

Abstract
Cytokines and chemokines are proteins that coordinate the immune response throughout the body. The dysregulation of cytokines and chemokines is a central feature in the development of neuroinflammation, neurodegeneration, and demyelination both in the central and peripheral nervous systems and in conditions of neuropathic pain. Pathological states within the nervous system can lead to activation of microglia. The latter may mediate neuronal and glial cell injury and death through production of proinflammatory factors such as cytokines and chemokines. These then help to mobilize the adaptive immune response. Although inflammation may induce beneficial effects such as pathogen clearance and phagocytosis of apoptotic cells, uncontrolled inflammation can result in detrimental outcomes via the production of neurotoxic factors that exacerbate neurodegenerative pathology. In states of prolonged inflammation, continual activation and recruitment of effector cells can establish a feedback loop that perpetuates inflammation and ultimately results in neuronal injury. A critical balance between repair and proinflammatory factors determines the outcome of a neurodegenerative process. This review will focus on how cytokines and chemokines affect neuroinflammation and disease pathogenesis in bacterial meningitis and brain abscesses, Lyme neuroborreliosis, human immunodeficiency virus encephalitis, and neuropathic pain.
1. Introduction
Cytokines are a class of small proteins that act as signaling molecules at picomolar or nanomolar concentrations to regulate inflammation and modulate cellular activities such as growth, survival, and differentiation [1]. Cytokines are an exceptionally large and diverse group of pro- or anti-inflammatory factors that are grouped into families based upon their structural homology or that of their receptors. Chemokines are a group of secreted proteins within the cytokine family whose generic function is to induce cell migration [2, 3]. These “chemotactic cytokines” are involved in leukocyte chemoattraction and trafficking of immune cells to locations throughout the body. Chemokines belong to two categories based on their biological activity, namely, the maintenance of homeostasis and the induction of inflammation [4]. Homeostatic chemokines are involved in immune surveillance and navigation of cells through hematopoiesis and are typically expressed constitutively. Inflammatory chemokines on the other hand are produced during infections or as a response to an inflammatory stimulus and facilitate an immune response by targeting cells of the innate and adaptive immune system. The binding of a cytokine or chemokine ligand to its cognate receptor results in the activation of the receptor, which in turn triggers a cascade of signaling events that regulate various cellular functions such as cell adhesion, phagocytosis, cytokine secretion, cell activation, cell proliferation, cell survival and cell death, apoptosis, angiogenesis, and proliferation [5].
In the field of neuroimmunology, the classical view that regarded the central nervous system (CNS) as an immune-privileged site by virtue of its shield, the blood brain barrier (BBB), has evolved to a view of significant CNS-immune system interactions [6]. Cytokines and chemokines are involved in the regulation of CNS-immune system interactions besides being important for the coordination of immune responses throughout the body. They are produced primarily not only by white blood cells or leukocytes but also by a variety of other cells as a response to various stimuli under both pathological and physiological conditions. In the nervous system, cytokines and chemokines function as neuromodulators and regulate neurodevelopment, neuroinflammation, and synaptic transmission. Cytokines and chemokines are crucial to the brain's immune function serving to maintain immune surveillance, facilitate leukocyte traffic, and recruit other inflammatory factors [7]. Upon stimulation by pathogens or abnormal cells, immune cells as well as cells of the nervous system such as microglia (the resident macrophages of the brain), astrocytes, oligodendrocytes, the myelinating cells of the CNS, and Schwann cells in the peripheral nervous system (PNS), endothelial cells of the brain microvasculature, and even neurons can release cytokines and chemokines as well as respond to them by way of cytokine and chemokine receptors [8–10]. Neuroinflammatory processes significantly affect both health and disease of the nervous system by regulating the development, maintenance, and sustenance of brain cells and their connections. In the steady state, microglia protect the nervous system by acting as scavengers of debris and microbial pathogens and by regulating the innate and adaptive immune responses. Pathological states within the nervous system including injury, ischemic stroke [11], and infection [12] can lead to activation of microglia. This in turn can cause release of inflammatory molecules that trigger astrocytes and cells of the immune system to respond to the injury [13]. In the disease state, activated microglia mediate neuronal and glial cell injury and death through production of proinflammatory factors like cytokines and chemokines, glutamate, and reactive oxygen species among others and help mobilize the adaptive immune response and cell chemotaxis, leading to transendothelial migration of immune cells across the BBB and even perpetuation of neuronal damage [14]. The central role of microglia in orchestrating neuroinflammation is described in Figure 1.
Figure 1.
