


Abstract
Bacterial DNA topoisomerase I (topoI) carries out relaxation of negatively supercoiled DNA through a series of orchestrated steps, DNA binding, cleavage, strand passage and religation. The N-terminal domain (NTD) of the type IA topoisomerases harbor DNA cleavage and religation activities, but the carboxyl terminal domain (CTD) is highly diverse. Most of these enzymes contain a varied number of Zn2+ finger motifs in the CTD. The Zn2+ finger motifs were found to be essential in Escherichia coli topoI but dispensable in the Thermotoga maritima enzyme. Although, the CTD of mycobacterial topoI lacks Zn2+ fingers, it is indispensable for the DNA relaxation activity of the enzyme. The divergent CTD harbors three stretches of basic amino acids needed for the strand passage step of the reaction as demonstrated by a new assay. We also show that the basic amino acids constitute an independent DNA-binding site apart from the NTD and assist the simultaneous binding of two molecules of DNA to the enzyme, as required during the catalytic step. Although the NTD binds to DNA in a site-specific fashion to carry out DNA cleavage and religation, the basic residues in CTD bind to non-scissile DNA in a sequence-independent manner to promote the crucial strand passage step during DNA relaxation. The loss of Zn2+ fingers from the mycobacterial topoI could be associated with Zn2+ export and homeostasis.
INTRODUCTION
Bacterial DNA topoisomerase I (topoI) relaxes negatively supercoiled DNA through a series of coordinated steps in an Mg2+ dependent manner (1,2). The enzyme binds and introduces a single-strand (ss) nick in the double-stranded (ds) DNA establishing a 5′-phosphotyrosine covalent bond with the hydroxyl group of the active site tyrosine. In the subsequent steps, the intact non-scissile strand is passed through the cleaved DNA gate, and resealing of the scissile DNA occurs by the second transesterification reaction to restore duplex DNA (1). The N-terminal domain (NTD) is highly conserved among all bacterial topoI harboring both metal coordination motif and the cleavage-religation domains (3–6). In contrast, the carboxyl terminal domain (CTD) is divergent and generally contains variable number of Zn2+ fingers ranging from 1 to 5 (7–10). Escherichia coli topoI (EctopoI) has three tetracysteine Zn2+-binding motifs in the CTD (10). Either removal of Zn2+ (11) or mutation in the second Zn2+ finger reduced the DNA relaxation activity of the enzyme (12). Moreover, in the absence of the Zn2+ finger domain, the core domain, Top 67 (13) fails to catalyze the DNA relaxation, indicating that the Zn2+ fingers are indispensable for the enzyme activity (14). In contrast, the topoI of the hyperthermophilic bacterium Thermotoga maritima contains a single Zn2+ finger motif, which is not essential for its DNA relaxation activity (15). As the Zn2+ finger motif is essential for the EctopoI catalyzed DNA relaxation and catenation/decatenation reactions, it is proposed that by binding to the DNA, it facilitates DNA movement into and out of the central cavity leading to the strand passage (1). However, it is yet unclear how the actual strand passage function is achieved in this enzyme, or in other bacterial topoisomerases that have a minimal component of a Zn2+ finger motif or are devoid of it.
The topoI from the Mycobacterium smegmatis (MstopoI) and Mycobacterium tuberculosis possesses several distinct properties compared with other bacterial topoI (7,16,17). The enzyme, with highly diverged CTD lack Zn2+ finger motif, recognizes both ssDNA as well as dsDNA with high affinity, and cleaves the DNA at specific-sites termed as strong topoI sites (STS) (7,18). Earlier studies showed that the NTD harbors the sequence-specific DNA binding, cleavage and religation activities but cannot catalyze relaxation of DNA (17). The CTD is devoid of any enzymatic activity but exhibits non-specific DNA binding. Importantly, the DNA relaxation activity is restored when the NTD is complemented with the CTD in vitro, implicating a role for the CTD in the strand passage step of DNA relaxation cycle (17).
Although typical DNA binding Zn2+ finger modules are absent, non-specific DNA binding and a likely role in the strand passage suggests the presence of alternate DNA-binding motifs in the CTD of MstopoI. Multiple sequence alignment of topoI from different mycobacterial species revealed three stretches of basic amino acids in the CTD (Supplementary Figure S1). We hypothesized that the stretches of basic amino acids could be involved in the non-specific DNA binding to mediate passage of the non-scissile strand through the cleaved DNA. The hypothesis was tested by deleting the basic stretches and evaluating the deletants, using a variety of assays. A new assay was developed to assess the strand passage and to elucidate the importance of basic stretches of CTD in the process. We demonstrate that the basic amino acid stretches are involved in binding the second DNA simultaneously when the scissile DNA is held by the enzyme, and the event is crucial for strand passage reaction of the enzyme. We also provide a plausible explanation for the selective loss of Zn2+ fingers in topoI from Actinobacteria.
MATERIALS AND METHODS
Bacterial strains and plasmids
Escherichia coli DH10B and E. coli BL21 (DE3) were used for cloning and overexpression of MstopoI and its mutants from pRSETA constructs, respectively. Supercoiled pUC18 plasmid DNA substrate for topoI activity assay was purified by Qiagen midiprep kits.
Enzymes and oligonucleotides
Restriction enzymes, T4 DNA ligase, T4 polynucleotide kinase, Klenow polymerase I and DNase I were purchased from New England Biolabs, USA. STS and non-STS oligonucleotides (Sigma Aldrich, USA) were resolved on 15% denaturing polyacrylamide gel, purified using NAP 10 columns (GE healthcare) and 5′-end labeled using T4 polynucleotide kinase and [γ-32P] ATP (6000 Ci/mmol). Labeled oligonucleotides were purified through Sephadex G-25 spin columns. Sequences of oligonucleotides used in this study are given in Supplementary Table S1.
