

Abstract
The multiple sclerosis (MS) patient population is highly heterogeneous in terms of disease course and treatment response. We used a transcriptional profile generated from peripheral blood mononuclear cells to define the structure of an MS patient population. Two subsets of MS subjects (MSA and MSB) were found among 141 untreated subjects. We replicated this structure in two additional groups of MS subjects treated with one of the two first-line disease-modifying treatments in MS: glatiramer acetate (GA) (n = 94) and interferon-β (IFN-β) (n = 128). One of the two subsets of subjects (MSA) was distinguished by higher expression of molecules involved in lymphocyte signaling pathways. Further, subjects in this MSA subset were more likely to have a new inflammatory event while on treatment with either GA or IFN-β (P = 0.0077). We thus report a transcriptional signature that differentiates subjects with MS into two classes with different levels of disease activity.
INTRODUCTION
Multiple sclerosis (MS) is thought to emerge when genetically susceptible individuals encounter environmental triggers that initiate an inflammatory reaction against self-antigens in the central nervous system. These events result in recurring episodes of inflammatory demyelination and, in many cases, a progressive neurodegenerative process (1). However, the prevalent syndromic definition of MS obscures extensive interindividual variation in terms of disease course, response to a given treatment, and distribution of deficits. Currently, although imaging may help to stratify risk of a second demyelinating event in the context of a clinically isolated demyelinating syndrome (CIS) (2), there are no validated molecular biomarkers that offer meaningful predictions for MS. As a result, in a clinical setting, one can only categorize subjects after prolonged observation of their disease course. Further, treatment decisions are based on study results from the highly selected subject populations used in clinical trials.
As illustrated by the success of natalizumab (an anti–VLA-4 antibody) and fingolimod (a sphingosine 1-phosphate receptor modulator), immune cells in the peripheral circulation play an important role in MS (3, 4). Sequestering these cells in the peripheral compartment reduces relapse rates and evidence of inflammatory demyelination on magnetic resonance imaging (MRI) in the brain. These and other studies suggest that sampling peripheral blood is likely to be informative in exploring the structure of the MS patient population. Here, we assess whether we can use such information to identify a priori subsets of subjects whose clinical course will differ over time.
Studies of the peripheral blood transcriptome in MS have been performed in many different ways; however, they have focused, for the most part, on identifying transcriptomic signatures of clinically defined classes of subjects, such as progressive versus relapsing subjects or subjects that respond to a given treatment. Here, similar to the work of Corvol and colleagues, which examined a population of subjects with CIS (5), we use an unsupervised clustering approach to explore whether there is more than one class of MS subjects that can be distinguished using transcriptomic data extracted from peripheral blood mononuclear cells (PBMCs). We define two subsets of MS that are present not only in our large collection of untreated subjects but also in two additional cohorts, one treated with glatiramer acetate (GA) and the other treated with interferon-β (IFN-β). Further, we report that the subset of subjects with higher expression of lymphocyte activation pathway genes is more likely to have another inflammatory event over the course of their disease.
RESULTS
Characteristics of the transcriptome in our three subject collections
In Table 1, we summarize the demographic and clinical characteristics of the subjects with demyelinating disease [relapsing-remitting (RR) MS or CIS] that were considered for this study. All subjects are participants in the Comprehensive Longitudinal Investigation of MS at the Brigham and Women’s Hospital (CLIMB), a prospective study of the early phase of demyelinating disease. Transcriptional profiles were generated from frozen PBMCs collected prospectively as part of CLIMB. There were some differences in the clinical characteristics among the three groups of subjects: (i) a slightly increased frequency of women in the untreated group; (ii) a higher number of CIS subjects in the untreated group; and (iii) a lower level of brain parenchymal fraction (BPF), a normalized measure of whole-brain volume, in the IFN-β–treated subjects, after correction for age at first symptom, sex, and disease duration (Table 1).
Table 1.
Demographic characteristics of subjects used in the study. Values are reported as means (SD). T2LV: T2 hyperintense lesion volume.
| GA (n = 94) | IFN-β (n = 128) | Untreated (n = 141) | Adjusted P value* | |
|---|---|---|---|---|
| Female, n (%) | 68 (73) | 95 (74) | 117 (85) | 0.0344 |
| Disease subtype (CIS/RR) | 7/87 | 9/119 | 32/109 | 0.0001 |
| Symptom duration at sampling | 6.12 (6.36) | 6.65 (6.36) | 7.48 (8.06) | 0.5644 |
| Age at first symptom | 34.23 (9.19) | 34.77 (9.30) | 33.35 (9.07) | 0.4841 |
| BPF | 0.885 (0.034) | 0.871 (0.036) | 0.880 (0.040) | 0.0178 |
| EDSS | 1.04 (1.01) | 1.19 (1.04) | 1.07 (1.09) | 0.5539 |
| T2LV** | 3.19 (2.58) | 3.65 (2.64) | 3.93 (4.75) | 0.1720 |
Adjusted for symptom duration, age at first symptom, and gender (unless outcome of interest). MRI measurements were captured in a time window of 6 months before or after sampling.
P value from model with natural log transformation of T2 lesion volume value.
