

Abstract
The precise regulation of gene transcription during metazoan development is controlled by a complex system of interactions between transcription factors, histone modifications and modifying enzymes and chromatin conformation. Developments in chromosome conformation capture technologies have revealed that interactions between regions of chromatin are pervasive and highly cell-type specific. The movement of enhancers and promoters in and out of higher-order chromatin structures within the nucleus are associated with changes in expression and histone modifications. However, the factors responsible for mediating these changes and determining enhancer:promoter specificity are still not completely known. In this review, we summarize what is known about the patterns of epigenetic and chromatin features characteristic of elements involved in long-range interactions. In addition, we review the insights into both local and global patterns of chromatin interactions that have been revealed by the latest experimental and computational methods.
INTRODUCTION
Gene regulation during differentiation and development is responsible for the precise coordination of processes, which determine cell fate and how the anatomical plan develops (1). Research in the past couple of decades has shed light on the regulation of some of the genes with the most complex expression patterns (2). The availability of the full sequences from multiple metazoan genomes, as well as an increasing number of high-throughput methods for detecting regulatory elements and monitoring their activity, has revolutionized our understanding of gene regulation. Much of the data produced by large-scale efforts to identify functional elements in the genome, such as ENCODE (3) and modENCODE (4), provide information on the epigenetic state and accessibility of chromatin along the entire genome in various, selected, biological contexts. These data sets contain information about the state of individual regulatory elements (5,6), providing the possibility to add a dynamic, functional layer to studies of regulation at the genomic level.
The various types of regulatory elements in the genome integrate and interpret information from a multitude of regulatory signals to tightly control the spatiotemporal expression of important developmental factors (7). Some genes are regulated by dozens, possibly hundreds, of distal regulatory elements found in large, megabase-sized regions surrounding them (8,9). The extreme distances between regulatory elements and their target genes open a series of questions about an additional aspect of long-range regulation: what is the mechanism by which these elements specifically select and communicate with their target genes, while preventing unwanted promiscuous communication with other genes in the region?
In this review, we give an overview of the current knowledge regarding regulatory elements involved in regulating gene expression, and how these elements communicate with each other during metazoan development. We discuss how the system of interactions between transcription factors (TFs), histone modifications and the 3D organization of chromatin result in the ability to generate complex spatiotemporal expression patterns.
ELEMENTS OF TRANSCRIPTIONAL CONTROL IN VERTEBRATES
Promoters and proximal elements
A promoter is the genomic region overlapping the transcription start sites (TSSs) of a gene. Because of the precision with which TSSs can be identified, especially using CAGE technology (10), this class of regulatory elements has been well studied in comparison with other classes. A promoter is typically described as comprising of two elements: a core promoter and a proximal (regulatory) region. The core promoter is the region near the TSS (including the TSS itself), required for the initiation of transcription and the recruitment of RNA polymerase (Pol) II at the TSS. The proximal promoter is defined as the region immediately upstream of the core promoter, and it typically contains several TF-binding sites (TFBSs), which serve as context-specific regulatory inputs to the core promoter (11).
The architecture of a gene’s promoter relates to both the function of the gene and how it is regulated. It has been proposed that there are at least three main functional classes of PolII promoters in metazoa (12). Type I promoters correspond to genes that display tissue-specific expression patterns, feature a sharp TSS with disordered nucleosomes and are not located close to CpG islands. Ubiquitously expressed genes typically have a Type II promoter. These promoters are located near CpG islands and have a broad TSS distribution with ordered nucleosomes. The promoters of developmentally regulated genes, Type III, typically feature a sharper TSS distribution than Type II promoters, are associated with repression by Polycomb proteins and typically have long and/or multiple CpG islands that extend into the body of the gene. We will discuss these classes further in the context of the differences in their epigenomic and histone properties and their propensity towards forming long-distance interactions. In addition to Type III promoters, developmentally regulated genes, such as developmental TFs, cell adhesion proteins and axon guidance mediators, have loci-specific features that set them aside from other genes (13).
The action of distal cis-acting elements
The regulatory content and architecture of the promoters is insufficient for explaining the diversity observed in gene expression patterns, especially the complex patterns of genes whose products are master regulators of development and differentiation. Most of the regulatory content of a metazoan genome lies outside of proximal promoters (14–16); enhancers are the most common and best understood subset of these elements (17). These sequences are up to several hundred base pairs in length and consist of multiple binding sites for many different TFs and chromatin regulators. The combinations of TFs bound at these elements permit the transcriptional control of their target gene in a dosage, spatial and temporal-specific manner. Traditionally, they are thought to act independently of both the distance and orientation relative to the promoter (18), although some have challenged this assumption (19).
Based on the existence of cooperativity between bound TFs and the spacing and order of TFBS within an enhancer (often referred to as an enhancer grammar), studies of cis-regulatory elements have found that the arrangement of TFBSs can be classified into one of three distinct architectures (20). The enhanceosome (21) architecture requires that the TFBSs follow a strict order and spacing to allow cooperative interactions between both neighbouring proteins and the bound chromatin. In the billboard model of enhancer function (22), TFs are recruited independently to the enhancer; therefore, the spacing and orientation of TFBSs within the enhancer are not important. An enhancer architecture that does not require a well-defined grammar but allows for cooperativity between TFs, called the TF collective model, has been proposed during studies of mesodermal specification (23). This model allows for turnover of the TFBSs within an enhancer while not affecting its activity.
Studies of the sparkling (spa) enhancer of Pax2 in Drosophila (24) have revealed that this enhancer seems to have features indicative of both the billboard and enhanceosome model. Rearrangements of its constitutive TFBSs result in the cell-type specificity of this enhancer being altered, suggesting that the structure of this enhancer is constrained by a number of short-range interactions between bound TFs, while still remaining evolutionarily labile (25). This study also identified a specific sequence at the 5′-end of the enhancer, which was necessary to enhance reporter gene transcription from a distance.
There does not seem to be a limitation to the location of enhancers relative to their target genes (26): in addition to intergenic regions (downstream as well as upstream), they can be located within the introns of the gene they regulate (27), within the introns of neighbouring genes (28), or in intergenic regions beyond neighbouring genes (29). Enhancers can communicate with their target promoter over large distances and over intermediate bystander genes: an enhancer of Shh, which is important for limb development, is located within an intron of the LMBR1 gene situated 1 Mb away (30), with the gene Rnf32 positioned between. Recently, it has been reported that sequences of coding exons can also act as enhancers for their own (31) or neighbouring genes (32,33). Even though the majority of enhancers are in cis to their target genes, and stay in cis through large-scale genomic events, such as whole-genome duplication (9,34), there is a handful of documented, but poorly understood, cases of trans-enhancers that activate the transcription of genes located on different chromosomes (35).
Recently, it has been suggested that genes important for cell identity are regulated by clusters of adjacent enhancers (36,37). These super-enhancers are bound by mediator, are much larger than normal enhancers, have a high density of TFBSs and binding sites for important TFs involved in regulating cell identity. These elements seem to be extremely sensitive to perturbations, i.e. inhibition of BRD4 in cancer cells was found to completely ablate its binding to a super-enhancer of MYC, leading to a significant decrease in the amount of MYC expression (37). Fragments of these elements were found to drive high levels of reporter gene expression. These findings suggest that the TFs bound to these elements are highly co-operative. However, it remains to be shown whether these domains function as a discrete unit or merely reflect enhancers that are clustered together for other reasons, e.g. chromatin accessibility or redundancy.
Promoter: enhancer interaction
There are several models of how distal elements communicate with their target promoters. In the tracking model, the transcription-initiating complex bound to an enhancer moves along the DNA until it reaches a promoter and initiates transcription (38); this model is highly unlikely to account for interactions between promoters and enhancers at megabase distances. In the facilitated tracking model, the tracking movement causes the chromatin to form a loop between the enhancer and promoter. The linking model proposes that enhancers and promoters communicate via intervening linking proteins, whose binding is mediated by enhancer activity (39). However, the simplest model with the most supporting evidence is that there is a direct physical interaction between enhancers and promoters brought about by chromatin looping (40). The mechanisms responsible for mediating this looping are not completely understood at this time, although cohesin and the CCCTC-binding factor (CTCF) are thought to play a prominent role (41).
EVOLUTIONARY FEATURES OF CIS-ACTING ELEMENTS OF DEVELOPMENTAL REGULATORY GENES
A subset of non-coding elements is highly conserved across vertebrates, in some cases being more conserved than the exons of genes that encode perfectly conserved polypeptides. The level of conservation of these sequences (42), their location within vertebrate genomes (43) and their distribution throughout the vertebrate lineage (44) suggested that these were candidates for regulatory elements important in the early stages of vertebrate development. A subset (>50%) of these conserved non-coding elements (CNEs) has been found to function as enhancers in vivo (44,45). The probability that a conserved sequence has enhancer activity is related to its level of conservation and the density of other conserved sequences in the surrounding locus (46,47). However, even developmental enhancers cannot always be identified using DNA sequence conservation methods (48), and their function can be conserved even when their sequence is not (49–51).
Even though many CNEs can function as enhancers, it is puzzling that at least in some cases, deletion of large clusters of CNEs yields viable mice (52) with no obvious deleterious phenotypic changes. The suggested interpretations for this are that some of these elements may be redundant (53), are essential only under specific conditions not found in a laboratory or only have phenotypes that are detectable over many generations.
Since their original discovery in vertebrate genomes, CNEs were found around orthologous genes in other metazoa (54) and recently in plants (55,56). With extremely rare exceptions (26), none of these CNEs in Drosophila or Caenorhabditis elegans are similar at sequence level to any of the vertebrate CNEs, suggesting the existence of equivalent elements in the common ancestor followed by parallel, slow turnover in independent lineages.
Long-range regulation imposes constraints on the organization of the genome (57) and its evolution (58). In Drosophila and vertebrates, developmental genes are associated with arrays of enhancers and CNEs (9,54). Maintaining the correct patterns of gene expression requires that regulatory elements are kept in cis with their target genes. This has led to the maintenance of large regions of conserved synteny over large evolutionary distances (9,54,59), referred to as genomic regulatory blocks (GRBs). These GRBs can extend over large distances and often span large-genomic regions of low-gene density, called gene deserts, or encompass one or more bystander genes in addition to the target gene (Figure 1). Misregulation of GRB target genes has been implicated in developmental disorders and abnormal phenotypes. Mutations in the gene deserts flanking GRB target genes [e.g. SOX9 (60) and PAX6 (49)] are involved in developmental disorders (61). More generally, mutations in distal regulatory elements have been implicated in a variety of diseases, including cancer (62), type II diabetes (63) and dyslexia (64). It is thought that variation within CNEs may have a role in a number of common human disorders (65), and it may help to explain why a large proportion of disease associated single-nucleotide polymorphisms identified using genome-wide association studies (GWAS) are located within non-coding regions (66,67).