Deletion of basic amino acid stretches
Strategy for the deletion of basic stretches and the primers used for the mutagenesis are depicted in Supplementary Figure S2 and Supplementary Table S1, respectively. Expression plasmid pPVN3 (17) encoding CTD (corresponding to amino acid residue 617–938) was used as a template for deletion mutagenesis. Basic stretch 1 (B1) was deleted by generating NarI restriction site on either side of B1 by site-directed mutagenesis (19), digestion with NarI followed by ligation. The truncated form of pPVN3 was sub-cloned into full-length topoI (pPVN123) using BamHI restriction enzyme. Basic stretch 2 (B2) was deleted by inverse PCR using the primers Rctd1 and Fctd3 (20).To delete stretch 3 (B3), primer Fctd1 and Rctd2 were designed to amplify the 879 bp DNA fragment, which lacks the B3. Double deletion mutants ΔB13 and ΔB23 were generated using the ΔB1and ΔB2 as templates followed by deletion of B3 as described earlier in the text. The mutations were confirmed by restriction digestion followed by sequencing of plasmid DNA.
Expression and purification of the proteins
Purification of proteins was carried out as described earlier (17). Briefly, pPVN123 encoding either MstopoI or its deletants was transformed into E. coli BL21 (DE3). Cultures grown at 37°C were induced with 0.3 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) at O.D.595nm = 0.6 for 3 h and harvested. The proteins were purified by two column chromatographic steps using Heparin Sepharose followed by SP Sepharose columns.
DNA relaxation assay
In all, 500 ng negatively supercoiled pUC18 DNA was incubated with varying amounts of the enzyme in 40 mM Tris–HCl (pH 8.0) containing 20 mM NaCl, 1 mM EDTA and 5 mM MgCl2 at 37°C for 30 min. The reactions were terminated with 0.1% SDS and heating at 75°C for 10 min. The reaction products were resolved on 1.2% agarose gel at 1 V/cm and visualized by staining with 0.5 μg/ml ethidium bromide after electrophoresis. One unit of enzyme is defined as the amount of enzyme that relaxes 50% of the 500 ng of supercoiled pUC18 in 30 min at 37°C.
Electrophoretic mobility shift assays
The 5′-end-labeled 32-mer oligonucleotide harboring STS was used as a specific single-stranded DNA substrate (Supplementary Table S1). The reaction mixture containing 40 mM Tris–HCl (pH 8.0), 20 mM NaCl, 1 mM EDTA and 0.1 pmol oligonucleotide was incubated with the varying concentrations of either WT or the deletants for 15 min on ice. The protein–DNA complexes were resolved from free DNA in 8% non-denaturing polyacrylamide gel electrophoresis (PAGE) at 4°C using 1× TBE buffer (Tris–Borate–EDTA). The bands were visualized by phosphorimager (model BAS 1800; Fujifilm), quantitated using Multi Gauge (Fujifilm) and analyzed by GraphPad Prism (Version 5.0) to determine affinity.
For Electrophoretic mobility shift assays (EMSA) with two DNA substrates of different sizes, 5′-end-labeled 32-mer STS DNA was incubated with 200 nM of the enzymes for 15 min at 4°C to form complex I. To this, increasing concentrations of 60-mer unlabeled non-STS oligonucleotide was added and incubated for additional 10 min. The protein–DNA complexes were resolved on 6% native PAGE at 4°C using 1× TBE (Tris–Borate–EDTA), visualized by phosphorimager (model BAS 1800; Fujifilm) and analyzed as aforementioned.
DNA cleavage and religation
The DNA cleavage was carried out by incubating the enzymes with 0.1 pmol of 32-mer oligonucleotide harboring STS in the cleavage buffer [40 mM Tris–HCl (pH 8.0), 20 mM NaCl and 1 mM EDTA] at 37°C for 30 min to yield a 5′-end-labeled 19-mer cleavage product. The reactions were terminated with 45% formamide dye and heating at 90°C for 2 min. Samples were resolved on 12% denaturing PAGE using 1× TBE as running buffer at 300 V, visualized and quantitated as described earlier in the text.
For intramolecular religation, after 15 min of cleavage reaction (as aforementioned), religation was initiated with 5 or 10 mM MgCl2, followed by incubation for further 15 min. The reaction products were resolved on 12% denaturing PAGE. To carry out intermolecular religation, 32-mer oligonucleotide harboring STS were incubated with WT or deletants to yield a non-covalenty held 19-mer and 13-mer covalent protein–DNA adduct. The reactions were carried out in the cleavage buffer at 37°C for 15 min. After the cleavage, 5′-end-labeled 11-mer pre-annealed to unlabeled 24-mer was added and incubated at room temperature for 5 min. Religation was initiated with 10 mM MgCl2 and incubated for 30 min at 37°C to yield a labeled 24-mer intermolecular religation product. The samples were processed as described previously (17,21).
Filter binding assay for strand passage
To test the strand passage, 500 nM enzyme was incubated with nick-translated radiolabeled pUC18 plasmid DNA for 15 min on ice in assay buffer [40 mM Tris–HCl (pH 8.0), 20 mM NaCl and 1 mM EDTA, 10% glycerol].The reaction was divided into two parts. In one part, to determine the initial counts, an anti-topoI clamp closing antibody 1E4F5 (22) was added to trap the plasmid DNA in the enzyme cavity. In the other part, 5 pmoles of 32-mer oligonucleotide harboring the STS was added to the reaction mix and incubated for 15 min at 37°C, followed by the addition of 10 mM MgCl2 and incubation for 30 min. At the end of the incubation, 1E4F5 antibody was added in these reactions. All the reactions were spotted on Millipore nitrocellulose filter (0.45 µm) pre-equilibrated with assay buffer, followed by washing with high salt buffer (40 mM Tris–HCl (pH 8.0), 1 M NaCl and 1 mM EDTA) thrice in a Millipore filtration apparatus equipped with vacuum pump to remove the unbound DNA, and the radioactivity retained on the filter was measured by liquid scientillation counter. Strand passage ability was calculated as the percentage reduction in the radioactive counts (cpm) after strand passage.