At the end of our quality control pipeline, 20,527 probe sets from 363 subjects passed our rigorous quality control measures. The first three principal components derived from these standardized data capture 41.4% of the variance in the data, and these three principal components are not associated with treatment category (PC1, P = 0.49; PC2, P = 0.16; PC3, P = 0.20). This is appreciated in Fig. 1A in which the distribution of PBMC RNA profiles from subjects of the three treatment categories (GA, IFN-β, and untreated) overlap. This figure shows that these major axes of transcriptome-wide variation are not significantly different among the three subject categories. When assessing individual probe sets for evidence of pairwise differential expression in the treatment groups, we find that there are no significant differences [false discovery rate (FDR) < 0.05] between the 94 GA-treated and 141 untreated subjects (Fig. 1B) (table S1). On the other hand, both of these groups reveal the expected altered expression of type 1 interferon response genes when compared to the 128 IFN-β–treated subjects: (i) for untreated versus IFN-β, 1649 probe sets are up-regulated and 1453 probe sets are down-regulated at an FDR < 0.05 in IFN-β subjects (table S2); (ii) for GA versus IFN-β, 568 probe sets are up-regulated and 840 probe sets are down-regulated at FDR < 0.05 in IFN-β–treated subjects (table S3). As illustrated in Fig. 1B, the differential expression patterns of the two comparisons (untreated and GA) with IFN-β overlap, and the direction of effect (up- or down-regulation) is the same in the two comparisons. The most up-regulated probe sets (targeting the IFI44L, IFIT1, IFI44, IFIT3, IFI6, OASL, MX1, IFIT2, OAS1, and OAS2 genes) quantify the RNA expression level of well-described interferon response genes (6–8) (table S4). Thus, whereas we find the expected transcriptional changes in PBMCs after treatment with IFN-β (9, 10), we see a lack of transcriptional changes in PBMCs after treatment with GA even though our large sample size gives us >90% power to identify an RNA expression difference between GA-treated and untreated subjects that is of the same magnitude as that seen in IFN-β–treated subjects relative to untreated subjects. This suggests that, at the patient population level, the effects of chronic GA treatment on transcription, which have not been explored deeply so far, (i) may be modest for any one gene, (ii) may be limited to a small subset of circulating cells, or (iii) may be limited to specific immunological compartments and are not reflected in peripheral blood. With this baseline assessment of the transcriptomic profiles of the three subject groups completed, we turned to analyses exploring the heterogeneity of the subject population.
Fig. 1.
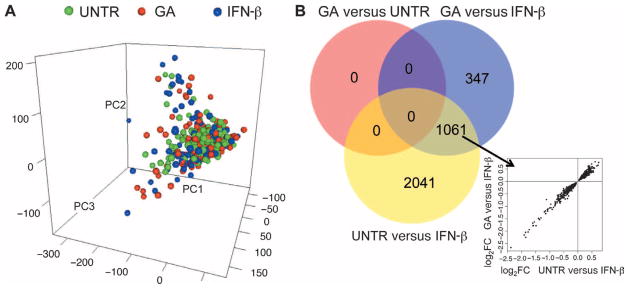
Transcriptomic data from MS subjects. (A) Distribution of the study subjects using the first three principal components calculated from the transcriptomic data. Each dot represents one individual, color-coded according to the treatment group to which they belong [green, untreated (UNTR); red, GA-treated; blue, IFN-β–treated]. (B) Venn diagram of the number of the differentially expressed probe sets in each pairwise comparison among the three treatment groups. The number of probe sets meeting a Benjamini-Hochberg threshold of FDR < 0.05 was reported. Between GA and untreated subsets, there were no probe sets meeting that threshold. The inset graph plots each of the 1061 probe sets shared by the two comparisons to IFN-β using the magnitude and direction of the differential expression in each comparison (x axis, untreated and IFN-β; y axis, GA and IFN-β) to illustrate the consistency of gene expression changes in PBMCs relative to the IFN-β subject group.
Unsupervised clustering identifies two untreated MS subgroups
To empirically determine the number of subsets of subjects present within our untreated group, we used an unsupervised clustering method, non-negative matrix factorization (NMF) (11). NMF allowed exploration of the structure of our subject population without any assumptions as to which genes may be important. It identified metagenes, or groups of coregulated genes, which were used to cluster subjects into a range of subsets. We tested a range of models (from two to five clusters of subjects) and used the cophenetic coefficient, a measure of how well a model fits the data, to identify the optimal k (k is the number of clusters). As illustrated in Fig. 2, the solution of k = 2 subsets is the one that returns the highest cophenetic coefficient for the untreated subjects (Fig. 2, A and B, and table S5A). We called the smaller subset (n = 58) MSA and the larger subset (n = 83) MSB. This structure in our 141 untreated subject population was not driven by the subset of 33 CIS subjects: There was no significant difference in the proportion of CIS subjects in MSA and MSB (table S6). Moreover, when the unsupervised clustering approach was repeated after excluding the CIS subjects, the same structure was identified, and there was a strong correlation between subject class assignments in the two independent classifications (with and without CIS subjects; P < 0.0001, Fisher’s exact test) (fig. S1).
Fig. 2.
