


Abstract
Many important model organisms for biomedical and evolutionary research have sequenced genomes, but occupy a phylogenetically isolated position, evolutionarily distant from other sequenced genomes. This phylogenetic isolation is exemplified for zebrafish, a vertebrate model for cis-regulation, development and human disease, whose evolutionary distance to all other currently sequenced fish exceeds the distance between human and chicken. Such large distances make it difficult to align genomes and use them for comparative analysis beyond gene-focused questions. In particular, detecting conserved non-genic elements (CNEs) as promising cis-regulatory elements with biological importance is challenging. Here, we develop a general comparative genomics framework to align isolated genomes and to comprehensively detect CNEs. Our approach integrates highly sensitive and quality-controlled local alignments and uses alignment transitivity and ancestral reconstruction to bridge large evolutionary distances. We apply our framework to zebrafish and demonstrate substantially improved CNE detection and quality compared with previous sets. Our zebrafish CNE set comprises 54 533 CNEs, of which 11 792 (22%) are conserved to human or mouse. Our zebrafish CNEs (http://zebrafish.stanford.edu) are highly enriched in known enhancers and extend existing experimental (ChIP-Seq) sets. The same framework can now be applied to the isolated genomes of frog, amphioxus, Caenorhabditis elegans and many others.
INTRODUCTION
DNA sequence conservation is often a hallmark of purifying selection, indicating an evolutionarily conserved function of the underlying genomic region. Comparative genomics can accurately locate conserved coding exons and has played an important role in completing gene catalogs by predicting novel genes and exons that had escaped transcriptome profiling and correcting previous gene structures (1,2). Genome comparisons also detected thousands of conserved non-genic elements (CNEs) and revealed that the majority of evolutionarily conserved DNA sequences do not code for proteins (3–6). Functional assays showed that a high fraction of these CNEs have specific cis-regulatory activity and act as enhancers in cell lines or during embryonic development (6–11). Furthermore, these CNEs are often associated with key developmental genes (12–14) and likely control the complex and highly regulated spatiotemporal expression patterns of these genes. In addition to being important in understanding cis-regulation, regulatory CNEs are implicated in human disease (15–17), and disruption of CNE-mediated gene regulation is linked to diseases such as Van Buchem disease, X-linked deafness, aniridia and preaxial polydactyly (18).
Comparative genomics has the most power to detect conservation if the aligned genomes cover a range of evolutionary distances (both closer and more distant species) (19). Furthermore, comparing multiple genomes is more powerful at capturing orthologous sequences than comparing a pair of genomes, especially for species with large evolutionary distances (19,20). Pairwise comparisons are also particularly susceptible to DNA contamination (inclusion of foreign DNA in genome assemblies), which mimics conservation in pairwise (but not multiple) species comparisons. Importantly, detecting CNEs is harder than detecting conserved genes using comparative genomics because coding regions typically have higher sequence conservation over large evolutionary distances (21). Coding genes can also be detected by protein-to-genome alignments.
Although human, mouse, Drosophila and other species are in the desirable situation of being accompanied by genomes of both evolutionarily close and distant species, many important genomes are phylogenetically isolated in that comparative genomics is restricted to using genomes of other species that are evolutionarily distant, operationally defined here as a distance exceeding 1 neutral substitution per site. Examples include zebrafish (see later in the text), frog, lamprey, amphioxus, sea urchin, hydra, sea anemone and sponges, which are all important models for developmental biology, regeneration, stem cell biology or evolutionary biology (22–27) (Figure 1). Even one of the most important model organisms, Caenorhabditis elegans, is separated from other sequenced nematodes by >1 neutral substitution per site (Figure 1), prompting the community to sequence evolutionarily closer species (29). Finally, some species of interest for evolutionary research, such as the coelacanth or the tuatara, have only one known surviving sister species in their order, and will thus remain in phylogenetic isolation indefinitely. This phylogenetic isolation hampers comparative analysis and results in poor genome annotation.
Figure 1.

Zebrafish is currently evolutionarily distant from all other available fish genomes. (A) Phylogeny with branch lengths and clade groupings (solid lines only). The ‘mousefish’, a desirable but currently unavailable teleost genome at human—mouse distance, is discussed in the text. Apart from zebrafish, frog (1.49 subs/site to chicken), lamprey (1.76 subs/site to zebrafish), amphioxus (>2.5 subs/site to lamprey) and C. elegans (1.07 subs/site to Caenorhabditis remanei) are also shown to have phylogenetically isolated genomes. Molecular distances were taken from the UCSC genome browser (28) for the hg18, braFlo1 and ce10 assemblies. (B) Evolutionary distances (neutral substitutions per site) between zebrafish (left) and human (right) to other sequenced species. In contrast to human, the zebrafish genome occupies a phylogenetic outgroup position with the closest sequenced teleosts at a distance of 1.25–1.41 subs/site, which exceeds the distance between the human and chicken genome (1.08 subs/site). (C) The portion of CNEs conserved to mouse that can be discovered in comparisons between human and evolutionarily more distant species can be used to estimate the fraction of zebrafish CNEs visible using the current availability of genomes.
Here, we present a general comparative genomics framework designed to improve alignments of genomes that are evolutionarily distant. We focus on comprehensively detecting CNEs in such genome alignments. Although the framework is general and can be applied to any eukaryotic genome, we use the zebrafish genome as a test case for the following three reasons. First, zebrafish (belonging to the order cypriniformes) is a phylogenetically isolated genome. Zebrafish is separated from other currently sequenced fish (belonging to the order percomorphs) by 1.25–1.41 neutral substitutions per site, exceeding the molecular distance between human and chicken (Figure 1). Second, zebrafish is one of the most important model organisms with a strong arsenal of experimental techniques for manipulating genes and genomic regions, and assaying expression patterns and cis-regulatory activity. A comprehensive and high-quality CNE resource is, therefore, of great value to the large zebrafish community that studies vertebrate development and models human disease (30,31). Third, existing resources that annotate CNEs in zebrafish based on pairwise genome comparisons (32–35) allow us to evaluate our approach.
Our approach uses highly sensitive local alignments to detect remote homologies, and it strictly controls noise and random alignments by quality filtering based on false discovery rates (FDRs).
We use the resulting multiple-genome alignment to comprehensively detect zebrafish CNEs (called zCNEs), requiring conservation between zebrafish and at least two additional species to avoid DNA contamination. We augment our set with non-assembled sequence reads from many additional species, while applying stringent filters to detect zCNEs embedded in regions with conserved synteny in at least one assembled species, which is a fundamental property of conserved cis-regulatory elements. Finally, we use novel methods (alignment transitivity and ancestral reconstruction) to annotate orthology between zebrafish and mammalian species. We show that zCNEs are highly enriched in validated enhancers and that they extend any existing computational or experimental (ChIP-Seq) set, while adding quality. To make this zCNE set available to the zebrafish community, we have created a resource with rich annotation at http://zebrafish.stanford.edu.
MATERIALS AND METHODS
Comparative genomics approach to obtain zCNEs
Our computational framework for aligning distant genomes comprises a number of steps that are shown in Figure 2 and detailed later in the text. The methodology we describe is general and readily applicable to any isolated genome with proper parameter tuning (see ‘Discussion’ section).
Figure 2.

Comparative genomics approach to detect CNEs in isolated genomes. (A) Several steps in this pipeline aim at detecting remote homologies. Still, we use strict filtering for alignment quality, synteny and conservative masking of potential genic sequences to achieve a high-quality CNE set. Total coverage in the zebrafish genome for each step is given as both fraction of the zebrafish genome and megabases (Mb). (B) The panels illustrate patching (highly sensitive alignment for a region bounded by up- and downstream aligning anchors in blue) and synteny filtering for chains and nets. Colored boxes are alignments, horizontal lines connect co-linear alignment blocks and different colors represent different chromosomes in the aligning species.