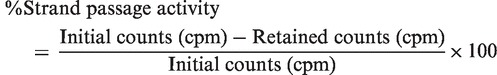 |
Overexpression of MstopoI Δ23 in M. smegmatis
MstopoI Δ23 and WT MstopoI fragments were amplified from their respective plasmid constructs by PCR and cloned into pMIND vector (23) using the NdeI and EcoRV restriction sites. The constructs were electroporated into the M. smegmatis mc2 155 cells and transformants were selected on Kanamycin (25 mg/ml) Middlebrook 7H9 agar plates. The WT and deletant topoI overexpression strains were inoculated in Middlebrook 7H9 broth supplemented with 0.2% glycerol and 0.05% Tween-80 and grown at 37°C with continuous shaking at 200 rpm. The growth was monitored at O.D. 595 nm at 2 h intervals for 48 h. The growth curves were plotted by the GraphPad Prism (version 5.0).
RESULTS
Basic amino acid stretches are essential for the DNA relaxation activity
Multiple sequence alignment showed that the NTD of MstopoI shares considerable sequence similarity with other eubacterial topoI (21). The NTD harbors the topoisomerase-primase domain for Mg2+ coordination and also the 5Y-CAP domain that contains the active site tyrosine needed for the transesterification reaction to establish the covalent phosphotyrosine adduct during the DNA cleavage (Figure 1B). Although the CTD of TmtopoI, EctopoI and Helicobacter pylori topoI harbor one, three and four Zn2+ finger motifs, respectively (10,15,24), the CTD of the mycobacterial topoI is devoid of the tetracysteine Zn2+ fingers (Figure 1A). However, the amino acid sequence analyses revealed the presence of three stretches of basic amino acids in the CTD of the enzyme, hinting at the possibility that these stretches could be involved in the enzyme function (Supplementary Figure S1). To assess the role of the basic amino acid stretches in enzyme activity, they were deleted individually as well as in combination (Supplementary Figure S2), and the deletants were purified for further analyses, as described in ‘Materials and Methods’ section. DNA relaxation ability of the MstopoI CTD deletants was analyzed; their specific activities are given in Figure 1D. Although deletants ΔB1and ΔB2 had ∼3.5-fold lower activity than WT enzyme, ΔB3 exhibited weaker (∼60-fold reduced) DNA relaxation activity. The deletants with two basic stretches deleted, ΔB13 and ΔB23 had highly reduced activity, and hence their specific activities could not be determined (Figure 1C). The pattern of DNA relaxation by ΔB1 and ΔB2 appeared to be distributive; DNA topoisomers of intermediate superhelical density were seen. To sum up, the data suggest that the basic residues play an important role in DNA relaxation reaction of the enzyme.
Figure 1.

The importance of basic stretches in DNA relaxation. (A) The pictorial representation of Zn2+ fingers in different topoisomerases; (B) Organization of various motifs in MstopoI. Conserved motifs are shown as dark boxes. NTD comprises the metal coordinating acidic triad DXDXE in the topoisomerase-primase domain and the CAP domain or the cleavage-religation domain comprising the active-site tyrosine. CTD contains three basic amino acid stretches shown in the form of shaded boxes (B1, B2 and B3). Different mutants generated for this study and their nomenclatures are given; (C) DNA relaxation activity of CTD deletants. In all, 500 ng negatively supercoiled pUC18 plasmid DNA was incubated with increasing concentration of the enzymes (as indicated in each panel) at 37°C for 30 min. (C) Control reaction with no enzyme. N and S indicate nicked and supercoiled form of plasmid DNA respectively; (D) Specific activity of the deletants.
DNA cleavage and religation activities of the deletants were unaffected
DNA relaxation reaction mediated by type IA topoisomerases uses two concerted transesterification reactions, DNA cleavage and religation. To assess the effect of deletions of basic amino acid stretches on DNA cleavage activity, assays were carried out with 5′-end-labeled, 32-mer STS harboring oligonucleotide. From these assays, it is evident that neither the efficiency of DNA cleavage nor the cleavage site preference was affected in the deletant enzymes (Supplementary Figure S3). Next, to evaluate the effect of the deletions in the CTD on the second transesterification reaction, intra-and inter-molecular religation assays were carried out as described in ‘Materials and Methods’ section (schematics in Supplementary Figure S4) (17). The deletants were proficient in catalyzing DNA religation, affirming that the basic stretches do not have a significant role at this step of the reaction (Supplementary Figure S4A and B).
Basic amino acid stretches are indispensable for strand passage
The data that the individual catalytical steps are not affected but the overall relaxation activity is compromised in the CTD deletants indicate the importance of basic stretches in some other step of enzyme action. As both the catalytic functions are confined to the NTD, the basic stretches in the CTD may be needed either directly or indirectly in the strand passage step, which precedes the second transesterification reaction to reseal the DNA. The crystal structure of EctopoI, EctopoIII and TmtopoI revealed that the ∼27 Å wide central cavity of the enzyme can accommodate either ssDNA or dsDNA (25–28). The sign inversion and the enzyme bridge model proposed for the catalysis mediated by type IA topoisomerases predict the entry and exit of non-scissile DNA into or from this enzyme cavity through the cleaved DNA gate (29,30). According to these models, the scissile ssDNA resides in the active site, which undergoes cleavage and religation, and the transfer strand remains in the central cavity of the enzyme. To assess the role of basic amino acid stretches in the strand passage step independent of cleavage and religation reactions, the strand passage ability was monitored by nitrocellulose-filter binding experiments.
MstopoI and the basic stretch deletants were incubated with radiolabeled relaxed pUC18 DNA so that the DNA occupies the enzyme cavity. Occupancy of pUC18 DNA was ensured by using an anti-topoI clamp closing monoclonal antibody, which would lock the topoI clamp (22), thus trapping plasmid DNA in the cavity of the enzyme. The 32-mer STS oligonucleotide was added to the complex to form enzyme-DNA gate (schematic in Figure 2A). Strand passage would result in the escape of the pUC18 DNA from the central cavity, which can be measured by filter binding assay. In assay with WT enzyme, labeled DNA retention decreased, indicating efficient strand passage. In contrast, the NTD of MstopoI did not show marked reduction in the retention of pUC18 DNA, implying that NTD is inefficient in strand passage unlike the full-length (MstopoI) enzyme. The results are in accordance with earlier findings that the NTD alone is not competent for strand passage activity (17). The mutant ΔB1 retained 80% of strand passage activity. In contrast, mutant ΔB3 was severely affected in strand passage and exhibited only 40% activity. The strand passage activity was completely abolished in ΔB13 and ΔB23 (Figure 2B and C). Notably, the extent of strand passage carried out by MstopoI and the deletants correlated well with their respective DNA relaxation activities.