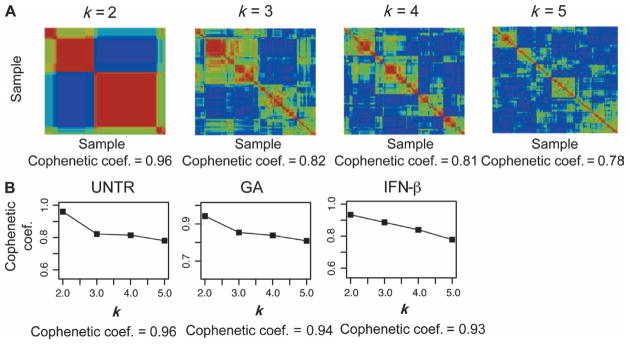
Two subsets of MS subjects in the untreated, GA, and IFN-β groups. (A) NMF consensus clustering results of mRNA expression array data from 141 untreated MS cases. Four models with two to five clusters of subjects (k = 2 to 5) were tested, and the results were summarized in the four consensus clustering plots that capture all pairwise comparisons for the 141 untreated subjects (one for each model). Each square is colored by the extent of correlation within the transcriptome of that subject pair: red, high correlation; yellow/green, moderate to low correlation; blue, no correlation. The cophenetic coefficient, a measure of how well a model fits the data, is reported below each plot, with 1.0 being maximal fit. (B) Plots summarizing the cophenetic coefficient for each of the four tested models (k = 2 to 5) in each of the three groups of subjects (untreated, GA, and IFN-β). In each case, the solution that maximizes the cophenetic coefficient is k = 2, meaning that the transcriptomic data are best explained by a model in which two subsets of subjects are present. The maximal cophenetic coefficient is reported below each plot.
Given that our three subject groups (GA, IFN-β, untreated) were not significantly different in the major axes of variation in the data, we attempted to replicate the observation of a two-subset population structure in the untreated subjects by deploying the NMF method in GA-treated and IFN-β–treated subjects. In both of the latter groups, the k = 2 subset solution is also the one that maximized the cophenetic coefficient (Fig. 2B) when testing models from k = 2 to k = 5. In untreated subjects, 205 probe sets were differentially expressed (FDR < 0.05) between the MSA and MSB subsets (fig. S2); thus, these 205 probe sets offer a transcriptional signature that can differentiate the two subsets of untreated subjects.
To further validate this signature and our hypothesis that the same two subsets of subjects with MS exist in the different treatment groups, we used a support vector machine (SVM) method (a form of supervised clustering) in which a predefined list of probe sets is used to separate subjects into two classes (12–14). With the SVM method and the 205 probe set list discriminating the untreated MSA and MSB subject subsets, we partitioned GA-treated subjects into two groups. As seen in Fig. 3A, we then compared the SVM-determined group assignments to the assignments determined by the unsupervised NMF method that uses the entire RNA data set. We observed that the structure of the population determined independently by each method was very similar (class assignment overlaps in 92.6% of subjects). Figure 3A more precisely displays the consistency of the two clustering strategies: Subjects that were classified differently in the two analyses were those subjects where the SVM method made a less confident assignment. Thus, taking the NMF-defined subject class (MSA versus MSB) in GA-treated subjects as our best estimate of the “correct” subject classification, we observed that the top differentially expressed genes between the untreated subgroups captured the structure of the GA-treated subject population. Also, the direction of the differential expression was the same: The same genes were over-expressed when comparing MSA versus MSB in either untreated or GA-treated subjects. Similarly, when we repeated this process with IFN-β–treated subjects, we were able to correctly predict the unsupervised, NMF-determined class of 90.6% of IFN-β–treated subjects with the SVM strategy and the expression signature determined in untreated subjects (Fig. 3B). As noted above, misclassified individuals were found among individuals for whom the SVM method returned a classification with lower confidence.
Fig. 3.

Top probe sets differentially expressed between MSA and MSB subsets in untreated subjects predict subject class in the GA- and IFN-β–treated subject groups. (A and B) The results of supervised SVM classification for GA (A) and IFN-β (B) using the untreated signature are shown. The confidence score for each SVM classification is plotted on the y axis. On the x axis, each subject is arrayed on the basis of the rank order of the likelihood of belonging to MSA after SVM classification. Each point represents one subject. The upper portion of each graph contains those classified as MSA by the SVM classifier; the lower portion contains MSB subjects. The unsupervised NMF-derived classification, which is used as the reference classification, is reported in color: blue, MSA; green, MSB. Thus, the misclassified subjects are readily apparent. The confusion matrix that compares the NMF and SVM classifications is reported in each graph; the number and percentage (in parenthesis) of subjects found in each cell of the confusion matrix is shown. A total of 94 GA-treated subjects and 128 IFN-β–treated subjects were classified. (C) The table reports the success of the classifier at different confidence thresholds for the SVM classification and the proportion of subjects (in parenthesis) that meet each threshold. Most of the misclassified individuals were the result of low confidence classifications.
