state FRET changes observed in the presence of HsRAD51 ultimately resulted in the disassembly of nucleosomes. Furthermore, the HsRAD51 concentration-dependent normalized FRET decay and the HsRAD51 concentration-dependent nucleosome fraction remaining as determined by gel shift analysis appear quantitatively identical (Figure 1F). Together, these results indicate that the reduction in FRET fluorescence by the 601D-linker nucleosome DNA substrate may be used as a direct measure of the rate of nucleosome disassembly (koff). Interestingly, HsRAD51 in the presence of Ca2+ or the ATP-cap substitution mutation [HsRAD51(D316K)] that naturally maintains an ATP-bound filament and enhances recombinase activity (21) displayed a reduced ability to disassemble nucleosomes (Figure 1F). These observations are consistent with the conclusion that HsRAD51 disassembles nucleosomes and that there appears to be a significant difference in the extent and/or kinetics of nucleosome disassembly between HsRAD51-Mg2+ and HsRAD51-Ca2+ or HsRAD51(D316K) (Figure 1F).
HsRAD51 unwraps the DNA from nucleosomes
There are two possible mechanisms for nucleosome disassembly: (i) sliding the HO along the DNA until it dissociates from an open end or (ii) unwrapping the DNA from the HO. These mechanisms produce different kinetic predictions for DNA substrates containing FRET pairs at the E or D regions of the nucleosome (Figure 1A and B). For example, the kinetics of Cy5 FRET fluorescence reduction during nucleosome sliding should be virtually identical for the E and D nucleosome substrates, as HO movement from these sites should occur simultaneously. In contrast, E FRET reduction should occur faster than D FRET reduction during nucleosome unwrapping, and the difference in rates depends on how rapidly the nucleosome is fully unwrapped.
We examined the Cy5 fluorescence kinetics by rapidly mixing HsRAD51 with nucleosomes (Figure 2). Representative fluorescence decay analysis of 601E-linker and 601D-linker nucleosome substrates in the presence of Mg2+ or Ca2+ are shown in Figure 2A–D. The reduction in FRET fluorescence at each concentration of HsRAD51 was fit to a single exponential decay. Visual inspection shows a clear difference in the kinetics and extent of the FRET decay curves between the 601E-linker and 601D-linker nucleosome substrates (compare Figure 2A with B and Figure 2C with D). These differences translated into different rates of FRET fluorescence decay (Figure 2E and F) that are consistent with a model in which the majority of nucleosomes are unwrapped before they are disassembled. An ∼20-fold difference between kunwrap and koff was consistently observed, regardless of the conditions (Figure 2G). These results support previous studies that demonstrated the entry–exit region of the nucleosome is capable of partial unwrapping independent of nucleosome disassembly (35,40). In the presence of HsRAD51 nucleosome, unwrapping seems to allow occupation of the entry–exit region by the NPF that ultimately results in FRET fluorescence decay. We determined that the rate of nucleosome disassembly was 4-fold faster with HsRAD51 in the presence of Mg2+ compared with HsRAD51 in the presence of Ca2+ or the HsRAD51(D316K) substitution (Figure 2H).
Figure 2.