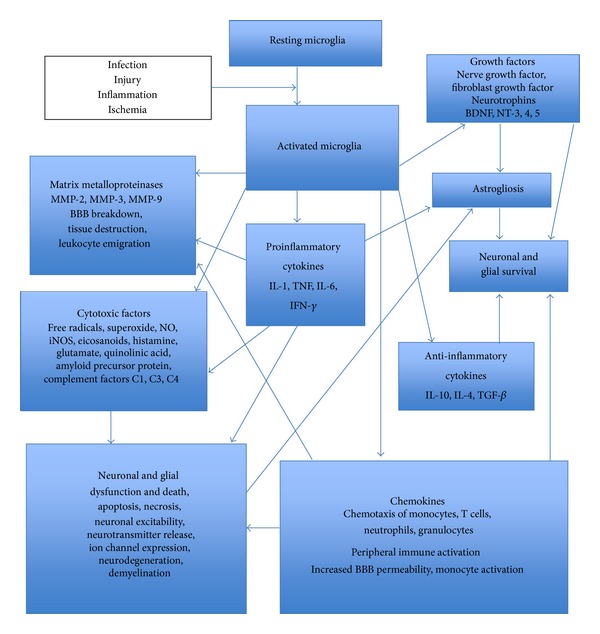
Central role of microglia in neuroinflammation.
In response to injury, neurons produce adhesion molecules and trophic factors that recruit microglial cells and astrocytes. The latter can participate in the ongoing process of damage and repair. In addition to glial cells, the microvasculature also participates in this process. Neurodegeneration is concomitant with astrogliosis, microgliosis, and microvasculature remodeling. Though the trophic factors released initially by astrocytes during astrogliosis aid in tissue repair, these factors amplify the inflammatory response, augment vascular permeability, and result in increased microglial activation and release of more cytokines and chemokines. In states of prolonged inflammation, continual activation and recruitment of effector cells can establish a feedback loop that perpetuates inflammation and ultimately results in neuronal injury [14]. Thus, a critical balance between repair and proinflammatory factors determines the rate of progression and outcome of a neurodegenerative process.
Understanding the role of proinflammatory cytokines in neurodegenerative diseases is complicated by the cytokines' dual roles in neuroprotection and neurodegeneration. For example, IL-6 has dual roles in brain injury and disease. It is produced during reactive astrogliosis as a response to neuronal damage, acting as a neurotrophin promoting neuronal survival, while elevated levels of IL-6 have also been adversely associated with several brain diseases [15].
Some cytokines like IL-1β and TNF induce neurotoxicity through elevated glutamate production that results in neuronal excitotoxic death [16]. The inactivation of IL-1β and TNF using neutralizing antibodies significantly reduced neuronal death in SK-N-SH cells induced by West Nile Virus [17]. Neuroinflammation and both cytotoxic and vasogenic edema were reduced in IL-1 type 1 receptor-deficient mice conferring neuroprotection in stroke [18]. IL-1β also promotes oligodendrocyte death through glutamate excitotoxicity [19]. IL-1β and TNF can cause death of oligodendrocytes in a calcium dependent manner [20]. Deletion of the TNF gene ameliorates neurodegeneration in Sandhoff disease (SD), a lysosomal storage disorder [21]. TNF acts as a neurodegenerative cytokine mediating astrogliosis and neuronal cell death in SD, suggesting TNF as a potential therapeutic target to attenuate neuropathogenesis [21]. On the contrary, aggravation of experimental autoimmune neuritis has been observed in TNF-α receptor 1 deficient-mice signifying an anti-inflammatory role for TNF in this mouse model [22]. TNF has been implicated in both neuronal death and survival and the level and time of expression determine its final effect on CNS damage or protection [23].
Evidence is emerging that chemokines play a role in the physiology of the nervous system, including neuronal migration, cell proliferation, and synaptic activity, besides mediating neuroinflammation. Chemokines are implicated in many diseases of the nervous system. Although their primary role is to induce inflammation through the recruitment of leukocytes by their chemotactic activity, they may also have direct effects on neuronal cells. Chemokines and their receptors are among the key players responsible for communication between neurons and inflammatory cells, and this crosstalk is crucial for normal neurological functioning. Evidence of major roles for chemokines and their receptors in diseases of the brain is accumulating and this system is a potential target for treatment of neurodegenerative diseases [24].
Chemokines may induce neuronal death directly through the activation of neuronal chemokine receptors or indirectly through the activation of microglial killing mechanisms. In addition, some chemokines have neuroprotective roles and function as pro- or anti-inflammatory mediators. For example, induction of neuronal MCP-1/CCL2 during mild impairment of oxidative metabolism caused by microglial recruitment/activation exacerbated neurodegeneration in thiamine-deficiency- (TD-) induced neuronal death, while CCL2-knockout (KO) mice were resistant to TD-induced neuronal death, suggesting that the chemokine CCL2 mediates microglial recruitment and neurodegeneration in this model [25]. However, in another system, CCL2 protected neurons from the toxic effects of glutamate and HIV-tat-induced apoptosis [26]. Interestingly, MCP-1-deficient mice showed reduced neuroinflammatory responses and increased peripheral inflammatory responses to peripheral endotoxin insult. These data demonstrate an important role for MCP-1 in regulation of brain inflammation after peripheral endotoxemia [27].