If we impose a confidence threshold >80% before we accept an SVM-determined classification, the accuracy becomes 98.8% for the 81 of 94 GA-treated subjects that have an SVM classification meeting this confidence threshold. Similarly, classification accuracy increases to 96.5% for the 114 of 128 IFN-β–treated subjects meeting the 80% confidence threshold (Fig. 3C). Thus, with a reasonable confidence threshold (>80% confidence), more than 86% of subjects treated with first-line disease-modifying agents (GA or IFN-β) could be classified to >96% accuracy with a signature derived from untreated subjects (Fig. 3C). These results validate the existence of the same two subsets of subjects (MSA and MSB) that can be differentiated by a single transcriptional signature in the three subject groups (GA, IFN-β, and untreated). In this signature, 98 probe sets were differentially expressed in the same direction in all three MSA versus MSB comparisons (table S7). Table S8 reports, by subject group, the probe sets differentially expressed for each MSA versus MSB comparison.
Functional annotation of the transcriptional signature
To search for patterns within the signature differentiating the MSA and MSB subsets identified by NMF, we used the Ingenuity Pathway tool (http://www.ingenuity.com) and its repertoire of known pathways. We focused this analysis on the pathways that are shared by the MSA versus MSB comparison in each of the three subject groups: Fig. S3 highlights those pathways that are enriched for genes differentiating the MSA and MSB subsets and meet a Benjamini-Hochberg P < 0.0005 (which is corrected for the testing of multiple comparisons) in each subject group. Seventeen pathways meet this criterion, but many more show robust enrichment below a Benjamini-Hochberg P < 0.05 threshold (table S9). Overall, many of the pathways have overlapping sets of genes and primarily involve adaptive immune functions. For example, the MSA subjects displayed, on average, greater expression of genes in the predefined “T cell receptor” and “B cell receptor” pathways (fig. S3); genes found in the NFAT (nuclear factor of activated T cells), ILK (integrin-linked kinase), PI3K (phosphatidylinositol 3-kinase), and EGF (epidermal growth factor) signaling pathways were also expressed more highly in the MSA subset. Thus, overall, it appears that MSA subjects may have a greater proportion of lymphocytes or perhaps more activated lymphocytes in their peripheral blood.
An increased likelihood for a demyelinating event in MSA subjects
Having defined two subsets of subjects with MS by NMF, we went on to assess whether this structure plays a role in disease course. First, we compared the demographic features of the two subsets of subjects in each treatment group to evaluate them for any differences that could confound our treatment response analysis. As outlined in table S5, A to C, none of the available clinical and paraclinical data appeared to be different between the two MS subsets after correcting for the testing of multiple hypotheses, except for a difference in disease duration at the time of sampling in GA-treated subjects. The following analysis therefore included a covariate for disease duration in addition to covariates for sex and age at symptom onset.
To maximize our power in our primary analysis of the role of the subject subsets in disease course, we included GA-and IFN-β–treated subjects that had information on new inflammatory events occurring after the time of blood sampling. The NMF-defined subject subsets were used in this analysis. Given their shared population structure, we merged GA- and IFN-β–treated subjects to assess the role of the subject subsets in disease course, independent of treatment. It is important to note that previous analysis of the extended CLIMB cohort from which our subjects were drawn revealed no significant difference in the likelihood of inflammatory events after the onset of GA or IFN-β treatment (15).
Our primary analysis therefore assessed the trajectories of disease activity in the merged MSA and MSB subsets of GA-and IFN-β–treated subjects after transcriptional profiling. Evidence of disease activity (the analysis end point) included (i) a clinical relapse, or (ii) new T2 hyperintense or gadolinium-enhanced lesions, or (iii) an expanded disability status scale (EDSS) increase of 1, sustained more than 6 months. Among the 56% of GA-treated subjects who reached one of these end points, 39.2% had a clinical relapse, 51.0% had a new lesion on MRI, and 9.8% had an increase in EDSS. For IFN-β–treated subjects who reached an end point (57% of subjects), the distribution included 46.7% with relapses, 38.7% with an MRI event, and 14.7% with an increase in EDSS. There was no significant difference in the distribution of these events between the two treatment categories (P = 0.37). After merging subjects on first-line disease-modifying treatments for our disease course analysis, we observed that subjects classified as MSA at the time of sampling were more likely to exhibit evidence of disease activity over time than MSB subjects (Cox proportional hazard ratio = 0.6, P = 0.0077). Specifically, this hazard ratio suggests that MSB subjects are 40% less likely to have a relapse than MSA subjects (Fig. 4). Secondarily, we assessed whether this association was driven by one of the two treatment groups. Given that there was no statistically significant difference in the trajectories of the MSA and MSB subject subsets when comparing GA- and IFN-β–treated subjects (P = 0.37, interaction term for an effect of treatment on the disease course associated with MSA), the role of the MSA and MSB subject subsets may be the same in the context of disease course while a subject is treated with either one of these two disease-modifying treatments. When the treatment groups were analyzed separately, we observed that, in GA-treated subjects, the MSA subset had more events than the MSB subset (P = 0.02) (fig. S4A). In IFN-β–treated subjects, the trend was the same, but the analysis returned a nonsignificant result (P = 0.17) (fig. S4B). Data on the trajectory of untreated subjects were unfortunately not available for comparison because most of these subjects were started on a disease-modifying treatment shortly after being sampled.
Fig. 4.