RAD51 unwraps the DNA from the histone core. (A) Representative FRET fluorescence decay of 601E-linker nucleosomes (12 nM) incubated with HsRAD51 at 0 (black), 50 (red), 100 (blue), 200 (green) and 500 nM (orange), in the presence of 130 mM KCl, ATP (250 µM) and 2 mM MgCl2 (Mg2+). (B) Representative FRET fluorescence decay of 601D-linker nucleosomes (12 nM) incubated with HsRAD51 at 0 (black), 50 (red), 100 (blue), 200 (green), 500 (orange) and 900 nM (purple) in the presence of 130 mM KCl, ATP (250 µM) and 2 mM Mg2+. (C) Representative FRET fluorescence decay of 601E-linker nucleosomes (12 nM) incubated with HsRAD51 at 0 (black), 50 (red), 100 (blue), 200 (green), 500 (orange) and 900 nM (purple) in the presence of 130 mM KCl, ATP (250 µM) and 2 mM CaCl2 (Ca2+). (D) Representative FRET fluorescence decay of 601D-linker nucleosomes (12 nM) incubated with HsRAD51 at 0 (black), 50 (red), 100 (blue), 200 (green) and 500 nM (orange), in the presence of 130 mM KCl, ATP (250 µM) and 2 mM Ca2+. (E) Graph of FRET fluorescence decay rates for 601E-linker nucleosomes versus HsRAD51 concentration: HsRAD51 with Mg2+ (black open circle), HsRAD51 with Ca2+ (blue open circle) and HsRAD51(D316K) with Mg2+ (red open circle). Each point represents the average and standard of deviation from at least two independent experiments. (F) Graph of FRET fluorescence decay rates for 601D-linker nucleosomes versus HsRAD51 concentration: HsRAD51 with Mg2+ (black open circle), HsRAD51 with Ca2+ (blue open circle) and HsRAD51(D316K) with Mg2+ (red open circle). Each point represents the average and standard of deviation from at least two independent experiments. (G) The rate of entry–exit unwrapping (kunwrap) divided by the rate of nucleosome disassembly (koff). kunwrap was determined from the FRET fluorescence decay of 601E-linker nucleosomes and koff was determined from the FRET fluorescence decay of 601D-linker nucleosomes. (H) Relative rate of disassembly determined by dividing the rate of HsRAD51 nucleosome disassembly in the presence of Mg2+ (koff•HsRAD51-Mg) by the rate of HsRAD51 nucleosome disassembly in the presence of Ca2+ (koff•HsRAD51-Ca) (Top blue) or the rate of HsRAD51(D316K) nucleosome disassembly (koff•HsRAD51(D316K)) (Bottom red).
HsRAD51 nucleosome disassembly does not require linker DNA
We examined nucleosome disassembly with DNA substrates that did not contain linker DNA. Representative steady-state HsRAD51-dependent FRET fluorescence decay of the 601D and 5SD nucleosome DNA substrates are shown in Figure 3A and B. The normalized FRET fluorescence decay was plotted and compared with the 601D-linker nucleosome substrate (Figure 3C). We found that the concentration of HsRAD51 required to disassemble half of the nucleosomes was virtually identical for nucleosomes that do not contain a linker and nucleosomes that do contain a linker (S0.5-601D = 370 nM ± 40 nM; S0.5-5SD= 400 nM ± 50 nM; S0.5-601D-linker = 340 nM ± 50 nM; Figure 3D). As the cellular concentration of HsRAD51 appears to be near 6 µM (41), there is likely to be sufficient protein to disassemble nucleosomes in vivo. Moreover, these results suggest that HsRAD51 may initiate the disassembly of wrapped DNA within a nucleosome independent of associated linker DNA regions. We also note that there is little or no difference in rate of nucleosome disassembly between the 601 and 5S rDNA NPS (Figure 3C). This observation suggests that relative localization stability is unlikely to significantly influence HsRAD51 catalyzed nucleosome disassembly.
Figure 3.

Nucleosome disassembly is independent of linker DNA and nucleosome positioning stability. (A) Representative fluorescence emission spectra of 601D nucleosomes (12 nM) incubated with HsRAD51 at 0 (black), 100 (blue) and 500 nM (orange) in the presence of 130 mM KCl, ATP (250 µM) and 2 mM Mg2+. (B) Representative fluorescence emission spectra of 5SD-linker nucleosomes (12 nM) incubated with HsRAD51 at 0 (black), 100 (blue), 200 (green) and 500 nM (orange) in the presence of 130 mM KCl and 2 mM Mg2+. (C) Normalized FRET efficiency of 601D (black open circle) and 5SD (black open circle) nucleosomes compared with 601D-linker nucleosomes (black open square) as a function of HsRAD51 concentration (see text). (D) Concentration of HsRAD51 (nM) at which 50% of nucleosomes are disassembled (S0.5). Values represent the average and standard of deviation from at least three independent experiments.
