


Abstract
Selenoproteins are proteins containing an uncommon amino acid selenocysteine (Sec). Sec is inserted by a specific translational machinery that recognizes a stem-loop structure, the SECIS element, at the 3′ UTR of selenoprotein genes and recodes a UGA codon within the coding sequence. As UGA is normally a translational stop signal, selenoproteins are generally misannotated and designated tools have to be developed for this class of proteins. Here, we present two new computational methods for selenoprotein identification and analysis, which we provide publicly through the web servers at http://gladyshevlab.org/SelenoproteinPredictionServer or http://seblastian.crg.es. SECISearch3 replaces its predecessor SECISearch as a tool for prediction of eukaryotic SECIS elements. Seblastian is a new method for selenoprotein gene detection that uses SECISearch3 and then predicts selenoprotein sequences encoded upstream of SECIS elements. Seblastian is able to both identify known selenoproteins and predict new selenoproteins. By applying these tools to diverse eukaryotic genomes, we provide a ranked list of newly predicted selenoproteins together with their annotated cysteine-containing homologues. An analysis of a representative candidate belonging to the AhpC family shows how the use of Sec in this protein evolved in bacterial and eukaryotic lineages.
INTRODUCTION
Selenoproteins are a class of proteins that contain the amino acid selenocysteine (Sec), known as the 21st amino acid in the genetic code. Sec is inserted co-translationally by recoding a UGA codon, which normally serves as a stop signal (1–4). Owing to this dual function of the UGA codon, selenoproteins are generally missed or mispredicted in genome projects, and their annotation has to be carried out with ad hoc developed tools. Since the beginning of the genomic era, a considerable effort has been placed at developing computational methods for selenoprotein prediction, including the detection and analysis of eukaryotic, archaeal and prokaryotic SECIS elements, and the identification of selenoproteins in genomes ab initio or by homology (5–17).
In this study, we present two new computational methods for selenoprotein prediction and analysis. SECISearch3 is a pipeline for predicting SECIS elements that significantly outperforms its predecessor SECISearch. Seblastian is a new method for the identification of selenoprotein genes in sequence databases that uses SECISearch3 and then identifies selenoprotein sequences upstream of the detected SECIS elements. Both services can be freely run through web servers at http://gladyshevlab.org/SelenoproteinPredictionServer and http://seblastian.crg.es.
Eukaryotic SECIS elements
SECIS elements are stem-loop structures that specify recoding of a UGA codon from its canonical translation termination function to a non-canonical one, Sec insertion. SECIS elements are completely different in eukaryotes, bacteria and archaea and may also be located in different regions of selenoprotein genes (18). Here, we focus on eukaryotic SECIS elements. These structures can be classified into two classes, type I and type II, differing in the presence of an additional helix in type 2 SECIS elements (19). The highest sequence conservation in SECIS elements is found in the core (or quartet), which forms a kink-turn motif through the non-canonical pairing of AG-GA. The core bears the conserved sequence UGAN/KGAW. Additionally, a stretch of conserved nucleotides are found in the apical loop, typically adenines (or cytosines in a few cases). The structural parts of SECIS elements are also found to be within specific length constraints [see (13) for a summary], although the precise definition of these boundaries has changed during the years, particularly with the analysis of these structures in newly sequenced eukaryotes. The distance between the Sec-UGA and the SECIS element varies substantially, e.g. from 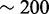 to
to 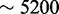 nt in mammalian selenoproteins. The minimum functional distance was tested in human embryonic kidney line 293 cells for deiodinase 1 (20), and it was found to be between 51 and 111 nt.
nt in mammalian selenoproteins. The minimum functional distance was tested in human embryonic kidney line 293 cells for deiodinase 1 (20), and it was found to be between 51 and 111 nt.
The original SECISearch
The most widely used method for SECIS prediction has been SECISearch (9). This method relies on sequence patterns (searched with PatScan http://blog.theseed.org/servers/2010/07/scan-for-matches.html) to identify initial hits in the query sequence, which are then processed and filtered. Several SECIS patterns were developed and optimized in the past 10 years. All patterns model a partition of the SECIS in helix1, core, loop1, helix2, conserved apical nucleotides, loop2 and optionally helix3 (only in type II SECIS elements). Thus, these criteria require the hits to have specific nucleotides in the core and in the apical nucleotides and to have stretches of nucleotides of a defined length that can pair to form the stems. The various patterns differ in the required conserved nucleotides and in the length and pairing rules allowed in stems. Currently, the patterns used by SECISearch are the following: strict, default, loose and loosest (loose+) (see Supplementary Material S1). The hits by PatScan are fed into RNAfold from the ViennaRNA package (21,22), which predicts their secondary structure and thermodynamic stability. This is used to filter out unstable structures. Finally, another filter analyzes the predicted secondary structure and the pattern-based partition of the candidate and filters out unlikely candidates with certain structural characteristics (e.g. Y-shaped or O-shaped). Although SECISearch has been extremely useful to selenoprotein research, it has some limitations. The main one is its dependence on sequence patterns. The patterns have been manually built to accommodate SECIS elements. As a result, whenever a species from a newly sequenced distant lineage is analyzed, the patterns had to be modified to optimize the searches. The current routine identifies a first set of bona fide selenoproteins by running SECISearch with the existing patterns or by homology to known selenoproteins with the tools such as Tblastn [or lately, with the more sophisticated Selenoprofiles (15)]. Then, a new pattern is developed that includes the bona fide selenoproteins while keeping the number of predictions under a manageable level, and the genome search is then done with this pattern. Another limitation of the original SECISearch is that is lacks the assignment of a score to the candidates.