The overexpression of CXCL10 or interferon gamma-induced protein 10 (IP-10) has been observed in several neurodegenerative diseases including multiple sclerosis (MS), Parkinson's disease (PD) HIV-associated dementia, and Alzheimer's disease (AD) [28–31]. CXCL10 elicits apoptosis in fetal neurons by elevating intracellular calcium levels [32]. On the contrary, the signaling of the neuronal chemokine fractalkine (CX3CL-1) and its receptor CX3CR1 has been shown to be neuroprotective, as they reduce the levels of neurotoxic substances like TNF and nitric oxide in activated microglia during neuroinflammation [33].
The chemokine IL-8 (CXCL8) that regulates neutrophil migration by signaling through the CXCR2 receptor is markedly elevated by brain injury and is associated with the propagation of secondary damage. Evaluating the function of CXCR2 in posttraumatic inflammation and secondary degeneration by examining Cxcr2-deficient (Cxcr2−/−) mice showed reduced tissue damage and neuronal loss in Cxcr2−/− mice compared to wild-type controls [34]. CXCR1, the receptor for MIP-2 (CXCL2) and CXCR2, has also been implicated in contributing directly to motor-neuron degeneration [35].
The chemokine CXCL1 or GRO1 that is upregulated in brain endothelium in the presence of IL-6 has been identified as a key regulator of granulocyte recruitment into the CNS. Though granulocytes generally exert a protective role in the CNS, they have been shown to be detrimental in experimental autoimmune encephalitis (EAE), the most common model of MS. Administering anti-CXCL1 antibodies attenuated EAE severity suggesting CXCL1 to be a new potential target for the treatment of neuroinflammatory conditions like MS [36].
The chemokine CXCL12 or stromal cell-derived factor 1 (SDF-1) is strongly chemotactic for lymphocytes and it modulates neurotransmission, neurotoxicity, and neuroglial interactions [37]. Growing evidence implicates enhanced expression of CXCL12 and its receptor CXCR4 in the pathogenesis of CNS disorders such as HIV-associated encephalopathy, brain tumor, stroke and MS, making them promising targets for pharmacological intervention [38]. CXCL12/CXCR4 have been shown to promote apoptotic death of dopaminergic neurons in a mouse model of PD [39]. CXCL12/CXCR4 have been suggested to be markers to grade CNS glioblastoma tumor progression. In glioblastoma, a CXCR4 antagonist (AMD3100) showed an inhibition of tumor growth [40]. Thus, several studies using cytokine/chemokine receptor antagonists and deletion mutant mice have provided exciting findings establishing a central role for cytokines and chemokines in mediating neuroinflammation and neurodegeneration and cytokines/chemokines and their receptors represent interesting therapeutic targets in this context.
Immune activation in the nervous system is associated with pathological conditions such as bacterial and viral infections, autoimmune diseases, and inflammatory neurodegenerative disorders including AD, PD, amyotrophic lateral sclerosis, MS [41–44], and Lyme neuroborreliosis (LNB) [45]. Peripheral neuropathies such as Guillain-Barré syndrome [46], PNS Lyme neuroborreliosis [47], demyelinating polyradiculoneuropathies [48], and conditions of neuropathic pain [10, 49] are also accompanied by inflammation. One underlying similarity in all these disease states is the cytokine and chemokine-driven inflammatory response. The dysregulation of cytokines and chemokines is a central feature in the development of neuroinflammation, neurodegeneration and demyelination in the CNS [43, 44], neuritis and axonal degeneration in the PNS [9], and conditions of neuropathic pain [10, 49]. Understanding the involvement of cytokines and chemokines in the pathogenesis of nervous system disorders is relevant for understanding brain pathophysiology and may lead to the development of targeted therapies to treat neurodegenerative diseases. This review will focus on how cytokines and chemokines affect neuroinflammation and disease pathogenesis in bacterial meningitis and brain abscesses, Lyme neuroborreliosis, human immunodeficiency virus encephalitis (HIVE), and neuropathic pain.