Figure 1.

(a) The genomic regulatory block model of transcriptional regulation. Genes involved in the regulation of developmental processes are themselves regulated by enhancers located in a variety of locations, both upstream and downstream. Developmentally regulated genes tend to have CpG islands (CGIs) that overlap with its promoter and extend into the body of the gene. Genes with Type II and III promoters typically feature a broad TSS distribution, as detected by CAGE. (b) Regulatory elements within the genome can be identified by distinct patterns of histone modifications and TF binding. Promoters are enriched for H3K4me3, with active promoters showing evidence of PolII binding. The presence of the repressive mark at promoters and depletion of H3K4me3 is associated with inactive repressed promoters. Promoters having both H3K4me3 and H3K27me3 are termed bivalent promoters; they are repressed but poised for activation. Both poised and active enhancers are marked by the histone modification H3K4me1 and show depletion of H3K4me3. In addition, active enhancers are marked by H3K27ac and the binding of P300, whereas poised enhancers lack H3K27ac and may be marked by H3K27me3.
FUNCTIONAL CHARACTERIZATION OF LONG-RANGE REGULATORY ELEMENTS
The enhancer potential of a putative regulatory sequence is determined by its ability to induce expression in reporter gene assays (68), where enhancer activity is visualized or quantitated based on the activity of a reporter gene. However, induction of reporter gene expression is often an inadequate proxy for the activities of regulatory elements in vivo: these elements can be active during narrow windows of development, the host cell could lack the cofactors necessary for enhancer activation, or the sequence context of the reporter gene can be too different from its native location. Historically identifying and validating enhancers has been a laborious process, although new high-throughput methods for interpreting enhancer function are now being developed (69–71).
Enhancer trap systems can be used to identify the genes and loci responsive to native enhancers in the genome (72). In Drosophila, enhancer traps have found that the gene closest to the insertion site is the gene whose expression pattern tends to be followed in a majority of cases (73). However, in vertebrates, this is often not the case (28), suggesting that the mechanisms of long-range enhancer:promoter communication get more complicated as distances between promoters and enhancers increase.
EPIGENETIC FEATURES OF REGULATORY ELEMENTS
The histone code hypothesis postulates that distinct combinatorial patterns of histone modifications are responsible for specifying function (74); reviewed in (75). Different classes of elements are marked by distinct patterns of histone modifications and TF binding (76) (Figure 1). These patterns modulate the interactions of TFs with chromatin, leading to differences in cell-type–specific transcription (77). They can be identified using chromatin immunoprecipitation (ChIP) coupled with either microarrays (ChIP-chip) or high-throughput sequencing (ChIP-seq). Several studies have identified signatures that are associated with specific genomic contexts and regulatory function (78,79). Many histone modifications are correlated with each other or mutually exclusive, the most obvious example being that two modifications cannot occur at the same position on the tail of the same histone, i.e. H3K27me3 and H3K27ac. However, there are difficulties in determining which modifications are causal and which are the consequence of regulatory binding or transcription without further biochemical studies. These modifications may act in a redundant manner or co-operatively (80), e.g. cross-talk between H3S10ph and H4K16ac has been found to lead to the precise control of transcriptional elongation (81).
We now review what is known about the epigenetic properties of promoters and enhancers, with an emphasis on developmental regulation and associated long-range interactions.
Chromatin modification states and DNA methylation at promoters
Active promoters are associated with the binding of PolII and TAF1, and their histones are marked with H3K4me3 (76). Results of ChIP experiments have provided further evidence for the different functional types of vertebrate promoters (Figure 1). In most vertebrate genes, the pattern of H3K4me3 deposition closely follows the distribution of CpG islands in the genome (82). In tissue-specific promoters, which commonly lack CpG islands, the H3K4me3 mark is generally limited to the 1–3 nucleosomes downstream of the TSS. In mammals, the H3K4me3 mark extends upstream of the gene start, in agreement with the finding that most CpG island promoters show bidirectional transcription (83–85). Studies in mouse stem cells have revealed that developmentally regulated promoters are associated with a combination of both repressive (H3K27me3) and active (H3K4me3) marks (86). These bivalent promoters are often associated with developmental TFs, which are silenced in embryonic stem cells (ESCs), but they are poised for either rapid activation or inactivation at later stages of differentiation. During this process, the marks associated with these promoters resolve to either repressive or active marks in terminally differentiated cells (87).
DNA methylation in vertebrate genomes can occur at cytosine or adenine nucleotides, but it is found preferentially at CpG dinucleotides. However, in vertebrate genomes, stretches of several hundred base pairs, which are enriched for these dinucleotides, called CpG islands, are frequently unmethylated. CpG islands overlap the majority of promoters, with high-CpG content promoters showing evidence of nucleosome deficiency, transient binding of PolII and small amounts of transcription initiation (88,89). Most regulatory regions, especially promoters, which contain CpG islands are unmethylated. However, during differentiation a number of them become methylated, which is responsible for their committed silencing (90–92). It is thought that methylation is responsible for creating a long-lasting state of repression, which is first preceded by the silencing of gene and deposition of repressive histone modifications, such as H3K27me3 (93). DNA methylation is associated with closed chromatin and is thought to prevent the binding of TFs and PolII to the promoter, thus preventing transcription (94,95). During differentiation, promoters associated with pluripotency factors in ESCs show evidence of preferential methylation (92). However, the majority of promoters that are methylated during differentiation show no evidence of transcription in ESCs and tend to be associated with Type I promoters (96), corresponding to tissue-specific genes not expressed in that specific lineage.
The chromatin and DNA methylation state at most promoters is invariant across different cell types (17). The promoters of developmental genes exhibit the highest level of variability, presumably influenced by regulation from multiple distal and proximal enhancers (97). Constitutively, active genes are typically characterized by an active, open chromatin state and are involved in interactions with a relatively small number of enhancers (98). Type I promoters are only marked by H3K4me3 when the gene is undergoing transcription and are predominantly regulated only by their core and proximal elements (99). Type I and Type III (developmental) promoters show changes in their epigenetic state during differentiation. The latter move from bivalent to an active or inactive state or from active/inactive to inactive/active state. It is unknown whether the changes in the modifications associated with a promoter are a consequence of enhancer:promoter interactions or actually play a role in facilitating them.
Chromatin modification states at enhancers
Results from ChIP experiments have been useful in identifying putative enhancer elements in the genome. Enhancers are associated with regions of lower nucleosomal density containing specific histone variants, such as H3.3 and H2A.Z (80,100). The presence of these variants contributes to nucleosome instability, allowing TFs to bind more easily and displace nucleosomes (101). These regions are characterized by many different histone modifications and bound by histone-modifying enzymes, such as CREB-binding protein (CBP) and P300 acetyltransferases (102). Enhancers are highly enriched for H3K4me1, H3K4me2 and H3K27ac marks, but they show little or no enrichment for the promoter-associated mark, H3K4me3 (Figure 1). The changes in histone modifications at enhancers play a major role in determining the differential binding of TFs to enhancer elements (77). These patterns are more variable compared with promoters and show good correlation with differences in expression of the target gene (17,103). A study over a large number of cell lines and tissues found that the majority of enhancers show high levels of tissue specificity (104), confirmed by combining the results from ChIP-seq and the mapping of DNase hypersensitive regions (105,106).
Constitutively open enhancers that bind glucocorticoid receptor have been found to be enriched for CpG dinucleotides, whereas enhancers that required chromatin remodelling for activation were not (107). The activation of distal regulatory elements is often accompanied by their demethylation (108,109). Active enhancers that have low levels of H3K27ac and H3K4me1 show higher levels of DNA methylation than enhancers with higher levels (110). These observations reflect the importance of methylation in regulating the cell-type specificity of enhancers and the binding of relevant histone modifiers and TFs.
It is thought that enhancers are initially marked by histone variants and H3K4me1/2 in ESCs, with the binding of P300 and the subsequent addition of H3K27ac leading to their activation. In addition, enhancers can exist in a poised state, marked by H3K4me1 but not H3K27ac. Also, at least a subset of poised developmental enhancers is marked by H3K27me3 (111). Enhancers are believed to be held in this state by Polycomb silencing. In human ESCs, Rada-Iglesias et al. (112) identified that active enhancers associated with pluripotency genes, many of which are at large distances from the gene, become inactivated during differentiation, and poised enhancers associated with genes involved in early development become activated. This activation is accomplished by the loss of H3K27me3 and the gain of H3K27ac. Thus, the epigenetic state of an enhancer can provide a measure of its transcription activation potential. Inactive enhancers lack H3K4me2 and have high levels of H3K9me2. In mouse ESCs, Zentner et al. (113) found evidence that other classes of enhancer states may exist, potentially specified by additional histone modifications.
The number of enhancer-related histone modifications is still increasing, and as such, the true extent of the enhancer complement of vertebrate genomes is unknown (80,114). Genome-wide profiling of P300, H3K27me3 and H3K27ac in mouse embryonic limb revealed that a significant percentage of experimentally validated enhancers was not identified by either mark (115). This suggests that additional marks play a role in specifying enhancer function and cell specificity. Enhancers containing nucleosomes marked by H4K16ac seem to be able to recruit factors that allow the release of promoter proximal paused PolII (81). H3K8ac is associated with active promoters, but has also found to mark distal elements (80,116) and is a potential indicator of active enhancers.
The epigenetic marks flanking an enhancer are capable of determining its cell-type specificity (117). Investigations of the epigenetic states of the enhancers and promoters of Mdc and IlI2b in fibroblasts and dendritic cells have found that enhancers flanked by H3K9me3 seem to be unable to drive expression of their target gene, despite being marked by H3K4me1 and being able to drive reporter expression in plasmids. It seems that this mechanism for repressing enhancer activity may be prevalent, as enhancers flanked by H3K9me3 seem to be functional when assayed over a large number of cell types, even though they do not drive expression in their native environment.
ORGANIZATION OF CHROMATIN AND ENHANCER:PROMOTER INTERACTIONS WITHIN THE NUCLEUS
The spatial organization of chromosomes within the nucleus is a major factor in determining gene expression. Active genes are typically localized in the interior of the nucleus, whereas silenced genes are associated with the peripheral regions. Within the nucleus, chromatin forms several substructures, which are involved in the repression or activation of genes that are co-localized within them.