Nucleosome disassembly occurs in the presence of ATP, but not ADP or ATPγS
We determined the adenosine nucleotide requirements of HsRAD51 nucleosome disassembly (Figure 4). We first determined that substitution of ATP with ADP or ATPγS did not result in dissociation of nucleosomes by gel analysis (Figure 4A and B). Moreover, we did not observe any FRET changes in the 601E nucleosomes in the presence of ADP (Figure 4C) or ATPγS (Figure 4D), nor the 601D nucleosomes in the presence of ADP (Figure 4E). Quantitative analysis of the gel and FRET results clearly shows that HsRAD51 is incapable of catalyzing nucleosome dissociation in the presence of ADP or ATPγS compared with ATP (Figure 4F). These findings are consistent with the HsRAD51 Mg2+/Ca2+ and HsRAD51(D316K) studies (Figures 1 and 2) and strongly support the conclusion that ATP hydrolysis is required for nucleosome disassembly. Taken as a whole, these observations are consistent with the idea that HsRAD51 may toggle between an active ATP-bound filament optimized for recombinase functions and an ATP hydrolysis-dependent form that is optimized for nucleosome disassembly functions.
Figure 4.

The adenosine nucleotide dependence of HsRAD51 nucleosome disassembly. (A) Gel analysis of 601E nucleosome disassembly in the presence of ADP. The 601E nucleosomes (10 nM) were incubated with indicated concentration of HsRAD51, 130 mM KCl, ADP (250 µM) and 2 mM MgCl2 (Mg2+). (B) Gel analysis of 601E nucleosome disassembly in the presence of ATPγS. The 601E nucleosomes (10 nM) were incubated with indicated concentration of HsRAD51, 130 mM KCl, ATPγS (250 µM) and 2 mM Mg2+. (C) Representative FRET fluorescence decay of 601E nucleosomes (12 nM) incubated with HsRAD51 at 0 (black), 50 (red), 100 (blue), 200 (green), 500 (orange) and 900 nM (purple) in the presence of 130 mM KCl, ADP (250 µM) and 2 mM Mg2+. (D) Representative FRET fluorescence decay of 601E nucleosomes (12 nM) incubated with HsRAD51 at 0 (black), 50 (red), 100 (blue), 200 (green), 500 (orange) and 900 nM (purple) in the presence of 130 mM KCl, ATPγS (250 µM) and 2 mM Mg2+. (E) FRET analysis of 601D nucleosome disassembly in the presence of ADP. The 601D nucleosomes (12 nM) were incubated with indicated concentration of HsRAD51, 130 mM KCl, ADP (250 µM) and 2 mM Mg2+. (F) Quantitative analysis of the calculated nucleosome fractions of 601D from gel and FRET nucleosome disassembly analysis (Panel A–E) compared with the nucleosome fraction of 601D determined by FRET in the presence of ATP (Figure 3C). Color-coded key is shown in the inset. Standard deviation was calculated from at least two independent experiments.
RecA and ScRAD51 are unable to disassemble nucleosomes in HsRAD51 conditions
A recent report showed that NPFs formed with either yeast Rad51 (ScRAD51) or RecA may slide and destabilize nucleosomes (26). We examined ScRad51 and RecA catalyzed nucleosome repositioning using our fluorophore labeled nucleosome substrates (Figure 5). We observed virtually no decay in Cy5 fluorescence from the entry–exit reporter (601E-linker) in representative analysis under conditions that were optimized for HsRAD51 catalyzed disassembly (130 mM KCl; Figure 5A and E). A modest decay of the entry–exit reporter (601E) was observed with RecA in conditions where the protein maintains substantial recombinase and ATPase activity (25 mM KCl; Figure 5C). However, we did not observe a change in Cy5 fluorescence from the dyad reporter under any of the conditions (601D-linker; Figure 5B, D and F). The lack of FRET decay with the 601D nucleosome reporter suggests that ScRad51 and RecA were significantly less efficient in disassembling nucleosomes under the conditions that we tested. We cannot rule out RecA or ScRad51 catalyzed nucleosome destabilization/repositioning, as we largely restricted our studies to HsRAD51 optimal conditions (with the exception of RecA optimal recombinase/ATPase conditions shown in Figure 5C and D), there was no linker DNA 5′ of the nucleosome on which to slide and the previous studies used histones isolated from cells to reconstitute nucleosomes.
Figure 5.