Pairwise whole-genome alignments
Our pipeline starts with pairwise alignments to the Zv9/danRer7 zebrafish genome assembly using the following genomes that broadly sample ∼450 My of vertebrate evolution: medaka (oryLat2), tetraodon (tetNig2), fugu (v5) (36), (fr3, http://www.fugu-sg.org/), stickleback (gasAcu1), lamprey (petMar1), cow (bosTau4), dog (canFam2), horse (equCab2), chicken (galGal3), human (hg19), elephant (loxAfr3), mouse (mm9), opossum (monDom5), platypus (ornAna1) and frog (xenTro2) (Figure 2, Steps 1 and 2). We built pairwise alignments to zebrafish using lastz (37) with the HoxD55 scoring matrix and sensitive parameter settings: H = 2000, Y = 3000, L = 3000 and K = 2000. The pairwise alignments were built using genomes that were hard-masked for repeats using UCSC Table Browser’s RepMask and Simple Repeats tracks (28).
Although sensitive alignment parameters are necessary to detect homologies between diverged sequences, they also produce dubious alignments. Therefore, we subsequently quality filtered the lastz alignments using threshold values determined by FDR calculations. Specifically, we created two randomized genomes obtained by di-nucleotide shuffling every entire zebrafish chromosome (global shuffling) as well as di-nucleotide shuffling 100-bp sliding windows in every zebrafish chromosome (local shuffling). The position and size of all assembly gaps was preserved in both cases. Then, we aligned both shuffled genomes against the real genome of stickleback, fugu, tetraodon, medaka and lamprey and estimated the FDR as the number of bases in the shuffled genome that align (false positives) divided by the number of bases in the real genome that align (true positives and false positives). We kept a local alignment if a sliding window of size ≥30 bp has ≥60% sequence identity and ≥1.8 bits entropy. These parameters give an FDR between 0.13 and 0.3 for global shuffling and an FDR between 0.15 and 0.34 for local shuffling (Supplementary Table S1) and were subsequently used for all real pairwise alignments. Here, sequence identity is calculated as the number of matches over the alignment length. Entropy is calculated as
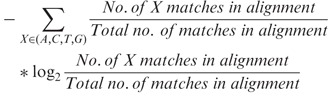 |
This entropy filter excludes unmasked tandem repeats or regions of low-sequence complexity that typically yield non-orthologous alignments with high-sequence identity but where only a subset of the four DNA bases predominately aligns.
Highly sensitive local alignments (patching)
To detect remote homologies between evolutionarily distant genomes, it would be desirable to run genome-wide alignments with highly sensitive parameters controlling the heuristics (such as seeding and seed-extension) of BLAST-based algorithms (37,38). However, the drastic increase in run-time prevents running whole-genome alignments with such parameters. Regulatory CNEs are expected to be in larger synteny blocks that have several clearly aligning regions. We exploited this property in a step termed ‘patching’ that uses highly sensitive seeding and seed-extension parameters to find new alignments within those unaligning genomic regions that are bounded by clearly aligning anchors upstream and downstream (Figure 2, Steps 3 and 4). To this end, we ‘chained’ filtered pairwise aligning blocks to get long co-linear alignments separated by unaligning sequence regions (39). For all pairs of alignment blocks, we tried to align the sequences between these aligning blocks using lastz (37) with the HoxD55 scoring matrix and highly sensitive parameters for seeding and seed-extension: K = 1500, L = 2300, M = 0 and W = 5. Any novel alignments were filtered as before requiring ≥60% sequence identity and ≥1.8 bits entropy in a window ≥30 bp. These parameters result in an FDR of 0.099 for medaka and 0.26 for fugu when we di-nucleotide shuffle the zebrafish sequence for all patched regions in the real chains (note that this is a stricter test than patching chains built for a shuffled zebrafish genome). Supplementary Table S2 lists how many bases were added to the chains. Patching ultimately allowed us to detect 1758 zCNEs covering 126 kb that we would otherwise have missed. Supplementary Figure S1 shows an example of a CNE only detected by patching.
Syntenic pairwise alignments
The additional filtered alignments found by patching, together with the filtered alignments from the genome-wide lastz step were chained (39) (Figure 2, Steps 5–7). As cis-regulatory CNEs typically maintain synteny with the nearby genes they regulate through evolution (40), we extracted syntenic pairwise alignments as long regions of co-linearity between zebrafish and another species. We kept only chains with either a score of ≥10 000 that spans ≥10 000 bp in both genomes or chains with a score of ≥50 000 that span ≥5000 bp in both genomes to also keep chains with strong alignments spanning only a shorter region. These parameters were empirically determined by inspecting the chains in the UCSC genome browser and looking for any genes in chains that were filtered out. Synteny filtering excluded many chains because the majority of unfiltered chains are short and low scoring (Supplementary Figure S2). As syntenic chains include both orthologous and paralogous alignments, we subjected these chains to ‘netting’ (39) to keep only the most likely orthologous alignments. The filtered nets were filtered again for the same score and span criteria, as nets might keep smaller regions from longer chains that represent paralogous alignments.
Di-nucleotide–shuffled genome
We used the globally and locally di-nucleotide–shuffled zebrafish genomes to test whether our alignment and filtering parameters would allow syntenic alignments between random sequences. This is necessary, as the genome-wide pairwise lastz alignments yielded FDRs of >0.25 for some species. We applied the entire pipeline (genome-wide lastz, alignment filtering, patching, chaining and filtering for syntenic chains) to the di-nucleotide–shuffled zebrafish genomes (proxy for a random sequence) against the real genomes of stickleback, fugu, tetraodon, medaka and lamprey. Despite our sensitive alignment parameters, we did not obtain a single chain that passed our synteny filters for any of these species for either the globally or locally shuffled genome, suggesting that our quality and synteny filters are appropriate.
Multiple alignment
The pairwise syntenic alignment nets are the input to multiz (41) to build a multiple alignment (Figure 2, Step 8). As multiple alignments become more fragmented, the more species are aligned (42), we build two multiple alignments: one that includes teleost species and lamprey but excludes all tetrapod species and one that includes only zebrafish and tetrapods. The phylogenetic tree for the teleosts and lamprey is ((danRer7:0.580406, (oryLat2:0.364485, (gasAcu1:0.30842, (tetNig2:0.201742, fr3:0.182553):0.217051):0.106429):0.308334):0.663203, petMar1:0.511293). The tree for the zebrafish and the tetrapods is ((((((((mm9:0.352605, hg19:0.142680):0.020666, (bosTau4:0.186713, (equCab2:0.107726, canFam2:0.150374):0.010431):0.032764):0.023276, loxAfr3:0.166583):0.232748, monDom5: 0.325899):0.072430, ornAna1:0.453916):0.109903, galGal3:0.474279):0.166150, xenTro2: 0.852482):0.300396, danRer7:0.886380). These distances were determined by the UCSC genome browser team using phyloFit (5) and 4-fold degenerate sites.
Using non-assembled DNA sequences
Although multiple species with high-quality genomes that cover the major tetrapod clades have been produced, the teleost clade is more sparsely sampled with whole-genome assemblies but holds a wealth of unassembled genomic DNA sequences (Figure 2, Step 9b). To use this unassembled genomic DNA, we downloaded all genomic non-mitochondrial DNA belonging to a non-tetrapod vertebrate species from both the NCBI trace archive and GenBank, excluding refseq_rna, nt and env_nt, as well as medaka, fugu, tetraodon, stickleback and lamprey, which are present in our alignment. This sequence data comprised 18.2 Gb in 21 117 826 sequences, with an average (median) length of 862 (908) bp. We extracted all regions ≥50 bp from the zebrafish genome that cover well-aligning windows from ≥1 species (Step 9a later in the text) and aligned those regions against the non-tetrapod vertebrate DNA sequences using lastz (HoxD55 matrix, K = 2000, L = 3000, M = 0 and W = 6). These new alignments include contributions from salmon, tilapia, seabass, the ghost shark (43) and many other species.
