


Abstract
Yeast pre-mRNA splicing initiates via formation of a complex comprising U1 snRNP bound at the 5′ splice site (5′SS) and the Msl5•Mud2 heterodimer engaged at the branchpoint (BP). Here, we present a mutational analysis of the U1 snRNA, which shows that although enlarging the 5′ leader between the TMG cap and the 3ACUUAC8 motif that anneals to the 5′SS is tolerated, there are tight constraints on the downstream spacer between 3ACUUAC8 and helix 1 of the U1 fold. We exploit U1 alleles with 5′ extensions, variations in the 3ACUUAC8 motif, downstream mutations and a longer helix 1 to discover new intra-snRNP synergies with U1 subunits Nam8 and Mud1 and the trimethylguanosine (TMG) cap. We describe novel mutations in U1 snRNA that bypass the essentiality of the DEAD-box protein Prp28. Structure-guided mutagenesis of Msl5 distinguished four essential amino acids that contact the BP sequence from nine other BP-binding residues that are inessential. We report new synthetic genetic interactions of the U1 snRNP with Msl5 and Mud2 and with the nuclear cap-binding subunit Cbc2. Our results fortify the idea that spliceosome assembly can occur via distinct genetically buffered microscopic pathways involving cross-intron-bridging interactions of the U1 snRNP•5′SS complex with the Mud2•Msl5•BP complex.
INTRODUCTION
The composition, function and dynamics of the pre-mRNA splicing machinery (the ‘spliceosome’) in the budding yeast Saccharomyces cerevisiae have been elucidated genetically and biochemically (1–3). The yeast spliceosome transits through assembly, activation, catalysis and disassembly steps that are programmed by the U1, U2, U4, U5 and U6 snRNPs and an army of proteins that interact with the snRNPs and the pre-mRNA. The first stage in spliceosome assembly entails the formation of a complex comprising the U1 snRNP bound at the intronic 5′ splice site (5′SS; consensus sequence: 5′-GUAUGU) and the Msl5•Mud2 protein heterodimer engaged at the intron branchpoint (BP) (consensus sequence: 5′-UACUAAC). Bridging interactions between U1 snRNP and Msl5•Mud2 serve to stabilize the complex and prepare a scaffold for recruitment of the U2 snRNP to the BP.
The yeast U1 snRNP consists of a trimethylguanosine (TMG)-capped 568-nt U1 snRNA, a toroidal Sm protein ring composed of seven subunits (present also in the U2, U4 and U5 snRNPs), and 10 additional protein subunits unique to the yeast U1 snRNP: Prp39, Prp40, Snu71, Snu56, Snp1, Mud1, Luc7, Prp42, Nam8 and Yhc1 (4–10). Yeast U1-specific subunits Snp1, Mud1 and Yhc1 are conserved in human U1 snRNP as U1-70K, U1-A and U1-C, respectively. The 5′ sequence of yeast U1 RNA—m2,2,7GpppAUACUUACC—is conserved in the much smaller human U1 snRNA (164 nt) and contains a hexanucleotide motif (underlined) complementary to the consensus 5′SS. This U1 5′ sequence anneals to the pre-mRNA to nucleate an early assembly complex. The U1 snRNP protein subunits interact with U1 RNA sequences and/or secondary structures, contact the pre-mRNA or contact other protein components of the spliceosome during its initial assembly (11–13). Additional non-snRNP elements aid in U1•pre-mRNA complex assembly, including the pre-mRNA m7G cap structure in association with the yeast nuclear cap-binding complex (CBC: a Sto1•Cbc2 heterodimer) (14). The U1 snRNP is ultimately displaced from the pre-mRNA•U1•U2-containing spliceosome at the point when the U5•U4•U6 tri-snRNP complex joins en route to forming a pre-mRNA•U2•U5•U6 spliceosome. Dissociation of U1 snRNP is thought to be triggered by the essential DEAD-box protein Prp28 (15,16), acting to disrupt the short U1:5′SS RNA duplex or remodel protein–RNA contacts at the 5′SS (or both).
Although yeast U1 RNA is essential for viability, substantial chunks of the U1 primary structure (more than half) are dispensable for cell growth (17,18). This situation inspired an elegant and highly successful genetic screen by the Rosbash laboratory for yeast mutations that caused synthetic lethality with an otherwise viable U1 snRNA mutation in which a large internal deletion was combined with a G27A change in the stem-loop-1 (SL1) sequence that interacts with U1-70K (19). The ‘Mutant-U1-Die’ or ‘MUD’ screen identified the genes encoding four of the U1-specific subunits: Mud1, Snu56/Mud10, Nam8/Mud15 and Prp42/Mud16 (5,19). The screen also identified the Cbc2/Mud13 subunit of nuclear CBC and the Mud2 subunit of the yeast BP-binding protein complex (Mud2 being the yeast homolog of mammalian U2AF65) (20,21). The findings that Mud1, Nam8, Cbc2 and Mud2 are themselves inessential for yeast vegetative growth immediately highlighted a network of genetically buffered functions centered around U1 snRNP during early spliceosome assembly. The Rosbash laboratory then bootstrapped the viable mud2Δ mutant to execute a ‘Mud2 synthetic lethal’ (MSL) screen and thereby identify Msl5, the yeast BP-binding protein, and Msl1, the U2B′′ subunit of the U2 snRNP (22,23).
Additional directed and genome-wide synthetic genetic array analyses have greatly expanded the network of mutational synergies involving: the U1 snRNP subunits Mud1, Nam8, Luc7 and Yhc1; the TMG cap structure of U1 snRNA (via inactivation of TMG synthase Tgs1); m7G cap binding by nuclear CBC; and the Msl5•Mud2 BP binding complex (24–32). Such genetic interactions among individually dispensable players (or otherwise benign mutations in essential factors) that act in a common pathway meet an operational definition of redundancy. Genetic redundancy does not necessitate that the synthetic interactor proteins or RNA elements perform the same task, but it rather suggests that spliceosome assembly can be accomplished or stabilized via different microscopic sub-pathways.
In the present study, we interrogate structure–function relations of the 5′ end of yeast U1 snRNA, by studying the effects of 5′ extensions; nucleobase changes in the 3ACUUAC8 sequence that pairs with the 5′SS; and insertions, deletions and nucleobase changes in the flanking 9CUU11 segment that interacts with U1-C and connects the U1•5′SS RNA duplex to helix 1 of the U1 RNA fold (4,8). We find that extending the segment upstream of the 3ACUUAC8 element has no effect on growth but is synthetically lethal or sick with mutations affecting TMG capping, m7G cap-binding by CBC, U1 snRNP protein Nam8 and the Msl5•Mud2 BP-binding complex. Insertions of 1 or 3 nt downstream of 3ACUUAC8 are viable, but display synthetic phenotypes; by contrast, insertions of ≥5 nucleotides are lethal, suggesting that the spacing between the U1•5′SS RNA duplex and helix 1 is important for U1 snRNP function. Changes that increase the length of helix 1 are benign per se but synthetic in combination with other agents of spliceosome assembly. Viable mutations within the 3ACUUAC8 element display allele-specific synergies. We also indentify novel mutations in U1 snRNA that bypass the essentiality of Prp28. Our results reveal a rich network of intramolecular and intermolecular genetic interactions of the yeast U1 snRNP, especially with the BP-binding protein Msl5.
MATERIALS AND METHODS
U1 expression plasmids and mutants
A 1.3-kb DNA segment bearing the SNR19 (U1) gene was amplified from S. cerevisiae genomic DNA by PCR using a forward primer that introduced an XhoI site 550 nt upstream of the transcription start site (+1) of U1 snRNA and a reverse primer downstream of a HindIII site at position +755. The DNA fragment was inserted into centromeric plasmids pRS316 (URA3) and pRS415 (LEU2). The resulting yeast expression plasmids p316-U1 and p415-U1 include the 568-nt U1 snRNA sequence plus 550 and 187 nt of upstream and downstream sequences. Mutations (insertions, deletions and single nucleotide changes) were introduced into the U1 plasmid by two-stage PCR overlap extension with mutagenic primers. The U1 genes were sequenced completely to confirm that no unwanted changes were acquired during amplification and cloning.
Msl5 expression plasmids and mutants
CEN LEU2 plasmids bearing wild-type and mutated MSL5 genes under the control of the native MSL5 promoter have been described (31). New missense mutations L169A, R172A, L176A, I189A, R190A, L256A and L259A in the KH-QUA2 domain and P97A, P98A, Y100A and Y100F in the PPxY motif were introduced into MSL5 by two-stage PCR overlap extension with mutagenic primers. The PCR products were digested and then inserted into the pRS415-based MSL5 expression plasmid (31). The MSL5 genes were sequenced completely to confirm that no unwanted changes were acquired during amplification and cloning. CEN HIS3 MSL5 plasmids were constructed by subcloning 2.2-kb MSL5 fragments (excised with XhoI and SacI from pRS415-MSL5) into pRS413.
Yeast strains and tests of U1 function in vivo
To develop a plasmid shuffle assay for gauging mutational effects on U1 function, we generated a U1Δ strain that relies for viability on maintenance of the wild-type U1 gene on a CEN URA3 plasmid, p316-U1. In brief, we first replaced the U1 locus from position +1 to +532 with a kanMX cassette in the BY4743 diploid strain and then transformed the heterozygous diploid with p316-U1. The diploid was sporulated, asci were dissected and haploid U1Δ Ura+ progeny were recovered. U1Δ (p316-U1) cells were resistant to G418 and unable to grow on medium containing 0.75 mg/ml 5-fluoroorotic acid (FOA). To assay the function of wild-type and mutated U1 alleles, U1Δ (p316-U1) cells were transfected with CEN LEU2 U1 plasmids. Individual Leu+ transformants were selected and streaked on agar medium containing FOA. The plates were incubated at 20, 30 or 37°C, and mutants that failed to form macroscopic colonies at any temperatures after 10 d were deemed lethal. Individual FOA-resistant colonies with viable U1 alleles were grown to mid-log phase in yeast extract, peptone, dextrose (YPD) broth and adjusted to the same A600 values. Aliquots (3 µl) of serial 10-fold dilutions were spotted to YPD agar plates, which were then incubated at temperatures ranging from 18 to 37°C.