Figure 2.

Assessment of strand passage. (A) Experimental design of filter-binding assay. Nick translated pUC18 DNA was incubated with 500 nM proteins at 37°C. The 32-mer oligonucleotide harboring STS was added to the enzyme–DNA complex to form cleaved DNA gate for the passage of pUC18 DNA. Radioactive counts (counts per minute) associated with each reaction were measured by filter binding assay; (B) Retention of pUC18 DNA. Graph showing the amount of total count and retained count associated with the enzymes. Total counts represent the counts (counts per minute) associated with the enzyme–pUC18 DNA complex. Retained counts (cpm) indicate remaining counts after the completion of strand passage reaction. WT + 32B represents the negative control in which 32B oligonucleotide was annealed with 32 mer and duplex DNA added to enzyme-pUC18 DNA complex. NTD alone used as a control did not show significant difference, suggesting that the decrease in the count is mediated by CTD; (C) Strand passage activity. Graph representing the strand passage activity of the WT and mutant enzymes. Strand passage activity of WT enzyme was normalized to 100%. The error bars represent standard deviation across three measurements. The plasmid DNA molecules, which entered the enzyme cavity, were captured by using a clamp closing anti-topoI monoclonal antibody, and the weakly bound salt sensitive complexes were removed by washing with the high salt buffer. The radioactive counts represent the salt resistant enzyme–DNA hetero-catenanes.
Basic amino acid stretches confer the non-specific DNA-binding property to the CTD
In an earlier study, we showed that the CTD binds to DNA in a non-specific manner (17). From the results described earlier in the text, it appeared that basic amino acids could confer DNA-binding property to the CTD. To test this, EMSA was carried out using 32-mer single-stranded STS oligonucleotide and various CTD deletant enzymes described earlier in the text. From the representative Kd values, it is apparent that the deletion of basic amino acid stretches reduced the DNA-binding affinity of the enzyme (Figure 3). Despite weaker binding of the mutants, their cleavage and religation activities were comparable with the WT enzyme (Supplementary Figures S3 and S4). From these data, one can surmise that the stretches in the CTD do not influence the binding of MstopoI to the scissile DNA. Instead, their role could be in holding the non-scissile (non-STS) complementary DNA required for strand passage. To assess their role in non-specific DNA capture, DNA-binding ability of deletants was determined with the non-STS DNA. As the binding of NTD to non-STS DNA is extremely low (17), the complex formed by the MstopoI with this DNA would mainly be by the contribution from the CTD. From the EMSA results (Figure 4A), it is evident that the ΔB1and ΔB3 were affected in non-specific DNA binding significantly, and the binding of ΔB13 and ΔB23 to DNA was completely abolished.
Figure 3.

DNA-binding ability of the MstopoI deletants. EMSAs were carried out by incubating 5′-end-labeled 32-mer oligonucleotide containing STS with varying concentrations of enzymes, as mentioned in each panel. DNA–protein complexes were quantitated, and DNA-binding affinity of WT and its deletants are represented in the form of their respective Kd value.
Figure 4.

Binding of WT and deletant enzymes with non-specific DNA. (A) 5′-end-labeled 32-mer non-STS oligonucleotide was incubated with 100 nM enzymes. Enzyme–DNA complexes were quantitated and represented in the form of graph (right panel). The extent of DNA binding by WT was normalized to 100%. Each value represents the mean of three independent experiments. Bars represent SD; (B) EMSA with the E. coli BL21 cell extracts expressing the intact and deletant CTD. The 5′-end-labeled 32-mer oligonucleotide was incubated with 2 µg and 5 µg of total cell extracts.
To further establish the role of the basic amino acid stretches in the DNA binding, EMSA was carried out by incubating the 5′-end-labeled non-specific 32-mer oligonucleotide with the CTD itself, or its deletants lacking the basic stretches. The deletants of CTD could not bind to either Hi-Trap Heparin or SP sepharose columns possibly owing to the absence of positively charged basic amino acid stretches. Hence, the EMSA was carried out with the crude cell lysates of E. coli BL21 cells, containing the different forms of CTD. DNA-binding results demonstrated that the mutant CTDs were highly compromised in the DNA-binding ability (Figure 4B). The reduced DNA binding of deletant CTDs correlates well with the altered DNA interaction of the MstopoI deletants. From these results, we conclude that the basic stretches contribute to the non-specific DNA binding by the CTD to guide the passage of non-scissile DNA through the enzyme-DNA gate.
The mutants are compromised in binding two DNA molecules
According to sign inversion model of DNA relaxation proposed for the mechanism of action of type IA topoisomerase, binding of two DNA molecules by the enzyme—scissile and the other transfer strand, is predicted (29). Indeed, Li et al. (31) suggested the existence of at least two independent DNA-binding sites in the type IA topoisomerase, which has not been experimentally verified yet. To evaluate MstopoI binding to two DNA, mobility shift assay was carried out with oligonucleotides of different sizes. First, 5′-end-labeled, 32-mer STS oligonucleotide incubated with MstopoI formed complex I (Figure 5). Incubation of complex I with increasing concentrations of unlabeled 60-mer non-STS DNA formed complex II (supershifted band in Figure 5), demonstrating that MstopoI can simultaneously bind two different oligonucleotides. Either NTD or CTD alone formed one complex with the DNA, indicating that each component contains one DNA-binding site. The complex formed with CTD could be competed out with 60-mer non-STS oligonucleotide, whereas the NTD–DNA complex was resistant to decay, confirming that the binding mediated by the CTD was non-specific, and the NTD specifically binds STS DNA, as previously demonstrated (17). Experiments with the deletants demonstrated that the deletion of basic amino acid stretches affected the formation of complex II to varying degrees. Deletant ΔB1 was marginally affected, whereas the deletants ΔB3 and ΔB23 failed to form complex II. Notably, as expected, results are in agreement with the DNA-binding experiments (STS and non-STS DNA-binding assay, Figures 3 and 4), confirming that the MstopoI can hold two DNA simultaneously by two distinct parts of the enzyme, viz. NTD and CTD. Further, CTD binds DNA through the basic amino acid stretches in a sequence-independent manner to mediate strand passage.