As these traces and GenBank sequences are too short to assess synteny, we required more stringent filtering parameters by keeping only those alignments that have a region that aligns with ≥75% identity and ≥1.8 bits entropy over ≥50 bp. These parameters were optimized by computing FDRs by di-nucleotide shuffling the zebrafish regions as done earlier in the text. Because of the long computation time, we restricted the FDR estimation to randomly selected 10% of the zebrafish regions (2444 CPU hours). These parameters give an overall FDR of 0.065 and per species FDRs ≤0.114 (Supplementary Table S3).
Conserved elements set
We defined conserved zebrafish elements as regions of ≥50 bp supported by good alignments to at least two other species (Figure 2, Step 9a and 10). To this end, we first obtained all pairwise-alignment regions that align with ≥65% identity and ≥1.8 entropy (as defined earlier in the text) over ≥30 alignment columns by extracting all regions from the multiple alignment for all pairs (zebrafish—another species) (Supplementary Table S4). Then, we obtained those zebrafish regions where the well-aligning windows from ≥2 species from the multiple alignments overlap basewise. Similarly, we obtained the regions where the well-aligning windows from only one species from the multiple alignments overlap basewise with ≥1 other species from non-assembled sequences (GenBank and traces). We merged adjacent regions if they were at most 15 bp apart in danRer7. For the final set of conserved regions, we required that the un-merged regions cumulatively cover ≥50 bp in danRer7. Thus, each zebrafish-conserved region is at least 50-bp long and is supported by at least 50 bp in well-aligning windows from at least two other species. We processed the two multiple alignments separately and merged the two sets of conserved elements.
Excluding genic conserved regions
To obtain a set of bona fide CNEs, we applied stringent filtering to exclude regions that are (potentially) protein coding or belong to gene UTRs or non-coding RNAs (Figure 2, Step 11). We built a set of ‘exclude regions’ by first combining exons from RefSeq transcripts, Ensembl transcripts, Uniprot protein mappings, zebrafish mRNAs and spliced ESTs downloaded from the UCSC genome browser (29). In addition, we added miRNAs from mirbase and Vega pseudogene regions (liftOver from danRer5) to the exclude regions. Second, as current gene annotations are likely incomplete for zebrafish, we used BlastX to find additional regions with protein homology. To this end, we added 50-bp flanks to all conserved elements, ran BlastX against the NCBI nr database (excluding predicted proteins, XP_) and added all regions detected with an E-value <0.01 to the exclude regions. Third, we used RNAcode with default parameters (44) and added all alignment regions that likely evolve under protein-coding constraints (P-value < 1E-5). We merged the set of exclude regions from the gene/ncRNA annotation tracks, BlastX and RNAcode and added 50-bp flanks to exclude splice site proximal regions that might harbor conserved splicing regulatory elements. We then excluded all these bases from the conserved elements and required the remaining conserved regions to be ≥50-bp long.
These stringent filters leave 16.9% (6.6 of 39.3 Mb in well-aligning windows) of all conserved elements as non-genic. A similar fraction of non-genic sequence is found in human–chicken (17.8%) or human–frog (15.7%) conserved sequences using the same filtering criteria.
Our final zCNE set comprises 54 533 elements covering 6 643 241 bp (0.47%) of the zebrafish genome.
Aligning zCNEs to their human and mouse orthologs
Non-genic regions conserved between related species are promising candidates for transcriptional enhancers with conserved expression patterns (7,9,11). For biomedical, evolutionary and developmental studies, it is desirable to comprehensively detect zCNE conservation between zebrafish and human/mouse. Using our syntenic multiple alignment (Figure 2, Step 8), we initially found 9373 CNEs that are conserved in human, 8757 zCNEs conserved in mouse and 11 516 zCNEs that are conserved in any tetrapod. Cognizant of the limitations of existing alignment tools, we set out to detect human/mouse conservation for additional zCNEs using two novel methods: alignment transitivity and ancestral reconstruction.
Annotating human and mouse ancestry using alignment transitivity
Transitivity infers orthology between zebrafish and human/mouse sequences that do not align directly but do share orthology to a common sequence in a third related species (Figure 3) (21,45). For each CNE, we obtained the genomic coordinates for any intermediate species from the multiple alignments (first transitive step). Then, we used the UCSC syntenic (orthology) liftOver chains in search of a syntenic alignment between the intermediate species and human/mouse, using liftOver with –minMatch = 0.7 (second transitive step). This procedure was repeated for all intermediate species (all tetrapods that align to zebrafish in our multiple alignments). Finally, as a quality-control measure, we only inferred zebrafish—human/mouse homology if the CNE mapped to the same location in the human/mouse genome for all available intermediate species.
Figure 3.

Transitivity can reveal orthology between distant genomes that is not directly visible. (A) Illustration of the transitivity principle. A zebrafish locus aligns to chicken but not directly to human. However, the chicken locus does align to human, allowing us to infer orthology and anchor an alignment between zebrafish and human. Conceptually, transitivity mimics a multiple alignment using the intermediate species as the reference species. (B) Sequence identity of zebrafish—human/mouse alignments, separating those alignments found only using transitivity (blue) and those directly aligning in the syntenic multiple alignments (gray), suggests that transitivity-inferred alignments also evolve under clear purifying selection. (C) An example where zebrafish has a weaker alignment to human that is not detected in the genome-wide pipeline. However, an anchored alignment using chicken as an intermediate species shows clear orthology between the diverged zebrafish and human sequence. (D) The CNE shown in (C) is in synteny with the PTPRE gene in all three species.
To assess the power of transitivity to detect the orthologous locus in human/mouse, we first used the 9373 (8757) zCNEs that directly align to human (mouse) as a test set, ignored these direct alignments and searched for transitivity-inferred alignments using the nine other tetrapod species. For 99.7% (99.6%) of these zCNEs, the coordinates found by transitivity overlap the human (mouse) coordinates from the syntenic multiple alignments, indicating very high power in uncovering orthology.
Applied to each of the 11 516 − 9373 (8757) = 2143 (2759) zCNEs that align to other tetrapods but not directly to human (mouse), transitivity identified an additional 1115 zCNEs conserved to human (52% of searched sequences) and an additional 1055 zCNEs (38%) conserved to mouse (Table 1). These zCNEs have a median pairwise sequence identity between zebrafish and human (mouse) of 68.7% (67.9%) (Figure 3B), a clear sign of previously undetected sequence homology between species separated by ≥1.8 subs. per site (Figure 1). Support for a transitive alignment came from two or more independent species for 96.6% of human elements and 97.3% of the mouse ones (Supplementary Figure S3). Chicken was the most useful intermediate species (Supplementary Figure S4), consistent with the observation that the bird lineage is more slowly evolving and has a low rate of genomic rearrangements (46).
Table 1.
The number and base pair (bp) coverage of zCNEs conserved between zebrafish and human/mouse obtained through our different processing steps
| Human |
Mouse |
|||
|---|---|---|---|---|
| Number | Genome coverage (bp) | Number | Genome coverage (bp) | |
| From multiple alignment | 9373 | 1 453 794 | 8757 | 1 386 647 |
| Detected by transitivity | 1115 | 143 380 | 1055 | 141 953 |
| Detected by ancestral reconstruction | 1262 | 146 303 | 1349 | 156 972 |
| Total | 11 573 | 1 769 804 | 10 989 | 1 707 381 |
Finding tetrapod orthologs using ancestral reconstruction
We also used ancestral sequence reconstruction (47,48) as an additional approach to reduce large evolutionary distances by aligning reconstructed percomorph ancestral zCNE sequences to reconstructed mammalian ancestral CNE sequences (Figure 4). As the evolutionary distance between the percomorph and mammalian ancestors is only 1.04 neutral subs. per site, much shorter than the distance of 1.8 (2.01) subs. per site between zebrafish and human (mouse) (Figure 4), we were hoping to detect additional alignments.
Figure 4.