Chromosome territories
Each chromosome occupies a distinct chromosomal territory (CT) within the nucleus. In mammalian cells, there is evidence that the relative position of a gene locus within its CT influences its ability to form either cis or trans interactions. Specific sequences on a given chromosome can loop out of their CT, leading to their subsequent upregulation. Activation of the HoxB gene cluster during differentiation coincides with its relocation away from its CT interior (118). Alterations in the localization of a gene with respect to its CT are believed to be controlled by the action of enhancers. The long-range enhancer of the Shh gene, required for limb bud development, is important in causing the Shh locus to extrude from its CT (119). Within the nucleus, highly expressed co-regulated genes associate together at discrete foci, called transcription factories (120,121). These compartments contain high concentrations of hyperphosphorylated PolII [as well as other TFs (122)], which allows the genes inside them to be efficiently transcribed. Movement of genes into or out of these factories results in their up- or downregulation, and it may explain why genes are transcribed in bursts (123). In some cases, genes found within transcription factories can be located on different chromosomes (124). The movement of genes within the nucleus likely plays a major role in the regulation of gene expression during differentiation.
Broader chromatin states and relationship with long-range regulation
The human genome shows large regions in which most genes are expressed at high levels, alternating with regions that mainly contain lowly expressed genes. At least half of the Drosophila genome consists of multi-gene chromatin domains, which can be large and include dozens of genes (125). These domains might be important for developmental synchronization of genes and are cell-type dependent. Broad regions of histone modifications may reflect both the transcriptional status of these domains and the mechanism by which they are silenced or expressed.
Large domains of chromatin (0.1–10 Mb) have been found to be in contact with the nuclear lamina (126) (Figure 2). These lamina-associated domains (LADs) are transcriptionally inactive, show enrichment for H3K9me2 and are depleted in active histone modifications. Large changes in the levels of PolII and H3K4me2 were observed at the boundaries of LADs. The interaction of a gene with the lamina directly leads to the silencing of transcription (127). Large domains of H3K9me2 (128), called LOCKS, overlap with LADs. These domains show changes during differentiation and are thought to be cell-type specific. This indicates that chromatin differentially associates with the lamina depending on the cell type. Genes that are not expressed in a specific cell type may be tethered to the lamina, whereas genes expressed in that specific cell type are not. This reorganization of the genome in the nucleus seems to be responsible for loss of competence during neuronal differentiation in Drosophila (129). The hunchback locus, which is responsible for determining cell fate, is first downregulated and then relocates towards the lamina leading to its permanent repression. During differentiation, genes that move away from the lamina are activated, as they become localized to the interior regions of the nucleus.
Figure 2.
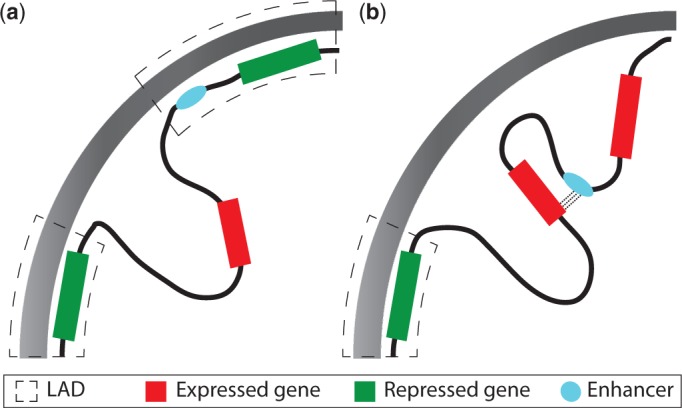
The organization and structure of chromatin within the nucleus is coupled with the regulation of gene expression (125). (a) Genes tethered to the nuclear lamina are silenced, whereas genes that are present in the centre of the nucleus are not. In addition, enhancers anchored to the lamina are restricted from interacting with their target promoters. (b) The movement of genes away from the lamina results in their upregulation, as they move to a more permissive transcriptional domain, and the movement of enhancers allows them to communicate properly and regulate the expression profile of their target gene.
Studies have shown that Polycomb and H3K27me3 can form continuous chromatin domains, which can be larger than 100 kb and show overlap with silenced genes and intergenic regions (130). These domains extensively and specifically interact with each other, with the majority of interactions between regions on the same chromosomal arm (131,132). Long-range contacts with sites devoid of Polycomb binding were found to be rare.
FACTORS INVOLVED IN MEDIATING LONG-RANGE INTERACTIONS
The mechanisms that modulate long-range interactions are currently poorly understood. Factors such as CTCF, cohesin, mediator and small RNAs (133) seem to play important roles in this process. Mediator and cohesin have been found to co-occur together at the promoters of active genes (134). In addition, there is evidence that a direct interaction between PolII and mediator is required for transcription (135), and that interactions between paused PolII and cohesin are required for transcriptional elongation to occur (136).
Regulatory elements referred to as insulators are able to block communication between enhancers and their cognate promoter. These elements are thought to function by blocking enhancer:promoter interactions (137), either by the insulator element binding to the enhancer or promoter and preventing them from interacting, or by partitioning the genome in a series of loop-like structures such that elements in one loop are not able to interact with elements in a different loop (138). CTCF, which binds at insulators, has been found to be involved in promoting and mediating long-range enhancer:promoter interactions (139), and it may be responsible for demarcating cell-type–specific regulatory regions (140). Along with CTCF, cohesin is also thought to play a major role in controlling 3D conformation (41). Recent work has proposed that CTCF may also play a role in regulating relatively short-range enhancer:promoter interactions (on the order of kilobases) (16) by altering local nucleosome configuration (141), whereas additional factors are required for long-distance communication.
It is suggested that CTCF, cohesin and mediator may be involved generally in the formation of chromatin structures, whereas specific TFs and their coactivators may be involved in controlling locus-specific looping interactions and structure. Through the use of a novel tethering assay, Deng et al. (142) identified that Ldb1 is an important TF involved in GATA-1-mediated looping in the β-globin locus. Ldb1 was attached to a zinc-finger protein, which allowed it to be artificially anchored to specific regions within the β-globin locus. In the absence of GATA-1, transcription of β-globin could be induced by attaching this construct to its promoter. This region was found to directly interact with elements at the locus control region, suggesting important roles for this factor in regulating long-range interactions.
Several studies have found that non-coding RNAs (ncRNAs) are important in mediating enhancer:promoter interactions. HOTTIP is a long intergenic non-coding RNA (lincRNA) located at the 5′ section of the HOXA locus (143), which is required for coordination of several distal HOX genes and is required for the maintenance of H3K4me3 at these promoters. It seems that looping brings HOTTIP into close proximity with its target genes and recruits WDR5-MLL to the locus. Knockdown of this lincRNA by siRNA does not alter the higher order structure of this locus; however, there is a reduction in the level of promoter-associated H3K4me3. A subset of ncRNAs called ncRNA-a (ncRNA-activating) seems to work by activating their neighbouring gene in cis. A number of these ncRNAs seem to bind with mediator, resulting in the expression of their target genes (144). Depletion of either the reported ncRNA-a transcripts or mediator subunits reduced looping between target genes and their regulatory elements, but did not completely remove it. This suggests that at least a subset of ncRNAs involved in long-range interactions work by stabilizing pre-existing chromatin structures rather than being directly involved in their formation.
ENHANCER TRANSCRIPTION AND ITS ROLE IN LONG-RANGE TRANSCRIPTIONAL REGULATION
Although transcription at enhancers was first observed >20 years ago (38), only recently has evidence been found that this phenomenon is both widespread and indicative of enhancer activation. In mouse motor neurons, Kim et al. (145) found that the transcriptional co-activator CBP recruits PolII to enhancers, leading to the bidirectional transcription of ncRNAs called enhancer RNAs (eRNAs). The level of eRNA expression was found to correlate well with expression at nearby genes. These eRNAs are produced bidirectionally from within a relatively small region (<2 kb), and not over the entire distance between the enhancer and promoter, providing further evidence that the tracking model of enhancer:promoter communication is incorrect. Transcription of eRNA did not occur unless there was a direct interaction between an enhancer and its cognate promoter, leading to the proposition that the PolII, which binds at an enhancer is transferred to its target promoter via looping. Analysis of Hi-C data generated by the ENCODE consortium has also found that enhancers involved in physical interactions are significantly more likely to be associated with transcription of eRNA (27).
In macrophages, a significant amount of PolII binding occurs extragenically (146), with 70% of these binding events located within putative enhancer regions. Transcription was confirmed at a number of these enhancers using quantitative polymerase chain reaction, and unlike the eRNAs reported by Kim et al., these transcripts were found to be polyadenylated. This study found that the number of non-transcribed enhancers was considerably larger than the number of transcribed ones, suggesting that they could belong to two different groups. Inhibition of RNA synthesis resulted in decreased acetylation at both the TSS and upstream regions.
Wang et al. (147) found that eRNA expression was better at indicating enhancer activation than TF binding and histone modification data. In addition to chromatin state and evolutionary conservation, the detection of eRNAs has been used to identify putative enhancers (148). It remains an open question whether these eRNAs are simply transcriptional noise or are functionally important. Recently, eRNAs produced by enhancers bound by p53 have been found to directly enhance transcription at multiple distant genes (149); siRNA knockdown of a number of these eRNAs resulted in a decrease in expression of their target genes, while maintaining existing chromatin interactions. Other classes of ncRNAs stabilize long-range enhancer:promoter interactions (150) or recruit chromatin remodellers (151), and eRNAs might perform a similar role.
DIRECT 3D INTERACTION INSIGHTS ON PROMOTER–ENHANCER INTERACTION
A single promoter can be involved in interactions with a single or multiple enhancers (152). Enhancers can either interact with a specific target gene or with many genes (153) (allowing the coordinated regulation of functionally related genes). Type I (tissue-specific) PolII promoters are typically controlled by regulatory elements in close proximity (99), whereas Type III (developmental) promoters are most often controlled by long-range regulation (13).
Evidence suggesting that physical interactions between enhancers and promoters are necessary has been found using a variety of approaches, including fluorescence in situ hybridisation (FISH) and chromosome conformation capture methods. By tagging specific sequences with fluorescent probes, FISH allows the identification of regions of the genome that are brought into close spatial proximity (154). Chromosome conformation capture methods (155–158) are useful for assessing the frequency of interactions between two genomic loci: either between two pre-selected loci (3C), one locus and the rest of the genome (4C, 3C-seq) or all interactions between multiple pre-selected elements (5C). Hi-C allows unbiased and genome-wide investigation of interactions, although currently its resolution is relatively low compared with other techniques (159,160). ChIA-PET (chromatin interaction assay with paired-end sequencing) makes it possible to identify all interactions mediated by a specific protein, allowing investigation of how specific TFs (and histone modifications) alter the structure of chromatin (161). Currently, the available evidence from 3C-based methods is suggestive of direct physical interactions between enhancers and promoters. However, recent work has identified that the results from FISH and different 3C-based methods are not always concordant (162), e.g. many of the loci predicted to interact by 5C were not found to co-localize in FISH experiments. The suggested reasons for these observations are that FISH is a more disruptive method leading to loss of some interactions during cell treatment, or that formaldehyde cross-linking in 3C-based methods does not always reflect average spatial distances between the interacting loci. Nevertheless, these technologies have provided insights into how promoters and enhancers communicate in 3D space and into the effects of chromosomal conformation on gene expression (159).