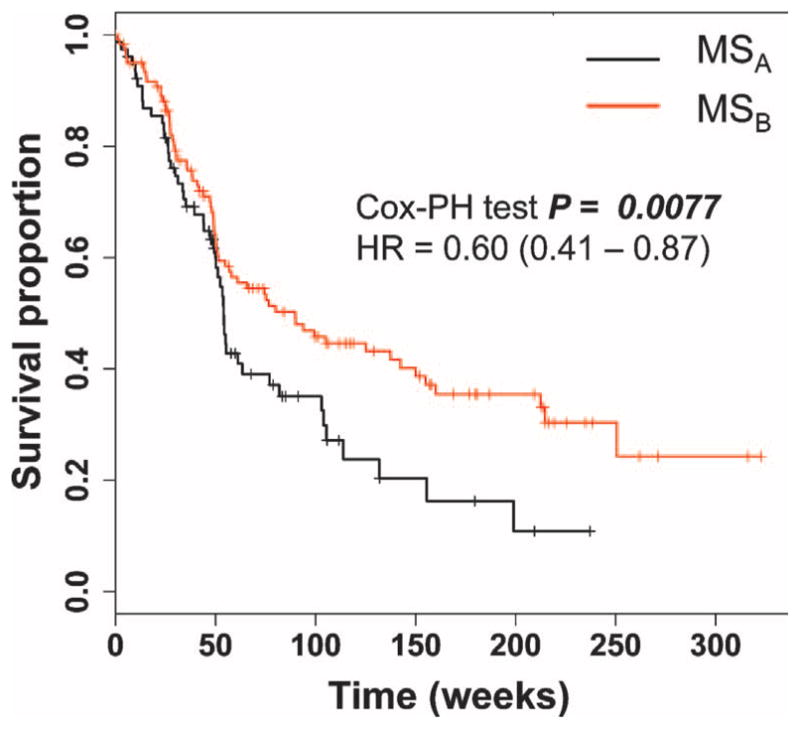
The MSA and MSB gene signatures are associated with disease outcome. The proportion of subjects in the MSA subgroup (black line) and in the MSB subgroup (red line) that were free of evidence of inflammatory demyelination was plotted. Subjects treated with either IFN-β or GA were pooled in this analysis. The results of the Cox proportional hazard (Cox-PH) models were adjusted for gender, disease duration from symptom onset, and treatment duration. The hazard ratio (HR) for the comparison of MSA versus MSB (with MSA as the reference group) is shown, along with its 95% confidence interval.
DISCUSSION
We have gathered a large set of transcriptomic data generated from MS patients and leveraged an unsupervised clustering method to identify two subsets of MS subjects (MSA and MSB) that can be distinguished by a transcriptional signature. We validated this architecture of the MS patient population in subjects treated with either GA or IFN-β, suggesting that the underlying architecture of the disease may not be fundamentally altered by these two treatments.
The MSA and MSB architecture of the MS subject population requires rigorous further validation in prospective cohorts. If this validation is achieved, we speculate that it may be useful to recognize this population architecture when performing studies of MS patients, particularly as the likelihood for further inflammatory events differs in the two subsets of MS patients. As virtually all MS patients are treated, we unfortunately do not have longitudinal clinical and MRI data on the untreated subject group and do not know whether there may be a possible differential disease course in the two subject subsets among individuals who remain untreated. However, looking at our data generated from subjects on chronic treatment, the population of MSA subjects had a more active disease course than the MSB subjects on either GA or IFN-β treatment, suggesting that the difference in disease course was not treatment-specific. Thus, our transcriptional signature may have uncovered information relating to the pathophysiology of MS, and MSA subjects may benefit from consideration of more aggressive treatments at an earlier stage of disease compared to MSB subjects. Further, although the two subsets appear to behave similarly in the context of GA and IFN-β treatment, it is possible that other interventions may have a different effect in the two subsets.
The nature of this transcriptional signature requires more detailed study; our initial pathway analysis highlighted lymphocyte activation pathways as being overrepresented among the genes that were differentially expressed between MSA and MSB. This observation fits with our existing knowledge of MS pathophysiology in which both B and T cells play an important role in the early, inflammatory phase of MS, which was the focus of this study (1). Whether this signature is driven by a higher frequency of lymphocytes among PBMCs of MSA subjects or by overexpression of specific molecules of activation pathways in lymphocytes remains to be determined. Investigation of specific cell populations is the next step. Such investigation will also need to sample subjects before starting therapy to integrate our results with expression signatures identified by other investigators, such as the higher expression of interferon response genes in pretreatment samples that appears to be associated with more disease activity after the initiation of IFN-β treatment (9, 10).
Our study has a number of limitations, including the lack of transcriptional profiles from longitudinal samples of our subjects. Thus, we cannot comment on the stability of our subject classification over time. Although there appear to be two MS states in our study (MSA and MSB), we do not know whether an individual subject fluctuates between these two states or remains in a single state over time. This important question needs to be explored in future longitudinal studies to determine how the molecular signature performs as a prognostic marker for predicting disease activity prospectively. The distinction is important because it would inform the frequency at which a subject’s state should be characterized; repeated sampling would be required if, for example, our transcriptional signature distinguishes active subjects with ongoing, acute episodes of inflammation from subjects who are quiescent at the time of sampling. None of our subjects had a clinical relapse at the time of sampling, but much of the disease activity in MS is asymptomatic and is probably imperfectly captured by MRI (16). Thus, whether our signature provides a surrogate marker for active, asymptomatic inflammation or captures an underlying difference in the pathophysiology of different subjects with MS, this information could be leveraged in the future to enhance patient care and drug development.