We also developed plasmid shuffle assays to test the mutational effects on U1 function in mud2Δ, tgs1Δ, nam8Δ, mud1Δ, swt21Δ, brr1Δ, cbc2-Y24A, swm2Δ and tgs1Δ cbc2-Y24A cells (7,27,29–32) using standard genetic manipulations of mating, sporulation and dissection. We thereby generated mud2Δ U1Δ (p316-U1), nam8Δ U1Δ (p316-U1), mud1Δ U1Δ (p316-U1), tgs1Δ U1Δ (p316-U1), swt21Δ U1Δ (p316-U1), swm2Δ U1Δ (p316-U1), brr1Δ U1Δ (p316-U1), cbc2-Y24A U1Δ (p316-U1) and tgs1Δ cbc2-Y24A U1Δ (p316-U1) cells, which were unable to grow on FOA-containing medium, unless they had been transformed with wild-type U1 or a functional U1 mutant allele on a CEN LEU2 plasmid before FOA selection. To investigate genetic interactions of U1 mutants with the essential MSL5 and PRP28 genes, we first generated heterozygous msl5Δ/MSL5 U1Δ/U1 and prp28Δ/PRP28 U1Δ/U1 diploids by crossing msl5Δ::natMX p316-MSL5 and prp28Δ::natMX p316-PRP28 cells with U1Δ::kanMX p316-U1 cells of the opposite mating type, selecting diploids on YPD medium containing G418 and clonNat and plating them to FOA-containing medium. The heterozygous diploids were then transformed with CEN URA3 U1 MSL5 (p316-U1-MSL5) or CEN URA3 U1 PRP28 (p316-U1-PRP28) plasmids. [In these plasmids, the U1 gene (−550 to +755) is arranged in a head-to-head configuration with the MSL5 gene or the PRP28 gene (−520 to +2120).] Ura+ heterozygous diploids were subjected to sporulation and tetrad dissection, after which haploid prp28Δ U1Δ (p316-U1-PRP28 and msl5Δ U1Δ (p316-U1-MSL5) progeny were recovered. These cells were unable to grow on FOA medium, but the double-deletion strains could be complemented by cotransformation with p(CEN LEU2 U1) plus either p(CEN HIS3 MSL5) or p(CEN HIS3 PRP28).
Primer extension analysis of U1 5′ ends
U1Δ cells bearing CEN LEU2 U1 plasmids were grown in YPD broth to mid-log phase. Cellular RNA was isolated using an RNeasy kit (Qiagen). Aliquots (20 µg of RNA) were used as templates for reverse transcriptase-catalyzed extension of 5′ 32P-labeled primers 5′-d(GAATGGAAACGTCAGCAAACAC) and 5′-d(CTTAAAAAGTCTCTTCCCGTC) that are complementary to U1 and U2 snRNA, respectively. The primer extension reactions were performed as described previously (33).
RESULTS
Effect of U1 5′ extensions that distance the cap from the 3ACUUAC8 sequence
Our interest in the 5′ end of yeast U1 snRNA was sparked by our earlier findings that (i) nuclear CBC became a stoichiometric subunit of the U1 snRNP in yeast tgs1Δ cells that lack the methyltransferase enzyme that converts m7G caps to TMG caps and (ii) the cold-sensitive (cs) growth phenotype of yeast tgs1Δ cells was rescued by mutations in the cap-binding pocket of nuclear CBC that have no effect on growth per se (7,32). Our inference was that ectopic binding of CBC to the residual m7G U1 snRNA cap was a key factor in the tgs1Δ cs phenotype and that reducing CBC affinity for the m7G U1 snRNA cap restored normal growth. Because the U1 snRNA cap is located just 2 nt upstream of the 3ACUUAC8 sequence that pairs with the pre-mRNA 5′SS, we considered that steric hindrance by CBC at the U1 5′ end might interfere with U1 function, in which case increasing the physical distance between CBC and the 3ACUUAC8 sequence might suppress the tgs1Δ cs defect.
To test this idea, we extended the 5′ sequence of the U1 gene by 5, 10, 15, 20, 25 or 30 nt, as depicted in Figure 1A. The wild-type (WT) and 5′-extended U1 alleles under the control of the native U1 promoter were placed on CEN LEU2 plasmids and tested for function in vivo by plasmid shuffle in a yeast strain deleted at the chromosomal U1 locus but bearing a WT U1 gene on a CEN URA3 plasmid. All of the 5′-extended U1 alleles supported growth of yeast U1Δ cells on medium containing FOA, a drug that selects against the CEN URA3 U1 plasmid. The U1 +5, +10, +15, +20, +25 and +30 strains grew as well as U1 WT cells at 20, 25, 30, 34 and 37°C, as gauged by spotting serial dilutions of the respective strains on YPD agar (Figure 1B). To assess whether the newly inserted sequences at the 5′ end of the U1 gene were transcribed into the U1 snRNA, we performed primer extension analysis on total RNA isolated from yeast cells bearing the WT and 5′-extended U1 alleles (Figure 1C). A 5′ 32P-labeled DNA oligonucleotide complementary to yeast U1 snRNA was extended by reverse transcriptase to yield a discrete cDNA corresponding in size to the distance (in nucleotides) from the primer 5′ end to the 5′ end of the U1 snRNA. A second 5′ 32P-labeled DNA oligonucleotide complementary to U2 snRNA was included in the reverse transcription reactions as a control. Denaturing polyacrylamide gel electrophoresis (PAGE) analysis of the primer extension products revealed that the 5′ ends of the U1 snRNAs were shifted serially ‘upstream’ by 5-nt intervals, as expected from the U1 DNA sequences. Thus, the DNA additions did not alter the site of transcription initiation directed by the 5′-flanking U1 gene promoter. The amounts of U1 cDNA synthesized from total RNAs derived from the WT and 5′-extended U1 yeast strains were similar, signifying that the steady-state levels of U1 snRNA were not affected by the 5′ leader sequences. The 5′ ends and steady-state levels of U2 snRNAs were unaltered by the U1 mutations.
Figure 1.

U1 snRNAs with extended 5′ ends are functional. (A) The DNA sequences are shown for the 5′ ends of wild-type U1 (WT) and the mutant variants +5, +10 and so forth, named according to the number of nucleotides inserted upstream of the 3ACTTAC8 segment (highlighted in gray) that pairs with the intron 5′SS. (B) The growth phenotypes of U1Δ p(CEN LEU2 U1) cells bearing the indicated U1 alleles were assessed as follows. Liquid cultures were grown to mid-log phase at 30°C and adjusted to the same A600. Aliquots (3 µl) of serial 10-fold dilutions of cells were spotted to YPD agar. The plates were incubated at the indicated temperatures and photographed after 2 d (30, 34 and 37°C), 3 d (25°C) or 4 d (20°C). (C) Primer extension analyses with 32P-labeled primers complementary to U1 snRNA (nt 161–182 ) and U2 snRNA (nt 140–160 ) was performed using as template total cellular RNA isolated from the indicated U1Δ p(CEN LEU2 U1) strains. The reaction products were analyzed by denaturing PAGE and visualized by autoradiography. The sizes (nt) of 32P-labeled marker DNAs that were analyzed in parallel are indicated at left.
Having shown that U1 snRNA tolerates installation of new RNA sequences upstream of the 3ACUUAC8 sequence, we constructed a tgs1Δ U1Δ p(CEN URA3 U1) strain to test how separating the U1 cap from the 5′SS interaction element would affect tgs1Δ. To our surprise, rather than alleviating the tgs1Δ cs phenotype, the U1 5′ extensions exacerbated the impact of losing the TMG cap. A gradient of increasing synthetic sickness with tgs1Δ was seen for the U1 +5, +25, +10 and +20 alleles (Figure 2). The +15 and +30 U1 alleles were lethal in the tgs1Δ background.
Figure 2.

Synthetic genetic interactions of the U1 5′ extensions. Plasmid shuffle assays were used to test whether 5′ extended U1 snRNAs are functional in various genetic backgrounds. Synthetic lethality was indicated by failure to form macroscopic colonies on FOA agar after 10 d at 20, 30 and 37°C. Cultures of viable FOA-resistant cells were grown in YPD broth at 37°C and the growth phenotypes were assessed by spotting serial 10-fold dilutions as described in Figure 1B. The U1 alleles are specified on the left and the genetic background for the test of mutational synergy is indicated on the right.
Synthetic genetic interactions of the U1 5′ extensions
A possible explanation for the synthetic lethality/sickness of tgs1Δ with the 5′-extended U1s is that the effect of ectopic binding of CBC to the U1 m7G cap in the tgs1Δ strain is more severe when the cap is distanced from the body of U1 by an otherwise benign 5′ RNA leader sequence (e.g. because the distance allows even freer access of CBC to the U1 cap at all growth temperatures). If this is the case, then we thought that the cbc2-Y24A mutation in the cap-binding pocket of nuclear CBC, which has no effect per se on yeast cell growth (Figure 2; cbc2-Y24A U1-WT) and which suppresses the tgs1Δ cs phenotype (Figure 2; tgs1Δ cbc2-Y24A U1-WT), might ameliorate the synergy between tgs1Δ and the 5′ U1 extensions. A necessary step en route to that issue was to query the effects of the U1 5′ extensions in a cbc2-Y24A background. This experiment revealed a gradient of synthetic sick and synthetic lethal interactions between cbc2-Y24A and the U1 mutant series, whereby the +5, +10, +20 and +25 extensions caused cs growth defects of increasing severity (reflected in the upward shift in the restrictive temperature) and the +15 and +30 extensions were lethal at all temperatures in the cbc2-Y24A background (Figure 2; cbc2-Y24A). Tyr24 in Cbc2 is the equivalent of Tyr20 in mammalian Cbc2 that forms a π–cation stack with the m7G nucleobase to confer high-affinity cap binding. The synergistic effects of cbc2-Y24A with the altered U1 snRNAs seen here resonate with the recently reported synthetic lethality or sickness of cbc2-Y24A with null mutations of proteins involved in early steps of spliceosome assembly (Nam8, Mud1, Swt21, Mud2, Ist3 and Brr1) and with otherwise benign mutations of Msl5, the essential BP-binding protein (32). These combinatorial effects highlight how the contributions to spliceosome assembly of CBC engaged at the 5′ pre-mRNA cap are genetically buffered by other components of the splicing machinery. The present results identify the U1 snRNA 5′ end as a novel component of this genetic network. In light of the aforementioned findings, it was not surprising that the growth phenotypes of the 5′-extended U1 alleles in the tgs1Δ cbc-Y24A background were virtually identical to those in the tgs1Δ background (Figure 2).