Figure 5.

MstopoI binds two DNA molecules. EMSAs were carried out by incubating 5′-end-labeled 32-mer oligonucleotide containing STS with proteins for 10 min on ice to form complex I. Further, 60-mer unlabeled oligonucleotide was incubated with the complex I for 15 min on ice to form complex II (as depicted in the schematic). Complexes were resolved on 6% native PAGE. (A) Complex formed by the WT MstopoI, NTD and CTD (as indicated in each panel); (B) EMSA with the WT and deletant enzymes. C: 32-mer with no enzyme; complex I: protein-32 mer complex; complex II: 32 mer-protein-60 mer complex.
Overexpression of deletant enzyme affects the growth of M. smegmatis
From the experiments described so far, it is evident that the mutants lacking the basic residues in the CTD are compromised only for strand passage but are competent in site-specific DNA binding, cleavage and religation. Indeed, DNA relaxation activity of the WT enzyme was suppressed in the presence of the deletant enzyme in vitro, indicating the competition between the two proteins for the same substrate DNA (Supplementary Figure S5). Hence, in principle, they should exert dominant negative effect on enzyme function when expressed in vivo. To understand the consequence of the deletions on intracellular topoI function, MstopoI ΔB23 was overexpressed in M. smegmatis, and the growth was monitored. Interestingly, the cells overexpressing the deletant topoI showed significant reduction in growth compared with the cells overexpressing the WT enzyme. The cells expressing the deletant showed, increased lag period (16 h) compared with WT (12 h), 1.6-fold reduced growth rate and were affected in attaining maximum growth density (Figure 6). The results suggest that the overexpression of a functionally compromised form of the enzyme can compete out the WT enzyme for DNA binding, affecting the DNA relaxation activity in vivo leading to the reduced growth. This dominant negative effect thus establishes the importance of the crucial residues of CTD in completion of the DNA relaxation cycle.
Figure 6.

Overexpression of the deletant enzyme results in growth defect. Exponential phase cultures of M. smegmatis mc2 155 and overexpression strains were diluted to O.D.595 nm = 0.05 in Middlebrook7H9 broth and incubated at 37°C with continuous shaking. The cultures were grown for 40 h, and the O.D.595nm was measured at every 2 h interval. The figure shows the mean O.D.595nmvalues obtained in three independent experiments. Bar represents the SD.
DISCUSSION
In this study, we demonstrate a new DNA-binding motif for strand passage function in a type IA topoisomerase that lacks Zn2+ fingers in the CTD. Three basic amino acid stretches found in the CTD of MstopoI confer non-specific DNA-binding property to the enzyme, rendering the CTD indispensable for the enzyme function. Basic residues, impart a critical role for the CTD in the essential strand passage step of the reaction, although they do not possess any enzymatic activity.
The sign inversion model proposed for type IA topoisomerase reaction mechanism advocates that both, the scissile strand and the intact complementary strand, are held simultaneously by the enzyme (1,29,30), suggesting the presence of two DNA-binding sites. EMSA with DNA molecules of different sizes established that MstopoI holds two DNA molecules simultaneously, and the deletants were compromised in binding to the second DNA molecule. The outcome of this experiment validates the enzyme bridge model for type IA topoisomerase, according to which, the topoI holds the two distinct DNA strands during DNA relaxation (30). In EctopoIII, it has been shown that the enzyme can concurrently bind dsDNA in the central hole and ssDNA in the ssDNA-binding groove, indicating that the enzyme has at least two DNA-binding sites (27,31). The present study with MstopoI reveals that one DNA-binding site lies in the NTD, and the other site resides in the basic stretches of the CTD. In an earlier study, we showed that the NTD bound preferentially to the ssDNA containing the specific sequence (STS DNA), whereas the CTD bound both the ssDNA as well as the dsDNA with comparable affinity (17). Based on all the observations, we suggest that the NTD accommodates and cleaves STS DNA, whereas the CTD holds non-scissile ssDNA (during DNA relaxation) as well as the dsDNA (during decatenation) for the strand passage.
The growth studies with topoI overexpression strains of M. smegmatis further established the importance of basic amino acid stretches. The reduced growth rate of MstopoIΔ23 overexpression strain can be the consequence of occupancy of deletant enzyme on the topoI-binding sites by competing out the native topoI from its site of action. Data from the in vitro studies showed that the deletants can bind to scissile DNA strand by virtue of their intact NTD to cleave and reseal the DNA. In a strain overexpressing MstopoIΔ23, the deletant would interfere with the WT topoI binding and carry out two transesterification steps, but in the absence of basic amino acid stretches, fail to hold the non-scissile DNA for the strand passage. This would result in futile cycles of cleavage and religation without any DNA relaxation activity. Basic amino acid stretches are thus indispensable for enzyme function both in vitro as well as in vivo.
We showed in an earlier study, that MstopoI carries out the DNA relaxation reaction in a processive fashion (7). However, what contributes to the processivity of the enzyme was not known. The DNA relaxation assay revealed that ΔB1 and ΔB2 generated topoisomers of intermediate superhelical density (partially relaxed) in contrast to WT MstopoI, which produced completely relaxed products, suggesting the distributive mode of enzyme action in the deletants (Figure 1C). The basic amino acids in the CTD might assist in capturing the plasmid molecules efficiently to catalyze the multiple DNA relaxation cycles leading to the complete relaxation of supercoiled substrate, thus enhancing the processivity of the enzyme. Positively charged lysine and arginine in the basic stretches could bind the negatively charged phosphate backbone of DNA. The deletion of these basic amino acids would reduce the charge interactions, thus lowering the affinity of the enzyme for substrate DNA, resulting in an enzyme that is distributive. Similarly, the processivity of the EctopoIII is governed by the positively charged amino acids in the CTD, and deletion of the basic amino acids rendered the enzyme distributive (32). Further, elucidation of the role of basic amino acid residues described here may provide an explanation for the apparent discrepancy in the results obtained when the Zn2+ fingers in the CTD were mutated in TmtopoI and EctopoI (12,15). Notably, positively charged amino acids were found flanking the single Zn2+ finger in TmtopoI (Supplementary Figure S6), whereas in EctopoI, there seems to be an under representation of basic amino acids neighboring the Zn2+ fingers. Thus, it appears that basic residues could have a role in TmtopoI-mediated DNA relaxation, although the Zn2+ finger is dispensable.