Saccharomyces cerevisae Rad51 and E. coli RecA are unable to disassemble nucleosomes. (A) Representative fluorescence emission spectra of 601 E-linker nucleosomes (12 nM) excited at 510 nM in the presence 130 mM KCl, ATP (250 µM) and 2 mM MgCl2 (Mg2+) with S. cerevisae Rad51 (yRad51) at 0 (black), 200 (green), 500 (orange) and 1000 nM (purple). (B) Representative fluorescence emission spectra of 601D-linker nucleosomes (12 nM) excited at 510 nM in the presence 130 mM KCl, ATP (250 µM) and 2 mM MgCl2 (Mg2+) with S. cerevisae Rad51 (yRad51) at 0 (black), 200 (green), 500 (orange) and 1000 nM (purple). (C) Representative fluorescence emission spectra of 601 E-linker nucleosomes (12 nM) excited at 510 nM in the presence 25 mM KCl, ATP (250 µM) and 2 mM Mg2+ with E. coli RecA (RecA) at 0 (black), 1 (purple), 2 (blue), 5 (red) and 10 µM (green). (D) Representative fluorescence emission spectra of 601D-linker nucleosomes (12 nM) excited at 510 nM in the presence 25 mM KCl, ATP (250 µM) and 2 mM MgCl2 (Mg2+) with E. coli RecA (RecA) at 0 (black), 1 (purple), 2 (blue), 5 (red) and 10 µM (green). (E) Representative fluorescence emission spectra of 601 E-linker nucleosomes (12 nM) excited at 510 nM in the presence 130 mM KCl, ATP (250 µM) and 2 mM Mg2+ with E. coli RecA (RecA) at 0 (black), 1 (purple), 2 (blue), 5 (red) and 10 µM (green). (F) Representative fluorescence emission spectra of 601D-linker nucleosomes (12 nM) excited at 510 nM in the presence 130 mM KCl, ATP (250 µM) and 2 mM MgCl2 (Mg2+) with E. coli RecA (RecA) at 0 (black), 1 (purple), 2 (blue), 5 (red) and 10 µM (green).
DISCUSSION
Our studies demonstrate that HsRAD51 may disassemble nucleosomes under physiological ionic conditions by a dynamic ‘occupation mechanism’ of DNA regions that are undergoing transient thermal unwrapping from the HO (Figure 6). This activity appears to be separate from its recombinase functions, as it is enhanced by conditions that support ATP hydrolysis. We observed a similar activity for the mismatch repair initiation heterodimer hMSH2-hMSH6 (42). Interestingly, unmodified nucleosome disassembly by HsRAD51 appears 3–4-fold more efficient at equivalent protein concentrations than hMSH2-hMSH6. Moreover, nucleosome disassembly by hMSH2-hMSH6 is dramatically stimulated by histone modifications and replacing the 601 with the 5S NPS, both of which destabilize the nucleosome and increase its mobility (42,43). This does not appear to be the case for HsRAD51 where nucleosomes reconstituted with 601 and 5S are disassembled equivalently, despite their significant stability differences (44). In addition, hMSH2-hMSH6 is at least 100-fold less efficient at nucleosome disassembly than HsRAD51 at physiological protein concentrations (koff•hMSH2-hMSH6•200 nM = 0.009 min−1; koff•HsRA51•1 µM = 0.15 min−1). These results may be an indicator of when and where these DNA repair components encounter nucleosomes. hMSH2-hMSH6 is largely associated with the replication fork (45,46) where partially assembled and/or acetylated nucleosomes occur that appear more easily disassembled (42). In contrast, HsRAD51 may catalyze HR genome-wide where a variety of chromatin structures, nucleosome types and modifications may occur.
Figure 6.
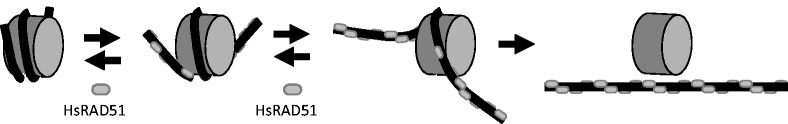
A model for HsRAD51 NPF catalyzed nucleosome disassembly. Nucleosome unwrapping by HsRAD51 occurs by gradual occupation of the entry–exit and dyad regions until the HO is destabilized.
Both ssDNA and dsDNA RAD51 NPFs occur in vivo and appear to be regulated by accessory factors (47). For example, BRCA2 has been shown to guide the assembly of a RAD51 ssDNA filament while excluding a dsDNA filament at the interface of the processed ssDNA tail during homologous pairing and strand exchange (48,49). In contrast, spontaneous ScRad51 foci form on yeast chromosomes in the absence of the SWI/SNF translocases ScRdh54, ScRad54 and ScUls1 that ultimately lead to chromosomal losses (25). These spontaneous foci were not due to dsDNA breaks or accumulated ssDNA, as they were not accompanied by RPA foci (25). Moreover, elevated expression of ScRAD51 is lethal in the absence of ScRdh54 (25). Taken as a whole, these results suggest that both ssDNA and dsDNA RAD51 NPFs occur in vivo, and that accessory proteins may dictate their timing, location and regulation.