2. Cytokines and Chemokines in Bacterial Meningitis and Brain Abscesses
Bacterial meningitis is among the top ten causes of death due to infectious agents worldwide. The major meningeal bacterial pathogens are Streptococcus pneumoniae, Neisseria meningitides, and Hemophilus influenzae, although other organisms are capable of causing disease in humans in an age-specific manner [50]. About 50% of patients surviving the infection present with neurological deficits [51]. The pathogen gains access to the bloodstream, penetrates the BBB, and replicates in the subarachnoid space. Immune cells from the peripheral circulation are attracted into the infected subarachnoid space by inflammatory mediators that are produced initially by ependymal cells, meningeal macrophages, and choroid plexus epithelium, followed by local microglia and recently emigrated leukocytes [52–54]. The pathological manifestations of meningitis include increased intracranial pressure, intense brain edema, impairments in cerebrospinal fluid (CSF) flow, seizures, and alterations in cerebral blood flow that may result in focal areas of ischemia and necrosis. During bacterial meningitis, the antibacterial response elicited by the host can be detrimental to neurons and glia in the CNS, due to the toxic effects of cytokines, chemokines, proteolytic enzymes, and oxidants produced locally at the site of infection, in addition to the direct damage caused by pathogens [55].
2.1. Streptococcus pneumoniae Meningitis
S. pneumoniae is the most frequent cause of bacterial meningitis [56, 57]. Effects from meningitis can range from memory deficits, hearing loss, hydrocephalus, cerebral palsy, and seizures. Since the pneumococci can cross the BBB, microglia may respond directly to intact bacteria or to pneumococcal cell wall antigens and produce a wide array of inflammatory mediators including TNF, IL-6, IL-12, keratinocyte-derived chemokine (CXCL1/KC), CCL2/MCP-1,CCL3/MIP-1α,CXCL2/MIP-2, and CCL5/RANTES, as well as soluble TNF-α receptor II, a TNF antagonist [58, 59]. The production of these inflammatory mediators is associated with the activation of the extracellular signal-regulated protein kinases ERK-1 and ERK-2 via a MAPK intracellular signaling pathway [58, 59].
The microglial-derived cytokine and chemokine profile represents a double-edged sword since it is effective at eliciting leukocyte recruitment into the CNS for the purpose of antibacterial defenses but at the same time can also contribute to inflammatory mediator-induced neuronal damage by apoptosis triggered, in part, by the inflammatory process via caspase activation [60]. This suggests that strategies to check microglial activation at a point where inflammation is no longer beneficial could reduce damage to surrounding normal parenchyma as a result of bystander destruction. It is suggested that neuronal damage in bacterial meningitis is caused by the dual effects of an overwhelming inflammatory response and the direct effects of bacterial toxins [61]. It is proposed that brain damage in bacterial meningitis leading to long-term neurologic sequelae and death involves several mechanisms. Bacterial invasion and the release of bacterial compounds promote inflammation, invasion of leukocytes, and stimulation of microglia. Leukocytes, macrophages, and microglia release free radicals, proteases, cytokines, and excitatory amino acids, eventually leading to energy failure and cell death. In addition, vasculitis, focal ischemia, and brain edema subsequent to an increase in CSF outflow resistance, breakdown of the BBB, and swelling of necrotic cells cause secondary brain damage.
2.2. Cytokines in the Pathogenesis of Pneumococcal Meningitis
The early response cytokines TNF, IL-1, and IL-6 are produced after pneumococcal recognition, which in turn induce upregulation of several adhesion factors on the vascular endothelium, mediating leukocyte influx [62]. An experimental rabbit model of pneumococcal meningitis has shown that the outcome of bacterial meningitis is related to the severity of inflammation in the subarachnoid space and that outcome can be improved by modulation of the inflammatory response [63]. Homologous antibodies to TNF, IL-1α and IL-1β inhibited leukocytosis and brain edema and moderately decreased BBB permeability in this model of meningitis [64]. A mouse model of S. pneumoniae meningitis that mimics several features of human disease describing meningeal inflammation and neuronal damage [65] and an infant mouse model of brain damage in pneumococcal meningitis that exhibits neuronal brain injury in the cortex and hippocampus that reflect the histomorphological findings in the human disease have been established [66]. The infant rat model of pneumococcal meningitis has also been very useful to study the pathogenesis of the disease [67, 68].
The use of KO mice has offered new insights into the role of cytokines involved in the inflammatory cascade during pneumococcal meningitis. Increased mortality and spatial memory deficits in TNF-α-deficient mice with experimental pneumococcal meningitis were observed suggesting that TNF plays a role in inflammation and hippocampal injury in bacterial meningitis [69]. Patients with pneumococcal meningitis show increased CSF-TNF-α, which correlates with severity of BBB disruption, disease severity, and neurological sequelae [70].
IL-1 is an important proinflammatory cytokine, which is upregulated in brain tissue after the induction of meningitis. Mortality was significantly higher and appeared earlier in the course of the disease in IL-1R (−/−) mice demonstrating that endogenous IL-1 is required for an adequate host defense in pneumococcal meningitis [71]. IL-18 gene-deficient mice showed enhanced defense and reduced inflammation during pneumococcal meningitis suggesting that endogenous IL-18 contributes to a detrimental inflammatory response during pneumococcal meningitis and that elimination of IL-18 may improve the outcome of this disease [72].