To delineate additional genetic interactions of the U1 5′ extensions, we constructed a series of yeast strains in which genes encoding inessential splicing factors were deleted in the U1Δ p(CEN URA3 U1) background, thereby allowing tests of synergy by plasmid shuffle. Although the ablation of U1 snRNP subunit Nam8 has no effect on yeast vegetative growth by itself, nam8Δ was lethal in combination with the U1 +30 allele and caused a strong cs growth defect in combination with the five other U1 5′ extension alleles, manifest as no growth at 25°C, minimal growth at 30°C and varying degrees of slow growth at 34°C (Figure 2). The U1 5′ extensions had less of an impact in the absence of U1 snRNP subunit Mud1, whereby all alleles except +30 were viable at 25°C in the mud1Δ background (Supplementary Figure S1). The U1 5′ extensions +15, +20 and +25 displayed cs synthetic sickness with a null mutation of the early splicing factor Swt21 that interacts genetically with CBC, Tgs1, Yhc1 and Prp28 (27,34); here, too the U1 +30 allele had the most impact, with feeble growth at 25 and 20°C in the swt21Δ background (Supplementary Figure S1). The U1 +20, +25, and +30 alleles grew poorly at 25 and 20°C in the absence of Brr1, a splicing factor implicated in snRNP biogenesis (35) (Supplementary Figure S1).
The strongest mutational synergies of the U1 5′ extensions were seen when the Mud2 subunit of the yeast heterodimeric BP-binding protein was ablated. Although mud2Δ cells grew normally with a WT U1 snRNA, the U1 +5 allele was barely viable in the mud2Δ background, and the +10, +15, +20, +25 and +30 alleles were lethal (Figure 2). By contrast, the U1 5′ extensions displayed little or no synergy with a null mutation in Swm2 (Supplementary Figure S1), so named because swm2Δ is synthetic with mud2Δ (29).
Effects of insertions and deletions immediately downstream of the 3ACUUAC8 sequence
The 5′ AUACUUACCU10 single-stranded segment of U1 snRNA precedes the folded U1 RNA tertiary structure that initiates at nucleotide U11 (i.e. a four-helix junction depicted in Figure 4A). The 8CCU10 segment connecting the 5′SS complementary motif to helix 1 interacts with the U1-C subunit in the U1 snRNP (4,8). Here, we queried the importance of the spacing between the 5′SS complementary motif and helix 1 by introducing insertions of 1–30 nt between C8 and C9 (Figure 3). The insertion alleles were tested for bioactivity by plasmid shuffle in the U1Δ strain. The [+1] and [+3] insert strains were viable at 18–37°C, albeit slower growing than WT U1 cells at low temperatures (Figure 3). By contrast, insertions of ≥5 nt between C8 and C9 were uniformly lethal (Figure 3). We performed primer extension analysis on RNA isolated from yeast cells with a wild-type chromosomal U1 gene that had been transformed with CEN plasmids bearing the series of U1 insertion alleles (Supplementary Figure S2). Denaturing PAGE analysis of the 5′ 32P-labeled primer extension products revealed that the 5′ ends of the plasmid-encoded [+1], [+3], [+5], [+10], [+20] and [+30] U1 snRNAs were shifted incrementally upstream compared with the endogenous wild-type U1 snRNA. The levels of radiolabeled cDNAs derived from the U1 snRNA insertion mutants were at least as high as the cDNA corresponding to endogenous wild-type U1 snRNA (Supplementary Figure S2), signifying that (i) the steady-state levels of the U1 snRNA were not affected by the inserted sequences and (ii) the lethality of the [+5], [+10], [+20] and [+30] U1 alleles was not attributable to a failure the produce the mutant snRNAs. We surmise that there are tight constraints on the length (and/or sequence) of the linker segment that interacts with U1-C. This theme was underscored as we systematically tested the viable [+1] and [+3] insert alleles for mutational synergies, which revealed that the [+1] and [+3] insertions were lethal in combination with tgs1Δ, cbc2-Y24A, nam8Δ, mud1Δ, swt21Δ and mud2Δ. Thus, even a single extra nucleotide sufficed to render U1 snRNA function dependent on otherwise inessential components of the U1 snRNP or the early spliceosome.
Figure 4.

Effects of deletions immediately downstream of 3ACUUAC8 and of extending the base-pairing potential of helix 1. (A) The primary and predicted secondary structures at the 5′ end of wild-type U1 RNA and the H1L mutant are shown. The 3ACUUAC8 segment that pairs with the intron 5′SS is highlighted in gray. Helix 1 is shaded orange. The mutation of 9CU10 to 9UA10 (colored red) extends helix 1 by two base pairs, which are shaded yellow. (B) Yeast strains bearing U1 WT, H1L or ΔU10 alleles in the indicated genetic backgrounds were spot-tested for growth at the temperatures specified. (C) The primary and predicted secondary structures of the ΔU10, ΔC9 and ΔC9U10 deletion mutants of U1 snRNA are shown. The ΔC9 and ΔC9U10 alleles failed to complement U1Δ and were deemed lethal. Genetic interactions of H1L and ΔU10 are indicated below the respective structures in (A) and (C).
Figure 3.

Effects of insertions immediately downstream of the 3ACUUAC8 sequence. The DNA sequences are shown for the 5′ ends of WT U1 and the mutant variants [+1], [+3], etc., named according to the number of nucleotides inserted between positions C8 and C9. The 3ACTTAC8 segment that pairs with the intron 5′SS is highlighted in gray. The [+5], [+10], [+20] and [+30] mutants failed to complement U1Δ in a plasmid shuffle assay and were deemed lethal. The viable U1-[+1] and U1-[+3] strains were spot-tested for growth at the indicated temperatures in parallel with WT cells as per Figure 1B, except that the plates were incubated for 5 d at 25, 20 and 18°C. The synthetic lethal interactions of the U1-[+1] and U1-[+3] mutants are indicated at the bottom of the figure.
We also tested the effects of shortening the linker segment, by deleting nucleotides C9 and U10, singly and in combination. The predicted 5′ structures of the deleted U1 RNAs are shown in Figure 4C. When tested by plasmid shuffle, the U1 ΔC9 and ΔC9U10 mutations were lethal. [Primer extension analysis showed that the mutant ΔC9U10 U1 snRNA was synthesized from the plasmid-borne ΔC9U10 gene in vivo and achieved steady-state levels at least as high as the chromosome-encoded wild-type U1 (Supplementary Figure S2).] By contrast, yeast U1-ΔU10 cells were viable and grew as well as WT cells at 37 and 34°C, but were slow growing at 25 and 20°C (Figure 4B). The U1-ΔU10 allele was synthetic lethal with tgs1Δ, cbc2-Y24A, nam8Δ, mud1Δ, swt21Δ and mud2Δ.
Effects of extending the base-pairing potential of helix 1
Helix 1 is conserved with respect to its presence and position in yeast and human U1 snRNAs but is not conserved at the level of primary structure. Whereas the 5′ strand of helix 1 in yeast U1 is 11UAAGAU16 (Figure 4A), the corresponding segment of human helix 1 is 12GCAGG16 (8). To probe the effects of increasing the length of yeast helix 1, we changed the 9CU10 dinucleotide to 9UA10, thereby extending the base-pairing potential to the sequence at the 3′ end of U1 RNA such that helix 1 might span 8 bp instead of 6 bp (Figure 4A). This U1 allele, named H1L (‘helix 1 long’), supported normal yeast growth at all temperatures tested (Figure 4B). Though benign per se, the H1L mutant displayed a broad range of synthetic genetic interactions with other splicing factors. At the most severe end of the spectrum, U1-H1L was synthetic lethal with mud2Δ. H1L was barely viable at 20–34°C in the mud1Δ background and did not grow at 37°C (Figure 4B). H1L synergized with tgs1Δ and cbc2-Y24A, resulting in feeble growth at 37 and 34°C and severe cs defects at the lower temperatures (Figure 4B). H1L caused slow growth at all temperatures in the nam8Δ and swt21Δ backgrounds (Figure 4B). These results show that increasing the size or stability of helix 1 enforces reliance on otherwise dispensable spliceosome components and assembly factors, especially Mud2.
Novel mutational effects in the 3ACUUAC8 sequence
The base-pairing interaction between the 5′ end of yeast U1 snRNA and the consensus 5′SS of yeast pre-mRNAs is shown in Figure 5. Early studies of the effects of nucleobase changes within this segment of yeast U1 RNA identified the following lethal pyrimidine-to-purine mutations: C4A, C4G, C8A and C8G (36,37). Pyrimidine-to-pyrimidine mutants C4U and C8U were viable but slow growing at 30°C (36). The U5A allele, which eliminates a U:U mismatch in the U1•5′SS duplex (Figure 5), was also viable but slow growing at 30°C (36). Here, we examined the effects of these known viable mutations, and of several novel nucleobase changes in the 3ACUUAC8 sequence, on the activity and genetic interactions of the U1 snRNA. The C8U mutant barely grew at 37 and 34°C and failed to grow at 25, 20 or 18°C (Figure 5A). The C4U strain grew best at 34°C and failed to grow at 25, 20 or 18°C (Figure 5A). We found that C4U was lethal at 34°C in combination with tgs1Δ, cbc2-Y24A, nam8Δ, mud1Δ, swt21Δ and mud2Δ. A new allele, U1-A7G, was viable but slow growing at 25, 30 and 34°C, but did not thrive at higher or lower temperatures (Figure 5A). A7G was inviable in combination with tgs1Δ, cbc2-Y24A, nam8Δ, mud1Δ, swt21Δ and mud2Δ.
Figure 5.