Nucleosome disassembly might be required at several stages of HR. The first is shortly after the introduction of a DSB where clearing the ends of nucleosomes would facilitate end processing before homologous pairing and strand exchange. As RAD51 generally appears at the DSB after end resection and the arrival of other repair proteins, this possibility seems relatively remote (50,51). Nucleosome disassembly by HsRAD51 might occur during replication fork collapse where the ssDNA NPF that protects against nuclease digestion (52) might extend into the adjacent dsDNA before repair and/or replication restart. One could also speculate that nucleosome disassembly might occur on the acceptor DNA during homologous pairing and strand exchange. In this case, the initiation of homologous pairing produces an ssDNA D-loop that may be bound by RPA, whereas the RAD51 remains bound to the heteroduplex as a dsDNA NPF (53,54). Extension of the crossover and heteroduplex dsDNA NPF might result in nucleosome disassembly by a similar ‘occupation mechanism’ of transiently unwrapped nucleosome DNA in the absence of other chromatin remodeling factors. Before or during these reactions, the ssDNA and dsDNA HsRAD51 NPF could be regulated by BRCA2 or the SWI/SNF translocases (25,48,49).
The central role of ATP hydrolysis in nucleosome disassembly is intriguing, as homologous pairing and strand exchange by RecA/RAD51 appear to require only ATP binding and largely occurs independent of ATP hydrolysis (55,56). Additional studies will be necessary to understand the physiological significance of the nucleosome disassembly activity and/or any regulation that might govern the separate HsRAD51 recombinase and nucleosome disassembly functions.
SUPPLMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Table 1 and Supplementary Figures 1–2.
FUNDING
American Heart Association Predoctoral Fellowship [0815460D] and a Pelotonia Fellowship (to J.A.N.); Career Award in Basic Biomedical Sciences from the Burroughs Wellcome Fund (to M.G.P.) and National Institutes of Health grants [GM083055 to M.G.P.] and [GM080176 to R.F.]. Funding for open access charge: [GM080176].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Wolf-Dietrich Heyer for kindly supplying ScRad51; Wolf-Dietrich Heyer, Jack Griffith and the Poirier and Fishel laboratories for helpful discussions. J.A.N., R.A., M.G.P. and R.F. conceived and designed the studies; J.A.N., R.A., M.K. and A.N. acquired the data; J.A.N., R.A., M.K., M.G.P. and R.F. analyzed and interpreted the results and wrote the manuscript.
REFERENCES
- 1.D'Andrea AD, Grompe M. The Fanconi anaemia/BRCA pathway. Nat. Rev. Cancer. 2003;3:23–34. doi: 10.1038/nrc970. [DOI] [PubMed] [Google Scholar]
- 2.Ellis NA, Groden J, Ye TZ, Straughen J, Lennon DJ, Ciocci S, Proytcheva M, German J. The Bloom's syndrome gene product is homologous to RecQ helicases. Cell. 1995;83:655–666. doi: 10.1016/0092-8674(95)90105-1. [DOI] [PubMed] [Google Scholar]
- 3.Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM, Ding W, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266:66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- 4.Savitsky K, Bar Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, Tagle DA, Smith S, Uziel T, Sfez S, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268:1749–1753. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- 5.Tavtigian SV, Simard J, Rommens J, Couch F, Shattuck-Eidens D, Neuhausen S, Merajver S, Thorlacius S, Offit K, Stoppa Lyonnet D, et al. The complete BRCA2 gene and mutations in chromosome 13q-linked kindreds. Nat. Genet. 1996;12:333–337. doi: 10.1038/ng0396-333. [DOI] [PubMed] [Google Scholar]