Ancestral reconstruction reveals additional CNE alignments between distant species. (A) Large evolutionary distances between zebrafish and human/mouse can be substantially reduced if (B) reconstructed ancestral sequences are aligned. The phylogenetic tree contains the species used to reconstruct the percomorph and mammalian ancestor. Species used as outgroups are in blue in (B). (C) Sequence identity of zebrafish–human alignments is shown for CNEs that align to human in our multiple alignment and for 1262 CNEs where ancestral reconstruction but not direct alignment detects conservation to human (630 align to a tetrapod but not human in our multiple alignment; 632 have no alignment to any vertebrate). Although alignments detected only using reconstruction have lower sequence identities, even values ∼50% indicate clear conservation between species separated by ≥1.8 neutral substitutions per site. (D) An example where conservation within teleosts and within tetrapods can be used to reconstruct the percomorph and mammalian ancestor of the CNE (1 and 2). The reconstructed ancestral sequences align with high enough sequence identity to detect orthology and anchor an alignment between the human and zebrafish CNEs not visible otherwise (3). The CNE shares conserved synteny with the same putative target gene (4). Blue background is identity to the ancestor in (1 and 2) and sequence identity in (3).
To this end, we first obtained a human-referenced tetrapod CNE set to reconstruct the mammalian ancestor of those CNEs. We downloaded vertebrate PhastCons (5) most-conserved elements for the human hg19 assembly. To get CNEs, we excluded all bases (padded 50 bp to their flanks to exclude conserved splicing signals) in coding gene tracks (knownGene, mgcGenes, ccdsGenes, ensGene, exoniphy and xenoRefGene), non-coding RNAs (evofold, wgRna, vegaPseudoGene and tRNAs), pseudogenes (pseudoYale and ucscRetroAli) and all repeats. The remaining conserved elements were merged if they were at most 15-bp apart, requiring that each element consists of ≥50 conserved bases after merging. We then filtered this set for CNEs, retaining only those that overlap with ≥80% at least one of frog, chicken, lizard or zebra finch (the species that we use as outgroups for reconstructing the mammalian ancestral sequence). We also required that each CNE is conserved in at least three of the following mammals: mouse, rat, guinea pig, cow, dog, horse and elephant. This results in 18.4 Mb (0.59% of the human genome) in 105 145 CNEs.
Next, we used prequel (48) with frog, chicken, lizard and zebra finch as outgroup species to reconstruct the most likely sequence of the mammalian ancestor for these 105 145 CNEs. Likewise, we used zebrafish as outgroup to reconstruct the percomorph ancestor for all zCNEs that have at least two percomorph species in the multiple alignments. Then, we used lastz (K = 1500, L = 3000, T = 0, M = 0 and W = 5) to align all mammalian ancestral sequences against all percomorph ancestral sequences. We kept those hits that have at least one window of ≥30 bp with sequence identity ≥65% and entropy ≥1.8 bits. These alignment and filtering parameters give an FDR of 0.04 when we di-nucleotide shuffle the reconstructed mammal ancestors.
To assess how often alignments between reconstructed ancestors detect the correct syntenic region in the human genome, we used the 2188 zCNEs that both align to human in our multiple alignment and have an alignment hit between the reconstructed ancestors. We found that the human coordinates obtained by reconstruction overlap the human coordinates from the syntenic multiple alignments for 1944 (88.8% of 2188) zCNEs, indicating correct and syntenic hits.
Applied to all zCNEs, we found tetrapod orthologs for 1262 (1349) zCNEs (Table 1) that did not align to human (mouse) in our multiple alignment. The median pairwise sequence identity between zebrafish and human of 63.5% (Figure 4C) is again indicative of homologous sequence between these diverged species. In all, 632 (50%) of these 1262 zCNEs had no previous alignment to human or to any other tetrapod.
Transitivity and ancestral reconstruction added 2200 (2232) CNEs covering 316 kb (321 kb) to the syntenic multiple alignment’s 9373 (8757) CNEs conserved between zebrafish and human (mouse) (Table 1). In total, 11 792 zCNEs (22% of 54 533) are conserved to human or mouse.
RESULTS
The comparative genomics approach detailed in the ‘Materials and Methods’ section (earlier in the text) results in a set of 54 533 zebrafish CNEs, of which 11 792 (22%) are conserved to human or mouse. Next, we turn to analyze our zCNE set and compare it with previous CNE annotation efforts, epigenetic enhancer marks and experimentally tested enhancers.
Comparison with previous zebrafish CNE sets
We first evaluated the comprehensiveness of our zCNE set by comparison with three previous sets obtained by pairwise genome comparisons: Ancora (34), CNEViewer (35) and ECR Browser (32). Briefly, Ancora and CNEViewer scan standard alignment nets provided by the UCSC genome browser (28) for conserved non-coding regions. CNEViewer uses a gene-centric screen that scans 500-kb regions around orthologous genes, whereas Ancora scans genome-wide. ECR Browser uses pairwise BLAST alignments between syntenic blocks and includes both conserved coding and non-coding elements. We did not compare with CONDOR (33), which contains CNEs aligning between fugu and human. As previous CNE sets contain some regions that are potentially coding (partially because of more recent gene annotations) or that arise from non-orthologous repeat alignments (Supplementary Table S5), we subjected these three CNE sets to exactly the same filters that we applied to remove repeats and genic regions from our own set to assure direct comparability (Supplementary Methods). Pairwise comparisons of these sets to our zCNEs show that 44–89% of our zCNE set is novel compared with these previous sets (Table 2a), despite applying our stringent synteny filter and requiring conservation between at least three species. Nearly 22% of zCNEs are not found in the union of all these previous resources (Table 2b).
Table 2.
Comparison of our zCNE set to previous zebrafish CNE sets
| Pairwise comparisona | |||||||
|---|---|---|---|---|---|---|---|
| Total (bp) | In both CNE sets | Unique in our zCNE set | Unique in other CNE set | ||||
| zCNEs all | 6 643 241 | ||||||
| zCNEs teleost + lamprey | 6 072 642 | ||||||
| zCNEs human | 1 769 804 | ||||||
| ECRBrowser | bp | % of other set | bp | % of our set | bp | % of other set | |
| Fugu | 4 014 503 | 2 509 862 | 62.52% | 3 562 780 | 58.67% | 1 504 641 | 37.48% |
| Human | 1 262 653 | 874 451 | 69.26% | 895 353 | 50.59% | 388 202 | 30.74% |
| ANCORA | |||||||
| Fugu | 4 085 372 | 2 870 306 | 70.26% | 3 202 336 | 52.73% | 1 215 066 | 29.74% |
| Tetraodon | 3 582 692 | 2 634 273 | 73.53% | 3 438 369 | 56.62% | 948 419 | 26.47% |
| Stickleback | 5 020 698 | 3 220 581 | 64.15% | 2 852 061 | 46.97% | 1 800 117 | 35.85% |
| Medaka | 4 745 417 | 2 945 151 | 62.06% | 3 127 491 | 51.50% | 1 800 266 | 37.94% |
| Human | 1 403 083 | 996 067 | 70.99% | 773 737 | 43.72% | 407 016 | 29.01% |
| CNEViewer | |||||||
| All CNEs | 563 113 | 416 675 | 73.99% | 1 353 129 | 76.46% | 146 438 | 26.01% |
| Syntenic CNEs | 248 105 | 196 911 | 79.37% | 1 572 893 | 88.87% | 51 194 | 20.63% |
| Union of all previous setsb | |||||||
| In zCNEs and other sets | Unique in our zCNE set | Unique in other sets | |||||
| Union of all previous resources | 10 765 678 | 5 204 466 | 48.34% | 1 438 775 | 21.66% | 5 561 212 | 51.66% |
| Breakdown of 5 561 212 bp unique to other sets | bp | % | |||||
| Do not align to zebrafish by our approach | 191 704 | 3.4% | |||||
| Are not syntenic according to our criteria | 3 714 572 | 66.8% | |||||
| Do not overlap well-aligning windows | 981 312 | 17.6% | |||||
| Overlap well-aligning windows but the region is <50 bp | 496 580 | 8.3% | |||||
| Overlap well-aligning window ≥50 bp but not supported by ≥2 species | 155 813 | 2.8% | |||||
| GREAT enrichment (top annotation term)c | |||||||
| # GREAT version 2.0.1 | In zCNEs and other sets | Unique in our zCNE set | Unique in other sets | ||||
| Ontology and term name | Binom FDR Q-Val | Binom fold enrichment | Binom FDR Q-Val | Binom fold enrichment | Binom FDR Q-Val | Binom fold enrichment | |
| GO molecular function | |||||||
| Sequence-specific DNA binding | 0 | 3.1 | 7 E-279 | 2.2 | n.d. | n.d. | |
| GO biological process | |||||||
| Regulation of transcription, DNA-dependent | 0 | 2.7 | 3 E-314 | 2.0 | n.d. | n.d. | |
| Wiki pathways | |||||||
| Nuclear receptors | 3 E-128 | 5.0 | 5 E-41 | 3.0 | n.d. | n.d. | |
| InterPro | |||||||
| Homeodomain-like | 0 | 3.7 | 5 E-199 | 2.5 | n.d. | n.d. | |
aTo exclude any differences because of our stringent filtering procedure, we applied the same filters for repeats and genic regions to the ECR Browser (pairwise zebrafish–fugu/human), Ancora (pairwise zebrafish–fugu/tetraodon/stickleback/medaka/human) and CNEViewer (pairwise zebrafish–human) sets (Supplementary Table S5). We used our set of CNEs built from the teleosts and lamprey multiple alignment (6 072 642 bp in 51 997 CNEs) to compare with pairwise zebrafish–teleost sets. We used our set of CNEs that are conserved to human (1 769 804 bp in 11 573 CNEs) to compare with pairwise zebrafish–human sets. We found that, despite our stringent synteny filter and requiring at least two other aligning species, our CNE set is substantially larger than any of these pairwise sets, as 44–89% of the bases in zCNEs are not contained in the other sets.