MATERIALS AND METHODS
New SECIS prediction methods
In the past several years, several programs have emerged for family-based prediction of RNA structures. To build a better tool for SECIS prediction, we tested three available methods: Infernal, Covels and Erpin. In most cases, we built our own SECIS models.
The program Infernal (Inference of RNA alignments) (23) ‘is an implementation of a special case of profile stochastic context-free grammars called covariance models (CMs). A CM is like a sequence profile, but it scores a combination of sequence consensus and RNA secondary structure consensus’. Infernal can be used to build a CM model from a secondary structure alignment and then search the model in nucleotide databases. To obtain a large set of SECIS elements for the alignment, we exploited an extensive collection of bona fide selenoprotein sequences predicted with Selenoprofiles (15). Initially, SECISearch was run on sequences downstream of all selenoprotein coding sequences. This set was used to build a first, very rough alignment, forcing the structural parts to be aligned (stem1, core, loop1, etc.) as shown in Supplementary Material S2. A consensus secondary structure was manually assigned to this alignment, based on the known pairings (part1 of helix1 with part2, and so on). The resulting secondary structure alignment was inspected with RALEE, a RNA alignment editor (24), to identify and extract the sequences satisfying the consensus secondary structure assigned, i.e. to obtain a subset of well-aligned sequences. This subset was used to build an Infernal model, and the Infernal program cmalign was used to align additional SECIS elements to the model. As this was a template-based alignment, the resulting quality was much superior. This procedure was used iteratively, inspecting manually the alignment to add or remove sequences, until we obtained our final model: a secondary structure alignment of 1122 SECIS elements from diverse eukaryotic lineages. We use this model with the Infernal program cmsearch as a new method to predict SECIS elements. Infernal computes two types of scores for each candidate: a bit-score, expressing how well it fits in the model, and an E-value, expressing how many alignments with the same or better bit-score are expected by chance searching the current target. We decided to use a bit-score-based filtering, for this is not dependent on the target size.
The program Covels (http://selab.janelia.org/software.html) is also based on variance models, but it does not model secondary structure explicitly. We built a Covels model as described in the program manual. For this purpose, 300 SECIS elements were manually aligned to produce the best results. Sequences were extracted from RefSeq NCBI database. Because our goal was to generate a ‘consensus model’, we did not consider here SECIS elements from organisms (such as Ostreococcus or Toxoplasma species) in which these structures have lineage-specific characteristics. In our study, we found that regions flanking the core lack the consensus (data not shown), therefore, including them in the model would lower the sensitivity. Thus, we included only the most functionally relevant part of their structure, beginning from the core. Like Infernal, Covels predictions include a bit-score that can be used for filtering. The recommended threshold value is 15. However, it should be taken into account that for SECIS elements not conforming to this model the score could be significantly lower.
The program Erpin (25) is another RNA motif search program. Given a secondary structure-based alignment, it infers a structural profile, which is then searched in the target database using a dynamic programming algorithm. Erpin also provides scores for the matches. In the case of Erpin, we found a SECIS model provided by the authors; therefore, we proceeded to the testing phase with this model. We noticed early on that a limitation of this program is that gaps are not allowed in the alignment model nor in the matches with the profile, thus any motif with insertions or deletions in respect to the model is missed.
RESULTS AND DISCUSSION
Testing SECIS prediction methods
To test the performance of the three methods and relate them to SECISearch, we first built a set of reliable SECIS elements from as diverse lineages as possible. The set contained 116 SECIS elements: 1 from Caenorhabditis elegans (11), 8 from Chlamydomonas reinhardtii (26), 5 from Toxoplasma gondii (27), 4 from Plasmodium falciparum (28), 4 from Dictyostelium purpureum, 3 from Drosophila melanogaster (8), 26 from Homo sapiens (9), 25 from Mus musculus and 40 from Danio rerio (see Supplementary Material S3 for details). We then evaluated all SECIS prediction methods when applied to the full genomes of these organisms. We computed an F-score (20) of the methods, which combines sensitivity and precision into a single measure, giving 20 times more importance to sensitivity (the desired trade-off in most SECISearch applications). Results are given in Table 1. When comparing the methods, Infernal with the score threshold of 20 was the best performer. Covels also performed well, with better sensitivity but additional false positives. SECISearch ranked third owing to the low values for sensitivity, and Erpin was the worst performer, owing to its low sensitivity. For all methods, SECIS elements from the non-metazoan eukaryotes were the hardest to predict (see Supplementary Material S3). We also tested the speed of the various methods. SECISearch was the quickest, although the time varied significantly depending on the pattern chosen. Erpin was the slowest, followed by Covels. It should be mentioned that Infernal can reduce its running time depending on the score threshold specified, owing to heuristics it adopts. In this case, a loose threshold was used (score 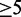 ); therefore, its speed was somewhat underestimated.