The anti-inflammatory cytokine IL-10 has been implicated in playing a role in modulating the immune response by downregulating TNF, IL-6, and keratinocyte-derived chemokine (KC), thereby reducing CSF pleocytosis in pneumococcal meningitis [71]. IL-10 has been shown to repress sepsis-associated hippocampal neuronal damage as a result of pneumococcal sepsis in mice overexpressing IL-10 [73]. Also, intravenously administered recombinant IL-10 reduced the levels of CSF pleocytosis, cerebral edema, and intracranial pressure in a rat model of pneumococcal meningitis [74]. In mice with S. pneumoniae-induced meningitis, a deletion of TGF-β receptor II on leukocytes is found to enhance recruitment of neutrophils to the site of infection and to promote bacterial clearance. The improved host defense against S. pneumoniae was associated with an almost complete prevention of meningitis-induced vasculitis, a major intracranial complication leading to brain damage. The data show that endogenous TGF-β suppresses host defense against pneumococcal infection in the CNS [75]. Activin A, a member of the TGF-β superfamily and a neuroprotectant that is expressed constitutively in the CSF, has been shown to be upregulated in patients during bacterial meningitis [76, 77]. Cotreatment with activin A and LPS showed increased microglial proliferation and negative regulation of NO, IL-1β, IL-6, and TNF in in vitro-cultured murine microglia [77].
2.3. Chemokines in the Pathogenesis of Pneumococcal Meningitis
Multiple chemokines have been reported to be upregulated in the CSF of patients with pneumococcal meningitis including CCL15, CXCL7, MIF, CCL8, CCL18, CCL20, CXCL5, CXCL-1, CXCL-8, CCL2, CCL3, and CCL4 [78–81]. In animal models of pneumococcal meningitis, additional chemokines have been identified by protein arrays for brain tissues, including CCL9, CXCL-2, XCL-1, CCL-1, CCL11, CCL12, CCL24, CCL25, CXCL4, CXCL10, CXCL12, CXCL13, and CXCL13 [82].
IL-8 (CXCL-8 was found to be chemotactic for neutrophils in the CSF of patients with bacterial meningitis [79]. IL-8 appears to regulate CSF pleocytosis in pneumococcal meningitis from the systemic compartment, similar to that seen for TNF, IL-10, and TGF-β [83]. Both MIP-1 (CCL3) and MIP-2 (CCL3) are produced by immune cells resident in the brain and attract monocytes and neutrophils from the bloodstream into the CSF in acute bacterial meningitis [84]. In vitro, antibodies against CCL2, CCL3, and CCL4 inhibited monocyte chemotactic properties of CSF from patients with pneumococcal meningitis [79]. The intracisternal inoculation of recombinant CCL3 and CCL4 induced BBB disruption, CSF leukocytosis, and cerebral edema in a rabbit model of pneumococcal meningitis [64]. Of the CXCL chemokines, ENA-78 (CXCL5) was found to be upregulated in patients with bacterial meningitis and exhibited neutrophil chemotactic properties together with IL-8 [80]. A study that evaluated the global response of the BBB to S. pneumoniae infection and the specific role of neuraminidase A (NanA), a pneumococcal protein described to promote CNS trophism, revealed that NanA was necessary and sufficient to activate host chemokine induction including IL-8, CXCL-1, and CXCL-2 [85]. In summary multiple chemokines have been reported to be upregulated in pneumococcal meningitis that primarily have a role in attracting leukocytes to the CSF, though the roles of many chemokines in the pathogenesis of the disease have not yet been investigated [86].
2.4. Staphylococcus aureus and Brain Abscesses
Abscesses in brain parenchyma develop as a consequence of local spread of pyogenic bacteria from the paranasal sinuses, middle ear, or oral cavity, via hematogenous dissemination from a systemic infection or by directly penetrating trauma to the head [87–92]. The most common etiologic agent of brain abscesses in humans is S. aureus, besides Streptococci [88]. These infections are characterized by extensive edema and tissue necrosis [89]. The activation of resident microglia is a hallmark of infection [90], in addition to the sequential progression to necrosis during brain abscess evolution. Microglial and astrocyte activation is evident immediately following the entry of bacteria into the CNS parenchyma and persists throughout abscess development in a mouse model [91]. The ensuing abscess formed at the site of infection may result in inflammation accompanied by edema, neuronal toxicity, seizures, and long-term cognitive loss [92]. This murine model has demonstrated that S. aureus not only induces brain abscesses but also elicits rapid and sustained expression of numerous proinflammatory cytokines and chemokines including IL-1β, TNF, IL-12 p40, CXCL2, CCL2, CCL3, and CCL4 [93–95]. Leukocyte recruitment elicited by microglia into the infected CNS facilitates bacterial clearance during abscess development. Microglia also exert S. aureus bactericidal activity. The organism is a potent inducer of numerous inflammatory molecules in microglia such as TNF, IL-1β, and CXCL1, among others [96, 97]. The necrotic damage associated with brain abscesses and other CNS infections is accompanied by release of endogenous host molecules that could potentially exacerbate parenchymal necrosis in addition to that mediated by unchecked microglial activation.