Effects of mutations in the 3ACUUAC8 sequence. (A) The base-pairing interaction between the U1 snRNA 3ACUUAC8 sequence and the consensus pre-mRNA 5′SS is shown. U1Δ p(CEN LEU2 U1) cells bearing the indicated U1 alleles were spot-tested for growth at the indicated temperatures. (B) Yeast strains bearing U1 WT, U5A or U5C alleles in the indicated genetic backgrounds were spot-tested for growth at the temperatures specified. Synthetic lethal interactions of U5A and U5C are indicated at bottom.
New and instructive findings emerged from our analysis of mutations U5A, U5G and U5C. In agreement with Siliciano and Guthrie (36), we found that U5A cells were viable but modestly slow growing at all temperatures tested (Figure 5A). The U5G mutant was also viable and had virtually the same growth effects as U5A at higher temperatures but was slightly more affected at cold temperatures (Figure 5A). The U5A and U5G phenotypes could be caused by installation of a purine per se or to the establishment of an extra A:U or G:U base pair with the 5′SS in lieu of the native U:U mispair. Thus, it is remarkable that the U5C mutant grew as well as the WT U1 strain at all temperatures (Figure 5A), signifying that a Py5:U mispair in the U1•5′SS RNA hybrid suffices for seemingly normal U1 snRNA activity. A corollary of this result is that the characteristic pseudouridine modification of the U5 nucleobase in U1 snRNA (38) (which will not occur in the U5C mutant) is not critical for U1 snRNA function in an otherwise wild-type background.
We surveyed whether and how the function of the U1 snRNA U5C variant was affected by deleting optional splicing factors and U1 snRNP subunits. The U1 U5C mutation was unaffected by swt21Δ and displayed a mild synthetic growth defect with mud1Δ at all temperatures tested (Figure 5B). U5C exacerbated the cs phenotype of tgs1Δ, pushing the restrictive temperature upwards (Figure 5B). Strong genetic interactions of U5C were seen with nam8Δ and cbc2-Y24A, whereby the U5C nam8Δ and U5C cbc2-Y24A strains grew slowly at 37 and 34°C and failed to grow at 30, 25 and 20°C (Figure 5B). These results fortify the case for a role of Nam8 and cap binding by CBC in U1 snRNP assembly at a non-consensus 5′SS (30,32,39). The U5C allele was unconditionally synthetic lethal in the mud2Δ background (Figure 5B).
The same synergy tests were performed for the U5A snRNA mutant. The slow growth phenotype of U5A was evident at all temperatures in the swt21Δ and tgs1Δ backgrounds (Figure 5B). The same was true of U5A nam8Δ, thereby highlighting a distinctive genetic interaction of nam8Δ with U5A versus U5C in which the loss of Nam8 exerted a stronger growth effect on U5C (Figure 5B). U5A was synthetically lethal with mud2Δ and cbc2-Y24A.
New genetic interactions of Msl5 with subunits of the U1 snRNP, Mud2 and Cbc2
Saccharomyces cerevisiae Msl5 (BP-binding protein) orchestrates spliceosome assembly by binding the intron BP sequence 5′-UACUAAC and establishing cross-intron-bridging interactions with other components of the splicing machinery (20,22,31,40–42). Msl5 (a 476-aa polypeptide) is essential for yeast vegetative growth. The central BP RNA-binding domain of Msl5—composed of KH and QUA2 modules (43)—is flanked by N- and C-terminal domains that have imputed functions in protein–protein interactions. By gauging the ability of Msl5 mutants to complement msl5Δ, we recently reported that the Msl5 N-terminal Mud2-binding domain (aa 35–68) and a downstream PPxY100 motif proposed to interact with WW-domain proteins (44) are inessential, as are a C-terminal proline-rich domain (aa 382–476) and two zinc-binding CxxCxxxxHxxxxC motifs (aa 273–286 and 299–312) (31). A subset of conserved BP RNA-binding amino acids in the central KH-QUA2 domain (aa 146–269) are essential pairwise (Ile189-Arg190; Leu256-Leu259) or in trios (Leu169-Arg172-Leu176), whereas other RNA-binding residues are dispensable (31). We have used our collection of viable Msl5 mutants to illuminate synthetic genetic interactions between Msl5 and Mud2, Nam8, Tgs1 and Cbc2 (31,32). The results suggested a network of important but functionally buffered protein–protein and protein–RNA interactions between the Msl5•Mud2 complex at the BP, U1 snRNP at the 5′SS and CBC at the pre-mRNA cap.
Here, we queried our collection of Msl5 mutants that have no growth defect per se for synthetic interactions with other components of the U1 snRNP not evaluated previously. We focused first on the Mud1 subunit. We found that mud1Δ was synthetically lethal with (i) an N-terminal truncation that eliminated part of the Mud2-binding site (aa 35–68); (ii) a triple-alanine mutation P97A-P98A-Y100A in the PPxY100 motif; and (iii) a C-terminal truncation that eliminated aa 402–425 (Supplementary Table S1). Otherwise benign double-alanine mutations in the Msl5 KH-QUA2 domain were either lethal (N163A-V165A) or severely sick (V195A-K196A, T265A-R267A and K252A-R253A) in the mud1Δ background (Supplementary Table S1). By contrast, there was no synthetic lethality/sickness of mud1Δ with the C273A–C276A and C299A–C302A alleles that respectively disrupt the proximal and distal zinc knuckle modules of Msl5 (Supplementary Table S1).
To better delineate the essential constituents of the BP RNA binding site of yeast Msl5, we tested the effects of single alanine substitutions within the three lethal alanine-cluster alleles identified previously (31). Tests of msl5Δ complementation showed that: (i) R172A, R190A and L259A were unconditionally lethal; (ii) L256A displayed a tight cs growth phenotype; and (iii) L169A, L176A and I189A grew normally on YPD agar at all temperatures (Table 1). The NMR structure of human SF1 (the ortholog of Msl5) bound to an RNA (5′-AUACUAACAA) containing the consensus yeast BP sequence (43) revealed an extensive network of atomic contacts between conserved amino acid side chains (many of them hydrophobic) and the RNA nucleobases and sugars, whereas the RNA phosphates are surface exposed and make relatively few protein contacts (Supplementary Figure S3). Given that six of the seven side chains mutated singly to alanine in Table 1 are identical in SF1 and Msl5 (Supplementary Figure S3), that the seventh Msl5 residue Leu169 is conserved as Ile in SF1, and that the RNA–protein contacts observed for these side chains in the SF1 structure are to a canonical yeast BP RNA recognition element, it is our presumption that the same RNA contacts are likely to be made by the equivalent Msl5 side chains. These contacts are listed in Table 1. (A complete list of the imputed BP RNA contacts made by all of the Msl5 residues that have been subjected to alanine scanning is provided in Supplementary Figure S4.) The striking finding is that only 4 of the 13 conserved side chains, and their imputed contacts to the BP (Supplementary Table S1), are essential for msl5Δ complementation.
Table 1.
Effect of KH-QUA2 domain mutations on Msl5 function in vivo
| MSL5 | Contacts in BBP•RNA structure: |
msl5Δ complementation |
|||
|---|---|---|---|---|---|
| Allele | pU1pA2pC3pU4pA5pA6pC7pA8pA9 | 18° | 25° | 30° | 37° |
| WT | +++ | +++ | +++ | +++ | |
| L169A | A6 base; A5 base | +++ | +++ | +++ | +++ |
| R172A | A6pC7 phosphate | – | – | – | – |
| L176A | A6 base N3, ribose O2′ | +++ | +++ | +++ | +++ |
| I189A | A6 base N1 N6 | +++ | +++ | +++ | +++ |
| R190A | A9 base | – | – | – | – |
| L256A | A2 ribose O2′; C3 base N3, ribose O4′ | – | – | + | ++ |
| L259A | U4 base C2 O2, ribose O4′ | – | – | – | – |
Complementation of msl5Δ by the indicated MSL5 alleles was assayed by plasmid shuffle (31). Individual Leu+ transformants were streaked on agar medium containing FOA. Growth was scored after incubation for 7 d at 18, 25, 30 or 37°C. Lethal mutants were those that failed to form colonies at any temperature. Individual FOA-resistant colonies with viable MSL5 alleles were grown to mid-log phase in YPD broth and adjusted to equivalent A600. Aliquots (3 µl) of serial 10-fold dilutions were spotted on YPD agar plates, which were then incubated at 18, 25, 30 and 37°C. Growth was scored as follows: (+++) colony size was indistinguishable from strains bearing wild-type MSL5; (++) slightly reduced colony size; (+) only pinpoint macroscopic colonies were formed; (–) no growth.
We tested the three newly identified fully viable MSL5 alleles for mutational synergies with mutations in other genes affecting early splicing events. L176A supported wild-type growth when combined with mud1Δ, nam8Δ, mud2Δ, tgs1Δ or cbc2-Y24A (Table 2). Thus, the inferred contacts of Leu176 with the BP adenosine (to the adenine N3 atom and the ribose O2′) are dispensable for yeast growth in all genetic backgrounds tested. By contrast, L169A and I189A were lethal in combination with mud1Δ, nam8Δ, mud2Δ, tgs1Δ or cbc2-Y24A (Table 2). Leu169 is imputed to intercalate between and make van der Waals contacts to the BP adenine and the upstream flanking adenine. Ile189 contacts the N1 and N6 atoms of the BP adenine. The emerging theme here is that the loss of U1 snRNP components, or weakening of CBC interactions with the mRNA cap, is buffered by cross-intron Msl5 contacts at the BP that, when weakened by mutations characterized presently, results in a failure of vegetative growth, presumably via insufficient splicing of one or more essential pre-mRNAs.
Table 2.
Synthetic interactions of Msl5 KH-QUA2 domain mutations
| MSL5 allele | Complementation |
||||
|---|---|---|---|---|---|
| mud1Δ | mud2Δ | nam8Δ | cbc2-Y24A | tgs1Δ | |
| WT | +++ | +++ | +++ | +++ | +++ |
| L169A | – | – | – | – | – |
| L176A | +++ | +++ | +++ | +++ | +++ |
| I189A | – | – | – | – | – |
Complementation of msl5Δ in the genetic backgrounds specified by otherwise functional MSL5 alleles was assayed by plasmid shuffle. L169A and I189A transformants failed to form FOA-resistant colonies at any temperature tested in each of the five strain backgrounds. By contrast, L176A supported growth on FOA and, by spot testing, +++ growth on YPD agar at all temperatures in all five strains.