Locus control regions and active chromatin hubs
In erythoid cells, where β-globin is expressed, these interactions form a compartment containing regulatory elements that has a high level of transcriptional activity (40). These chromatin structures depend on sequence-specific TFs bound to both the enhancer and promoter and thus can explain the specificity observed in enhancer:promoter interactions. Knock out of TFs responsible for regulating β-globin expression (EKLF and GATA-1) result in the loss of interactions between the gene and its enhancers, leading to loss of the overall hub-like structure (163,164). Interactions between regulatory elements separated by large genomic distances have been observed at high frequencies at other loci, leading to the proposition that chromatin looping generally results in the formation of hub-like structures. These active chromatin hubs (ACH) are responsible for bringing enhancers and promoters into close spatial proximity as well as providing an environment that is transcriptionally permissive (Figure 3a). The contents of the β-globin ACH changes during differentiation, with the ACH lacking certain elements in progenitor cells, which are found there later during differentiation (167). At the Myb locus during erythoid proliferation, intergenic enhancers, the Myb promoter and its first intron are brought together to form an ACH (168). The ACH leads to high concentrations of PolII and TFs being present around Myb. The first intron of this gene contains a site for regulating transcriptional elongation. Interactions between this element and distal enhancers lead to the generation of full-length transcripts. This structure is lost when cells terminally differentiate, coincident with the loss of expression of Myb and a reduction in TF binding at regulatory elements.
Figure 3.

Chromatin looping is responsible for forming higher-order hub-like structures within the nucleus. (a) An active chromatin hub (ACH) is a structure that allows enhancers and promoters to come into close spatial proximity with each other (40). This structure has a high concentration of chromatin remodellers and PolII, which allows stable/high levels of transcription. Interactions between promoters and enhancers and represented by dashed lines. (b) The recruitment of genes and enhancers to a repressive chromatin hub (RCH) results in their downregulation (165). This structure potentially prevents enhancers from communicating with their cognate promoter by looping them out and preventing them from interacting. This structure may also restrict the amount of PolII from binding to gene promoters. (c and d) During development, genes in the Hox locus show a linear movement from a repressive chromatin structure to a region where they are expressed (166). This movement allows enhancers to interact with targets that were previously held in the repressive domain.
Chromatin can form structures that prevent enhancer: promoter communication and repress transcription
Chromatin hub-like structures have also been found to play a role in repressing gene expression (165). Studies of the GATA-4 locus in undifferentiated Tera-2 cells have revealed the existence of a 3D structure consisting of multiple chromatin loops (Figure 3b), termed a pre-repressive chromatin hub (pre-RCH), which is lost when these cells differentiate. This structure was found to comprise several H3K27me3-enriched elements and was maintained by Polycomb, suggesting that Polycomb may repress genes through the formation and maintenance of these structures. In undifferentiated cells, this structure results in GATA-4 being held in a poised state, and its loss during differentiation was accompanied with an increase in the expression of GATA-4. This structure may function by either preventing enhancer:promoter interactions or by restricting the access of PolII to the GATA-4 promoter.
Coordinated developmental control of Hox loci by long-range regulation
In vertebrates, Hox genes are typically organized into clusters and are transcribed sequentially; with the first gene in the cluster being expressed in the anterior part of the organism. This collinear expression pattern results from changes in both the histone modification patterns within a Hox cluster and its overall 3D conformation (118). Within a Hox cluster, active and inactive genes are separated into distinct domains labelled by distinct histone modifications, H3K4me3 and H3K27me3, respectively. It is thought that Hox genes are expressed sequentially because of their highly regulated movement out of chromosome territories and the decondensation of associated chromatin (169). Studies of the HoxD locus using 4C (166) have found that when all genes within a cluster are not expressed, these genes form a single 3D structure that represses transcription. This led the authors to propose a model whereby as genes are expressed they progressively migrate from this structure and cluster into a transcriptionally active structure, leading to a bimodal organization of chromatin at a single Hox cluster (Figure 3c and d). This movement is associated with changes in histone modifications and suggests that the observed collinear expression may be the result of a stepwise movement of genes from an RCH-like structure to an ACH-like structure.
An examination of the HoxD locus, important for limb development, using chromosome conformation capture techniques has revealed that this locus has a tissue-specific conformation involving interactions between genes and enhancers located within the adjacent gene deserts (152). The active section of gene cluster seems to be in contact with several enhancers concurrently, each of which seem to be important for some aspect of limb development. The concept of a regulatory archipelago proposed by Montavon et al. (152) is basically the same as the previously proposed GRB model of transcriptional regulation (9). These findings suggests that the 3D conformation of loci changes during differentiation as a result of interactions between promoters and enhancers, resulting in the creation of cell-type–specific patterns of gene expression and organization. Indeed, the most recent results suggest that the 3D conformation at developmental loci is both the most dynamic and most divergent across mammals (170).
Hi-C provides information on the global patterns of long-range interactions within the genome
Hi-C has provided further evidence for the presence of CTs and found that eukaryotic genomes are organized into functional domains (A and B compartments) that are important for controlling DNA transcription (159). By applying a hidden Markov model to Hi-C data, Dixon et al. (160) were able to partition the genome into megabase-sized domains of chromatin, called topological domains. Elements within these domains preferentially interacted with other elements in the same domain. The boundaries of these domains were found to be associated with known elements displaying barrier activity and correlated well with known CTCF-binding sites.
Hi-C has enabled the calculation of the probability that two randomly chosen loci interact (159). This contact probability follows a power-law distribution and suggests that chromatin is organized globally in a fractal globule structure. However, the exponent that describes this distribution has been found to vary (171), and recent studies have found that the contact probability plateaus as the genomic distance increases (172). These findings suggest that this model cannot adequately describe the observed patterns of chromatin folding. The strings and binders model (173) has been proposed, which not only recapitulates this structure but is also more related to the known underlying biology of protein-mediated chromatin folding.
5C confirms non-linear arrangement of enhancers and their target genes
By examining 5C data from three different cell lines, Sanyal et al. (27) found evidence that large numbers of looping interactions were cell-type specific and tended to occur between active functional elements. Long-range interactions between enhancer:promoter pairs were found in domains enriched for both H3K9ac and H3K27ac. Additionally, only 7% of identified interactions were between a distal element and it’s nearest TSS (this increased to 22% when only considering active TSSs), which shows the flaw in studies that assign enhancers and other distal elements to the nearest TSS.
Chromatin folding mediated by transcription factors reveals an extensive network of interactions between genomic elements
ChIA-PET has revealed insights into how these interactions are mediated by TFs, allowing the generation of genome-wide chromatin interactomes. In the initial ChIA-PET experiments, estrogen-receptor (ER)-α was used as bait to identify interactions between regions of the genome, which it was mediating (161). As expected, most interactions occur between elements located on the same chromosome, rather than interchromosomally. A positive association was found between chromatin interactions and the activation of genes involved in those interactions. In addition to identifying interactions between distal sites and promoters, a number of interactions were found between distal sites, hinting at the possibility of widespread enhancer:enhancer interactions. Li et al. (154) used PolII as bait in ChIA-PET experiments and identified that promoters are involved in three distinct types of interactions; either with the body of the gene, with distal elements or with other promoters. Expression of genes regulated by promoters involved in interactions with other promoters was found to be highly correlated. This may explain how transcription of tissue-specific and housekeeping genes is co-ordinated. Long-range interactions between distal elements and promoters were found to be highly cell-type specific, and promoters were involved in interactions at different distances depending on the cell line under investigation. To investigate enhancer:promoter interactions, Chepelev et al. (174) used the enhancer mark H3K4me2 as bait and identified >6000 potential interactions. Promoters that were found to interact with the same enhancer showed evidence of tissue-specific co-expression.
ChIA-PET has also revealed that CTCF-mediated interactions correlate with distinct domains of histone modifications in the genome (175). The interactions between CTCF-bound sites and promoters seem to be more cell-type invariant when compared with enhancer:promoter interactions, as is CTCF binding. As such, insulators involved in enhancer-blocking may function to mediate cell-type–specific long-range interactions. However, recently a number of looping interactions were found to skip sites bound by both CTCF and cohesin, which may suggest that additional factors are required (27).
This form of interaction data can be easily represented as a network, with an edge present between two nodes (each of which corresponds to a region of chromatin), when they have been found to interact. A large connected component (containing ∼40% of the elements involved) has been identified within the network of PolII-mediated chromatin interactions (176). This network exhibits a hierarchical structure and has a scale-free degree distribution. Investigation of communities of strongly connected elements within this network revealed that these were involved in functional compartmentalization. The majority of these chromatin communities were conserved between cell lines, indicating that cell-type specificity may be defined by long-range transient interactions or by small differences in the content and organization of these communities.
CONCLUSION
The identification of the different factors and histone modifications involved in long-range regulation will help elucidate how specific enhancers target specific promoters without binding to intervening promoters.
It does not seem that there are any features specific to enhancers involved in long-range regulation compared with those involved with regulation over shorter distances. Indeed, it seems that the ability to respond to long-range interactions depends on the promoter architecture of a gene and the state of intervening insulators. The development of new genome-wide techniques has provided a way to systematically identify regions that are targeted by specific proteins and co-localize in nuclear space. The results from ChIP-seq and chromosome conformation capture assays are, however, averaged over a cell population, and they do not provide any information on the temporal dynamics and cell-to-cell variation of long-range interactions. Improvements in the temporal resolution of experiment techniques should help determine how chromatin moves during the cell cycle and how this affects and which genes are upregulated and downregulated at specific stages. Studies of long-range interactions are also limited by the relatively low resolution of Hi-C. As this improves, we expect that new findings will follow regarding the global patterns of interactions between individual regulatory elements. Examination of this in multiple cell types and species will enable us to understand how the constraints on 3D conformation affect the arrangement and conservation of regulatory elements.
ACKNOWLEDGEMENTS
Thanks to Anja Barešić, Petar Glažar, Vanja Haberle and Elizabeth Ing-Simmons for their comments.
FUNDING
Medical Research Council UK (to N.H. and B.L.); Norwegian Research Council (YFF) (to B.L.). Funding for open access charge: MRC Clinical Sciences Centre.
Conflict of interest statement. None declared.