Based on the present findings, we propose a model for the mechanism of strand passage by MstopoI (Figure 7). According to this model, MstopoI binds scissile DNA in a site-specific manner by virtue of the NTD, whereas the CTD simultaneously interacts with the non-scissile DNA. On encountering the STS DNA region, the NTD cleaves the DNA to form the 5′-phosphotyrosine covalent adduct, followed by the opening of the DNA gate. Basic stretches in the CTD guide the non-scissile DNA from the central cavity to the DNA gate, leading to the strand passage, followed by resealing of the DNA ends, thus completing one cycle of DNA relaxation. After the catalysis, MstopoI may remain bound to the same DNA molecule through the basic amino acids, thus, enhancing the processivity of the enzyme.
Figure 7.

Strand passage mechanism of topoI. TopoI-mediated relaxation is proposed to occur by recognizing and binding to a single-stranded region within the supercoiled substrate (A and B), followed by nicking of the scissile strand. The enzyme undergoes conformational changes resulting in opening of the DNA gate (C). The complementary non-scissile strand held by the basic amino acid stretches in the CTD is passed through the opened DNA gate (D). Subsequently, two ends of the DNA are brought closer for religation (E) leading to the relaxation of supercoiled DNA. The enzyme releases the DNA or begins another catalytic cycle. Basic amino acid stretches are shown in cyan.
Sequence analyses indicated that topoI from all the Actinomycetes lack Zn2+ finger (data not shown). The Zn2+ fingers in topoI from this group could have been eliminated for two possible reasons. First, an enzyme without the Zn2+ finger may tolerate extreme conditions of pH and oxidative stresses, which otherwise could affect the structural integrity of the Zn2+ motif (33,34). Indeed, the mycobacterial topoI is catalytically competent even at broad pH range and high temperatures (7). Similarly, another non-Zn2+ finger enzyme EctopoIII also been shown to be active at high temperatures (35). These features may provide an edge to some of the mycobacteria, which encounter low pH and oxidative conditions prevailing inside the host macrophages (36,37). Another advantage of replacing the Zn2+ motif in topoI could be related to the intracellular scarcity of Zn2+ ions. The free Zn2+ ions have been shown to cause metallotoxicity, leading to the death of mycobacteria (38,39). Studies with various mycobacteria have revealed the protective mechanisms used by them to overcome the Zn2+ cytotoxicity (38,39). Mycobacterium tuberculosis uses the CtpC (P-type ATPases) transporters to pump out the Zn2+ from the cell, thus circumventing the host defense system that exploits the Zn2+ ions to kill the pathogen (38). Similarly, other mycobacteria also endure the diverse inhospitable conditions in external environment including the Zn2+ contaminated habitats (40–42). To withstand the Zn2+ toxicity, environmental mycobacteria exploit cation diffusion facilitator family protein zitA, which provide resistance against Zn2+ toxicity. The mycobacterial genome codes for more number of P-type ATPases compared with E. coli genome implying their role in overcoming the cidal effect of Zn2+ (43). Thus, it appears that these organisms have developed one or other mechanism to pump out the Zn2+ ions from the cell, inducing paucity of the Zn2+ ions in the cellular milieu of mycobacteria, eventually leading to underrepresentation of Zn2+ metalloproteins. UvrA (a component of nucleotide excision repair pathway) of M. smegmatis and M. tuberculosis can function without Zn2+ finger unlike their counterpart in E. coli (44,45). Another Zn2+-dependent enzyme in most bacteria, i.e. Cu-Zn superoxide dismutase appears to have evolved as a Zn2+-independent enzyme in mycobacteria (46). Similarly, Tau subunit of DNA polymerase III of mycobacteria seems to have dispensed of Zn2+-binding module. Thus, it is apparent that many important enzymes in Actinomycetes, including topoI, carry out their cellular functions in Zn2+-independent fashion to cope up with the Zn2+ scarcity arising owing to export and homeostasis of the metal ion.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Table 1 and Supplementary Figures 1–6.
FUNDING
Shyama Prasad Mukherjee senior research fellowship of Council of Scientific and Industrial Research, Government of India (to W.A.); J.C. Bose fellowship of Department of Science and Technology and Centre for Excellence grant from Department of Biotechnology, Government of India (to V.N.). Funding for open access charge: J.C. Bose fellowship of Department of Science and Technology.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank the members of VN laboratory for valuable suggestions and critical reading of the manuscript. Rupesh Kumar is acknowledged for his assistance in preparing the model for the topoI reaction mechanism.