); therefore, its speed was somewhat underestimated.
Table 1.
Testing SECIS prediction methods
| TP | FP | Sn (%) | Pr (%) | FP/Mb | F-score(20) | Speed (min/Mb) | TP after filtering | FP after filtering | |
|---|---|---|---|---|---|---|---|---|---|
| Covels.5 | 114 | 1 747 455 | 98.3 | 0.007 | 224.54 | 0.026 | 33.51 | 107 | *201482 |
| Covels.10 | 108 | 188 466 | 93.1 | 0.057 | 24.22 | 0.184 | 101 | 35945 | |
| Covels.15 | 104 | 16 691 | 89.7 | 0.619 | 2.15 | 0.660 | 97 | 4152 | |
| Infernal.10 | 106 | 166 085 | 91.4 | 0.064 | 21.34 | 0.200 | 6.92 | 105 | 50814 |
| Infernal.15 | 98 | 9383 | 84.5 | 1.034 | 1.21 | 0.703 | 97 | 5697 | |
| Infernal.20 | 86 | 485 | 74.1 | 15.061 | 0.06 | 0.734 | 85 | 393 | |
| Secisearch.strict | 65 | 20 694 | 56.0 | 0.313 | 2.66 | 0.388 | 0.14 | 60 | 10557 |
| Secisearch.def | 86 | 110 532 | 74.1 | 0.078 | 14.20 | 0.220 | 0.18 | 76 | 42719 |
| Secisearch.loose | 79 | 262 710 | 68.1 | 0.030 | 33.76 | 0.102 | 3.18 | 64 | *54775 |
| Secisearch.looser | 84 | 2 689 478 | 72.4 | 0.003 | 345.59 | 0.012 | 2.62 | 66 | *542199 |
| Erpin.25 | 70 | 225 801 | 60.3 | 0.031 | 29.01 | 0.103 | 75.37 | ||
| Erpin.35 | 58 | 3754 | 50.0 | 1.522 | 0.48 | 0.463 | |||
| Erpin.45 | 43 | 48 | 37.1 | 47.253 | 0.01 | 0.371 |
The test set consisted of 116 SECIS elements from nine species (see Supplementary Material S3). For Covels, Infernal and Erpin, various score thresholds were considered; different patterns were considered for SECISearch. The two last columns show the effect of the SECISearch3 filter (see text). Erpin is not shown, as it is not included in SECISearch3.
For the methods indicated with a star (asterisk), the number of false positives after filtering was estimated by running the filter only on a subset of the total predictions, to save computational time. TP, number of true positives; FP, number of false positives; Sn, sensitivity (recall); Pr, precision; FP/Mb, average number of false positives per Mb of input sequence; F-score(20), F-score computed with beta = 20; Speed, total run time divided by the total input sequence length (∼8 Gb); TP after filtering, true positives passing the SECIS filter; FP after filtering, false positives passing the SECIS filter.
SECISearch3
Given these results, we built a pipeline that combined the predictions of Infernal, Covels and the original SECISearch. We call the new program SECISearch3 (see Figure 1). The Infernal model is central to the program. It is used not only as a prediction method but also to derive the secondary structure of the predictions by Covels and SECISearch, ensuring consistency. The redundant predictions are then removed, and a procedure of structural refinement is executed. This process compensates for structural inconsistencies owing to the template-based structure assignment of Infernal, particularly improving the pairing near insertions and in the boundaries of helixes and loops. After refinement, the thermodynamic stability of the structure is predicted with RNAeval from the Vienna package (21,22). At this point, all predictions are also assigned a score by the Covels model.
Figure 1.

Workflow of the SECISearch3 program.
Next, a filtering procedure is applied to the candidate SECIS elements. The candidates are discarded if they have any of following features (see the SECISearch filtering section in Supplementary Material S4): core is not included in the prediction, no GA-AG in the core, apical loop is too short or too long, helix2 is too short or too long, too much bending (computed as the difference in number of insertions on the two sides of helix2) and the free energy is too high. The effect of this filter is shown in Table 1 (right column): although true positives remain stable, the number of false positives significantly decreases following the filtering.