Another limitation of our study is the lack of data relating to disease course in our untreated subjects; as a result, we cannot assess whether GA and IFN-β treatments altered the natural history of disease activity in MSA and MSB subjects. It is possible that the difference in disease trajectories may be more pronounced in the untreated state. In addition, although the use of a single RNA detection platform and cellular substrate (PBMCs) enhanced our discovery effort, we have not addressed whether the signature is transportable to other settings that more closely approximate a clinical laboratory. Finally, our focus on the early, inflammatory phase of MS does not address the pressing need to understand the pathophysiology of progressive disease, that is, typically, the more disabling phase of MS. It is unclear, at this time, whether the architecture of the early MS patient population that we describe persists in the progressive stage and whether the MSA and MSB classification affects the timing or likelihood of entering the progressive phase of MS.
Overall, we report a transcriptional signature that distinguishes a subset of MS patients with more active disease. Stratifying MS subjects into meaningful subsets in this manner has potential for personalizing patient care and for enhancing our understanding of this disease.
MATERIALS AND METHODS
Patients and samples
Three hundred and sixty-three subjects with demyelinating disease and frozen PBMC samples were selected from the CLIMB (17) at the Partners Multiple Sclerosis Center in Boston. At the time of sampling, 315 subjects (86.77%) were diagnosed with RRMS per McDonald criteria (18) and 48 subjects (13.22%) were diagnosed with CIS. Only patients who were untreated or treated with one of the two first-line disease-modifying treatments, IFN-β and GA, at the time of sampling were part of the study. The untreated subjects (i) had never been treated with cytotoxic, monoclonal, or investigational agents and (ii) had been sampled either before their first treatment or at least 6 weeks after their last treatment. The disease-modifying treatment groups were limited to subjects that (i) had never been treated with cytotoxic, monoclonal, or investigational agents and (ii) had been treated for at least 3 months at the time of sampling. Finally, we excluded all samples collected within 4 weeks of a methylprednisolone pulse.
Computational methods
Unsupervised clustering
NMF consensus clustering was used to uncover the model that best fit each of the three data sets. It was performed with the algorithm proposed by Brunet et al. in 2004 (11). Briefly, NMF is an unsupervised learning algorithm (19, 20) that identifies molecular patterns in gene expression data. Rather than separating gene clusters on the basis of distance computation (Euclidean), NMF detects context-dependent patterns of gene expression in complex biological systems.
First, we reduce the dimensionality of the expression data from thousands of genes to a few metagenes by applying NMF consensus as implemented in GenePattern version 4 (14). We computed multiple factorizations of our expression matrix A. The number of classes k that we tested on the basis of the expected heterogeneity of our MS data set was between 2 and 5. In NMF, we repeated the clustering process many times to assess its stability and robustness and to define consensus membership. We have tested 20 and 40 initial clustering attempts per k to define the factorization of matrix A (20 is a standard default setting and 40 matrices require more computation time but may provide more robust results). Matrix A derives from the orthogonal combination of W × H, where W and H are the dimensionality-reduced matrices obtained by number of genes and number of experiments, respectively, per number of classes that are tested to assess heterogeneity of the data set (k = 2 to 5) (11). k = 2 was always the most stable model when testing either 20 or 40 initial different W × H matrices. In each of the three classes, only one sample was differently classified when we compare the classifications generated with n = 20 and n = 40 permutations of the matrix. We therefore selected the results based on the 40 matrices for each model (k = 2 to 5).
Supervised SVM clustering
To validate the structure of the MS patient population observed in untreated subjects, we used SVM (as implemented in GenePattern version 4, namely, with SVM R algorithm in e1071 package) (14) and the set of 205 probe sets identified by a linear regression model (Limma, R package) (21) as significantly differentially expressed between the untreated MSA and MSB subgroups to predict class membership of each individual in the GA- and IFN-β–treated subject groups. For each subject, the class defined by NMF (unsupervised) was used as the reference “true” class in our predictive analyses. We used the Prediction Results Viewer tool (version 4.5) written in Java and available in the GenePattern toolkit to visualize the results.
Supplementary Material
Acknowledgments
We thank Affymetrix Inc. for generation of the microarray data.
Funding: P.L.D.J. is a Harry Weaver Scholar of the National MS Society. L.O. was supported by a training/ research fellowship Fondazione Italiana Sclerosi Multipla (Cod. 2008/B/5). This work was supported, in part, by R01 NS067305.
Footnotes
www.sciencetranslationalmedicine.org/cgi/content/full/4/153/153ra131/DC1
Methods
Fig. S1. NMF clustering of untreated subjects after excluding CIS subjects.
Fig. S2. Venn diagram illustrating the differentially expressed (DE) probe sets between MSA and MSB in untreated as well as GA- and IFN-β–treated subjects.