REFERENCES
- 1.Champoux JJ. DNA topoisomerases: structure, function, and mechanism. Annu. Rev. Biochem. 2001;70:369–413. doi: 10.1146/annurev.biochem.70.1.369. [DOI] [PubMed] [Google Scholar]
- 2.Gellert M. DNA topoisomerases. Annu. Rev. Biochem. 1981;50:879–910. doi: 10.1146/annurev.bi.50.070181.004311. [DOI] [PubMed] [Google Scholar]
- 3.Aravind L, Leipe DD, Koonin EV. Toprim – a conserved catalytic domain in type IA and II topoisomerases, DnaG-type primases, OLD family nucleases and RecR proteins. Nucleic Acids Res. 1998;26:4205–4213. doi: 10.1093/nar/26.18.4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen SJ, Wang JC. Identification of active site residues in Escherichia coli DNA topoisomerase I. J. Biol. Chem. 1998;273:6050–6056. doi: 10.1074/jbc.273.11.6050. [DOI] [PubMed] [Google Scholar]
- 5.Zhu CX, Roche CJ, Papanicolaou N, DiPietrantonio A, Tse-Dinh YC. Site-directed mutagenesis of conserved aspartates, glutamates and arginines in the active site region of Escherichia coli DNA topoisomerase I. J. Biol. Chem. 1998;273:8783–8789. doi: 10.1074/jbc.273.15.8783. [DOI] [PubMed] [Google Scholar]
- 6.Lynn RM, Bjornsti MA, Caron PR, Wang JC. Peptide sequencing and site-directed mutagenesis identify tyrosine-727 as the active site tyrosine of Saccharomyces cerevisiae DNA topoisomerase I. Proc. Natl Acad. Sci. USA. 1989;86:3559–3563. doi: 10.1073/pnas.86.10.3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bhaduri T, Bagui TK, Sikder D, Nagaraja V. DNA topoisomerase I from Mycobacterium smegmatis. An enzyme with distinct features. J. Biol. Chem. 1998;273:13925–13932. doi: 10.1074/jbc.273.22.13925. [DOI] [PubMed] [Google Scholar]
- 8.Serre MC, Duguet M. Enzymes that cleave and religate DNA at high temperature: the same story with different actors. Prog. Nucleic Acid Res. Mol. Biol. 2003;74:37–81. doi: 10.1016/s0079-6603(03)01010-9. [DOI] [PubMed] [Google Scholar]
- 9.Bouthier de la Tour C, Kaltoum H, Portemer C, Confalonieri F, Huber R, Duguet M. Cloning and sequencing of the gene coding for topoisomerase I from the extremely thermophilic eubacterium, Thermotoga maritima. Biochim. Biophys. Acta. 1995;1264:279–283. doi: 10.1016/0167-4781(96)83596-2. [DOI] [PubMed] [Google Scholar]
- 10.Tse-Dinh YC, Beran-Steed RK. Escherichia coli DNA topoisomerase I is a zinc metalloprotein with three repetitive zinc-binding domains. J. Biol. Chem. 1988;263:15857–15859. [PubMed] [Google Scholar]
- 11.Tse-Dinh YC. Zinc (II) coordination in Escherichia coli DNA topoisomerase I is required for cleavable complex formation with DNA. J. Biol. Chem. 1991;266:14317–14320. [PubMed] [Google Scholar]
- 12.Zhu CX, Qi HY, Tse-Dinh YC. Mutation in Cys662 of Escherichia coli DNA topoisomerase I confers temperature sensitivity and change in DNA cleavage selectivity. J. Mol. Biol. 1995;250:609–616. doi: 10.1006/jmbi.1995.0402. [DOI] [PubMed] [Google Scholar]
- 13.Lima CD, Wang JC, Mondragon A. Crystallization of a 67 kDa fragment of Escherichia coli DNA topoisomerase I. J. Mol. Biol. 1993;232:1213–1216. doi: 10.1006/jmbi.1993.1474. [DOI] [PubMed] [Google Scholar]
- 14.Ahumada A, Tse-Dinh YC. The role of the Zn(II) binding domain in the mechanism of E. coli DNA topoisomerase I. BMC Biochem. 2002;3:13. doi: 10.1186/1471-2091-3-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Viard T, Lamour V, Duguet M, Bouthier de la Tour C. Hyperthermophilic topoisomerase I from Thermotoga maritima. A very efficient enzyme that functions independently of zinc binding. J. Biol. Chem. 2001;276:46495–46503. doi: 10.1074/jbc.M107714200. [DOI] [PubMed] [Google Scholar]
- 16.Godbole AA, Leelaram MN, Bhat AG, Jain P, Nagaraja V. Characterization of DNA topoisomerase I from Mycobacterium tuberculosis: DNA cleavage and religation properties and inhibition of its activity. Arch. Biochem. Biophys. 2012;528:197–203. doi: 10.1016/j.abb.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 17.Jain P, Nagaraja V. Indispensable, functionally complementing N and C-terminal domains constitute site-specific topoisomerase I. J. Mol. Biol. 2006;357:1409–1421. doi: 10.1016/j.jmb.2006.01.079. [DOI] [PubMed] [Google Scholar]
- 18.Bhaduri T, Sikder D, Nagaraja V. Sequence specific interaction of Mycobacterium smegmatis topoisomerase I with duplex DNA. Nucleic Acids Res. 1998;26:1668–1674. doi: 10.1093/nar/26.7.1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sarkar G, Sommer SS. The “megaprimer” method of site-directed mutagenesis. Biotechniques. 1990;8:404–407. [PubMed] [Google Scholar]
- 20.Hemsley A, Arnheim N, Toney MD, Cortopassi G, Galas DJ. A simple method for site-directed mutagenesis using the polymerase chain reaction. Nucleic Acids Res. 1989;17:6545–6551. doi: 10.1093/nar/17.16.6545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bhat AG, Leelaram MN, Hegde SM, Nagaraja V. Deciphering the distinct role for the metal coordination motif in the catalytic activity of Mycobacterium smegmatis topoisomerase I. J. Mol. Biol. 2009;393:788–802. doi: 10.1016/j.jmb.2009.08.064. [DOI] [PubMed] [Google Scholar]
- 22.Leelaram MN, Bhat AG, Godbole AA, Bhat RS, Manjunath R, Nagaraja V. Type IA topoisomerase inhibition by clamp closure. FASEB J. 2013;27:3030–3038. doi: 10.1096/fj.12-226118. [DOI] [PubMed] [Google Scholar]