Knowledge of the staging of brain abscess in humans is based on findings of CT and MRI scans [98]. During the last decade, an experimental brain abscess model in rats and mice has been established by direct intracerebral injection of live S. aureus, [90, 92, 99, 100]. Rodent models mimic accurately the natural course of brain abscess development in humans and have been investigated intensely to understand the mechanisms in disease pathogenesis and the possible treatment modalities.
Brain abscess is typified by a sequential series of pathological changes that have been elucidated in experimental rodent models [90, 92, 99–101]. Briefly, the early stage cerebritis occurs from day 1 to day 3 and is marked by neutrophil accumulation, tissue necrosis, and edema. Astrocyte and microglial activation are seen at this stage and persist throughout abscess development, accompanied by the induction of proinflammatory cytokines and chemokines [90, 94]. From day 4 to day 9 (intermediate or late cerebritis), predominant macrophage and lymphocyte infiltrates are commonly seen. The last or capsule stage is seen from day 10 onwards and is characterized by the formation of a well-vascularized abscess wall that in turn helps to sequester the lesion and protects the surrounding brain parenchyma from additional damage.
However, the immune response that is essential for abscess formation also destroys surrounding normal brain tissue. There is a prolonged expression of IL-1β, TNF, and macrophage inflammatory protein-2 (MIP-2/CXCL2), concomitant with a chronic disruption of the BBB in mice with S. aureus-induced brain abscess. These changes correlated with the continued presence of infiltrating neutrophils and macrophages/microglia. These observations suggest that the excessive tissue damage that often results from brain abscess may be mediated, in part, by the perpetuation of antibacterial immune responses that are not down-regulated in a timely manner [91]. Similarly, human brain abscess lesions have been found to encompass a large area of the brain, often spreading well beyond the initial focus of infection [102].
On the other hand, cytokines like IL-1β, TNF, and IL-6 may exert beneficial effects on the establishment of host antibacterial immune responses. A study that examined the relative importance of IL-1β, TNF, and IL-6 in experimental brain abscess using cytokine KO mice showed that IL-1 and TNF play a major role in directing the ensuing antibacterial response, as bacterial burdens were significantly higher in both IL-1 and TNF-α-KO mice compared to wild-type mice which correlated with enhanced mortality rates in KO mice [91].
Neutrophils that are the major peripheral cell infiltrate associated with brain abscesses are the source of proinflammatory cytokines such as TNF that serve to amplify the antibacterial immune response [103]. Neutrophils can also exert bactericidal activity through the production of reactive oxygen intermediates and nitrogen intermediates and hydrolytic enzymes that can directly destroy bacteria; however, the continuous release of cytokines and bactericidal products by neutrophils can also contribute to tissue damage [103]. CXCR2 ligands, namely, MIP2 (CXCL2) and KC (CXCL2) are the major ligands required for chemotactic signaling of neutrophils into brain abscesses as demonstrated by CXCR2 KO mice studies [92]. Impaired neutrophil influx into evolving brain abscesses in antibody-mediated neutrophil-depleted mice and CXCR2 KO mice resulted in exacerbated disease accompanied by elevated bacterial burdens compared to wild-type mice [92, 104]. In addition, chemokines such as CCL1, CCL2, CCL3, and CCL4 were also detected in evolving brain abscesses that most probably contribute to the influx of lymphocytes and monocytes and the establishment of the adaptive immune response [92].
Using a minocycline-resistant strain of S. aureus to dissect the antibiotic's bacteriostatic versus immune modulatory effects in a mouse experimental brain abscess model, minocycline was found to significantly reduce mortality rates within the first 24 hours following bacterial exposure [105]. This protection was associated with a transient decrease in the expression of several proinflammatory mediators, including IL-1β and CCL2. Minocycline was also capable of protecting the brain parenchyma from necrotic damage as evidenced by significantly smaller abscesses in minocycline-treated mice. In addition, minocycline exerted anti-inflammatory effects when administered as late as 3 days following S. aureus infection, which correlated with a significant decrease in brain abscess size. Finally, minocycline was capable of partially attenuating S. aureus-dependent microglial and astrocyte activation. This study suggests that minocycline may afford additional therapeutic benefits extending beyond its antimicrobial activity for the treatment of CNS infectious diseases typified by a pathogenic inflammatory component through its ability to balance beneficial versus detrimental inflammation.