bCompared with the union of all previous sets, zCNEs still add 1.4 Mb (22% of our set). Other sets contain 5.6 Mb that are not in our zCNE set for reasons listed in the table.
cCNEs that are in both our zCNE and other sets as well as CNEs that are unique to our set show the expected enrichments for transcription factors using the zebrafish GREAT webserver http://great.stanford.edu. In contrast, these enrichments were not detected (n.d.) for CNEs unique to other sets. Top enrichment is shown. Size-matched sets were compared to assure equal statistical power of GREAT.
The union of all three previous sets contained 5.6 Mb (52%) that is not in our zCNE set, mostly because of lack of synteny and lack of good alignments (Table 2b gives a breakdown of these 5.6 Mb). Furthermore, the regions unique to other sets lack the functional enrichments for transcription factors that are typical for CNEs (49), in contrast to CNEs detected in our and other sets or elements unique in our zCNE set (Table 2c). This shows that the combination of doing sensitive pairwise alignments, requiring multiple species rather than single species support and using non-assembled DNA sequence reads substantially improved the quality and comprehensiveness of the zebrafish CNE set.
zCNEs are highly enriched in overlapping epigenetic enhancer marks and active enhancers
To further assess the quality of our approach, we intersected our zCNEs with three sets of functional gene regulation data, in search of enriched overlaps. We computed fold enrichment and an empirical P-value by randomly placing the same number of zCNEs in the sequenced portion of the zebrafish genome 105 times and comparing how often randomly placed elements overlap these three sets. First, we used monomethylation of histone 3 at lysine 4 (H3K4me1), which marks active and poised enhancers (50). A recent study identified 65 585 H3K4me1-marked genomic regions in whole zebrafish at 24 h post-fertilization (24 hpf) (51). We found that 14 008 of 54 533 (25.7%) zCNEs overlap these H3K4me1 hotspots (6.9-fold enriched, empirical P < 10−5, observed standard deviations above mean or ‘Z-score’: 272). Second, the combination of H3K4me1/me3 and H3K27ac (histone H3 lysine 27 acetylation) marks from four different zebrafish developmental stages (dome, epiboly, 24 hpf and 48 hpf) were used in (52) to define putative distal regulatory elements. In all, 12 791 of 54 533 (23.5%) zCNEs overlap this set (5.8-fold enriched, empirical P < 10−5, Z-score: 230). Third, we mapped 331 human developmental enhancers tested using mouse transgenics in the VISTA database (53) to their orthologous zebrafish coordinates (using UCSC’s liftOver with –minMatch 0.1). In all, 510 (4.4%) of the 11 573 human-conserved zCNEs overlap these 331 VISTA enhancers (169-fold enriched, empirical P < 10−5, Z-score: 292). These enrichments suggest that zCNEs offer promising cis-regulatory candidate regions identified by conservation.
To compare the potential of novel and previously detected zCNEs to act as developmental enhancers, we again used the large number of human regions tested for enhancer activity using mouse transgenics in the VISTA database (53). To this end, we divided our human-conserved zCNEs into (i) those zCNE regions where human-conservation was only detected by us and (ii) those zCNE regions where human-conservation was also detected by at least one of Ancora, CNEViewer or ECR Browser, and restricted the analysis to regions ≥50 bp. Of the zCNE regions where human-conservation was only detected by our approach, 170 overlapped regions tested in VISTA, and 105 of these (62%, Z-score: 84) were positive for enhancer function. For comparison of the zCNE regions where human-conservation was detected by others and us, 988 were tested and 572 were positive (58%, Z-score: 370). This suggests that novel human-conserved zCNE regions are as likely to function as developmental enhancers.
zCNEs are not subsumed by available epigenetic enhancer marks
The union of all H3K4me1, H3K4me3 or H3K27ac marks obtained by two studies for four different developmental stages (51,52) together covers 227 Mb (16%) of the zebrafish genome. Despite covering a large portion of the genome, we found that 45% (24 520 of 54 533) zCNEs do not overlap these epigenetic enhancer marks. Thus, these zCNEs likely highlight novel cis-regulatory elements with evolutionary importance.
CNE-dense genomic regions highlight key developmental genes
It is well known that CNE clusters are often associated with transcription factors and developmental genes having complex expression patterns (12–14,49,54). CNE-dense genomic regions can highlight such important genes, including some that are less characterized. We computed the percentage of bases in zCNEs that overlap sliding windows of size 100 kb with a 5 kb offset across the entire zebrafish genome, after excluding the number of bases in assembly gaps from each window. Using the regulatory domain concept of GREAT (55), we determined the gene(s) whose regulatory domain overlaps the midpoint of each window (often the gene/s with the closest transcription start site 5′ and 3′).
All top 10 windows overlap transcription factors (Table 3), with the densest window (18.8% of bases in zCNEs) found next to the neuronal differentiation gene foxp2 (56). Interestingly, two top non-overlapping windows are associated with znf536, a poorly studied zinc-finger transcription factor that is also involved in neuronal differentiation (57) and may be linked to a case of ataxia telangiectasia (58). The full Supplementary Table S7 likely highlights additional less characterized genes with complex regulation and key developmental roles.
Table 3.
The 10 genomic regions with the highest zCNE density are all associated with transcription factors
| Locus danRer7 |
% Bases in zCNEs | Putative target gene(s) | ||
|---|---|---|---|---|
| chr | Window start | Window end | ||
| chr4 | 5805000 | 5905000 | 18.83% | foxp2 |
| chr24 | 23860000 | 23960000 | 17.92% | zfhx4 |
| chr23 | 29215000 | 29315000 | 16.89% | casz1 |
| chr7 | 28480000 | 28580000 | 16.74% | sox6 |
| chr7 | 47940000 | 48040000 | 15.50% | znf536 |
| chr7 | 47810000 | 47910000 | 15.40% | ccne & znf536 |
| chr9 | 31905000 | 32005000 | 15.12% | dachd |
| chr7 | 37245000 | 37345000 | 14.33% | irx5a |
| chr7 | 70460000 | 70560000 | 14.21% | zfhx3 |
| chr12 | 44145000 | 44245000 | 13.99% | ebf3 |
Each window is 100 kb. The nearest gene(s) is listed. See Supplementary Table S7 for a longer table.