- 6.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 7.Lobrich M, Jeggo PA. The impact of a negligent G2/M checkpoint on genomic instability and cancer induction. Nat. Rev. Cancer. 2007;7:861–869. doi: 10.1038/nrc2248. [DOI] [PubMed] [Google Scholar]
- 8.Symington LS, Gautier J. Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 2011;45:247–271. doi: 10.1146/annurev-genet-110410-132435. [DOI] [PubMed] [Google Scholar]
- 9.Brugmans L, Kanaar R, Essers J. Analysis of DNA double-strand break repair pathways in mice. Mutat. Res. 2007;614:95–108. doi: 10.1016/j.mrfmmm.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 10.Krogh BO, Symington LS. Recombination proteins in yeast. Annu. Rev. Genet. 2004;38:233–271. doi: 10.1146/annurev.genet.38.072902.091500. [DOI] [PubMed] [Google Scholar]
- 11.Pfeiffer P, Goedecke W, Kuhfittig-Kulle S, Obe G. Pathways of DNA double-strand break repair and their impact on the prevention and formation of chromosomal aberrations. Cytogenet. Genome Res. 2004;104:7–13. doi: 10.1159/000077460. [DOI] [PubMed] [Google Scholar]
- 12.Andrews AJ, Luger K. Nucleosome structure(s) and stability: variations on a theme. Annu. Rev. Biophys. 2011;40:99–117. doi: 10.1146/annurev-biophys-042910-155329. [DOI] [PubMed] [Google Scholar]
- 13.San Filippo J, Sung P, Klein H. Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 2008;77:229–257. doi: 10.1146/annurev.biochem.77.061306.125255. [DOI] [PubMed] [Google Scholar]
- 14.West SC. Molecular views of recombination proteins and their control. Nat. Rev. Mol. Cell Biol. 2003;4:435–445. doi: 10.1038/nrm1127. [DOI] [PubMed] [Google Scholar]
- 15.Kowalczykowski SC. Biochemical and biological function of Escherichia coli RecA protein: behavior of mutant RecA proteins. Biochimie. 1991;73:289–304. doi: 10.1016/0300-9084(91)90216-n. [DOI] [PubMed] [Google Scholar]
- 16.Ogawa T, Yu X, Shinohara A, Egelman EH. Similarity of the yeast RAD51 filament to the bacterial RecA filament. Science. 1993;259:1896–1899. doi: 10.1126/science.8456314. [DOI] [PubMed] [Google Scholar]
- 17.Sung P, Robberson DL. DNA strand exchange mediated by a RAD51-ssDNA nucleoprotein filament with polarity opposite to that of RecA. Cell. 1995;82:453–461. doi: 10.1016/0092-8674(95)90434-4. [DOI] [PubMed] [Google Scholar]
- 18.Bugreev DV, Mazin AV. Ca2+ activates human homologous recombination protein Rad51 by modulating its ATPase activity. Proc. Natl Acad. Sci. USA. 2004;101:9988–9993. doi: 10.1073/pnas.0402105101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Y, Stasiak AZ, Masson JY, McIlwraith MJ, Stasiak A, West SC. Conformational changes modulate the activity of human RAD51 protein. J. Mol. Biol. 2004;337:817–827. doi: 10.1016/j.jmb.2004.02.022. [DOI] [PubMed] [Google Scholar]
- 20.Shim KS, Schmutte C, Yoder K, Fishel R. Defining the salt effect on human RAD51 activities. DNA Repair. 2006;5:718–730. doi: 10.1016/j.dnarep.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 21.Amunugama R, He Y, Willcox S, Forties RA, Shim KS, Bundschuh R, Luo Y, Griffith J, Fishel R. RAD51 protein ATP cap regulates nucleoprotein filament stability. J. Biol. Chem. 2012;287:8724–8736. doi: 10.1074/jbc.M111.239426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alexiadis V, Kadonaga JT. Strand pairing by Rad54 and Rad51 is enhanced by chromatin. Genes Dev. 2002;16:2767–2771. doi: 10.1101/gad.1032102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sinha M, Peterson CL. A Rad51 presynaptic filament is sufficient to capture nucleosomal homology during recombinational repair of a DNA double-strand break. Mol. Cell. 2008;30:803–810. doi: 10.1016/j.molcel.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Z, Fan HY, Goldman JA, Kingston RE. Homology-driven chromatin remodeling by human RAD54. Nat. Struct. Mol. Biol. 2007;14:397–405. doi: 10.1038/nsmb1223. [DOI] [PubMed] [Google Scholar]