3. Cytokines and Chemokines in Lyme Neuroborreliosis
Lyme borreliosis the most frequently reported vector-borne disease in the USA is caused by the spirochete Borrelia burgdorferi (Bb) [106]. It is transmitted through Ixodes ticks and is also prevalent in Europe and Asia [106, 107]. Lyme neuroborreliosis (LNB), the form of Lyme disease that affects the nervous system, manifests in about 15% of Lyme disease patients and affects both CNS and PNS [108–110]. Patients with CNS involvement complain of severe headaches, flu-like symptoms, fatigue, memory loss, learning disability, and/or depression. Infection of the PNS with Bb may result in facial nerve paralysis or palsy, pain in the back and limbs, and movement disorders.
Clinically, LNB may manifest as meningitis typically characterized by lymphocytic pleocytosis in the CSF, meningoradiculitis (a.k.a. Bannwarth's syndrome), cranial neuritis, encephalopathy, peripheral neuropathy, and, less commonly, encephalitis and encephalomyelitis. Radiculitis, or inflammation in the dorsal roots, is the most common manifestation of untreated Lyme borreliosis in humans [110]. LNB patients may also experience a wide array of neurological symptoms as a result of white matter inflammation that results in a subacute MS-like manifestation [111, 112]. Perivascular and vascular inflammatory processes may also occur in CNS LNB and several case reports of seizures or stroke have been attributed to neurologic Lyme disease [113–115].
Reports from human cases of LNB often include lymphocyte and plasma cell infiltration in the meninges and perivascularly in the nerve roots, dorsal root ganglia (DRG), and demyelination in the brain and spinal cord [113, 116–120]. Typically, PNS-Lyme disease is associated with transverse myelitis and patchy multifocal axonal loss with epineural perivascular inflammatory infiltrates or perineuritis [121–125]. The primary findings of axonal degeneration and regeneration and multifocal nerve lesions showing perivascular inflammatory cellular infiltrates have been documented in almost all patients with Lyme-associated peripheral neuropathy [123–127]. The results of these studies suggest that immune mediated neuronal and glial cell damage could be involved in the neuropathogenesis of LNB.
It is suggested that adherence of the spirochete to the endothelium lining of blood vessel walls leads to the release of inflammatory mediators [128]. This could in turn alter the permeability of the BBB and ensue entry of Bb into the CNS [128, 129]. The perivascular mononuclear cell infiltrates observed in the cerebral cortex during Bb infection consist predominantly of T-helper cells [130] and are associated with a focal increase in microglial cells and infiltration of lymphocytes and plasma cells in the leptomeninges [131].
Elevated levels of the proinflammatory cytokines IL-6, IL-8, IL-12, IL-18, and interferon γ [71–74] and the chemokines interferon-inducible T-cell chemoattractant (I-TAC), CCL2, CXCL-11, and CXCL13 [132–139] have been reported in the CSF of patients with LNB. The amount of IL-6 in human serum and CSF has been shown to correlate with disease activity in neurologic Lyme disease [132]. The chemokine CXCL13, which is known to attract B-lymphocytes, is also elevated in other instances of neuroinflammation [140]. CXCL13 expression in the CSF precedes the intrathecal production of Bb-specific antibodies [141] and may account for the high proportion of B-lymphocytes and plasma cells in the CSF of LNB patients, suggesting a role for the humoral immune response in Lyme neuroborreliosis [142].
Increased production of the neuromodulator quinolinic acid, an excitotoxin and N-methyl-D aspartate (NMDA) agonist, has been demonstrated in the CSF of patients with neurologic Lyme disease [143]. As the NMDA receptor mediates synaptic function and is involved in learning, memory, and synaptic plasticity [144], its dysregulation mediated by Bb-induced inflammation may contribute to the neurologic and cognitive deficits seen in many Lyme disease patients by mechanisms such as glutamate-mediated excitotoxicity [145].
The rhesus macaque is the preferred animal model for studying neurologic Lyme disease, as it exhibits most of the signs of Lyme disease seen in humans, both in the PNS and CNS [146, 147]. Using the monkey model, investigators have been able to examine the effect of Bb infection on neural tissue and its relationship to the adaptive and acquired immune responses. Since Bb itself does not produce any known endotoxin [148], damage to neural cells may occur in part due to bacterial lipoproteins that are present on the spirochetal surface or are released or shed by live or dead organisms. Lipidated outer-surface protein A (L-OspA), a prototype Bb lipoprotein, has been shown to induce IL-6, cell proliferation, and concomitant apoptosis in rhesus astrocytes in vitro [149, 150]. Bb-infected rhesus monkeys also showed astrogliosis in the frontal cortex [149]. In the presence of Bb, primary cultures of astrocytes or microglia have been shown to produce IL-6, IL-8, and the macrophage inflammatory proteins CCL3 and CCL4 [151]. Human neurons cocultured with Bb and rhesus microglia undergo apoptosis in the presence of proinflammatory mediators chiefly produced by the microglia [152].