Properties of human-conserved zCNEs
To specifically explore the properties of the human-conserved zCNEs, we first compared the distribution of expression patterns of VISTA enhancers (53). Compared with all positively tested VISTA enhancers, the enhancers that overlap human-conserved zCNEs drive expression more frequently in the central nervous system and less frequently in the limb (Supplementary Figure S5). This is consistent with previous observations that genomic regions bound by the enhancer-associated protein p300 in forebrain and midbrain are more conserved than similar regions bound in limb (59). Second, we used GREAT (55) to identify functional roles enriched among the human-conserved zCNEs. GREAT detected a strong enrichment of such zCNEs next to transcription factors (data not shown), validating a known association of deeply conserved CNEs (12,49). Interestingly, GREAT also detected enrichment of several signaling pathways such as Id, Notch and TGF-β (Supplementary Table S6), highlighting specific functions of the human–zebrafish conserved CNEs.
DISCUSSION
CNEs are prime candidates for harboring biologically important cis-regulatory function, but such CNEs are hard to detect for phylogenetically isolated genomes. Here, we developed a general framework to align isolated genomes and to comprehensively detect CNEs in them. We applied our framework to zebrafish, an important model organism with an isolated genome, and showed that, despite the rigor of our methods, our zCNE set improved on both the quality and the comprehensiveness of previous sets. Part of our improvement stems from using additional sequence data, such as unassembled DNA. Improved methodology, in particular refraining from relying on pairwise alignments alone, however, makes an important contribution. Patching allowed us to detect 1758 zCNEs that we would miss otherwise, and alignment transitivity and ancestral reconstruction used to annotate orthology between zebrafish and mammalian species revealed such deep conservation for an additional 2200 zCNEs (Table 1). The high-sequence identity between these alignments, once we are able to obtain them, suggests that we have uncovered orthologous sequence that has been missed by the heuristics of the BLAST-derived search tool.
Our framework will be highly useful to align genomes of multiple other species that are important for biomedical or evolutionary research or where an attractive genetic toolkit is complemented only by genomes of distantly related species. This is currently the case for frog, amphioxus, sea urchin, hydra, C. elegans and other species, which have already been sequenced. Although the methodology described in Figure 2 is general and readily applicable to these other isolated genomes, some parameter settings may need to be changed according to the general principles we describe here. The parameters for quality-filtering sensitive pairwise alignments can be adjusted by computing FDRs to a new randomized reference genome. The parameters to extract syntenic chains and nets likely depend on the genome size (and gene size), as well as the molecular distance between the species. These parameters can be determined empirically by (i) inspecting alignments between orthologous genes that are filtered out and importantly (ii) assuring that no chain between a randomized genome passed these filters. Finally, the minimal length of the CNEs depends on the molecular distance between the species as often only a small core of enhancers is conserved between distant species (60,61).
Zebrafish, with its high-throughput in vivo enhancer assays, is a great system to experimentally explore the expression patterns driven by CNEs. To allow easy access to our zCNE set, we have created a web portal at http://zebrafish.stanford.edu (Figure 5) to display our CNEs together with human and mouse coordinates (where orthology exists), conservation in other species, neighboring genes with contextual links to ZFIN (62), and the UCSC (28) and Ensembl (63) genome browsers. The portal also allows searching and downloading zCNEs near specific genes or genomic regions. The zCNE set was also added to ZFIN’s GBrowse (62). Because zebrafish is distant to the other species in our study, we recommend that zCNEs are padded up to 200 bp on either side before testing for cis-regulatory function, as human comparison with similarly distant chicken or frog often highlights only the core of human–mouse conserved elements (61).
Figure 5.

Top: UCSC genome browser-like representation of the Sox5 locus shows clusters of zCNEs (blue), many of which align to human or mouse (red). Bottom: a screenshot of the details page that is available for each zCNE.
Despite our improved framework to detect CNEs between distantly related species, CNEs that came under selection during more recent evolution (after the zebrafish–percomorph split) can only be detected when evolutionarily closer species are sequenced. We used comparisons between human, mouse, chicken and frog to determine which fraction of human CNEs would be missed if only distant species are available and used this to estimate how large the set of evolutionarily younger zebrafish CNEs is. Our baseline is the 73.9 Mb in human CNEs [PhastCons elements (5) from the hg19 genome assembly filtered for CNEs as aforementioned] that align between human and mouse (Figure 1C). If only species at the human–chicken (human–frog) molecular distance were available to detect conservation in the human genome, only 27.2% (12.1%) of these 73.9 Mb would have been found (Figure 1C). A linear regression of the human–chicken and human–frog distance (1.076 and 1.62 neutral subs. per site, respectively) indicates that at the current zebrafish–medaka distance (1.25 subs. per site), only 22.3% of zebrafish CNEs have been found. By selecting and sequencing a fish at a comparable molecular distance with that between human and mouse (which we dub as ‘mousefish’ in Figure 1), we would expect to reveal, in addition to the 6.6 Mb or 22.3% in our zCNE set, 23 Mb of additional conserved non-genic sequence. Molecular phylogenies indicate that species belonging to the family Cyprinidae may lie at the desired ‘mousefish’ distance, although molecular divergence estimates vary (64,65). Comparison with evolutionary closer genomes, available in future (66), will likely reveal the conservation of many additional functional zebrafish elements that are currently not annotated as such (67).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–7, Supplementary Figures 1–5 and Supplementary Methods.
FUNDING
German Research Foundation [Hi 1423/2-1] and Human Frontier Science Program [LT000896/2009-L to M.H.]; NSF [DGE-1147470 to J.H.N. and H.G.]; Stanford Graduate Fellowship and Bio-X Stanford Interdisciplinary Graduate Fellowship (to A.M.W.); NIH [R01HG005058 and R01HD059862] and NSF Center for Science of Information (CSoI) [CCF-0939370 to G.B.]. GB is a Packard Fellow and a Microsoft Faculty Fellow. Funding for open access charge: NIH [R01HG005058 to G.B.] (Stanford University).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors are grateful to the UC Santa Cruz genome browser team for software and genome annotations, Doug Howe and Monte Westerfield for adding the zCNE set to ZFIN’s GBrowse, Deborah Ritter from Jeffrey Chuang’s Laboratory for kindly providing the CNEViewer data, Shoa Clarke for early zebrafish analysis work and William Talbot, Romain Madelaine, Nadav Ahituv and members of the Bejerano laboratory for helpful discussions.