REFERENCES
- 1.Pearson JC, Lemons D, McGinnis W. Modulating Hox gene functions during animal body patterning. Nat. Rev. Genet. 2005;6:893–904. doi: 10.1038/nrg1726. [DOI] [PubMed] [Google Scholar]
- 2.Armit C, Venkataraman S, Richardson L, Stevenson P, Moss J, Graham L, Ross A, Yang Y, Burton N, Rao J, et al. eMouseAtlas, EMAGE, and the spatial dimension of the transcriptome. Mamm. Genome. 2012;23:514–524. doi: 10.1007/s00335-012-9407-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Celniker SE, Dillon LAL, Gerstein MB, Gunsalus KC, Henikoff S, Karpen GH, Kellis M, Lai EC, Lieb JD, MacAlpine DM, et al. Unlocking the secrets of the genome. Nature. 2009;459:927–930. doi: 10.1038/459927a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.ENCODE Project Consortium. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.May D, Blow MJ, Kaplan T, McCulley DJ, Jensen BC, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, et al. Large-scale discovery of enhancers from human heart tissue. Nat. Genet. 2012;44:89–93. doi: 10.1038/ng.1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wilson NK, Foster SD, Wang X, Knezevic K, Schütte J, Kaimakis P, Chilarska PM, Kinston S, Ouwehand WH, Dzierzak E, et al. Combinatorial transcriptional control in blood stem/progenitor cells: genome-wide analysis of ten major transcriptional regulators. Cell Stem Cell. 2010;7:532–544. doi: 10.1016/j.stem.2010.07.016. [DOI] [PubMed] [Google Scholar]
- 8.Nobrega MA, Ovcharenko I, Afzal V, Rubin EM. Scanning human gene deserts for long-range enhancers. Science. 2003;302:413. doi: 10.1126/science.1088328. [DOI] [PubMed] [Google Scholar]
- 9.Kikuta H, Laplante M, Navratilova P, Komisarczuk AZ, Engström PG, Fredman D, Akalin A, Caccamo M, Sealy I, Howe K, et al. Genomic regulatory blocks encompass multiple neighboring genes and maintain conserved synteny in vertebrates. Genome Res. 2007;17:545–555. doi: 10.1101/gr.6086307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, Oyama R, Ravasi T, Lenhard B, Wells C, et al. The transcriptional landscape of the mammalian genome. Science. 2005;309:1559–1563. doi: 10.1126/science.1112014. [DOI] [PubMed] [Google Scholar]
- 11.Juven-Gershon T, Hsu J-Y, Kadonaga JT. Perspectives on the RNA polymerase II core promoter. Biochemical Society Transactions. 2006;34(Pt. 6):1047–1050. doi: 10.1042/BST0341047. [DOI] [PubMed] [Google Scholar]
- 12.Lenhard B, Sandelin A, Carninci P. Metazoan promoters: emerging characteristics and insights into transcriptional regulation. Nat. Rev. Genet. 2012;13:233–245. doi: 10.1038/nrg3163. [DOI] [PubMed] [Google Scholar]
- 13.Akalin A, Fredman D, Arner E, Dong X, Bryne JC, Suzuki H, Daub CO, Hayashizaki Y, Lenhard B. Transcriptional features of genomic regulatory blocks. Genome Biol. 2009;10:R38. doi: 10.1186/gb-2009-10-4-r38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gerstein MB, Kundaje A, Hariharan M, Landt SG, Yan K-K, Cheng C, Mu XJ, Khurana E, Rozowsky J, Alexander R, et al. Architecture of the human regulatory network derived from ENCODE data. Nature. 2012;489:91–100. doi: 10.1038/nature11245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yip KY, Cheng C, Bhardwaj N, Brown JB, Leng J, Kundaje A, Rozowsky J, Birney E, Bickel P, Snyder M, et al. Classification of human genomic regions based on experimentally determined binding sites of more than 100 transcription-related factors. Genome Biol. 2012;13:R48. doi: 10.1186/gb-2012-13-9-r48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maston GA, Evans SK, Green MR. Transcriptional regulatory elements in the human genome. Annu. Rev. Genomics Hum. Genet. 2006;7:29–59. doi: 10.1146/annurev.genom.7.080505.115623. [DOI] [PubMed] [Google Scholar]
- 19.Marinić M, Aktas T, Ruf S, Spitz F. An integrated holo-enhancer unit defines tissue and gene specificity of the Fgf8 regulatory landscape. Dev. Cell. 2013;24:530–542. doi: 10.1016/j.devcel.2013.01.025. [DOI] [PubMed] [Google Scholar]
- 20.Spitz F, Furlong EEM. Transcription factors: from enhancer binding to developmental control. Nat. Rev. Genet. 2012;13:613–626. doi: 10.1038/nrg3207. [DOI] [PubMed] [Google Scholar]
- 21.Panne D. The enhanceosome. Curr. Opin. Struct. Biol. 2008;18:236–242. doi: 10.1016/j.sbi.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 22.Kulkarni MM, Arnosti DN. Information display by transcriptional enhancers. Development. 2003;130:6569–6575. doi: 10.1242/dev.00890. [DOI] [PubMed] [Google Scholar]
- 23.Junion G, Spivakov M, Girardot C, Braun M, Gustafson EH, Birney E, Furlong EEM. A transcription factor collective defines cardiac cell fate and reflects lineage history. Cell. 2012;148:473–486. doi: 10.1016/j.cell.2012.01.030. [DOI] [PubMed] [Google Scholar]
- 24.Swanson CI, Evans NC, Barolo S. Structural rules and complex regulatory circuitry constrain expression of a Notch- and EGFR-regulated eye enhancer. Dev. Cell. 2010;18:359–370. doi: 10.1016/j.devcel.2009.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Swanson CI, Schwimmer DB, Barolo S. Rapid evolutionary rewiring of a structurally constrained eye enhancer. Curr. Biol. 2011;21:1186–1196. doi: 10.1016/j.cub.2011.05.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vavouri T, McEwen GK, Woolfe A, Gilks WR, Elgar G. Defining a genomic radius for long-range enhancer action: duplicated conserved non-coding elements hold the key. Trends Genet. 2006;22:5–10. doi: 10.1016/j.tig.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 27.Sanyal A, Lajoie BR, Jain G, Dekker J. The long-range interaction landscape of gene promoters. Nature. 2012;489:109–113. doi: 10.1038/nature11279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kikuta H, Fredman D, Rinkwitz S, Lenhard B, Becker TS. Retroviral enhancer detection insertions in zebrafish combined with comparative genomics reveal genomic regulatory blocks - a fundamental feature of vertebrate genomes. Genome Biol. 2007;8(Suppl. 1):S4. doi: 10.1186/gb-2007-8-s1-s4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abbasi AA, Paparidis Z, Malik S, Goode DK, Callaway H, Elgar G, Grzeschik K-H. Human GLI3 intragenic conserved non-coding sequences are tissue-specific enhancers. PLoS One. 2007;2:e366. doi: 10.1371/journal.pone.0000366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lettice LA, Heaney SJH, Purdie LA, Li L, de Beer P, Oostra BA, Goode D, Elgar G, Hill RE, de Graaff E. A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum. Mol. Genet. 2003;12:1725–1735. doi: 10.1093/hmg/ddg180. [DOI] [PubMed] [Google Scholar]
- 31.Lampe X, Samad OA, Guiguen A, Matis C, Remacle S, Picard JJ, Rijli FM, Rezsohazy R. An ultraconserved Hox-Pbx responsive element resides in the coding sequence of Hoxa2 and is active in rhombomere 4. Nucleic Acids Res. 2008;36:3214–3225. doi: 10.1093/nar/gkn148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Birnbaum RY, Clowney EJ, Agamy O, Kim MJ, Zhao J, Yamanaka T, Pappalardo Z, Clarke SL, Wenger AM, Nguyen L, et al. Coding exons function as tissue-specific enhancers of nearby genes. Genome Res. 2012;22:1059–1068. doi: 10.1101/gr.133546.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dong X, Navratilova P, Fredman D, Drivenes O, Becker TS, Lenhard B. Exonic remnants of whole-genome duplication reveal cis-regulatory function of coding exons. Nucleic Acids Res. 2010;38:1071–1085. doi: 10.1093/nar/gkp1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McEwen GK, Woolfe A, Goode D, Vavouri T, Callaway H, Elgar G. Ancient duplicated conserved noncoding elements in vertebrates: a genomic and functional analysis. Genome Res. 2006;16:451–465. doi: 10.1101/gr.4143406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bateman JR, Johnson JE, Locke MN. Comparing enhancer action in cis and in trans. Genetics. 2012;191:1143–1155. doi: 10.1534/genetics.112.140954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, Young RA. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–334. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tuan D, Kong S, Hu K. Transcription of the hypersensitive site HS2 enhancer in erythroid cells. Proc. Natl Acad. Sci. USA. 1992;89:11219–11223. doi: 10.1073/pnas.89.23.11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bulger M, Groudine M. Looping versus linking: toward a model for long-distance gene activation. Genes Dev. 1999;13:2465–2477. doi: 10.1101/gad.13.19.2465. [DOI] [PubMed] [Google Scholar]
- 40.Tolhuis B, Palstra R-J, Splinter E, Grosveld F, de Laat W. Looping and interaction between hypersensitive sites in the active beta-globin locus. Mol. Cell. 2002;10:1453–1465. doi: 10.1016/s1097-2765(02)00781-5. [DOI] [PubMed] [Google Scholar]
- 41.Degner SC, Verma-Gaur J, Wong TP, Bossen C, Iverson GM, Torkamani A, Vettermann C, Lin YC, Ju Z, Schulz D, et al. CCCTC-binding factor (CTCF) and cohesin influence the genomic architecture of the Igh locus and antisense transcription in pro-B cells. Proc. Natl Acad. Sci. USA. 2011;108:9566–9571. doi: 10.1073/pnas.1019391108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bejerano G, Pheasant M, Makunin I, Stephen S, Kent WJ, Mattick JS, Haussler D. Ultraconserved elements in the human genome. Science. 2004;304:1321–1325. doi: 10.1126/science.1098119. [DOI] [PubMed] [Google Scholar]
- 43.Sandelin A, Bailey P, Bruce S, Engström PG, Klos JM, Wasserman WW, Ericson J, Lenhard B. Arrays of ultraconserved non-coding regions span the loci of key developmental genes in vertebrate genomes. BMC Genomics. 2004;5:99. doi: 10.1186/1471-2164-5-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Woolfe A, Goodson M, Goode DK, Snell P, McEwen GK, Vavouri T, Smith SF, North P, Callaway H, Kelly K, et al. Highly conserved non-coding sequences are associated with vertebrate development. PLoS Biol. 2005;3:e7. doi: 10.1371/journal.pbio.0030007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pennacchio LA, Ahituv N, Moses AM, Prabhakar S, Nobrega MA, Shoukry M, Minovitsky S, Dubchak I, Holt A, Lewis KD, et al. In vivo enhancer analysis of human conserved non-coding sequences. Nature. 2006;444:499–502. doi: 10.1038/nature05295. [DOI] [PubMed] [Google Scholar]