New genetic interactions of Msl5 with the 5′ end of U1 snRNA
Viable U1 snRNA mutants were tested for synthetic interactions with otherwise viable Msl5 mutations by constructing a U1Δ msl5Δ double knockout strain with a resident CEN URA3 U1 MSL5 plasmid to sustain growth. This strain was co-transformed with various combinations of CEN HIS3 MSL5 and CEN LEU2 U1 plasmids, and individual His+ Leu+ transformants were tested for growth on FOA at 20, 30 and 37°C. Allelic pairs that failed to support growth on FOA at any temperature were deemed synthetic lethal (Table 3). FOA-resistant survivors were then grown in YPD liquid culture and tested for growth by spotting serial dilutions on YPD agar at 20, 25, 30, 34 and 37°C. The results are compiled in Table 3, wherein +++ signifies growth indistinguishable from wild-type U1 MSL5 cells, ++ denotes smaller colony size and + indicates pinpoint colonies. Several of the allelic combinations elicited ts or cs growth failures; these are denoted as such in Table 3 next to the growth scores that applied to the permissive temperatures for these strains.
Table 3.
Synergies of U1 snRNA mutations with Msl5 mutations
| U1 snRNA |
MSL5 |
|||||||
|---|---|---|---|---|---|---|---|---|
| 55–476 | 69–476 | 97AAxA100 | L169A | L176A | I189A | V195 K196A | K252R | |
| WT | +++ | ++ | +++ | +++ | +++ | +++ | +++ | +++ |
| +5 | +++ | lethal | lethal | ++ /csa | +++ | + | +++ /csb | +++ /csc |
| +15 | +++ /tsd | lethal | lethal | lethal | +++ | lethal | +++ /cse | +++ /csc |
| +20 | +++ /tsd | lethal | lethal | + /csf | +++ | lethal | +++ /cse | ++ /csa |
| +30 | +++ /tsd | lethal | lethal | lethal | +++ | lethal | +++ /cse | ++ /csa |
| U5C | +++ /tsd | lethal | lethal | lethal | +++ | lethal | ++ /csa | ++ /csa |
| H1L | +++ /tsd | lethal | ++ /csg | lethal | +++ | lethal | ++ /csg | + |
| ΔU10 | lethal | lethal | lethal | lethal | + | lethal | lethal | lethal |
| [+1] | + /tsh | lethal | lethal | lethal | ++ | lethal | lethal | lethal |
Complementation of msl5Δ in the U1 genetic backgrounds specified was assayed by plasmid shuffle. Allelic combinations that failed to form colonies at any temperature are denoted as lethal. Viable strains were tested for growth on YPD agar and scored as described in Table 1. ts or cs phenotypes were as follows:
a++ at 34–37°C.
b+++ at 25–37°C.
c+++ at 34–37°C.
d+++ at 18–30°C.
e+++ at 30–37°C.
f+ at 34–37°C.
g++at 30–37°C.
h+ at 18–25°C.
The U1 5′ extensions, the U5C mutation and the H1L, ΔU10 and [+1] changes distal to the 5′SS complementarity segment were uniformly synthetic lethal with MSL5-(69–476), an N-terminal truncation that disrupts the interaction of the BP binding protein with Mud2/U2AF65 (42,45,46) (Table 3). Indeed, the synergies of MSL5-(69-476) with the U1 snRNA mutations phenocopied those of mud2Δ (Figures 2–5). By contrast, MSL5-(55–476), which deletes only part of the Mud2-binding interface, permitted +++ growth of yeast bearing the 5′ extensions, U5C and H1L variants of U1 snRNA at low temperatures but did not support growth at 34–37°C (Table 3).
Lesions L169A and I189A in the RNA-binding site of Msl5 were synthetically lethal with most of the U1 mutant alleles surveyed and, where the allelic pairs were viable, the cells were sick and cold-sensitive (Table 3). Other changes in the KH-QUA2 domain of Msl5 displayed less severe synthetic interactions with the U1 mutations. For example, V195A K196A and K252R elicited cs defects in combination with the U1 5′ extensions but displayed +++ or ++ growth at higher temperatures. The same V195A K196A and K252R alleles were lethal in combination with the ΔU10 and [+1] U1 alleles (Table 3). At the other end of the spectrum, MSL5-L176A did not synergize with any of the 5′ extensions or H1L, but did display synthetic sickness with [+1] and ΔU10(Table 3).
Dissecting the genetic interactions of the Msl5 PPxY100 motif
The msl5-(P97A-P98A-Y100A) allele was synthetically lethal in combination with the 5′ extensions, U5C, ΔU10 and [+1] mutations in U1 snRNA (Table 3; 97AAxA100). The U1-H1L msl5-(P97A-P98A-Y100A) strain was viable but slow growing and cold-sensitive (Table 3). The PPxY100 motif was suggested to mediate a cross-intron-bridging interaction of Msl5 with the Prp40 subunit of the U1 snRNP (22). Prp40 is a 583-aa protein containing two tandem WW modules at the N-terminus (aa 1–70) and four FF domains dispersed downstream (44,47,48). The isolated tandem WW domain binds in vitro to a synthetic peptide PSPPPVYDA corresponding to Msl5 residues 94–102 (44). A key question is whether the synthetic lethality of P97A-P98A-Y100A with mutations at the 5′ end of U1 snRNA is causally related to the loss of the imputed binding of the PPxY motif to the Prp40 WW domain. If this is the case, then deletion of the WW domain in Prp40 might phenocopy the mutational synergies of msl5-(P97A-P98A-Y100A). We put this idea to the test by replacing the chromosomal PRP40 locus with a truncated allele, prp40-(77-583) (henceforth named prp40-ΔWW), which cleanly subtracts the tandem WW modules. In agreement with the recent report by Görnemann et al. (49), we found that the prp40-ΔWW strain grew as well as the PRP40 control strain on YPD agar at all temperatures (not shown). We proceeded to construct a U1Δ prp40-ΔWW p(CEN URA3 U1) strain suitable for plasmid shuffle of U1 snRNA variants. The instructive findings were that the +5, +15, +20, +30 and U5C alleles of U1 that were synthetic lethal with msl5-(P97A-P98A-Y100A) were all viable in combination with prp40-ΔWW (Supplementary Figure S5). The prp40-ΔWW U1-U5C strain grew well at all temperatures. prp40-ΔWW strains with U1 5′ extensions grew well at 37, 34 and 30°C but were slow growing at 20°C. The prp40-ΔWW U1-H1L strain grew slowly at all temperatures. These results are not compatible with a model whereby the severe synergies of the U1 snRNA 5′ mutations with msl5-(P97A-P98A-Y100A) are mediated exclusively (or predominantly) via the Prp40 WW domain. We surmise that the PPxY100 motif of Msl5 plays an important, albeit genetically buffered, role in pre-mRNA splicing that has naught to do with the Prp40 WW domain.
To better understand how the PPxY motif functions, we tested the effects of single mutations P97A, P98A and Y100A on Msl5 activity in several genetic backgrounds for which the msl5-(P97A-P98A-Y100A) triple-mutant was lethal (e.g. mud1Δ, nam8Δ, cbc2-Y24A and tgs1Δ) or sick (mud2Δ). Initial control experiments verified that the single mutations in PPxY had no impact on complementation of msl5Δ (not shown). Complementation tests for mutational synergies revealed that the P97A and P98A alleles had no genetic interactions with mud1Δ, nam8Δ, cbc2-Y24A, tgs1Δ or mud2Δ (Figure 6A). By contrast, the Y100A allele was synthetically lethal with mud1Δ and nam8Δ and synthetic sick with mud2Δ, effectively phenocopying the synthetic defects of the 97AAxA100 triple-mutant in these backgrounds (Figure 6A). Y100A was also synthetically sick with cbc2-Y24A and tgs1Δ (Figure 6A), as evinced by smaller colony size at 34 and 37°C and cold-sensitivity (Figure 6B). These results implicate the loss of Tyr100 as principally responsible for the synthetic interactions of the Msl5 97AAxA100 mutant with proteins involved in spliceosome assembly.
Figure 6.

Genetic interactions of the Msl5 PPxY100 motif. (A) Complementation of msl5Δ by the indicated MSL5 alleles was assayed by plasmid shuffle. Individual Leu+ transformants were streaked on agar medium containing FOA. Growth was scored after incubation for 7 d at 18, 25, 30 or 37°C. Lethal mutants were those that failed to form colonies at any temperature. Individual FOA-resistant colonies with viable MSL5 alleles were grown to mid-log phase in YPD broth and adjusted to equivalent A600. Serial 10-fold dilutions were spotted on YPD agar plates, which were then incubated at 25, 30, 34 and 37°C. Growth was scored as follows: (+++) colony size indistinguishable from strains bearing wild-type MSL5 at all temperatures; (++) slightly reduced colony size; (cs) pinpoint colonies at 25°C. (B) Spot testing of the growth of MSL5 WT versus Y100A in the indicated strain backgrounds at the temperatures specified.
Tyr100 has been identified as a site of Msl5 phosphorylation in vivo (50). To probe whether the synthetic phenotypes of msl5-Y100A are related to the absence of tyrosine phosphorylation, we replaced Tyr100 with phenylalanine. The Y100F allele displayed no synthetic defects in combination with mud1Δ, nam8Δ, cbc2-Y24A, tgs1Δ or mud2Δ (Figure 6A). We conclude that the genetically buffered essential functions of Tyr100 are not related to its phosphorylation status.
We proceeded to test the effects of P97A, P98A, Y100A and Y100F in tandem with several U1 alleles with which the 97AAxA100 triple-mutant was either synthetic lethal (i.e. +5, +20, +30, U5C and [+1]) or sick (H1L). P97A and Y100F sustained +++ growth in each U1 mutant background at 20, 30, 34 and 37°C (not shown). P98A supported ++ growth at 20–37°C in the +5, +20, +30, U5C and H1L strains; the msl5-P98A U1-[+1] strain grew +++ at 30, 34 and 37°C and ++ at 20 and 25°C (not shown). By contrast, msl5-Y100A was sick in combination with U1-[+1] and +30. The msl5-Y100A U1-[+1] strain formed pinpoint colonies at 37 and 30°C and failed to grow at 25 or 20°C (Supplementary Figure S6). The msl5-Y100A U1 +30 strain also grew slowly at high temperature and did not grow at low temperature (Supplementary Figure S6). Y100A displayed a cs growth defect in combination with the +20 5′ extension but had little or no synthetic phenotype with U1-H1L and +5 (Supplementary Figure S6). Thus, the loss of Tyr100 sufficed to synergize with otherwise benign mutations of the U1 snRNA, albeit as a forme fruste of the impact of 97AAxA100 in these mutant U1 backgrounds.