REFERENCES
- 1.Stark A, Lin MF, Kheradpour P, Pedersen JS, Parts L, Carlson JW, Crosby MA, Rasmussen MD, Roy S, Deoras AN, et al. Discovery of functional elements in 12 Drosophila genomes using evolutionary signatures. Nature. 2007;450:219–232. doi: 10.1038/nature06340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lindblad-Toh K, Garber M, Zuk O, Lin MF, Parker BJ, Washietl S, Kheradpour P, Ernst J, Jordan G, Mauceli E, et al. A high-resolution map of human evolutionary constraint using 29 mammals. Nature. 2011;478:476–482. doi: 10.1038/nature10530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- 4.Hillier LW, Miller W, Birney E, Warren W, Hardison RC, Ponting CP, Bork P, Burt DW, Groenen MA, Delany ME, et al. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature. 2004;432:695–716. doi: 10.1038/nature03154. [DOI] [PubMed] [Google Scholar]
- 5.Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, Thurman RE, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Woolfe A, Goodson M, Goode DK, Snell P, McEwen GK, Vavouri T, Smith SF, North P, Callaway H, Kelly K, et al. Highly conserved non-coding sequences are associated with vertebrate development. PLoS Biol. 2005;3:e7. doi: 10.1371/journal.pbio.0030007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 9.Visel A, Prabhakar S, Akiyama JA, Shoukry M, Lewis KD, Holt A, Plajzer-Frick I, Afzal V, Rubin EM, Pennacchio LA. Ultraconservation identifies a small subset of extremely constrained developmental enhancers. Nat. Genet. 2008;40:158–160. doi: 10.1038/ng.2007.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shin JT, Priest JR, Ovcharenko I, Ronco A, Moore RK, Burns CG, MacRae CA. Human-zebrafish non-coding conserved elements act in vivo to regulate transcription. Nucleic Acids Res. 2005;33:5437–5445. doi: 10.1093/nar/gki853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Navratilova P, Fredman D, Hawkins TA, Turner K, Lenhard B, Becker TS. Systematic human/zebrafish comparative identification of cis-regulatory activity around vertebrate developmental transcription factor genes. Dev. Biol. 2009;327:526–540. doi: 10.1016/j.ydbio.2008.10.044. [DOI] [PubMed] [Google Scholar]
- 12.Bejerano G, Pheasant M, Makunin I, Stephen S, Kent WJ, Mattick JS, Haussler D. Ultraconserved elements in the human genome. Science. 2004;304:1321–1325. doi: 10.1126/science.1098119. [DOI] [PubMed] [Google Scholar]
- 13.Sandelin A, Bailey P, Bruce S, Engstrom PG, Klos JM, Wasserman WW, Ericson J, Lenhard B. Arrays of ultraconserved non-coding regions span the loci of key developmental genes in vertebrate genomes. BMC Genomics. 2004;5:99. doi: 10.1186/1471-2164-5-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vavouri T, Walter K, Gilks WR, Lehner B, Elgar G. Parallel evolution of conserved non-coding elements that target a common set of developmental regulatory genes from worms to humans. Genome Biol. 2007;8:R15. doi: 10.1186/gb-2007-8-2-r15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ragvin A, Moro E, Fredman D, Navratilova P, Drivenes O, Engstrom PG, Alonso ME, de la Calle Mustienes E, Gomez Skarmeta JL, Tavares MJ, et al. Long-range gene regulation links genomic type 2 diabetes and obesity risk regions to HHEX, SOX4, and IRX3. Proc. Natl Acad. Sci. USA. 2010;107:775–780. doi: 10.1073/pnas.0911591107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wasserman NF, Aneas I, Nobrega MA. An 8q24 gene desert variant associated with prostate cancer risk confers differential in vivo activity to a MYC enhancer. Genome Res. 2010;20:1191–1197. doi: 10.1101/gr.105361.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Visel A, Rubin EM, Pennacchio LA. Genomic views of distant-acting enhancers. Nature. 2009;461:199–205. doi: 10.1038/nature08451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kleinjan DA, Lettice LA. Long-range gene control and genetic disease. Adv. Genet. 2008;61:339–388. doi: 10.1016/S0065-2660(07)00013-2. [DOI] [PubMed] [Google Scholar]
- 19.Miller W, Makova KD, Nekrutenko A, Hardison RC. Comparative genomics. Annu. Rev. Genomics Hum. Genet. 2004;5:15–56. doi: 10.1146/annurev.genom.5.061903.180057. [DOI] [PubMed] [Google Scholar]
- 20.Margulies EH, Chen CW, Green ED. Differences between pair-wise and multi-sequence alignment methods affect vertebrate genome comparisons. Trends Genet. 2006;22:187–193. doi: 10.1016/j.tig.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 21.Clarke SL, VanderMeer JE, Wenger AM, Schaar BT, Ahituv N, Bejerano G. Human developmental enhancers conserved between deuterostomes and protostomes. PLoS Genet. 2012;8:e1002852. doi: 10.1371/journal.pgen.1002852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sodergren E, Weinstock GM, Davidson EH, Cameron RA, Gibbs RA, Angerer RC, Angerer LM, Arnone MI, Burgess DR, Burke RD, et al. The genome of the sea urchin Strongylocentrotus purpuratus. Science. 2006;314:941–952. doi: 10.1126/science.1133609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Putnam NH, Butts T, Ferrier DE, Furlong RF, Hellsten U, Kawashima T, Robinson-Rechavi M, Shoguchi E, Terry A, Yu JK, et al. The amphioxus genome and the evolution of the chordate karyotype. Nature. 2008;453:1064–1071. doi: 10.1038/nature06967. [DOI] [PubMed] [Google Scholar]
- 24.Hellsten U, Harland RM, Gilchrist MJ, Hendrix D, Jurka J, Kapitonov V, Ovcharenko I, Putnam NH, Shu S, Taher L, et al. The genome of the Western clawed frog Xenopus tropicalis. Science. 2010;328:633–636. doi: 10.1126/science.1183670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Putnam NH, Srivastava M, Hellsten U, Dirks B, Chapman J, Salamov A, Terry A, Shapiro H, Lindquist E, Kapitonov VV, et al. Sea anemone genome reveals ancestral eumetazoan gene repertoire and genomic organization. Science. 2007;317:86–94. doi: 10.1126/science.1139158. [DOI] [PubMed] [Google Scholar]
- 26.Chapman JA, Kirkness EF, Simakov O, Hampson SE, Mitros T, Weinmaier T, Rattei T, Balasubramanian PG, Borman J, Busam D, et al. The dynamic genome of Hydra. Nature. 2010;464:592–596. doi: 10.1038/nature08830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Srivastava M, Simakov O, Chapman J, Fahey B, Gauthier ME, Mitros T, Richards GS, Conaco C, Dacre M, Hellsten U, et al. The Amphimedon queenslandica genome and the evolution of animal complexity. Nature. 2010;466:720–726. doi: 10.1038/nature09201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dreszer TR, Karolchik D, Zweig AS, Hinrichs AS, Raney BJ, Kuhn RM, Meyer LR, Wong M, Sloan CA, Rosenbloom KR, et al. The UCSC Genome Browser database: extensions and updates 2011. Nucleic Acids Res. 2012;40:D918–D923. doi: 10.1093/nar/gkr1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Piano F, Cherbas P. A Proposal for Comparative Genomics in Support the modENCODE Project. 2008. http://www.genome.gov/Pages/Research/Sequencing/SeqProposals/ modENCODE_ComparativeGenomics_WhitePaper.pdf. [Google Scholar]
- 30.Lieschke GJ, Currie PD. Animal models of human disease: zebrafish swim into view. Nat. Rev. Genet. 2007;8:353–367. doi: 10.1038/nrg2091. [DOI] [PubMed] [Google Scholar]
- 31.Becker TS, Rinkwitz S. Zebrafish as a genomics model for human neurological and polygenic disorders. Dev. Neurobiol. 2012;72:415–428. doi: 10.1002/dneu.20888. [DOI] [PubMed] [Google Scholar]