- 46.Prabhakar S, Poulin F, Shoukry M, Afzal V, Rubin EM, Couronne O, Pennacchio LA. Close sequence comparisons are sufficient to identify human cis-regulatory elements. Genome Res. 2006;16:855–863. doi: 10.1101/gr.4717506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Engström PG, Fredman D, Lenhard B. Ancora: a web resource for exploring highly conserved noncoding elements and their association with developmental regulatory genes. Genome Biol. 2008;9:R34. doi: 10.1186/gb-2008-9-2-r34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McGaughey DM, Vinton RM, Huynh J, Al-Saif A, Beer MA, McCallion AS. Metrics of sequence constraint overlook regulatory sequences in an exhaustive analysis at phox2b. Genome Res. 2008;18:252–260. doi: 10.1101/gr.6929408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Griffin C, Kleinjan DA, Doe B, van Heyningen V. New 3’ elements control Pax6 expression in the developing pretectum, neural retina and olfactory region. Mech. Dev. 2002;112:89–100. doi: 10.1016/s0925-4773(01)00646-3. [DOI] [PubMed] [Google Scholar]
- 50.Fisher S, Grice EA, Vinton RM, Bessling SL, McCallion AS. Conservation of RET regulatory function from human to zebrafish without sequence similarity. Science. 2006;312:276–279. doi: 10.1126/science.1124070. [DOI] [PubMed] [Google Scholar]
- 51.Hare EE, Peterson BK, Iyer VN, Meier R, Eisen MB. Sepsid even-skipped enhancers are functionally conserved in Drosophila despite lack of sequence conservation. PLoS Genet. 2008;4:e1000106. doi: 10.1371/journal.pgen.1000106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ahituv N, Zhu Y, Visel A, Holt A, Afzal V, Pennacchio LA, Rubin EM. Deletion of ultraconserved elements yields viable mice. PLoS Biol. 2007;5:e234. doi: 10.1371/journal.pbio.0050234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hong J-W, Hendrix DA, Levine MS. Shadow enhancers as a source of evolutionary novelty. Science. 2008;321:1314. doi: 10.1126/science.1160631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Engström PG, Ho Sui SJ, Drivenes O, Becker TS, Lenhard B. Genomic regulatory blocks underlie extensive microsynteny conservation in insects. Genome Res. 2007;17:1898–1908. doi: 10.1101/gr.6669607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Baxter L, Jironkin A, Hickman R, Moore J, Barrington C, Krusche P, Dyer NP, Buchanan-Wollaston V, Tiskin A, Beynon J, et al. Conserved noncoding sequences highlight shared components of regulatory networks in dicotyledonous plants. Plant Cell. 2012;24:3949–3965. doi: 10.1105/tpc.112.103010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kritsas K, Wuest SE, Hupalo D, Kern AD, Wicker T, Grossniklaus U. Computational analysis and characterization of UCE-like elements (ULEs) in plant genomes. Genome Res. 2012;22:2455–2466. doi: 10.1101/gr.129346.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goode DK, Snell P, Smith SF, Cooke JE, Elgar G. Highly conserved regulatory elements around the SHH gene may contribute to the maintenance of conserved synteny across human chromosome 7q36.3. Genomics. 2005;86:172–181. doi: 10.1016/j.ygeno.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 58.Irimia M, Tena JJ, Alexis M, Fernandez-Miñan A, Maeso I, Bogdanovic O, de la Calle Mustienes E, Roy SW, Gómez-Skarmeta JL, Fraser HB. Extensive conservation of ancient microsynteny across metazoans due to cis-regulatory constraints. Genome Res. 2012;22:2356–2367. doi: 10.1101/gr.139725.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Maeso I, Irimia M, Tena JJ, González-Pérez E, Tran D, Ravi V, Venkatesh B, Campuzano S, Gómez-Skarmeta JL, Garcia-Fernàndez J. An ancient genomic regulatory block conserved across bilaterians and its dismantling in tetrapods by retrogene replacement. Genome Res. 2012;22:642–655. doi: 10.1101/gr.132233.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Benko S, Fantes JA, Amiel J, Kleinjan D-J, Thomas S, Ramsay J, Jamshidi N, Essafi A, Heaney S, Gordon CT, et al. Highly conserved non-coding elements on either side of SOX9 associated with Pierre Robin sequence. Nat. Genet. 2009;41:359–364. doi: 10.1038/ng.329. [DOI] [PubMed] [Google Scholar]
- 61.D’haene B, Attanasio C, Beysen D, Dostie J, Lemire E, Bouchard P, Field M, Jones K, Lorenz B, Menten B, et al. Disease-causing 7.4 kb cis-regulatory deletion disrupting conserved non-coding sequences and their interaction with the FOXL2 promotor: implications for mutation screening. PLoS Genet. 2009;5:e1000522. doi: 10.1371/journal.pgen.1000522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wasserman NF, Aneas I, Nobrega MA. An 8q24 gene desert variant associated with prostate cancer risk confers differential in vivo activity to a MYC enhancer. Genome Res. 2010;20:1191–1197. doi: 10.1101/gr.105361.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ragvin A, Moro E, Fredman D, Navratilova P, Drivenes O, Engström PG, Alonso ME, de la Calle Mustienes E, Gómez-Skarmeta JL, Tavares MJ, et al. Long-range gene regulation links genomic type 2 diabetes and obesity risk regions to HHEX, SOX4, and IRX3. Proc. Natl Acad. Sci. USA. 2010;107:775–780. doi: 10.1073/pnas.0911591107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dennis MY, Paracchini S, Scerri TS, Prokunina-Olsson L, Knight JC, Wade-Martins R, Coggill P, Beck S, Green ED, Monaco AP. A common variant associated with dyslexia reduces expression of the KIAA0319 gene. PLoS Genet. 2009;5:e1000436. doi: 10.1371/journal.pgen.1000436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Knight JC. Resolving the variable genome and epigenome in human disease. J. Intl. Med. 2012;271:379–391. doi: 10.1111/j.1365-2796.2011.02508.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Visel A, Rubin EM, Pennacchio LA. Genomic views of distant-acting enhancers. Nature. 2009;461:199–205. doi: 10.1038/nature08451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Long Q, Meng A, Wang H, Jessen JR, Farrell MJ, Lin S. GATA-1 expression pattern can be recapitulated in living transgenic zebrafish using GFP reporter gene. Development. 1997;124:4105–4111. doi: 10.1242/dev.124.20.4105. [DOI] [PubMed] [Google Scholar]
- 69.Melnikov A, Murugan A, Zhang X, Tesileanu T, Wang L, Rogov P, Feizi S, Gnirke A, Callan CG, Kinney JB, et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat. Biotechnol. 2012;30:271–277. doi: 10.1038/nbt.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Patwardhan RP, Hiatt JB, Witten DM, Kim MJ, Smith RP, May D, Lee C, Andrie JM, Lee S-I, Cooper GM, et al. Massively parallel functional dissection of mammalian enhancers in vivo. Nat. Biotechnol. 2012;30:265–270. doi: 10.1038/nbt.2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Arnold CD, Gerlach D, Stelzer C, Boryn LM, Rath M, Stark A. Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science. 2013;339:1074–1077. doi: 10.1126/science.1232542. [DOI] [PubMed] [Google Scholar]
- 72.Parinov S, Kondrichin I, Korzh V, Emelyanov A. Tol2 transposon-mediated enhancer trap to identify developmentally regulated zebrafish genes in vivo. Dev. Dyn. 2004;231:449–459. doi: 10.1002/dvdy.20157. [DOI] [PubMed] [Google Scholar]
- 73.Bellen HJ, Levis RW, Liao G, He Y, Carlson JW, Tsang G, Evans-Holm M, Hiesinger PR, Schulze KL, Rubin GM, et al. The BDGP gene disruption project: single transposon insertions associated with 40% of Drosophila genes. Genetics. 2004;167:761–781. doi: 10.1534/genetics.104.026427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 75.Rando OJ. Combinatorial complexity in chromatin structure and function: revisiting the histone code. Curr. Opin. Genet. Dev. 2012;22:148–155. doi: 10.1016/j.gde.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 77.Lupien M, Eeckhoute J, Meyer CA, Wang Q, Zhang Y, Li W, Carroll JS, Liu XS, Brown M. FoxA1 translates epigenetic signatures into enhancer-driven lineage-specific transcription. Cell. 2008;132:958–970. doi: 10.1016/j.cell.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kharchenko PV, Alekseyenko AA, Schwartz YB, Minoda A, Riddle NC, Ernst J, Sabo PJ, Larschan E, Gorchakov AA, Gu T, et al. Comprehensive analysis of the chromatin landscape in Drosophila melanogaster. Nature. 2011;471:480–485. doi: 10.1038/nature09725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim T-K, Koche RP, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh T-Y, Peng W, Zhang MQ. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008;40:897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zippo A, Serafini R, Rocchigiani M, Pennacchini S, Krepelova A, Oliviero S. Histone crosstalk between H3S10ph and H4K16ac generates a histone code that mediates transcription elongation. Cell. 2009;138:1122–1136. doi: 10.1016/j.cell.2009.07.031. [DOI] [PubMed] [Google Scholar]
- 82.Thomson JP, Skene PJ, Selfridge J, Clouaire T, Guy J, Webb S, Kerr ARW, Deaton A, Andrews R, James KD, et al. CpG islands influence chromatin structure via the CpG-binding protein Cfp1. Nature. 2010;464:1082–1086. doi: 10.1038/nature08924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Seila AC, Calabrese JM, Levine SS, Yeo GW, Rahl PB, Flynn RA, Young RA, Sharp PA. Divergent transcription from active promoters. Science. 2008;322:1849–1851. doi: 10.1126/science.1162253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Core LJ, Waterfall JJ, Lis JT. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 2008;322:1845–1848. doi: 10.1126/science.1162228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Preker P, Nielsen J, Kammler S, Lykke-Andersen S, Christensen MS, Mapendano CK, Schierup MH, Jensen TH. RNA exosome depletion reveals transcription upstream of active human promoters. Science. 2008;322:1851–1854. doi: 10.1126/science.1164096. [DOI] [PubMed] [Google Scholar]
- 86.Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 87.Cui K, Zang C, Roh T-Y, Schones DE, Childs RW, Peng W, Zhao K. Chromatin signatures in multipotent human hematopoietic stem cells indicate the fate of bivalent genes during differentiation. Cell Stem Cell. 2009;4:80–93. doi: 10.1016/j.stem.2008.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Illingworth RS, Gruenewald-Schneider U, Webb S, Kerr ARW, James KD, Turner DJ, Smith C, Harrison DJ, Andrews R, Bird AP. Orphan CpG islands identify numerous conserved promoters in the mammalian genome. PLoS Genet. 2010;6 doi: 10.1371/journal.pgen.1001134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fenouil R, Cauchy P, Koch F, Descostes N, Cabeza JZ, Innocenti C, Ferrier P, Spicuglia S, Gut M, Gut I, et al. CpG islands and GC content dictate nucleosome depletion in a transcription-independent manner at mammalian promoters. Genome Res. 2012;22:2399–2408. doi: 10.1101/gr.138776.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mohn F, Weber M, Rebhan M, Roloff TC, Richter J, Stadler MB, Bibel M, Schübeler D. Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol. Cell. 2008;30:755–766. doi: 10.1016/j.molcel.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 91.Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766–770. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Borgel J, Guibert S, Li Y, Chiba H, Schübeler D, Sasaki H, Forné T, Weber M. Targets and dynamics of promoter DNA methylation during early mouse development. Nat. Genet. 2010;42:1093–1100. doi: 10.1038/ng.708. [DOI] [PubMed] [Google Scholar]