- 23.Blokpoel MC, Murphy HN, O'Toole R, Wiles S, Runn ES, Stewart GR, Young DB, Robertson BD. Tetracycline-inducible gene regulation in mycobacteria. Nucleic Acids Res. 2005;33:e22. doi: 10.1093/nar/gni023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suerbaum S, Brauer–Steppkes T, Labigne A, Cameron B, Drlica K. Topoisomerase I of Helicobacter pylori: juxtaposition with a flagellin gene (flaB) and functional requirement of a fourth zinc finger motif. Gene. 1998;210:151–161. doi: 10.1016/s0378-1119(98)00065-1. [DOI] [PubMed] [Google Scholar]
- 25.Hansen G, Harrenga A, Wieland B, Schomburg D, Reinemer P. Crystal structure of full length topoisomerase I from Thermotoga maritima. J. Mol. Biol. 2006;358:1328–1340. doi: 10.1016/j.jmb.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 26.Lima CD, Wang JC, Mondragon A. Three-dimensional structure of the 67K N-terminal fragment of E. coli DNA topoisomerase I. Nature. 1994;367:138–146. doi: 10.1038/367138a0. [DOI] [PubMed] [Google Scholar]
- 27.Changela A, DiGate RJ, Mondragon A. Crystal structure of a complex of a type IA DNA topoisomerase with a single-stranded DNA molecule. Nature. 2001;411:1077–1081. doi: 10.1038/35082615. [DOI] [PubMed] [Google Scholar]
- 28.Changela A, DiGate RJ, Mondragon A. Structural studies of E. coli topoisomerase III-DNA complexes reveal a novel type IA topoisomerase-DNA conformational intermediate. J. Mol. Biol. 2007;368:105–118. doi: 10.1016/j.jmb.2007.01.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brown PO, Cozzarelli NR. A sign inversion mechanism for enzymatic supercoiling of DNA. Science. 1979;206:1081–1083. doi: 10.1126/science.227059. [DOI] [PubMed] [Google Scholar]
- 30.Dekker NH, Rybenkov VV, Duguet M, Crisona NJ, Cozzarelli NR, Bensimon D, Croquette V. The mechanism of type IA topoisomerases. Proc. Natl Acad. Sci. USA. 2002;99:12126–12131. doi: 10.1073/pnas.132378799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Z, Mondragon A, DiGate RJ. The mechanism of type IA topoisomerase-mediated DNA topological transformations. Mol. Cell. 2001;7:301–307. doi: 10.1016/s1097-2765(01)00178-2. [DOI] [PubMed] [Google Scholar]
- 32.Zhang HL, DiGate RJ. The carboxyl-terminal residues of Escherichia coli DNA topoisomerase III are involved in substrate binding. J. Biol. Chem. 1994;269:9052–9059. [PubMed] [Google Scholar]
- 33.Jakob U, Eser M, Bardwell JC. Redox switch of hsp33 has a novel zinc-binding motif. J. Biol. Chem. 2000;275:38302–38310. doi: 10.1074/jbc.M005957200. [DOI] [PubMed] [Google Scholar]
- 34.Larabee JL, Hocker JR, Hanas JS. Cys redox reactions and metal binding of a Cys2His2 zinc finger. Arch. Biochem. Biophys. 2005;434:139–149. doi: 10.1016/j.abb.2004.10.024. [DOI] [PubMed] [Google Scholar]
- 35.DiGate RJ, Marians KJ. Identification of a potent decatenating enzyme from Escherichia coli. J. Biol. Chem. 1988;263:13366–13373. [PubMed] [Google Scholar]
- 36.Schnappinger D, Ehrt S, Voskuil MI, Liu Y, Mangan JA, Monahan IM, Dolganov G, Efron B, Butcher PD, Nathan C, et al. Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: insights into the phagosomal environment. J. Exp. Med. 2003;198:693–704. doi: 10.1084/jem.20030846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Triccas JA, Gicquel B. Life on the inside: probing Mycobacterium tuberculosis gene expression during infection. Immunol. Cell Biol. 2000;78:311–317. doi: 10.1046/j.1440-1711.2000.00934.x. [DOI] [PubMed] [Google Scholar]
- 38.Botella H, Peyron P, Levillain F, Poincloux R, Poquet Y, Brandli I, Wang C, Tailleux L, Tilleul S, Charriere GM, et al. Mycobacterial p(1)-type ATPases mediate resistance to zinc poisoning in human macrophages. Cell Host Microbe. 2011;10:248–259. doi: 10.1016/j.chom.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grover A, Sharma R. Identification and characterization of a major Zn(II) resistance determinant of Mycobacterium smegmatis. J. Bacteriol. 2006;188:7026–7032. doi: 10.1128/JB.00643-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dean-Ross D, Cerniglia CE. Degradation of pyrene by Mycobacterium flavescens. Appl. Microbiol. Biotechnol. 1996;46:307–312. doi: 10.1007/s002530050822. [DOI] [PubMed] [Google Scholar]
- 41.Leys NM, Ryngaert A, Bastiaens L, Wattiau P, Top EM, Verstraete W, Springael D. Occurrence and community composition of fast-growing Mycobacterium in soils contaminated with polycyclic aromatic hydrocarbons. FEMS Microbiol. Ecol. 2005;51:375–388. doi: 10.1016/j.femsec.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 42.Norton CD, LeChevallier MW, Falkinham JO., 3rd Survival of Mycobacterium avium in a model distribution system. Water Res. 2004;38:1457–1466. doi: 10.1016/j.watres.2003.07.008. [DOI] [PubMed] [Google Scholar]
- 43.Rensing C, Fan B, Sharma R, Mitra B, Rosen BP. CopA: An Escherichia coli Cu(I)-translocating P-type ATPase. Proc. Natl Acad. Sci. USA. 2000;97:652–656. doi: 10.1073/pnas.97.2.652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Visse R, de Ruijter M, Ubbink M, Brandsma JA, van de Putte P. The first zinc-binding domain of UvrA is not essential for UvrABC-mediated DNA excision repair. Mutat. Res. 1993;294:263–274. doi: 10.1016/0921-8777(93)90009-6. [DOI] [PubMed] [Google Scholar]
- 45.Wang J, Mueller KL, Grossman L. A mutational study of the C-terminal zinc-finger motif of the Escherichia coli UvrA protein. J. Biol. Chem. 1994;269:10771–10775. [PubMed] [Google Scholar]
- 46.Battistoni A, Pacello F, Mazzetti AP, Capo C, Kroll JS, Langford PR, Sansone A, Donnarumma G, Valenti P, Rotilio G. A histidine-rich metal binding domain at the N terminus of Cu,Zn-superoxide dismutases from pathogenic bacteria: a novel strategy for metal chaperoning. J. Biol. Chem. 2001;276:30315–30325. doi: 10.1074/jbc.M010527200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.