Bypass of the essentiality of Prp28 by U1 snRNA mutations
Yeast Prp28 is an essential pre-mRNA splicing factor and a member of the DEAD-box family of nucleic acid-dependent NTPases (51,52). Although Prp28 has been imputed to be an RNA helicase, the catalytic activities inherent to yeast Prp28 have not been characterized. Nonetheless, Prp28 is implicated genetically in displacement of the U1 snRNP from the 5′SS during the transition from a pre-mRNA•U1•U2-containing spliceosome to a pre-mRNA•U2•U5•U6 spliceosome (15,16,34). Initial insights came from the observation that the deleterious effects of hyper-stabilizing the U1•5′SS base-paring interaction were genetically enhanced by a cs mutant of prp28, but not by conditional mutants of six other helicase-like splicing factors (16). In turn, the prp28-cs phenotype could be suppressed by an extra U1 snRNA gene with a C4U change that weakens its base-pairing to the consensus 5′SS (16). The case was fortified by the finding that the essentiality of Prp28 for vegetative growth could be bypassed by mutations in the essential U1 snRNP subunits Yhc1, Prp42 and Snu71, but not by deletions of the inessential subunits Nam8 and Mud1 (15,34). Deletion of the splicing factor Swt21 also rescued the lethality of prp28Δ (34). These results highlighted that the need for Prp28 during U1 snRNP ejection from the early spliceosome is alleviated by certain alterations that are presumed to weaken U1•5′SS contacts. This idea was consolidated by the demonstration that ectopic expression of mutant U1 snRNAs C4U, U5C or C8U (on top of a wild-type U1 snRNA) could also restore viability to prp28Δ (15).
Here, we surveyed our collection of viable U1 snRNA mutants for Prp28 bypass, by constructing a U1Δ prp28Δ double-knockout strain with a resident CEN URA3 U1 PRP28 plasmid to sustain growth. This strain was transformed with CEN LEU2 U1 plasmids, and the transformants were screened for growth on FOA at 18–37°C such that only U1 variants that bypass the Prp28 requirement will give rise to FOA-resistant colonies. The important distinction between this and prior tests of U1 bypass of Prp28 is that our assay mandates that the U1 variants bypass prp28Δ when they are the only source of U1 snRNA in the cell. Four U1 snRNA mutants passed this test: C4U, U5C, ΔU10 and [+3]. The viable U1Δ prp28Δ cells bearing these bypass U1 alleles were tested for growth on YPD agar at 20, 25, 30, 34 and 37°C (Figure 7).
Figure 7.
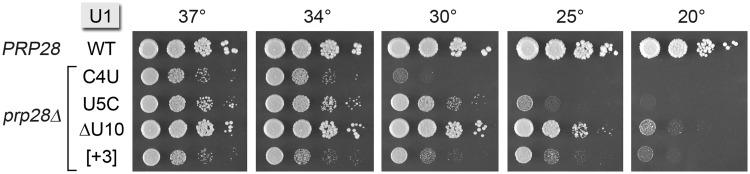
U1 mutations bypass the requirement for the essential PRP28 gene. Yeast prp28Δ U1Δ cells harboring the indicated U1 allele—WT, C4U, U5C, ΔU10 or [+3]—on a CEN LEU2 plasmid and either wild-type PRP28 (CEN HIS3) or an empty CEN HIS3 plasmid (prp28Δ) were grown in liquid cultures at 37°C to mid-log phase. The cultures were adjusted to A600 of 0.1, and aliquots of serial dilutions were spotted to YPD agar. Photographs of the plates after incubation for 2 d (30, 34, 37°C), 4d (25°C) or 5 d (20°C) are shown.
The hierarchy of bypass strengths was noteworthy. The ΔU10 variant of U1 snRNA was the best of the Prp28 bypass suppressors, insofar as ΔU10 prp28Δ cells grew about as well as wild-type cells at 37, 34 and 30°C, as gauged by colony size (Figure 7). The progressive cs growth defect of the ΔU10 prp28Δ strain at 25 and 20°C (Figure 7) was exactly what was seen for the ΔU10 PRP28 mutant strain (Figure 4, ΔU10 WT). Thus, ΔU10 appears to be fully independent of Prp28.
U5C was the next best of the Prp28 bypass suppressors. U5C prp28Δ cells grew at 37, 34 and 30°C, albeit slower than the wild-type strain, and were unable to grow at 20°C (Figure 7). Because the U5C snRNA supported apparently normal growth at all temperatures in a PRP28 background (Figure 5A), we surmise that the bypass effect of U5C is cold-sensitive. This result suggests that higher temperatures help destabilize already weakened U1-U5C•5′SS pairing and thereby permits U1 snRNP ejection without the assistance of Prp28. However, when the pairing is more stable at lower temperatures, Prp28 is still required.
Although C4U appeared to be less effective in Prp28 bypass when comparing growth of C4U prp28Δ to the wild-type strain (Figure 7), bear in mind that C4U per se elicits the same phenotype in a PRP28 background (Figure 5). (Side-by-side comparisons of the C4U PRP28 and C4U prp28Δ strains revealed no difference in growth; not shown.)
The [+3] insertion between the 5′SS complementarity motif and the first hairpin restored slow growth to prp28Δ cells at 37, 34, 30 and 25°C but did not bypass at 20°C (Figure 6). Because the [+3] insertion did not cause a strong cs phenotype in the PRP28 background (Figure 3 and side-by-side growth comparisons not shown), we conclude that the [+3] bypass effect is cold-sensitive, à la U5C.
DISCUSSION
The present study reveals structure–function relationships at the 5′ end of the yeast U1 snRNA and their contributions to an expansive network of genetic interactions among proteins and RNA elements that drive spliceosome assembly. We show that the 5′ dinucleotide leader between the TMG cap and the conserved 3ACUUAC8 motif can be enlarged by 30 nt without compromising cell growth. By contrast, there are tight functional constraints on the downstream dinucleotide spacer between 3ACUUAC8 and helix 1 of the U1 snRNA fold, whereby insertions of ≥5 nt are lethal. We exploited our collection of biologically active U1 alleles with 5′ extensions, variations in the 3ACUUAC8 sequence, short downstream insertions/mutations and a longer H1 helix to discover new intra-snRNP synergies with the inessential U1 components Nam8, Mud1 and the TMG cap. We also found new synergies with the non-snRNP splicing factor complexes Msl5•Mud2 and CBC that bind the intron BP and pre-mRNA 5′ cap, respectively. These results fortify the idea that spliceosome assembly can be facilitated by any of several genetically buffered microscopic pathways involving cross-intron-bridging interactions of the U1 snRNP•5′SS complex with the Mud2•Msl5•BP complex and cross-exon-bridging interactions of the CBC•m7GpppRNA complex with the U1•5′SS complex. Although elimination or weakening of one set of interactions by deletions of inessential components or missense mutations in essential components can be tolerated at the level of vegetative growth, the effects of simultaneous subtraction of functionally overlapping microscopic pathways are seen as synthetic lethality or sickness.
The composition of the U1 snRNP is much more complex in budding yeast than in mammals, with respect to the size of the U1 RNA (568 versus 164 nt) and the number of U1-specific protein subunits (10 versus 3). The larger yeast U1 RNA may include binding sites for some of the yeast-specific U1 subunit proteins, which are Prp39, Prp40, Snu71, Snu56, Luc7, Prp42 and Nam8. Mammalian cells have putative homologs of Luc7 (hLuc7), Nam8 (TIA-1) and Prp40 (FBP11) (1,13,53), but they are not subunits of the U1 snRNP. By assimilating these seven yeast proteins as stoichiometric subunits, the yeast U1 snRNP can serve as a recipient of splicing regulation (via signals in the pre-mRNA or interactions with other splicing factors) that, in the case of mammalian splicing, are conveyed by an armada of trans-acting splicing factors that influence 5′SS usage and/or formation of the U1•5′SS complex (54,55), most of which have no equivalents in budding yeast.
The present study is informative regarding the cross-intron contacts between the U1•5′SS and Mud2•Msl5•BP complexes, via our analysis of the effects of mutations in the BP-binding protein Msl5. Because Msl5 is essential in budding yeast, and because mutational analyses of the yeast intron BP have focused almost exclusively on base-pairing interactions with the U2 snRNP (56–58), there is little known about the necessity and function of Msl5•BP interactions in vivo. Here, we extended our initial structure-guided mutagenesis of the RNA-binding site of Msl5 to consolidate the following points. First, that alanine mutations of only four of the Msl5 amino acids that contact the consensus BP are unconditionally or conditionally lethal: Arg172, Arg190, Leu256 and Leu259. By contrast, alanine mutations of nine other Msl5 amino acids that contact the consensus BP elicit no growth phenotypes. However, we found allele-specific synergies of otherwise benign RNA-binding site mutations with mutations in the U1 snRNA, U1 snRNP subunits Mud1 and Nam8, Tgs1, Mud2 and Cbc2. These results discriminate essential from optional BP RNA contacts and imply that the effects of hypomorphic Msl5 changes that weaken BP binding are buffered by interactions with the U1 snRNP that are mediated, at least in part, via the Mud2 subunit of the Msl5•Mud2 heterodimer.