Fig. S3. Pathways enriched for genes that are up-regulated in the MSA subgroup in each of the three treatment groups.
Fig. S4. Assessment of the MSA and MSB gene signature for association with disease outcome in GA- and IFN-β–treated subjects.
Table S1. List of differentially expressed probe sets between GA and untreated groups.
Table S2. List of differentially expressed probe sets between untreated and IFN-β groups.
Table S3. List of differentially expressed probe sets between GA and IFN-β groups.
Table S4. One thousand and sixty-one probe sets differentially expressed between GA versus IFN-β and untreated versus IFN-β groups.
Table S5A. Demographic and clinical characteristics of untreated individuals in MSA and MSB.
Table S5B. Demographic and clinical characteristics of GA individuals in MSA and MSB.
Table S5C. Demographic and clinical characteristics of IFN-β individuals in MSA and MSB.
Table S6. Distribution of CIS and RR individuals in MSA and MSB subgroups.
Table S7. Ninety-eight probe sets differentially expressed in all three MSA versus MSB comparisons for untreated, GA, and IFN-β groups.
Table S8. Complete list of probe sets differentially expressed in each of the three MSA versus MSB comparisons.
Table S9. List of enriched pathways in MSA across the three groups of subjects.
Author contributions: L.O. and P.L.D.J. designed the study; they also selected subjects for inclusion into the study. Affymetrix Inc. produced the RNA data. L.O. completed the quality control analysis as well as the feature selection, NMF, SVM, and pathway analyses. B.T.K. performed the treatment response analysis; M.K. assisted with RNA data analysis; P.T. provided the pathway enrichment code and feedback on its application; L.O. and P.L.D.J. wrote the paper; D.A.H., H.L.W., G.J.B., S.J.K., and J.P.M. critically helped with data interpretation.
Competing interests: P.L.D.J. and G.J.B. are consultants for TEVA and Biogen Idec. G.J.B. is also a consultant for EMD Serono and Bayer Pharmaceuticals. The other authors declare that they have no competing financial interests.
Data and materials availability: The microarray data discussed in this publication have been deposited in the Gene Expression Omnibus (GEO; www.ncbi.nlm.nih.gov/geo/info/linking.html) of the National Center for Biotechnology Information and are accessible through GEO series accession number GSE16214.
REFERENCES AND NOTES
- 1.Hauser SL, Oksenberg JR. The neurobiology of multiple sclerosis: Genes, inflammation, and neurodegeneration. Neuron. 2006;52:61–76. doi: 10.1016/j.neuron.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 2.Brex PA, O’Riordan JI, Miszkiel KA, Moseley IF, Thompson AJ, Plant GT, Miller DH. Multisequence MRI in clinically isolated syndromes and the early development of MS. Neurology. 1999;53:1184–1190. doi: 10.1212/wnl.53.6.1184. [DOI] [PubMed] [Google Scholar]
- 3.Polman CH, O’Connor PW, Havrdova E, Hutchinson M, Kappos L, Miller DH, Phillips JT, Lublin FD, Giovannoni G, Wajgt A, Toal M, Lynn F, Panzara MA, Sandrock AW. AFFIRM Investigators, A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006;354:899–910. doi: 10.1056/NEJMoa044397. [DOI] [PubMed] [Google Scholar]
- 4.Kappos L, Radue EW, O’Connor P, Polman C, Hohlfeld R, Calabresi P, Selmaj K, Agoropoulou C, Leyk M, Zhang-Auberson L, Burtin P FREEDOMS Study Group. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362:387–401. doi: 10.1056/NEJMoa0909494. [DOI] [PubMed] [Google Scholar]
- 5.Corvol JC, Pelletier D, Henry RG, Caillier SJ, Wang J, Pappas D, Casazza S, Okuda DT, Hauser SL, Oksenberg JR, Baranzini SE. Abrogation of T cell quiescence characterizes patients at high risk for multiple sclerosis after the initial neurological event. Proc Natl Acad Sci USA. 2008;105:11839–11844. doi: 10.1073/pnas.0805065105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baranzini SE, Mousavi P, Rio J, Caillier SJ, Stillman A, Villoslada P, Wyatt MM, Comabella M, Greller LD, Somogyi R, Montalban X, Oksenberg JR. Transcription-based prediction of response to IFNβ using supervised computational methods. PLoS Biol. 2005;3:e2. doi: 10.1371/journal.pbio.0030002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Singh MK, Scott TF, LaFramboise WA, Hu FZ, Post JC, Ehrlich GD. Gene expression changes in peripheral blood mononuclear cells from multiple sclerosis patients undergoing β-interferon therapy. J Neurol Sci. 2007;258:52–59. doi: 10.1016/j.jns.2007.02.034. [DOI] [PubMed] [Google Scholar]