Lastly, the remaining candidates are assigned a grade (A, B or C). We included this procedure after inspecting and grading manually hundreds of SECIS elements trying to incorporate our extensive experience with these structures. The grade depends on several characteristics: the presence of conserved unpaired nucleotides in the apical loop, the bending coefficient for helix2, the Covels score, the presence of mismatches or insertion in key positions (just before or just after the core, or in any two consecutive positions along helix2). For details, see the SECIS grading section in Supplementary Material S4. SECISearch3 may generate graphical output of publication quality: the program RNAplot from the RNAfold package is used with custom settings to highlight the key SECIS features (see Figure 2). We designed SECISearch3 to be as flexible as possible. Any combination of the prediction methods (or any single method) can be run. This allows balancing the trade-off between sensitivity and speed. For example, Covels should be avoided for large databases but may be used to find unusual candidate SECIS elements in relatively small databases. As default settings, we recommend to use the Infernal model with a score threshold of 10, prioritizing sensitivity.
Figure 2.
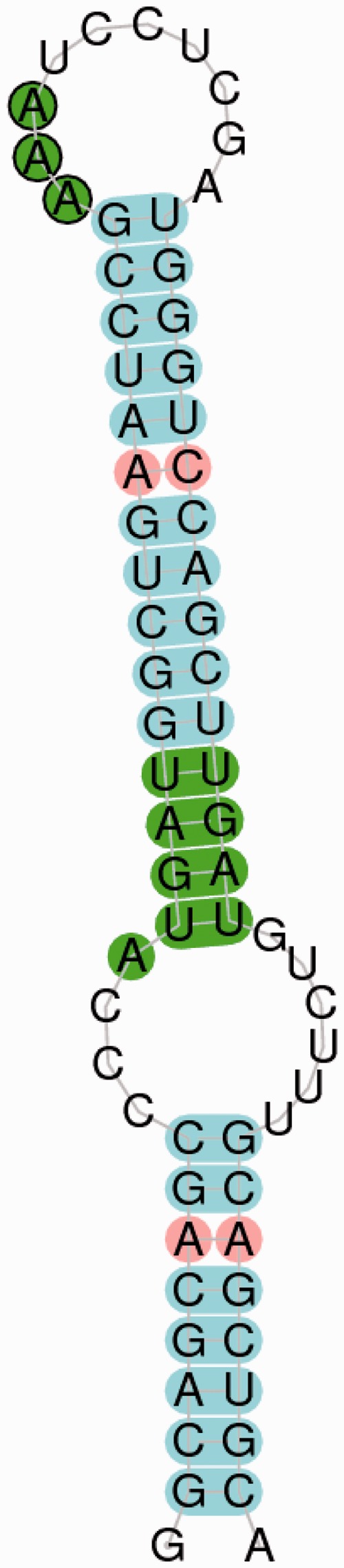
Example of SECISearch3 generated image: SECIS type I of human SelN. The core and the unpaired conserved nucleotides of the SECIS element are highlighted in green, and mismatches in red. SECISearch3 uses internally RNAplot.
Seblastian
Based on SECISearch3, we build a new method for selenoprotein gene prediction and analysis: Seblastian. This pipeline automatizes a process that we used to carry out to predict selenoproteins in newly sequenced species (Figure 3). First, all potential SECIS elements are predicted in a target sequence (a genome, for instance), and then the sequences upstream of each SECIS candidate are examined for selenoprotein coding potential. To search for selenoprotein-coding sequences, we use homology information: the sequence upstream of each SECIS is run with Blastx (29) against a comprehensive protein database (Genbank NCBI nr). As Blastx is used to make a gene prediction on the nucleotide sequence, we refer to the proteins annotated in the database as queries and to the nucleotide sequence as the target. The Blastx output is parsed, and, mostly, two types of blast alignments are considered: (i) those in which a Sec in a query protein is aligned with a UGA in the target sequence and (ii) those in which a cysteine in a query is aligned with a UGA in the target. This procedure yields two conceptually different classes of output candidates: known selenoproteins and new selenoprotein homologues of known proteins. The second category includes the candidate selenoproteins for which sequence homologues exist, but none of them is a selenoprotein (i.e. known protein family, undiscovered selenoprotein family). As the absolute majority of known selenoproteins possess cysteine homologues (30,31), Seblastian is effectively able to predict new selenoproteins. In practice, other types of blast alignments are also kept to ensure maximum sensitivity: for example, all blast hits in which the query has a Sec in its sequence are kept, even if it is not aligned to a UGA in the target sequence. Blast alignments are then filtered, and those with the same query and likely to belong to the same gene are joined. Here, the concept of colinearity is used: if blast hit A is found in the target downstream of blast hit B, and also the portion of the query aligned in blast hit A is downstream of that in blast hit B, they will be joined. A set of joined blast hits constitutes a possibly multiexonic gene prediction.
Figure 3.

Workflow of the Seblastian program.