The N-terminal domains of Msl5 and human SF1 are responsible for interactions with the C-terminal RRM3 domains of Mud2 and U2AF65, respectively. Initial structural studies identified an SF1 peptide segment (13PSKKRKRSRWNQD25) that bound U2AF65, with the principal contact point being the insertion of a conserved SF1 tryptophan side chain (Trp22 in SF1; Trp35 in Msl5) into a hydrophobic pocket of U2AF65 (59) (see Figure 8). Although this SF1 segment is conserved in yeast Msl5, mutational studies showed that the Msl5 W35A change did not phenocopy the synthetic genetic interactions of mud2Δ (31). Moreover, compound mutations of the conserved Mud2 amino acids that, in U2AF65, comprise the SF1 peptide-binding site did not affect Mud2 function in vivo in genetic backgrounds where Mud2 is essential (31). These results suggested that the Mud2•Msl5 interface might be more complex than that seen in the U2AF65•SF1 peptide structure. The detailed anatomy of the SF1•USAF65 interface (and by extension the Mud2•Msl5 interface) has since been clarified by X-ray and NMR structures of a complex between SF1-(20-128) and the U2AF65 RRM3 (45,46). RRM3 interacts with a newly defined domain module of SF1, composed of two long α-helices that form an antiparallel coiled coil (Figure 8A). The SF1 α-helices include several side chains that contact U2AF65 directly. Because these side chains are conserved in yeast Msl5 (highlighted in Figure 8B), we presume they comprise a homologous interface with Mud2. The salient point we take from the new SF1–U2AF65 structure is that the equivalent of the Msl5 97PPxY100 motif (84EPxY87 in SF1) is located within the loop that connects the two α-helices and is not a component of the interface with Mud2/U2AF65 (Figure 8A). Our studies here show that the aromatic Tyr100 residue of the Msl5 loop plays an important role in vivo that is revealed when the other splicing factors or the U1 snRNA are absent or mutated. The earlier suggestion from in vitro studies that the PPxY peptide of Msl5 mediates Msl5 binding to the WW domains of Prp40 is not consistent with our findings here that deletion of the WW domain in Prp40 did not phenocopy the mutational synergies of msl5-(P97A-P98A-Y100A) with U1 alleles (Supplementary Figure S4). Also, we noted that a prp40-ΔWW mud1Δ strain displayed wild-type growth at all temperatures (not shown), in contrast to the unconditional synthetic lethality of msl5-(P97A-P98A-Y100A) mud1Δ (Supplementary Table S1). The location of Tyr100 on an exposed surface of the loop distant from the putative Mud2-Msl5 interface (Figure 8A) suggests that Tyr100 mediates the interaction of Msl5 with another component of the yeast splicing apparatus, either a domain of Mud2 or Msl5 not present in the SF1-U2AF65 structure, or a component of the U1 snRNP, or yet another splicing factor.
Figure 8.

Disposition of PxY100 in the structure of SF1 bound to U2AF65. (A) Stereo view of the crystal structure (pdb 4FXW) of the complex between SF1-(20-128) (colored cyan) and the U2AF65 RRM3 domain (colored gray). Selected SF1 side chains are shown a stick models and are numbered according to their positions in yeast Msl5. (B) Alignment of the amino acid sequences of the N-terminal domains of Msl5 and SF1, corresponding to the SF1 structure shown in (A). Positions of side chain identity/similarity are indicated by •. The two α-helices comprising the coiled-coil of SF1 are indicated by cylinders below the alignment. The PPxY motif in the connecting loop is denoted by a bracket.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Table 1 and Supplementary Figures 1–6.
FUNDING
National Institutes of Health (NIH) [GM52470 to S.S.] and [GM50288 to B.S.]. S.S. is an American Cancer Society Research Professor. B.S. is a recipient of a Hearst Foundation Award. Funding for open access charge: NIH [GM52470].
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.Fabrizio P, Dannenberg J, Dube P, Kastner B, Stark H, Urlaub H, Lührmann R. The evolutionarily conserved core design of the catalytic activation step of the yeast spliceosome. Mol. Cell. 2009;36:593–608. doi: 10.1016/j.molcel.2009.09.040. [DOI] [PubMed] [Google Scholar]
- 2.Warlocki Z, Odenwälder P, Schitzova J, Platzmann F, Stark H, Urlaub H, Ficner R, Fabrizio P, Lührmann R. Reconstitution of both steps of Saccharomyces cerevisiae splicing with purified spliceosomal components. Nat. Struct. Mol. Biol. 2009;16:1237–1243. doi: 10.1038/nsmb.1729. [DOI] [PubMed] [Google Scholar]
- 3.Hoskins AA, Friedman LJ, Gallagher SS, Crawford DJ, Anderson EG, Wombacher R, Ramirez N, Cornish VW, Gelles J, Moore MJ. Ordered and dynamic assembly of single spliceosomes. Science. 2011;331:1289–1295. doi: 10.1126/science.1198830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tang J, Abovich N, Fleming MJ, Séraphin B, Rosbash M. Identification and characterization of a yeast homolog of U1 snRNP-specific protein C. EMBO J. 1997;13:4082–4091. doi: 10.1093/emboj/16.13.4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gottschalk A, Tang J, Puig O, Salgado J, Neubauer G, Colot HV, Mann M, Séraphin B, Rosbash M, Lührmann R. A comprehensive biochemical and genetic analysis of the yeast U1 snRNP reveals five novel proteins. RNA. 1998;4:374–393. [PMC free article] [PubMed] [Google Scholar]
- 6.Fortes P, Bilbao-Cortés D, Fornerod M, Rigaut G, Raymond W, Séraphin B, Mattaj IW. Luc7p, a novel yeast U1 snRNP protein with a role in 5′ splice site recognition. Genes Dev. 1999;13:2425–2438. doi: 10.1101/gad.13.18.2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schwer B, Erdjument-Bromage H, Shuman S. Composition of yeast snRNPs and snoRNPs in the absence of trimethylguanosine caps reveals nuclear cap binding protein as a gained U1 component implicated in the cold-sensitivity of tgs1Δ cells. Nucleic Acids Res. 2011;39:6715–6728. doi: 10.1093/nar/gkr279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pomeranz Krummel DA, Oubridge C, Leung AKW, Li J, Nagai K. Crystal structure of human spliceosomal U1 snRNP at 5.5 Å resolution. Nature. 2009;458:475–480. doi: 10.1038/nature07851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weber G, Trowitzsch S, Kastner B, Lührmann R, Wahl MC. Functional organization of the Sm core in the crystal structure of human U1 snRNP. EMBO J. 2010;29:4172–4184. doi: 10.1038/emboj.2010.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leung AKW, Nagai K, Li J. Structure of the spliceosomal U4 snRNP core domain and its implications for snRNP biogenesis. Nature. 2011;473:536–539. doi: 10.1038/nature09956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van der Feltz C, Anthony K, Brilot A, Pomeranz Krummel DA. Architecture of the spliceosome. Biochemistry. 2012;51:3321–3333. doi: 10.1021/bi201215r. [DOI] [PubMed] [Google Scholar]
- 12.Zhang D, Rosbash M. Identification of eight proteins that cross-link to pre-mRNA in the yeast commitment complex. Genes Dev. 1999;13:581–592. doi: 10.1101/gad.13.5.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Puig O, Bragado-Nilsson E, Koski T, Séraphin B. The U1 snRNP-associated factor Luc7p affects 5′ splice site selection in yeast and human. Nucleic Acids Res. 2007;35:5874–5885. doi: 10.1093/nar/gkm505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lewis JD, Görlich D, Mattaj IW. A yeast cap binding protein complex (yCBC) acts at an early step in pre-mRNA splicing. Nucleic Acids Res. 1996;24:3332–3336. doi: 10.1093/nar/24.17.3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen JY, Stands L, Staley JP, Jackups RR, Latus LJ, Chang TH. Specific alterations of U1-C protein or U1 small nuclear RNA can eliminate the requirement of Prp28p, an essential DEAD box splicing factor. Mol. Cell. 2001;7:227–232. doi: 10.1016/s1097-2765(01)00170-8. [DOI] [PubMed] [Google Scholar]
- 16.Staley JP, Guthrie C. An RNA switch at the 5′ splice site requires ATP and the DEAD box protein Prp28p. Mol. Cell. 1999;3:55–64. doi: 10.1016/s1097-2765(00)80174-4. [DOI] [PubMed] [Google Scholar]
- 17.Siliciano PG, Kivens WJ, Guthrie C. More than half of yeast U1 snRNA is dispensable for growth. Nucleic Acids Res. 1991;19:6367–6372. doi: 10.1093/nar/19.23.6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liao X, Kretzner L, Séraphin B, Rosbash M. Universally conserved and yeast-specfic U1 snRNA sequences are important but not essential for U1 snRNP function. Genes Dev. 1990;4:1766–1774. doi: 10.1101/gad.4.10.1766. [DOI] [PubMed] [Google Scholar]
- 19.Liao XC, Tang J, Rosbash M. An enhancer screen identifies a gene that encodes the yeast U1 snRNP A protein: implications for snRNP protein function in pre-mRNA splicing. Genes Dev. 1991;7:419–428. doi: 10.1101/gad.7.3.419. [DOI] [PubMed] [Google Scholar]
- 20.Abovich N, Liao XC, Rosbash M. The yeast MUD2 protein: an interaction with PRP11 defines a bridge between commitment complexes and U2 snRNP addition. Genes Dev. 1994;8:843–854. doi: 10.1101/gad.8.7.843. [DOI] [PubMed] [Google Scholar]
- 21.Colot HV, Stutz F, Rosbash M. The yeast splicing factor Mud13p is a commitment complex component and corresponds to CBP20, the small subunit of the nuclear cap-binding complex. Genes Dev. 1996;10:1699–708. doi: 10.1101/gad.10.13.1699. [DOI] [PubMed] [Google Scholar]
- 22.Abovich N, Rosbash M. Cross-intron bridging interactions in the yeast commitment complex are conserved in mammals. Cell. 1997;89:403–412. doi: 10.1016/s0092-8674(00)80221-4. [DOI] [PubMed] [Google Scholar]