- 93.Ng K, Pullirsch D, Leeb M, Wutz A. Xist and the order of silencing. EMBO Rep. 2007;8:34–39. doi: 10.1038/sj.embor.7400871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Boyes J, Bird A. DNA methylation inhibits transcription indirectly via a methyl-CpG binding protein. Cell. 1991;64:1123–1134. doi: 10.1016/0092-8674(91)90267-3. [DOI] [PubMed] [Google Scholar]
- 95.Bogdanovic O, Veenstra GJC. DNA methylation and methyl-CpG binding proteins: developmental requirements and function. Chromosoma. 2009;118:549–565. doi: 10.1007/s00412-009-0221-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Isagawa T, Nagae G, Shiraki N, Fujita T, Sato N, Ishikawa S, Kume S, Aburatani H. DNA methylation profiling of embryonic stem cell differentiation into the three germ layers. PLoS One. 2011;6:e26052. doi: 10.1371/journal.pone.0026052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mikkelsen TS, Xu Z, Zhang X, Wang L, Gimble JM, Lander ES, Rosen ED. Comparative epigenomic analysis of murine and human adipogenesis. Cell. 2010;143:156–169. doi: 10.1016/j.cell.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Soler E, Andrieu-Soler C, Boer Ed, Bryne JC, Thongjuea S, Rijkers E, Demmers J, Ijcken Wv, Grosveld F. A systems approach to analyze transcription factors in mammalian cells. Methods. 2011;53:151–162. doi: 10.1016/j.ymeth.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 99.Roider HG, Lenhard B, Kanhere A, Haas SA, Vingron M. CpG-depleted promoters harbor tissue-specific transcription factor binding signals–implications for motif overrepresentation analyses. Nucleic Acids Res. 2009;37:6305–6315. doi: 10.1093/nar/gkp682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Barski A, Cuddapah S, Cui K, Roh T-Y, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 101.Jin C, Felsenfeld G. Nucleosome stability mediated by histone variants H3.3 and H2A.Z. Genes Dev. 2007;21:1519–1529. doi: 10.1101/gad.1547707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457:854–858. doi: 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ernst J, Kellis M. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat. Biotechnol. 2010;28:817–825. doi: 10.1038/nbt.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhu J, Adli M, Zou JY, Verstappen G, Coyne M, Zhang X, Durham T, Miri M, Deshpande V, De Jager PL, et al. Genome-wide chromatin state transitions associated with developmental and environmental cues. Cell. 2013;152:642–654. doi: 10.1016/j.cell.2012.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Blow MJ, McCulley DJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, et al. ChIP-Seq identification of weakly conserved heart enhancers. Nat. Genet. 2010;42:806–810. doi: 10.1038/ng.650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Xi H, Shulha HP, Lin JM, Vales TR, Fu Y, Bodine DM, McKay RDG, Chenoweth JG, Tesar PJ, Furey TS, et al. Identification and characterization of cell type-specific and ubiquitous chromatin regulatory structures in the human genome. PLoS Genet. 2007;3:e136. doi: 10.1371/journal.pgen.0030136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wiench M, John S, Baek S, Johnson TA, Sung M-H, Escobar T, Simmons CA, Pearce KH, Biddie SC, Sabo PJ, et al. DNA methylation status predicts cell type-specific enhancer activity. EMBO J. 2011;30:3028–3039. doi: 10.1038/emboj.2011.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Xu J, Pope SD, Jazirehi AR, Attema JL, Papathanasiou P, Watts JA, Zaret KS, Weissman IL, Smale ST. Pioneer factor interactions and unmethylated CpG dinucleotides mark silent tissue-specific enhancers in embryonic stem cells. Proc. Natl Acad. Sci. USA. 2007;104:12377–12382. doi: 10.1073/pnas.0704579104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sérandour AA, Avner S, Percevault F, Demay F, Bizot M, Lucchetti-Miganeh C, Barloy-Hubler F, Brown M, Lupien M, Métivier R, et al. Epigenetic switch involved in activation of pioneer factor FOXA1-dependent enhancers. Genome Res. 2011;21:555–565. doi: 10.1101/gr.111534.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Schmidl C, Klug M, Boeld TJ, Andreesen R, Hoffmann P, Edinger M, Rehli M. Lineage-specific DNA methylation in T cells correlates with histone methylation and enhancer activity. Genome Res. 2009;19:1165–1174. doi: 10.1101/gr.091470.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Nalt Acad. Sci. USA. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470:279–283. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zentner GE, Tesar PJ, Scacheri PC. Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res. 2011;21:1273–1283. doi: 10.1101/gr.122382.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, Buchou T, Cheng Z, Rousseaux S, Rajagopal N, et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. 2011;146:1016–1028. doi: 10.1016/j.cell.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cotney J, Leng J, Oh S, Demare LE, Reilly SK, Gerstein MB, Noonan JP. Chromatin state signatures associated with tissue-specific gene expression and enhancer activity in the embryonic limb. Genome Res. 2012;22:1069–1080. doi: 10.1101/gr.129817.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Roh T-Y, Cuddapah S, Cui K, Zhao K. The genomic landscape of histone modifications in human T cells. Proc. Natl Acad. Sci. USA. 2006;103:15782–15787. doi: 10.1073/pnas.0607617103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhu Y, van Essen D, Saccani S. Cell-type-specific control of enhancer activity by H3K9 trimethylation. Mol. Cell. 2012;46:408–423. doi: 10.1016/j.molcel.2012.05.011. [DOI] [PubMed] [Google Scholar]
- 118.Chambeyron S, Bickmore WA. Chromatin decondensation and nuclear reorganization of the HoxB locus upon induction of transcription. Genes Dev. 2004;18:1119–1130. doi: 10.1101/gad.292104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Amano T, Sagai T, Tanabe H, Mizushina Y, Nakazawa H, Shiroishi T. Chromosomal dynamics at the Shh locus: limb bud-specific differential regulation of competence and active transcription. Dev. Cell. 2009;16:47–57. doi: 10.1016/j.devcel.2008.11.011. [DOI] [PubMed] [Google Scholar]
- 120.Dorier J, Stasiak A. The role of transcription factories-mediated interchromosomal contacts in the organization of nuclear architecture. Nucleic Acids Res. 2010;38:7410–7421. doi: 10.1093/nar/gkq666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Cook PR. A model for all genomes: the role of transcription factories. J. Mol. Biol. 2010;395:1–10. doi: 10.1016/j.jmb.2009.10.031. [DOI] [PubMed] [Google Scholar]
- 122.Schoenfelder S, Sexton T, Chakalova L, Cope NF, Horton A, Andrews S, Kurukuti S, Mitchell JA, Umlauf D, Dimitrova DS, et al. Preferential associations between co-regulated genes reveal a transcriptional interactome in erythroid cells. Nat. Genet. 2010;42:53–61. doi: 10.1038/ng.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chubb JR, Trcek T, Shenoy SM, Singer RH. Transcriptional pulsing of a developmental gene. Curr. Biol. 2006;16:1018–1025. doi: 10.1016/j.cub.2006.03.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Osborne CS, Chakalova L, Mitchell JA, Horton A, Wood AL, Bolland DJ, Corcoran AE, Fraser P. Myc dynamically and preferentially relocates to a transcription factory occupied by Igh. PLoS Biol. 2007;5:e192. doi: 10.1371/journal.pbio.0050192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.de Wit E, Braunschweig U, Greil F, Bussemaker HJ, van Steensel B. Global chromatin domain organization of the Drosophila genome. PLoS Genet. 2008;4:e1000045. doi: 10.1371/journal.pgen.1000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, Eussen BH, de Klein A, Wessels L, de Laat W, et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature. 2008;453:948–951. doi: 10.1038/nature06947. [DOI] [PubMed] [Google Scholar]
- 127.Finlan LE, Sproul D, Thomson I, Boyle S, Kerr E, Perry P, Ylstra B, Chubb JR, Bickmore WA. Recruitment to the nuclear periphery can alter expression of genes in human cells. PLoS Genet. 2008;4:e1000039. doi: 10.1371/journal.pgen.1000039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wen B, Wu H, Shinkai Y, Irizarry RA, Feinberg AP. Large histone H3 lysine 9 dimethylated chromatin blocks distinguish differentiated from embryonic stem cells. Nat. Genet. 2009;41:246–250. doi: 10.1038/ng.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kohwi M, Lupton JR, Lai S-L, Miller MR, Doe CQ. Developmentally regulated subnuclear genome reorganization restricts neural progenitor competence in Drosophila. Cell. 2013;152:97–108. doi: 10.1016/j.cell.2012.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Pauler FM, Sloane MA, Huang R, Regha K, Koerner MV, Tamir I, Sommer A, Aszodi A, Jenuwein T, Barlow DP. H3K27me3 forms BLOCs over silent genes and intergenic regions and specifies a histone banding pattern on a mouse autosomal chromosome. Genome Res. 2009;19:221–233. doi: 10.1101/gr.080861.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Tolhuis B, Blom M, Kerkhoven RM, Pagie L, Teunissen H, Nieuwland M, Simonis M, de Laat W, van Lohuizen M, van Steensel B. Interactions among Polycomb domains are guided by chromosome architecture. PLoS Genet. 2011;7:e1001343. doi: 10.1371/journal.pgen.1001343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bantignies F, Roure V, Comet I, Leblanc B, Schuettengruber B, Bonnet J, Tixier V, Mas A, Cavalli G. Polycomb-dependent regulatory contacts between distant Hox loci in Drosophila. Cell. 2011;144:214–226. doi: 10.1016/j.cell.2010.12.026. [DOI] [PubMed] [Google Scholar]
- 133.Chu C, Qu K, Zhong FL, Artandi SE, Chang HY. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol. Cell. 2011;44:667–678. doi: 10.1016/j.molcel.2011.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kagey MH, Newman JJ, Bilodeau S, Zhan Y, Orlando DA, van Berkum NL, Ebmeier CC, Goossens J, Rahl PB, Levine SS, et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010;467:430–435. doi: 10.1038/nature09380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Soutourina J, Wydau S, Ambroise Y, Boschiero C, Werner M. Direct interaction of RNA polymerase II and mediator required for transcription in vivo. Science. 2011;331:1451–1454. doi: 10.1126/science.1200188. [DOI] [PubMed] [Google Scholar]