Seblastian then attempts to improve the gene structure predictions by running the program Exonerate (32). As query, the full sequence of the nr protein in the blast alignment is used. As target, we use the region in the same blast alignment, properly extended: to ensure an optimal choice of the target boundaries, we use the cyclic Exonerate routine (15). At this point, the Exonerate and Blastx predictions for each candidate are compared, and only the best one is kept.
Finally, all candidates must pass a filter (see Seblastian filtering section in Supplementary Material S4). This requires the gene predictions to have the SECIS element properly positioned (downstream from the coding sequence) and not possess pseudogene-like features such as frameshifts or in-frame stop codons apart from the candidate Sec-UGA. Also, candidates are required to possess a convincing pattern of conservation on both sides of the Sec-UGA. Although the vast majority of selenoproteins contain a single Sec, Seblastian procedures and filters were designed to accept also candidates with multiple Sec residues, such as selenoprotein P.
Testing Seblastian
We benchmarked Seblastian using the same data set used for testing SECIS prediction methods. For SECISearch3, we chose Infernal with the score threshold of 15. Our test set was thus limited to the SECIS elements that this method is able to predict. Two separate benchmarks were executed for known selenoproteins and for new selenoproteins.
For known selenoproteins, we ran Seblastian using a modified version of the nr protein database, containing only the protein sequences with at least 1 Sec. This database was also depleted of all sequences coming from any of the tested species, to simulate a run on a newly sequenced species. For new selenoproteins, we used again nr but removed all selenoproteins, thus simulating the situation as if all selenoprotein families were undiscovered (Table 2). The search for known selenoproteins worked well, with sensitivity of 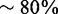 and specificity >90%. We analyzed in detail the false positives for known selenoproteins, as none were expected, as these predictions must feature a good alignment between a candidate and a known selenoprotein, with a Sec to UGA alignment. There were five false positives, all in mouse. All were similar in sequence to one of two known selenoproteins in the same species, either SelK or GPx4, but they all were intronless and with no evidence of transcription. These are recently retrotransposed pseudogenes, so similar to real selenoproteins that it is actually desirable that our method finds them. There were 19 false negatives, caused by a variety of reasons. For example, Drosophila SelK was missed because all other SelK proteins annotated in nr were too distant to give good Blastx alignments. This small selenoprotein is known to show poor homology even among closely related organisms. Drosophila SPS2 was processed as a candidate, but it was discarded during filtering owing to the presence of in-frame stop codons. These in reality reside in an intron of the gene, but they were included in the coding sequence prediction owing to spurious similarity with a portion of the selected query (SPS2 from Saccoglossus kowalevskii).
and specificity >90%. We analyzed in detail the false positives for known selenoproteins, as none were expected, as these predictions must feature a good alignment between a candidate and a known selenoprotein, with a Sec to UGA alignment. There were five false positives, all in mouse. All were similar in sequence to one of two known selenoproteins in the same species, either SelK or GPx4, but they all were intronless and with no evidence of transcription. These are recently retrotransposed pseudogenes, so similar to real selenoproteins that it is actually desirable that our method finds them. There were 19 false negatives, caused by a variety of reasons. For example, Drosophila SelK was missed because all other SelK proteins annotated in nr were too distant to give good Blastx alignments. This small selenoprotein is known to show poor homology even among closely related organisms. Drosophila SPS2 was processed as a candidate, but it was discarded during filtering owing to the presence of in-frame stop codons. These in reality reside in an intron of the gene, but they were included in the coding sequence prediction owing to spurious similarity with a portion of the selected query (SPS2 from Saccoglossus kowalevskii).
Table 2.
Testing Seblastian
| Species | Selenoproteinsa | Known selenoproteins |
New selenoproteins |
||
|---|---|---|---|---|---|
| Sn (%) | Pr (%) | Sn (%) | Pr (%) | ||
| Caenorhabditis elegans | 1 | 100.00 | 100.00 | 0.00 | 0.00 |
| Chlamydomonas reinhardtii | 3 | 33.33 | 100.00 | 0.00 | 0.00 |
| Danio rerio | 32 | 65.63 | 100.00 | 9.38 | 27.27 |
| Drosophila melanogaster | 3 | 33.33 | 100.00 | 66.67 | 66.67 |
| Homo sapiens | 25 | 96.00 | 100.00 | 40.00 | 21.28 |
| Mus musculus | 24 | 91.67 | 81.48 | 33.33 | 7.84 |
| Toxoplasma gondii | 3 | 66.67 | 100.00 | 33.33 | 100.00 |
| Dictyostelium purpureum | 1 | 100.00 | 100.00 | 0.00 | 0.00 |
| Plasmodium falciparum | 2 | 100.00 | 100.00 | 0.00 | 0.00 |
| Global | 94 | 79.79 | 93.75 | 25.53 | 14.63 |
The testing was split for known and new selenoproteins, as described in the text.
aTo test Seblastian independently of SECISearch3, we considered here only the selenoproteins whose SECIS elements were correctly predicted by Infernal with the score threshold of 15. Thus, the number of selenoproteins reported here do not necessarily represent the complete selenoproteome of the species (see Supplementary Material S3 for full sets).