- 25.Shah PP, Zheng X, Epshtein A, Carey JN, Bishop DK, Klein HL. Swi2/Snf2-related translocases prevent accumulation of toxic Rad51 complexes during mitotic growth. Mol. Cell. 2010;39:862–872. doi: 10.1016/j.molcel.2010.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dupaigne P, Lavelle C, Justome A, Lafosse S, Mirambeau G, Lipinski M, Pietrement O, Le Cam E. Rad51 polymerization reveals a new chromatin remodeling mechanism. PLoS One. 2008;3:e3643. doi: 10.1371/journal.pone.0003643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Polach KJ, Widom J. Restriction enzymes as probes of nucleosome stability and dynamics. Methods Enzymol. 1999;304:278–298. doi: 10.1016/s0076-6879(99)04017-3. [DOI] [PubMed] [Google Scholar]
- 28.Tombline G, Fishel R. Biochemical characterization of the human RAD51 protein. I. ATP hydrolysis. J. Biol. Chem. 2002;277:14417–14425. doi: 10.1074/jbc.M109915200. [DOI] [PubMed] [Google Scholar]
- 29.Sung P. Catalysis of ATP-dependent homologous DNA pairing and strand exchange by yeast RAD51 protein. Science. 1994;265:1241–1243. doi: 10.1126/science.8066464. [DOI] [PubMed] [Google Scholar]
- 30.Simon M, North JA, Shimko JC, Forties RA, Ferdinand MB, Manohar M, Zhang M, Fishel R, Ottesen JJ, Poirier MG. Histone fold modifications control nucleosome unwrapping and disassembly. Proc. Natl Acad. Sci. USA. 2011;108:12711–12716. doi: 10.1073/pnas.1106264108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luger K, Rechsteiner TJ, Richmond TJ. Expression and purification of recombinant histones and nucleosome reconstitution. Methods Mol. Biol. 1999;119:1–16. doi: 10.1385/1-59259-681-9:1. [DOI] [PubMed] [Google Scholar]
- 32.Shimko JC, North JA, Bruns AN, Poirier MG, Ottesen JJ. Preparation of fully synthetic histone H3 reveals that acetyl-lysine 56 facilitates protein binding within nucleosomes. J. Mol. Biol. 2011;408:187–204. doi: 10.1016/j.jmb.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Manohar M, Mooney AM, North JA, Nakkula RJ, Picking JW, Edon A, Fishel R, Poirier MG, Ottesen JJ. Acetylation of histone H3 at the nucleosome dyad alters DNA-histone binding. J. Biol. Chem. 2009;284:23312–23321. doi: 10.1074/jbc.M109.003202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clegg RM. Fluorescence resonance energy transfer and nucleic acids. Methods Enzymol. 1992;211:353–388. doi: 10.1016/0076-6879(92)11020-j. [DOI] [PubMed] [Google Scholar]
- 35.Li G, Widom J. Nucleosomes facilitate their own invasion. Nat. Struct. Mol. Biol. 2004;11:763–769. doi: 10.1038/nsmb801. [DOI] [PubMed] [Google Scholar]
- 36.Lowary PT, Widom J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol. 1998;276:19–42. doi: 10.1006/jmbi.1997.1494. [DOI] [PubMed] [Google Scholar]
- 37.Rhodes D. Structural analysis of a triple complex between the histone octamer, a Xenopus gene for 5S RNA and transcription factor IIIA. EMBO J. 1985;4:3473–3482. doi: 10.1002/j.1460-2075.1985.tb04106.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Richmond TJ, Davey CA. The structure of DNA in the nucleosome core. Nature. 2003;423:145–150. doi: 10.1038/nature01595. [DOI] [PubMed] [Google Scholar]
- 39.North JA, Javaid S, Ferdinand MB, Chatterjee N, Picking JW, Shoffner M, Nakkula RJ, Bartholomew B, Ottesen JJ, Fishel R, et al. Phosphorylation of histone H3(T118) alters nucleosome dynamics and remodeling. Nucleic Acids Res. 39:6465–6474. doi: 10.1093/nar/gkr304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Polach KJ, Widom J. Mechanism of protein access to specific DNA sequences in chromatin: a dynamic equilibrium model for gene regulation. J. Mol. Biol. 1995;254:130–149. doi: 10.1006/jmbi.1995.0606. [DOI] [PubMed] [Google Scholar]