- 8.Goertsches RH, Zettl UK, Hecker M. Sieving treatment biomarkers from blood gene-expression profiles: A pharmacogenomic update on two types of multiple sclerosis therapy. Pharmacogenomics. 2011;12:423–432. doi: 10.2217/pgs.10.190. [DOI] [PubMed] [Google Scholar]
- 9.Comabella M, Lünemann JD, Río J, Sánchez A, López C, Julià E, Fernández M, Nonell L, Camiña-Tato M, Deisenhammer F, Caballero E, Tortola MT, Prinz M, Montalban X, Martin R. A type I interferon signature in monocytes is associated with poor response to interferon-β in multiple sclerosis. Brain. 2009;132:3353–3365. doi: 10.1093/brain/awp228. [DOI] [PubMed] [Google Scholar]
- 10.Rudick RA, Rani MR, Xu Y, Lee JC, Na J, Shrock J, Josyula A, Fisher E, Ransohoff RM. Excessive biologic response to IFNβ is associated with poor treatment response in patients with multiple sclerosis. PLoS One. 2011;6:e19262. doi: 10.1371/journal.pone.0019262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brunet JP, Tamayo P, Golub TR, Mesirov JP. Metagenes and molecular pattern discovery using matrix factorization. Proc Natl Acad Sci USA. 2004;101:4164–4169. doi: 10.1073/pnas.0308531101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rifkin R, Mukherjee S, Tamayo P, Ramaswamy S, Yeang CH, Angelo M, Reich M, Poggio T, Lander ES, Golub TR, Mesirov JP. An analytical method for multiclass molecular cancer classification. SIAM Rev. 2003;45:706–723. [Google Scholar]
- 13.Evgeniou T, Pontil M, Poggio T. Regularization networks and support vector machines. Adv Comput Math. 2000;13:1–50. [Google Scholar]
- 14.Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, Mesirov JP. GenePattern 2.0. Nat Genet. 2006;38:500–501. doi: 10.1038/ng0506-500. [DOI] [PubMed] [Google Scholar]
- 15.Gross R, Healy BC, Cepok S, Chitnis T, Khoury SJ, Hemmer B, Weiner HL, Hafler DA, De Jager PL. Population structure and HLA DRB1 1501 in the response of subjects with multiple sclerosis to first-line treatments. J Neuroimmunol. 2011;233:168–174. doi: 10.1016/j.jneuroim.2010.10.038. [DOI] [PubMed] [Google Scholar]
- 16.Khoury SJ, Guttmann CR, Orav EJ, Hohol MJ, Ahn SS, Hsu L, Kikinis R, Mackin GA, Jolesz FA, Weiner HL. Longitudinal MRI in multiple sclerosis: Correlation between disability and lesion burden. Neurology. 1994;44:2120–2124. doi: 10.1212/wnl.44.11.2120. [DOI] [PubMed] [Google Scholar]
- 17.Gauthier SA, Glanz BI, Mandel M, Weiner HL. A model for the comprehensive investigation of a chronic autoimmune disease: The multiple sclerosis CLIMB study. Autoimmun Rev. 2006;5:532–536. doi: 10.1016/j.autrev.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 18.Polman CH, Reingold SC, Edan G, Filippi M, Hartung HP, Kappos L, Lublin FD, Metz LM, McFarland HF, O’Connor PW, Sandberg-Wollheim M, Thompson AJ, Weinshenker BG, Wolinsky JS. Diagnostic criteria for multiple sclerosis: 2005 revisions to the “McDonald Criteria”. Ann Neurol. 2005;58:840–846. doi: 10.1002/ana.20703. [DOI] [PubMed] [Google Scholar]
- 19.Lee DD, Seung HS. Learning the parts of objects by non-negative matrix factorization. Nature. 1999;401:788–791. doi: 10.1038/44565. [DOI] [PubMed] [Google Scholar]
- 20.Lee DD, Seung HS. Algorithms for non-negative matrix factorization. Adv Neural Inf Process Syst. 2001;13:556–562. [Google Scholar]
- 21.Smyth GK. Limma: Linear models for microarray data. In: Gentleman R, Carey V, Dudoit S, Irizarry R, Huber W, editors. Bioinformatics and Computational Biology Solutions using R and Bioconductor. Springer; New York: 2005. pp. 397–420. [Google Scholar]
- 22.De Jager PL, Chibnik LB, Cui J, Reischl J, Lehr S, Simon KC, Aubin C, Bauer D, Heubach JF, Sandbrink R, Tyblova M, Lelkova P, Havrdova E, Pohl C, Horakova D, Ascherio A, Hafler DA, Karlson EW Steering Committee of the BENEFIT Study; Steering Committee of the BEYOND Study; Steering Committee of the LTF Study; Steering Committee of the CCR1 Study. Integration of genetic risk factors into a clinical algorithm for multiple sclerosis susceptibility: A weighted genetic risk score. Lancet Neurol. 2009;8:1111–1119. doi: 10.1016/S1474-4422(09)70275-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fraley C, Raftery AE. Model-based clustering, discriminant analysis, and density estimation. J Am Stat Assoc. 2002;97:611–631. [Google Scholar]
- 24.Fraley C, Raftery AE. Technical Report no 504. Department of Statistics; Washington: 2006. MCLUST Version 3 for R: Model Mixture Modeling and Model-Based Clustering. [Google Scholar]
- 25.Jolliffe IT. Principal Component Analysis. 2. Springer; New York: 2002. [Google Scholar]
- 26.Therneau TM, Grambsch PM. Modeling Survival Data: Extending the Cox Model. 1. Springer; New York: 2000. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.