Sn, sensitivity (recall); Pr, precision.
The method was also able to find new selenoproteins. Across all eukaryotes, we estimate that Seblastian alone would have identified at least 25% of all known selenoproteins. We believe that this is a remarkable result, given the difficulty of de novo prediction of selenoproteins. Indeed, for known selenoproteins, a Blastx alignment between an annotated Sec and a UGA is unlikely to happen by chance, and thus it is a sufficient argument to call a selenoprotein gene. For new selenoproteins, any cysteine of any query is a candidate Sec position. Thus, many false positives arise. Possible false candidates are real genes with sequencing errors occurring in cysteine positions, pseudogenes with a single in-frame UGA in a cysteine position, or non-coding repetitive stretches of sequence matching our criteria just by chance. Therefore, we need to apply the filters described earlier in the text to maintain false positives to a manageable level, even though this procedure would miss some true candidates.
New selenoprotein candidates
We ran Seblastian on a number of genomes of non-metazoan eukaryotes, which normally represent the most challenging cases. In addition, we expected that some selenoproteins remain undiscovered in some of these lineages, based on the previous searches with other eukaryotic genomes (27,28,33). Seblastian yielded a ranked set of 186 selenoprotein predictions in 25 species. Although we expect a portion of them to be false positives, we also believe that the set includes bona fide novel selenoproteins. We implemented a procedure to assign a score to the predicted selenoproteins. The score takes into account the SECIS-coding sequence distance, the Covels score and the grade of the SECIS element, the Blastx E-value, the presence of a redox box motif including the candidate Sec, the similarity with other Seblastian candidates, and the matches with EST and protein databases. The new selenoprotein candidates, the species list and a more detailed explanation of the scoring procedure can be found in Supplementary Material S5.
The best scoring candidate was found in the choanoflagellate Monosiga brevicollis and showed homology to AhpC. This is a thioredoxin-like protein family (like many known selenoproteins), and its distant homolog was previously detected as a selenoprotein in Bacteria. Recently, an AhpC-like selenoprotein was also predicted in some sponges (17), but it was thought to be limited to this lineage. Using Selenoprofiles, we built a profile alignment with the AhpC selenoproteins in Bacteria, choanoflagellates and Porifera, including also a number of metazoan cysteine homologues. We used our new profile to scan a collection of eukaryotic and prokaryotic genomes and detected AhpC selenoproteins in a wide range of lineages, including protists and basal metazoans. In Figure 4, we present an alignment of the Sec-containing domain of AhpC selenoproteins, along with some metazoan cysteine homologues. Among the Selenoprofiles AhpC predictions, we also found our second best scoring Seblastian candidate, in the Emiliana huxleyi genome.
Figure 4.

AhpC selenoproteins. Two selenoprotein candidates in our Seblastian predictions were found in M.brevicollis and E.huxleyi, here framed in orange. The figure shows them aligned with other AhpC selenoproteins predicted using Selenoprofiles in eukaryotes (top) and prokaryotes (bottom). Some metazoan cysteine homologues are also shown on the top. The Sec is found in the highlighted redox box UXXC, present also in vertebrates as CXXC. For the full alignment and further details regarding the search for AhpC proteins, see Supplementary Material S6.
Given the conservation of these genes, the thioredoxin fold, the cysteine homology and the presence of SECIS elements in most of eukaryotic candidates, the finding leaves no doubt that this is a true selenoprotein. For details and data on the analysis on AhpC, see Supplementary Material S6. It may seem controversial that our best new selenoprotein candidate was already described in literature as a eukaryotic selenoprotein, in Porifera. However, this eukaryotic selenoprotein family was novel to Seblastian, as no Porifera AhpC selenoprotein was yet annotated in the nr database. Bacterial homologues were annotated, but their phylogenetic distance exceeds the detection power of our method. The example of AhpC supports the quality of Seblastian predictions. Further use of this tool should be instrumental in finding new selenoproteins, both in our current ranked set and in future runs, as more and more species are sequenced.
A webserver for SECISearch3 and Seblastian
We created a web server to allow users world-wide to upload any nucleotide sequence and run SECISearch3 and/or Seblastian (Figure 5). It is hosted both at http://gladyshevlab.org/SelenoproteinPredictionServer and at http://seblastian.crg.es. The user can choose to run Seblastian or just SECISearch3 and can also control the main options of the programs. For example, the SECIS prediction methods can be chosen and their stringency can be set, the SECIS filter can be toggled and so forth. An important option for Seblastian is whether the search is done for known selenoproteins or new ones. In the first case, Blastx is run only against a reduced version of nr containing only selenoproteins, which greatly reduces the computational time. Once ready, results can be inspected directly on a web page or downloaded as fasta or gff files. Until today, selenoprotein prediction was a task typically carried out by only a few experts in the field. This web server allows for the first time any user, even with little expertise in bioinformatics, to perform reliable selenoprotein predictions on any nucleotide sequence of interest.