- 32.Ovcharenko I, Nobrega MA, Loots GG, Stubbs L. ECR Browser: a tool for visualizing and accessing data from comparisons of multiple vertebrate genomes. Nucleic Acids Res. 2004;32:W280–W286. doi: 10.1093/nar/gkh355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Woolfe A, Goode DK, Cooke J, Callaway H, Smith S, Snell P, McEwen GK, Elgar G. CONDOR: a database resource of developmentally associated conserved non-coding elements. BMC Dev. Biol. 2007;7:100. doi: 10.1186/1471-213X-7-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Engstrom PG, Fredman D, Lenhard B. Ancora: a web resource for exploring highly conserved noncoding elements and their association with developmental regulatory genes. Genome Biol. 2008;9:R34. doi: 10.1186/gb-2008-9-2-r34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Persampieri J, Ritter DI, Lees D, Lehoczky J, Li Q, Guo S, Chuang JH. cneViewer: a database of conserved non-coding elements for studies of tissue-specific gene regulation. Bioinformatics. 2008;24:2418–2419. doi: 10.1093/bioinformatics/btn443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kai W, Kikuchi K, Tohari S, Chew AK, Tay A, Fujiwara A, Hosoya S, Suetake H, Naruse K, Brenner S, et al. Integration of the genetic map and genome assembly of fugu facilitates insights into distinct features of genome evolution in teleosts and mammals. Genome Biol. Evol. 2011;3:424–442. doi: 10.1093/gbe/evr041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schwartz S, Kent WJ, Smit A, Zhang Z, Baertsch R, Hardison RC, Haussler D, Miller W. Human-mouse alignments with BLASTZ. Genome Res. 2003;13:103–107. doi: 10.1101/gr.809403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 39.Kent WJ, Baertsch R, Hinrichs A, Miller W, Haussler D. Evolution's cauldron: duplication, deletion, and rearrangement in the mouse and human genomes. Proc. Natl Acad. Sci. USA. 2003;100:11484–11489. doi: 10.1073/pnas.1932072100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Akalin A, Fredman D, Arner E, Dong X, Bryne JC, Suzuki H, Daub CO, Hayashizaki Y, Lenhard B. Transcriptional features of genomic regulatory blocks. Genome Biol. 2009;10:R38. doi: 10.1186/gb-2009-10-4-r38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blanchette M, Kent WJ, Riemer C, Elnitski L, Smit AF, Roskin KM, Baertsch R, Rosenbloom K, Clawson H, Green ED, et al. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 2004;14:708–715. doi: 10.1101/gr.1933104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wenger AM, Clarke SL, Guturu H, Chen J, Schaar BT, McLean CY, Bejerano G. PRISM offers a comprehensive genomic approach to transcription factor function prediction. Genome Res. 2013;23:889–904. doi: 10.1101/gr.139071.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Venkatesh B, Kirkness EF, Loh YH, Halpern AL, Lee AP, Johnson J, Dandona N, Viswanathan LD, Tay A, Venter JC, et al. Survey sequencing and comparative analysis of the elephant shark (Callorhinchus milii) genome. PLoS Biol. 2007;5:e101. doi: 10.1371/journal.pbio.0050101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Washietl S, Findeiss S, Muller SA, Kalkhof S, von Bergen M, Hofacker IL, Stadler PF, Goldman N. RNAcode: robust discrimination of coding and noncoding regions in comparative sequence data. RNA. 2011;17:578–594. doi: 10.1261/rna.2536111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Taher L, McGaughey DM, Maragh S, Aneas I, Bessling SL, Miller W, Nobrega MA, McCallion AS, Ovcharenko I. Genome-wide identification of conserved regulatory function in diverged sequences. Genome Res. 2011;21:1139–1149. doi: 10.1101/gr.119016.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dalloul RA, Long JA, Zimin AV, Aslam L, Beal K, Blomberg Le A, Bouffard P, Burt DW, Crasta O, Crooijmans RP, et al. Multi-platform next-generation sequencing of the domestic turkey (Meleagris gallopavo): genome assembly and analysis. PLoS Biol. 2010;8:9. doi: 10.1371/journal.pbio.1000475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blanchette M, Green ED, Miller W, Haussler D. Reconstructing large regions of an ancestral mammalian genome in silico. Genome Res. 2004;14:2412–2423. doi: 10.1101/gr.2800104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hubisz MJ, Pollard KS, Siepel A. PHAST and RPHAST: phylogenetic analysis with space/time models. Brief Bioinform. 2011;12:41–51. doi: 10.1093/bib/bbq072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lowe CB, Kellis M, Siepel A, Raney BJ, Clamp M, Salama SR, Kingsley DM, Lindblad-Toh K, Haussler D. Three periods of regulatory innovation during vertebrate evolution. Science. 2011;333:1019–1024. doi: 10.1126/science.1202702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl Acad. Sci. USA. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aday AW, Zhu LJ, Lakshmanan A, Wang J, Lawson ND. Identification of cis regulatory features in the embryonic zebrafish genome through large-scale profiling of H3K4me1 and H3K4me3 binding sites. Dev. Biol. 2011;357:450–462. doi: 10.1016/j.ydbio.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bogdanovic O, Fernandez-Minan A, Tena JJ, de la Calle-Mustienes E, Hidalgo C, van Kruysbergen I, van Heeringen SJ, Veenstra GJ, Gomez-Skarmeta JL. Dynamics of enhancer chromatin signatures mark the transition from pluripotency to cell specification during embryogenesis. Genome Res. 2012;22:2043–2053. doi: 10.1101/gr.134833.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Visel A, Minovitsky S, Dubchak I, Pennacchio LA. VISTA Enhancer Browser—a database of tissue-specific human enhancers. Nucleic Acids Res. 2007;35:D88–D92. doi: 10.1093/nar/gkl822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lowe CB, Bejerano G, Haussler D. Thousands of human mobile element fragments undergo strong purifying selection near developmental genes. Proc. Natl Acad. Sci. USA. 2007;104:8005–8010. doi: 10.1073/pnas.0611223104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Scharff C, Petri J. Evo-devo, deep homology and FoxP2: implications for the evolution of speech and language. Philos. Trans. R Soc. Lond. B Biol. Sci. 2011;366:2124–2140. doi: 10.1098/rstb.2011.0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Qin Z, Ren F, Xu X, Ren Y, Li H, Wang Y, Zhai Y, Chang Z. ZNF536, a novel zinc finger protein specifically expressed in the brain, negatively regulates neuron differentiation by repressing retinoic acid-induced gene transcription. Mol. Cell. Biol. 2009;29:3633–3643. doi: 10.1128/MCB.00362-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bartsch O, Schindler D, Beyer V, Gesk S, van't Slot R, Feddersen I, Buijs A, Jaspers NG, Siebert R, Haaf T, et al. A girl with an atypical form of ataxia telangiectasia and an additional de novo 3.14 Mb microduplication in region 19q12. Eur. J. Med. Genet. 2012;55:49–55. doi: 10.1016/j.ejmg.2011.08.001. [DOI] [PubMed] [Google Scholar]
- 59.Blow MJ, McCulley DJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, et al. ChIP-Seq identification of weakly conserved heart enhancers. Nat. Genet. 2010;42:806–810. doi: 10.1038/ng.650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McEwen GK, Goode DK, Parker HJ, Woolfe A, Callaway H, Elgar G. Early evolution of conserved regulatory sequences associated with development in vertebrates. PLoS Genet. 2009;5:e1000762. doi: 10.1371/journal.pgen.1000762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Prabhakar S, Poulin F, Shoukry M, Afzal V, Rubin EM, Couronne O, Pennacchio LA. Close sequence comparisons are sufficient to identify human cis-regulatory elements. Genome Res. 2006;16:855–863. doi: 10.1101/gr.4717506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bradford Y, Conlin T, Dunn N, Fashena D, Frazer K, Howe DG, Knight J, Mani P, Martin R, Moxon SA, et al. ZFIN: enhancements and updates to the Zebrafish Model Organism Database. Nucleic Acids Res. 2011;39:D822–D829. doi: 10.1093/nar/gkq1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Flicek P, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fairley S, Fitzgerald S, et al. Ensembl 2012. Nucleic Acids Res. 2012;40:D84–D90. doi: 10.1093/nar/gkr991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mayden RL, Tang KL, Conway KW, Freyhof J, Chamberlain S, Haskins M, Schneider L, Sudkamp M, Wood RM, Agnew M, et al. Phylogenetic relationships of Danio within the order Cypriniformes: a framework for comparative and evolutionary studies of a model species. J. Exp. Zool. B Mol. Dev. Evol. 2007;308:642–654. doi: 10.1002/jez.b.21175. [DOI] [PubMed] [Google Scholar]
- 65.Kong X, Wang X, Gan X, Li J, He S. Phylogenetic relationships of Cyprinidae (Teleostei: Cypriniformes) inferred from the partial S6K1 gene sequences and implication of indel sites in intron 1. Sci. China C Life Sci. 2007;50:780–788. doi: 10.1007/s11427-007-0076-3. [DOI] [PubMed] [Google Scholar]
- 66.Bernardi G, Wiley EO, Mansour H, Miller MR, Orti G, Haussler D, O'Brien SJ, Ryder OA, Venkatesh B. The fishes of Genome 10K. Mar. Genomics. 2012;7:3–6. doi: 10.1016/j.margen.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 67.McGaughey DM, Vinton RM, Huynh J, Al-Saif A, Beer MA, McCallion AS. Metrics of sequence constraint overlook regulatory sequences in an exhaustive analysis at phox2b. Genome Res. 2008;18:252–260. doi: 10.1101/gr.6929408. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.