- 136.Schaaf CA, Kwak H, Koenig A, Misulovin Z, Gohara DW, Watson A, Zhou Y, Lis JT, Dorsett D. Genome-wide control of RNA Polymerase II activity by cohesin. PLoS Genet. 2013;9:e1003382. doi: 10.1371/journal.pgen.1003382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Raab JR, Kamakaka RT. Insulators and promoters: closer than we think. Nat. Rev. Genet. 2010;11:439–446. doi: 10.1038/nrg2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mishiro T, Ishihara K, Hino S, Tsutsumi S, Aburatani H, Shirahige K, Kinoshita Y, Nakao M. Architectural roles of multiple chromatin insulators at the human apolipoprotein gene cluster. EMBO J. 2009;28:1234–1245. doi: 10.1038/emboj.2009.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Phillips JE, Corces VG. CTCF: master weaver of the genome. Cell. 2009;137:1194–1211. doi: 10.1016/j.cell.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Botta M, Haider S, Leung IXY, Lio P, Mozziconacci J. Intra- and inter-chromosomal interactions correlate with CTCF binding genome wide. Mol. Syst. Biol. 2010;6:426. doi: 10.1038/msb.2010.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Fu Y, Sinha M, Peterson CL, Weng Z. The insulator binding protein CTCF positions 20 nucleosomes around its binding sites across the human genome. PLoS Genet. 2008;4:e1000138. doi: 10.1371/journal.pgen.1000138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Deng W, Lee J, Wang H, Miller J, Reik A, Gregory PD, Dean A, Blobel GA. Controlling long-range genomic interactions at a native locus by targeted tethering of a looping factor. Cell. 2012;149:1233–1244. doi: 10.1016/j.cell.2012.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y, Lajoie BR, Protacio A, Flynn RA, Gupta RA, et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011;472:120–124. doi: 10.1038/nature09819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lai F, Orom UA, Cesaroni M, Beringer M, Taatjes DJ, Blobel GA, Shiekhattar R. Activating RNAs associate with Mediator to enhance chromatin architecture and transcription. Nature. 2013;494:497–501. doi: 10.1038/nature11884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kim T-K, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S, et al. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465:182–187. doi: 10.1038/nature09033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.De Santa F, Barozzi I, Mietton F, Ghisletti S, Polletti S, Tusi BK, Muller H, Ragoussis J, Wei C-L, Natoli G. A large fraction of extragenic RNA pol II transcription sites overlap enhancers. PLoS Biol. 2010;8:e1000384. doi: 10.1371/journal.pbio.1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wang D, Garcia-Bassets I, Benner C, Li W, Su X, Zhou Y, Qiu J, Liu W, Kaikkonen MU, Ohgi KA, et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature. 2011;474:390–394. doi: 10.1038/nature10006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Melgar MF, Collins FS, Sethupathy P. Discovery of active enhancers through bidirectional expression of short transcripts. Genome Biol. 2011;12:R113. doi: 10.1186/gb-2011-12-11-r113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Melo CA, Drost J, Wijchers PJ, van de Werken H, de Wit E, Oude Vrielink JAF, Elkon R, Melo SA, Léveillé N, Kalluri R, et al. eRNAs are required for p53-dependent enhancer activity and gene transcription. Mol. Cell. 2013;49:524–535. doi: 10.1016/j.molcel.2012.11.021. [DOI] [PubMed] [Google Scholar]
- 150.Ørom UA, Derrien T, Beringer M, Gumireddy K, Gardini A, Bussotti G, Lai F, Zytnicki M, Notredame C, Huang Q, et al. Long noncoding RNAs with enhancer-like function in human cells. Cell. 2010;143:46–58. doi: 10.1016/j.cell.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kanhere A, Viiri K, Araújo CC, Rasaiyaah J, Bouwman RD, Whyte WA, Pereira CF, Brookes E, Walker K, Bell GW, et al. Short RNAs are transcribed from repressed polycomb target genes and interact with polycomb repressive complex-2. Mol. Cell. 2010;38:675–688. doi: 10.1016/j.molcel.2010.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Montavon T, Soshnikova N, Mascrez B, Joye E, Thevenet L, Splinter E, de Laat W, Spitz F, Duboule D. A regulatory archipelago controls Hox genes transcription in digits. Cell. 2011;147:1132–1145. doi: 10.1016/j.cell.2011.10.023. [DOI] [PubMed] [Google Scholar]
- 153.Lower KM, Hughes JR, De Gobbi M, Henderson S, Viprakasit V, Fisher C, Goriely A, Ayyub H, Sloane-Stanley J, Vernimmen D, et al. Adventitious changes in long-range gene expression caused by polymorphic structural variation and promoter competition. Proc. Natl Acad. Sci. USA. 2009;106:21771–21776. doi: 10.1073/pnas.0909331106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Li G, Ruan X, Auerbach RK, Sandhu KS, Zheng M, Wang P, Poh HM, Goh Y, Lim J, Zhang J, et al. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell. 2012;148:84–98. doi: 10.1016/j.cell.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Dekker J, Rippe K, Dekker M, Kleckner N. Capturing chromosome conformation. Science. 2002;295:1306–1311. doi: 10.1126/science.1067799. [DOI] [PubMed] [Google Scholar]
- 156.Simonis M, Klous P, Splinter E, Moshkin Y, Willemsen R, de Wit E, van Steensel B, de Laat W. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C) Nat. Genet. 2006;38:1348–1354. doi: 10.1038/ng1896. [DOI] [PubMed] [Google Scholar]
- 157.Dostie J, Richmond TA, Arnaout RA, Selzer RR, Lee WL, Honan TA, Rubio ED, Krumm A, Lamb J, Nusbaum C, et al. Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Res. 2006;16:1299–1309. doi: 10.1101/gr.5571506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Stadhouders R, Kolovos P, Brouwer R, Zuin J, van den Heuvel A, Kockx C, Palstra R-J, Wendt KS, Grosveld F, van Ijcken W, et al. Multiplexed chromosome conformation capture sequencing for rapid genome-scale high-resolution detection of long-range chromatin interactions. Nat. Protoc. 2013;8:509–524. doi: 10.1038/nprot.2013.018. [DOI] [PubMed] [Google Scholar]
- 159.Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Fullwood MJ, Liu MH, Pan YF, Liu J, Xu H, Mohamed YB, Orlov YL, Velkov S, Ho A, Mei PH, et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature. 2009;462:58–64. doi: 10.1038/nature08497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Dostie J, Bickmore WA. Chromosome organization in the nucleus - charting new territory across the Hi-Cs. Curr. Opin. Genet. Dev. 2012;22:125–131. doi: 10.1016/j.gde.2011.12.006. [DOI] [PubMed] [Google Scholar]
- 163.Drissen R, Palstra R-J, Gillemans N, Splinter E, Grosveld F, Philipsen S, de Laat W. The active spatial organization of the beta-globin locus requires the transcription factor EKLF. Genes Dev. 2004;18:2485–2490. doi: 10.1101/gad.317004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Vakoc CR, Letting DL, Gheldof N, Sawado T, Bender MA, Groudine M, Weiss MJ, Dekker J, Blobel GA. Proximity among distant regulatory elements at the beta-globin locus requires GATA-1 and FOG-1. Mol. Cell. 2005;17:453–462. doi: 10.1016/j.molcel.2004.12.028. [DOI] [PubMed] [Google Scholar]
- 165.Tiwari VK, McGarvey KM, Licchesi JDF, Ohm JE, Herman JG, Schübeler D, Baylin SB. PcG proteins, DNA methylation, and gene repression by chromatin looping. PLoS Biol. 2008;6:2911–2927. doi: 10.1371/journal.pbio.0060306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Noordermeer D, Leleu M, Splinter E, Rougemont J, de Laat W, Duboule D. The dynamic architecture of Hox gene clusters. Science. 2011;334:222–225. doi: 10.1126/science.1207194. [DOI] [PubMed] [Google Scholar]
- 167.Palstra R-J, Tolhuis B, Splinter E, Nijmeijer R, Grosveld F, de Laat W. The beta-globin nuclear compartment in development and erythroid differentiation. Nat. Genet. 2003;35:190–194. doi: 10.1038/ng1244. [DOI] [PubMed] [Google Scholar]
- 168.Stadhouders R, Thongjuea S, Andrieu-Soler C, Palstra R-J, Bryne JC, van den Heuvel A, Stevens M, de Boer E, Kockx C, van der Sloot A, et al. Dynamic long-range chromatin interactions control Myb proto-oncogene transcription during erythroid development. EMBO J. 2012;31:986–999. doi: 10.1038/emboj.2011.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Morey C, Da Silva NR, Perry P, Bickmore WA. Nuclear reorganisation and chromatin decondensation are conserved, but distinct, mechanisms linked to Hox gene activation. Development. 2007;134:909–919. doi: 10.1242/dev.02779. [DOI] [PubMed] [Google Scholar]
- 170.Chambers EV, Bickmore WA, Semple CA. Divergence of mammalian higher order chromatin structure is associated with developmental loci. PLoS Comput. Biol. 2013;9:e1003017. doi: 10.1371/journal.pcbi.1003017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Sexton T, Yaffe E, Kenigsberg E, Bantignies F, Leblanc B, Hoichman M, Parrinello H, Tanay A, Cavalli G. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell. 2012;148:458–472. doi: 10.1016/j.cell.2012.01.010. [DOI] [PubMed] [Google Scholar]
- 172.Kalhor R, Tjong H, Jayathilaka N, Alber F, Chen L. Genome architectures revealed by tethered chromosome conformation capture and population-based modeling. Nat. Biotechnol. 2012;30:90–98. doi: 10.1038/nbt.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Barbieri M, Chotalia M, Fraser J, Lavitas L-M, Dostie J, Pombo A, Nicodemi M. Complexity of chromatin folding is captured by the strings and binders switch model. Proc. Natl Acad. Sci. USA. 2012;109:16173–16178. doi: 10.1073/pnas.1204799109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Chepelev I, Wei G, Wangsa D, Tang Q, Zhao K. Characterization of genome-wide enhancer-promoter interactions reveals co-expression of interacting genes and modes of higher order chromatin organization. Cell Res. 2012;22:490–503. doi: 10.1038/cr.2012.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Handoko L, Xu H, Li G, Ngan CY, Chew E, Schnapp M, Lee CWH, Ye C, Ping JLH, Mulawadi F, et al. CTCF-mediated functional chromatin interactome in pluripotent cells. Nat. Genet. 2011;43:630–638. doi: 10.1038/ng.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Sandhu KS, Li G, Poh HM, Quek YLK, Sia YY, Peh SQ, Mulawadi FH, Lim J, Sikic M, Menghi F, et al. Large-scale functional organization of long-range chromatin interaction networks. Cell Rep. 2012;2:1207–1219. doi: 10.1016/j.celrep.2012.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]