Figure 5.

Two snapshots of the SECISearch3/Seblastian web server. On the left, the input form. On the right, the output page displayed when submitting the human GPx2 sequence.
CONCLUSION
We describe two new computational methods for selenoprotein prediction and analysis: SECISearch3 and Seblastian. The former is a major improvement of SECISearch and is currently the best method to predict eukaryotic SECIS elements. The latter is a new method to predict selenoproteins in nucleotide sequences, which is built based on SECIS prediction. Seblastian is able to predict known selenoproteins as well as new selenoprotein homologues of known proteins, provided that they have at least one cysteine homologue. We ran Seblastian on the available protist genomes, where we expect a number of selenoproteins to be still undiscovered, and we provided a list of ranked selenoprotein candidates. An analysis of a representative candidate selenoprotein AhpC is used to illustrate the predictions and evolution of new selenoprotein families. Both SECISearch3 and Seblastian are public and can be run on a dedicated web server at http://gladyshevlab.org/SelenoproteinPredictionServer or http://seblastian.crg.es.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Materials 1–6.
FUNDING
Funding for open access charge: NIH [GM061603].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Sean Eddy for his assistance over building an Infernal model for eukaryotic SECIS.
REFERENCES
- 1.Hoffmann PR, Berry MJ. Selenoprotein synthesis: a unique translational mechanism used by a diverse family of proteins. Thyroid. 2005;15:769–775. doi: 10.1089/thy.2005.15.769. [DOI] [PubMed] [Google Scholar]
- 2.Allmang C, Wurth L, Krol A. The selenium to selenoprotein pathway in eukaryotes: more molecular partners than anticipated. Biochim. Biophys. Acta. 2009;1790:1415–1423. doi: 10.1016/j.bbagen.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 3.Hatfield D, Carlson B, Xu X, Mix H, Gladyshev V. Selenocysteine incorporation machinery and the role of selenoproteins in development and health. Prog. Nucleic Acid Res. Mol. Biol. 2006;81:97–142. doi: 10.1016/S0079-6603(06)81003-2. [DOI] [PubMed] [Google Scholar]
- 4.Squires J, Berry M. Eukaryotic selenoprotein synthesis: mechanistic insight incorporating new factors and new functions for old factors. IUBMB Life. 2008;60:232–235. doi: 10.1002/iub.38. [DOI] [PubMed] [Google Scholar]
- 5.Driscoll DM, Chavatte L. Finding needles in a haystack. In silico identification of eukaryotic selenoprotein genes. EMBO Rep. 2004;5:140–141. doi: 10.1038/sj.embor.7400080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kryukov GV, Kryukov VM, Gladyshev VN. New mammalian selenocysteine-containing proteins identified with an algorithm that searches for selenocysteine insertion sequence elements. J. Biol. Chem. 1999;274:33888–33897. doi: 10.1074/jbc.274.48.33888. [DOI] [PubMed] [Google Scholar]
- 7.Lescure A, Gautheret D, Carbon P, Krol A. Novel selenoproteins identified in silico andin Vivo by using a conserved RNA structural motif. J. Biol. Chem. 1999;274:38147. doi: 10.1074/jbc.274.53.38147. [DOI] [PubMed] [Google Scholar]
- 8.Castellano S, Morozova N, Morey M, Berry M, Serras F, Corominas M, Guigó R. In silico identification of novel selenoproteins in the Drosophila melanogaster genome. EMBO Rep. 2001;2:697. doi: 10.1093/embo-reports/kve151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kryukov G, Castellano S, Novoselov S, Lobanov A, Zehtab O, Guigo R, Gladyshev V. Characterization of mammalian selenoproteomes. Science. 2003;300:1439. doi: 10.1126/science.1083516. [DOI] [PubMed] [Google Scholar]
- 10.Kryukov G, Gladyshev V. The prokaryotic selenoproteome. EMBO Rep. 2004;5:538. doi: 10.1038/sj.embor.7400126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taskov K, Chapple C, Kryukov GV, Castellano S, Lobanov AV, Korotkov KV, Guigó R, Gladyshev VN. Nematode selenoproteome: the use of the selenocysteine insertion system to decode one codon in an animal genome? Nucleic Acids Res. 2005;33:2227–2238. doi: 10.1093/nar/gki507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li M, Huang Y, Xiao Y. A method for identification of selenoprotein genes in archaeal genomes. Genomics Proteomics Bioinformatics. 2009;7:62–70. doi: 10.1016/S1672-0229(08)60034-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chapple CE, Guigó R, Krol A. SECISaln, a web-based tool for the creation of structure-based alignments of eukaryotic SECIS elements. Bioinformatics. 2009;25:674–675. doi: 10.1093/bioinformatics/btp020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang L, Liu Q, Ni J. In silico identification of the sea squirt selenoproteome. BMC Genomics. 