- 41.Essers J, Hendriks RW, Wesoly J, Beerens CE, Smit B, Hoeijmakers JH, Wyman C, Dronkert ML, Kanaar R. Analysis of mouse Rad54 expression and its implications for homologous recombination. DNA Repair. 2002;1:779–793. doi: 10.1016/s1568-7864(02)00110-6. [DOI] [PubMed] [Google Scholar]
- 42.Javaid S, Manohar M, Punja N, Mooney A, Ottesen JJ, Poirier MG, Fishel R. Nucleosome remodeling by hMSH2-hMSH6. Mol. Cell. 2009;36:1086–1094. doi: 10.1016/j.molcel.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.North JA, Javaid S, Ferdinand MB, Chatterjee N, Picking JW, Shoffner M, Nakkula RJ, Bartholomew B, Ottesen JJ, Fishel R, et al. Phosphorylation of histone H3(T118) alters nucleosome dynamics and remodeling. Nucleic Acids Res. 2011;39:6465–6474. doi: 10.1093/nar/gkr304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thastrom A, Lowary PT, Widlund HR, Cao H, Kubista M, Widom J. Sequence motifs and free energies of selected natural and non-natural nucleosome positioning DNA sequences. J. Mol. Biol. 1999;288:213–229. doi: 10.1006/jmbi.1999.2686. [DOI] [PubMed] [Google Scholar]
- 45.Hombauer H, Campbell CS, Smith CE, Desai A, Kolodner RD. Visualization of eukaryotic DNA mismatch repair reveals distinct recognition and repair intermediates. Cell. 2011;147:1040–1053. doi: 10.1016/j.cell.2011.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hombauer H, Srivatsan A, Putnam CD, Kolodner RD. Mismatch repair, but not heteroduplex rejection, is temporally coupled to DNA replication. Science. 2011;334: 1713–1716. doi: 10.1126/science.1210770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Symington LS, Heyer WD. Some disassembly required: role of DNA translocases in the disruption of recombination intermediates and dead-end complexes. Genes Dev. 2006;20:2479–2486. doi: 10.1101/gad.1477106. [DOI] [PubMed] [Google Scholar]
- 48.Jensen RB, Carreira A, Kowalczykowski SC. Purified human BRCA2 stimulates RAD51-mediated recombination. Nature. 2010;467:678–683. doi: 10.1038/nature09399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thorslund T, McIlwraith MJ, Compton SA, Lekomtsev S, Petronczki M, Griffith JD, West SC. The breast cancer tumor suppressor BRCA2 promotes the specific targeting of RAD51 to single-stranded DNA. Nat. Struct. Mol. Biol. 2010;17:1263–1265. doi: 10.1038/nsmb.1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Petrini JH. At the end, remodeling leads to eviction. Nat. Struct. Mol. Biol. 2005;12:1028–1029. doi: 10.1038/nsmb1205-1028. [DOI] [PubMed] [Google Scholar]
- 51.Tsukuda T, Fleming AB, Nickoloff JA, Osley MA. Chromatin remodelling at a DNA double-strand break site in Saccharomyces cerevisiae. Nature. 2005;438:379–383. doi: 10.1038/nature04148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 2011;145:529–542. doi: 10.1016/j.cell.2011.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cox MM. The bacterial RecA protein as a motor protein. Annu. Rev. Microbiol. 2003;57:551–577. doi: 10.1146/annurev.micro.57.030502.090953. [DOI] [PubMed] [Google Scholar]
- 54.Kowalczykowski SC, Eggleston AK. Homologous pairing and DNA strand-exchange proteins. Annu. Rev. Biochem. 1994;63:991–1043. doi: 10.1146/annurev.bi.63.070194.005015. [DOI] [PubMed] [Google Scholar]
- 55.Menetski JP, Bear DG, Kowalczykowski SC. Stable DNA heteroduplex formation catalyzed by the Escherichia coli RecA protein in the absence of ATP hydrolysis. Proc. Natl Acad. Sci. USA. 1990;87:21–25. doi: 10.1073/pnas.87.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morrison C, Shinohara A, Sonoda E, Yamaguchi Iwai Y, Takata M, Weichselbaum RR, Takeda S. The essential functions of human Rad51 are independent of ATP hydrolysis. Mol. Cell. Biol. 1999;19:6891–6897. doi: 10.1128/mcb.19.10.6891. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.