- 23.Tang J, Abovich N, Rosbash M. Identification and characterization of a yeast gene encoding the U2 small nuclear ribonucleoprotein particle B” protein. Mol. Cell. Biol. 1996;16:2787–2795. doi: 10.1128/mcb.16.6.2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fortes P, Kufel J, Fornerod M, Polycarpou-Schwarz M, Lafontaine D, Tollervey D, Mattaj IW. Genetic and physical interaction involving the yeast nuclear cap-binding complex. Mol. Cell. Biol. 1999;19:6543–6553. doi: 10.1128/mcb.19.10.6543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tong AH, Evangelista M, Parsons AB, Xu H, Bader GD, Page N, Robinson M, Raghibizadeh S, Hogue CW, Bussey H. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science. 2001;294:2364–2368. doi: 10.1126/science.1065810. [DOI] [PubMed] [Google Scholar]
- 26.Wilmes GM, Bergkessel M, Bandyopadhyay S, Shales M, Braberg H, Cagney G, Collins SR, Whitworth GB, Kress TL, Weissman JS. A genetic interaction map of RNA-processing factors reveals links between Sem1/Dss1-containing complexes and mRNA export and splicing. Mol. Cell. 2008;32:735–746. doi: 10.1016/j.molcel.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hausmann S, Zheng S, Costanzo M, Brost RL, Garcin D, Boone C, Shuman S, Schwer B. Genetic and biochemical analysis of yeast and human cap trimethylguanosine synthase: functional overlap of TMG caps, snRNP components, pre-mRNA splicing factors, and RNA decay pathways. J. Biol. Chem. 2008;283:31706–31718. doi: 10.1074/jbc.M806127200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, Ding H, Koh JL, Toufighi K, Mostafavi S, et al. The genetic landscape of a cell. Science. 2010;327:425–431. doi: 10.1126/science.1180823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chang J, Schwer B, Shuman S. Mutational analyses of trimethylguanosine synthase (Tgs1) and Mud2: proteins implicated in pre-mRNA splicing. RNA. 2010;16:1018–1031. doi: 10.1261/rna.2082610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qiu ZR, Schwer B, Shuman S. Determinants of Nam8-dependent splicing of meiotic pre-mRNAs. Nucleic Acids Res. 2011;39:3427–3445. doi: 10.1093/nar/gkq1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chang J, Schwer B, Shuman S. Structure-function analysis and genetic interactions of the yeast branchpoint binding protein Msl5. Nucleic Acids Res. 2012;40:4539–4552. doi: 10.1093/nar/gks049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qiu ZR, Chico L, Chang J, Shuman S, Schwer B. Genetic interactions of hypomorphic mutations in the m7G cap binding pocket of yeast nuclear cap binding complex: an essential role for Cbc2 in meiosis via splicing of MER3 pre-mRNA. RNA. 2012;18:1996–2011. doi: 10.1261/rna.033746.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schwer B, Mao X, Shuman S. Accelerated mRNA decay in conditional mutants of yeast mRNA capping enzyme. Nucleic Acids Res. 1998;26:2050–2057. doi: 10.1093/nar/26.9.2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hage R, Tung L, Du H, Stands L, Rosbash M, Chang TH. A targeted bypass screen identifies Ynl187p, Prp42p, Snu71p, and Cbp80p for stable U1 snRNP/pre-mRNA interaction, Mol. Cell. Biol. 2009;29:3941–3952. doi: 10.1128/MCB.00384-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Noble SM, Guthrie C. Transcriptional pulse-chase analysis reveals a role for a novel snRNP-associated protein in the manufacture of spliceosomal snRNPs. EMBO J. 1996;15:4368–4379. [PMC free article] [PubMed] [Google Scholar]
- 36.Siliciano PG, Guthrie C. 5′ splice site selection in yeast: genetic alterations in base-pairing with U1 reveal additional requirements. Genes Dev. 1988;2:1258–1267. doi: 10.1101/gad.2.10.1258. [DOI] [PubMed] [Google Scholar]
- 37.Séraphin B, Kretzner L, Rosbash M. A U1 snRNA:pre-mRNA base pairing interaction is required early in yeast spliceosome assembly but does not uniquely define the 5′ cleavage site. EMBO J. 1988;7:2533–2538. doi: 10.1002/j.1460-2075.1988.tb03101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Massenet S, Motorin Y, Lafontaine DLJ, Hurt EC, Grosjean H, Branlant C. Pseudouridine mapping in the Saccharomyces cerevisiae spliceosomal U small RNAs (snRNAs) reveals that pseudouridine synthase Pus1p exhibits a dual substrate specificity for U2 snRNA and tRNA. Mol. Cell. Biol. 1999;19:2142–2154. doi: 10.1128/mcb.19.3.2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Spingola M, Ares M. A yeast intronic splicing enhancer and Nam8p are required for Mer1p-activated splicing. Mol. Cell. 2000;6:329–338. doi: 10.1016/s1097-2765(00)00033-2. [DOI] [PubMed] [Google Scholar]
- 40.Rain JC, Rafi Z, Legrain P, Krämer A. Conservation of functional domains involved in RNA binding and protein-protein interactions in human and Saccharomyces cerevisiae pre-mRNA splicing factor SF1. RNA. 1998;4:551–565. doi: 10.1017/s1355838298980335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rutz B, Seraphin B. Transient interaction of BBP/ScF1 and Mud2 with the splicing machinery affects the kinetics of spliceosome assembly. RNA. 1999;5:819–831. doi: 10.1017/s1355838299982286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Q, Zhang L, Lynn B, Rymond BC. A BBP-Mud2p heterodimer mediates branchpoint recognition and influences splicing substrate abundance in budding yeast. Nucleic Acids Res. 2008;36:2787–2798. doi: 10.1093/nar/gkn144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu Z, Luyten I, Bottomley MJ, Messias AC, Houngninou-Molango S, Sprangers R, Zanier K, Krämer A, Sattler M. Structural basis for recognition of the intron branch site RNA by splicing factor 1. Science. 2001;294:1098–1102. doi: 10.1126/science.1064719. [DOI] [PubMed] [Google Scholar]
- 44.Wiesner S, Stier G, Sattler M, Macias MJ. Solution structure and ligand recognition of the WW domain pair of the yeast splicing factor Prp40. J. Mol. Biol. 2002;324:807–822. doi: 10.1016/s0022-2836(02)01145-2. [DOI] [PubMed] [Google Scholar]
- 45.Zhang Y, Madl T, Bagdiul I, Kern T, Kang HS, Zou P, Mäusbacher N, Sieber SA, Krämer A, Sattler M. Structure, phosphorylation and U2AF65 binding of the N-terminal domain of splicing factor 1 during 3′-splice site recognition. Nucleic Acids Res. 2013;41:1343–1354. doi: 10.1093/nar/gks1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang W, Maucuer A, Gupta A, Manceau V, Thickman KR, Bauer WJ, Kennedy SD, Wedekind JE, Green MR, Kielkopf CL. Structure of phosphorylated SF1 bound to U2AF65 in an essential splicing factor complex. Structure. 2013;21:197–208. doi: 10.1016/j.str.2012.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gasch A, Wiesner S, Martin-Malpartida P, Ramirez-Espain X, Ruiz L, Macias MJ. The structure and Prp40 FF1 domain and its interaction with the crn-TPR1 motif of Clf1 gives a new insight into the binding mode of FF domains. J. Biol. Chem. 2006;281:356–364. doi: 10.1074/jbc.M508047200. [DOI] [PubMed] [Google Scholar]
- 48.Bonet R, Ruiz L, Morales B, Macias MJ. Solution structure of the fourth FF domain of yeast Prp40 splicing factor. Proteins. 2009;77:1000–1003. doi: 10.1002/prot.22547. [DOI] [PubMed] [Google Scholar]
- 49.Görnemann J, Barrandon C, Hujer K, Rutz B, Rigaut G, Kotovic KM, Faux C, Neugebauer KM, Séraphin B. Cotranscriptional spliceosome assembly and splicing are independent of the Prp40p WW domain. RNA. 2011;17:2119–2129. doi: 10.1261/rna.02646811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bodenmiller B, Wanka S, Kraft C, Urban J, Campbell D, Pedrioli PG, Gerrits B, Picotti P, Lam H, Vitek O, et al. Phosphoproteomic analysis reveals interconnected system-wide responses to perturbations of kinases and phosphatases in yeast. Sci. Signal. 2010;3:rs4. doi: 10.1126/scisignal.2001182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Strauss EJ, Guthrie C. PRP28, a DEAD-box protein, is required for the first step of mRNA splicing in vitro. Nucleic Acids Res. 1994;22:3187–3193. doi: 10.1093/nar/22.15.3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chang TH, Latus LJ, Liu Z, Abbott JM. Genetic interactions of conserved regions in the DEAD-box protein Prp28p. Nucleic Acids Res. 1997;24:5033–5040. doi: 10.1093/nar/25.24.5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Förch P, Puig O, Kedersha N, Martínez C, Granneman S, Séraphin B, Anderson P, Valcárcel J. The aopoptosis-promoting factor TIA-1 is a regulator of alternative pre-mRNA splicing. Mol. Cell. 2000;6:1089–1098. doi: 10.1016/s1097-2765(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 54.Gabut M, Chaudry S, Blencowe BJ. The splicing regulatory machinery. Cell. 2008;133:192. doi: 10.1016/j.cell.2008.03.010. [DOI] [PubMed] [Google Scholar]
- 55.Roca X, Krainer AR, Eperon IC. Pick one, but be quick: 5′ splice sites and the problems of too many choices. Genes Dev. 2013;27:129–144. doi: 10.1101/gad.209759.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Parker R, Siliciano PG, Guthrie C. Recognition of the TACTAAC box during mRNA splicing in yeast involves base pairing to the U2-like snRNA. Cell. 1987;49:229–239. doi: 10.1016/0092-8674(87)90564-2. [DOI] [PubMed] [Google Scholar]
- 57.Smith DJ, Konarska MM, Query CC. Insights into branch nucleophile positioning and activation from an orthogonal pre-mRNA splicing system in yeast. Mol. Cell. 2009;34:333–343. doi: 10.1016/j.molcel.2009.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Perriman R, Ares M. Invariant U2 snRNA nucleotide from a stem loop to recognize the intron early in splicing. Mol. Cell. 2010;38:416–427. doi: 10.1016/j.molcel.2010.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Selenko P, Gregorovic G, Sprangers R, Stier G, Rhani Z, Krämer A, Sattler M. Structural basis for the molecular recognition between human splicing factors U2AF65 and SF1/BBP. Mol. Cell. 2003;11:965–976. doi: 10.1016/s1097-2765(03)00115-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.