2010;11:289. doi: 10.1186/1471-2164-11-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mariotti M, Guigó R. Selenoprofiles: profile-based scanning of eukaryotic genome sequences for selenoprotein genes. Bioinformatics. 2010;26:2656–2663. doi: 10.1093/bioinformatics/btq516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gobler CJ, Berry DL, Dyhrman ST, Wilhelm SW, Salamov A, Lobanov AV, Zhang Y, Collier JL, Wurch LL, Kustka AB, et al. Niche of harmful alga Aureococcus anophagefferens revealed through ecogenomics. Proc. Natl Acad. Sci. USA. 2011;108:4352–4357. doi: 10.1073/pnas.1016106108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang L, Ni J, Liu Q. Evolution of selenoproteins in the metazoan. BMC Genomics. 2012;13:446. doi: 10.1186/1471-2164-13-446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krol A. Evolutionarily different RNA motifs and RNA-protein complexes to achieve selenoprotein synthesis. Biochimie. 2002;84:765–774. doi: 10.1016/s0300-9084(02)01405-0. [DOI] [PubMed] [Google Scholar]
- 19.Grundner-Culemann E, Martin GW, Harney JW, Berry MJ. Two distinct SECIS structures capable of directing selenocysteine incorporation in eukaryotes. RNA. 1999;5:625–635. doi: 10.1017/s1355838299981542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martin GW, Harney JW, Berry MJ. Selenocysteine incorporation in eukaryotes: insights into mechanism and efficiency from sequence, structure, and spacing proximity studies of the type 1 deiodinase SECIS element. RNA. 1996;2:171–182. [PMC free article] [PubMed] [Google Scholar]
- 21.Hofacker I, Fontana W, Stadler P, Bonhoeffer S, Tacker M, Schuster P. Fast folding and comparison of RNA secondary structures. Monatshefte für Chemie. 1994;125:167–188. [Google Scholar]
- 22.Hofacker IL. Vienna RNA secondary structure server. Nucleic Acids Res. 2003;31:3429–3431. doi: 10.1093/nar/gkg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nawrocki EP, Kolbe DL, Eddy SR. Infernal 1.0: inference of RNA alignments. Bioinformatics. 2009;25:1335–1337. doi: 10.1093/bioinformatics/btp157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Griffiths-Jones S. RALEE–RNA ALignment editor in Emacs. Bioinformatics. 2005;21:257–259. doi: 10.1093/bioinformatics/bth489. [DOI] [PubMed] [Google Scholar]
- 25.Gautheret D, Lambert A. Direct RNA motif definition and identification from multiple sequence alignments using secondary structure profiles. J. Mol. Biol. 2001;313:1003–1011. doi: 10.1006/jmbi.2001.5102. [DOI] [PubMed] [Google Scholar]
- 26.Novoselov S, Rao M, Onoshko N, Zhi H, Kryukov G, Xiang Y, Weeks D, Hatfield D, Gladyshev V. Selenoproteins and selenocysteine insertion system in the model plant cell system, Chlamydomonas reinhardtii. EMBO J. 2002;21:3681. doi: 10.1093/emboj/cdf372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Novoselov S, Lobanov A, Hua D, Kasaikina M, Hatfield D, Gladyshev V. A highly efficient form of the selenocysteine insertion sequence element in protozoan parasites and its use in mammalian cells. Proc. Natl Acad. Sci. USA. 2007;104:7857–7862. doi: 10.1073/pnas.0610683104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lobanov A, Delgado C, Rahlfs S, Novoselov S, Kryukov G, Gromer S, Hatfield D, Becker K, Gladyshev V. The plasmodium selenoproteome. Nucleic Acids Res. 2006;34:496. doi: 10.1093/nar/gkj450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Altschul S, Madden T, Schaffer A, Zhang J, Zhang Z, Miller W, Lipman D. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fomenko DE, Xing W, Adair BM, Thomas DJ, Gladyshev VN. High-throughput identification of catalytic redox-active cysteine residues. Science. 2007;315:387–389. doi: 10.1126/science.1133114. [DOI] [PubMed] [Google Scholar]
- 31.Fomenko DE, Gladyshev VN. Comparative genomics of thiol oxidoreductases reveals widespread and essential functions of thiol-based redox control of cellular processes. Antioxidants Redox Signal. 2012;16:193–201. doi: 10.1089/ars.2011.3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Slater GS, and Birney E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics. 2005;6:31. doi: 10.1186/1471-2105-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gobler CJ, Lobanov AV, Tang YZ, Turanov AA, Zhang Y, Doblin M, Taylor GT, SañudoWilhelmy SA, Grigoriev IV, Gladyshev VN. The central role of selenium in the biochemistry and ecology of the harmful pelagophyte, Aureococcus anophagefferens. ISME J. 2013;7:1333–1343. doi: 10.1038/ismej.2013.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.