In an ex vivo stimulation of monkey brain frontal cortex tissue explants with live Bb, IL-6, IL-8, IL-1β, and CXCL13 were visualized in glial cells, with concomitant oligodendrocyte and neuronal apoptosis [153], suggesting that the glial inflammatory response to Bb could contribute to cell death. In addition, microarray analyses of tissue RNA revealed altered transcription of multiple genes that regulate the immune response as well as apoptosis [153]. When live Bb was inoculated into the CNS of rhesus macaques via the cisterna magna [154], within one week after inoculation there was a monocytic and lymphocytic pleocytosis and increased expression of IL-6, IL-8, CCL2, and CXCL13 in the CSF. Histopathological changes consistent with acute neurologic Lyme disease, showing leptomeningitis and radiculitis, as well as satellite glial cell and neuronal apoptosis in the DRG were also observed. IL-6 was produced by both astrocytes and neurons of spinal cord tissue and by neurons in the DRG. The chemokines CXCL13 and CCL2 were detected in microglia of the spinal cord. CCL2 was also detected in endothelial cells in the periventricular area of the brain. Other investigators have also confirmed the production of IL-6 and CXCL13 in B. burgdorferi-infected rhesus and human tissues [155–157].
Patients with chronic and recurrent neurologic Lyme disease who have persistent symptoms even after treatment are plagued primarily by pain, fatigue, and cognitive dysfunction. Elevated levels of IL-6 can cause symptoms of fatigue and malaise, common to many infectious and neurodegenerative diseases. IL-6 is pyrogenic, promotes B cell differentiation, stimulates the synthesis of acute phase reactants, and can also contribute to pain by increasing the sensitivity of nerve endings [158]. It is possible that IL-6 mediates the pain response in the sensory neurons of the DRG in LNB as well, since it was seen in DRG neurons of infected rhesus macaques [155, 158].
In a recent study, live Bb was shown to elicit the production of cytokines and chemokines, particularly IL-6, IL-8, and CCL2, as well as to induce apoptosis in human oligodendrocytes cultured in vitro [159], by activating the enzyme caspase-3 [160]. Oligodendrocytes could therefore contribute to the elevated levels of cytokines and chemokines detected in the CSF of patients with LNB. Importantly, in the presence of the anti-inflammatory drug dexamethasone, a reduction in the amount of proinflammatory mediators, and a significant reduction in the Bb-induced oligodendrocyte apoptosis was observed [159]. This outcome is a strong indication that inflammation plays a role in mediating oligodendrocyte apoptosis, which could be mediated in part by the direct action of the spirochetes on oligodendrocytes or via inflammation mediated by Bb in oligodendrocytes.
Oligodendrocytes in brain tissue are especially vulnerable to demyelination as they are located immediately adjacent to the subarachnoid space, in the region known as the subpial space [161]. Oligodendrocytes are known to express receptors for various cytokines and chemokines [162]. Since inflammatory lesions are commonly found in the meninges in LNB, the myelitis that is seen in LNB may be in part due to oligodendrocyte dysfunction. These cells could be damaged by the inflammatory process initiated by the oligodendrocytes themselves, with participation of other glial cells, in addition to inflammatory mediators produced by the perivascular cellular infiltrates that are often present in CNS infection. As oligodendrocytes are vital for the survival and optimal function of neurons [162], oligodendrocyte damage could contribute to neuronal dysfunction and death and result in the impairment of CNS functions seen in patients with LNB. Caspase-mediated oligodendrocyte cell death has also been documented in inflammatory demyelinating diseases such as MS [163]. Cytokines and chemokines play a central role in inflammation, demyelination, and neurodegeneration in the CNS during inflammatory neurodegenerative diseases such as MS, which shows similar clinical signs as those shown by LNB [164].
The chemokine CCL2 reported in Lyme neuroborreliosis is of particular importance in mediating inflammation in neurodegenerative diseases [165, 166]. It is an important mediator in many neuroinflammatory and neurodegenerative brain diseases characterized by neuronal degeneration [167]. CCL2 has been found to be upregulated in actively demyelinating MS plaques [168], and its expression is increased in experimental autoimmune encephalomyelitis [169]. CCL2 modulates microglial activation and proliferation, thereby contributing to the inflammatory response mounted in the CNS [170]. Importantly, CCL2 levels are elevated in the CSF of patients with LNB [136], and high levels of CCL2 have been found in the CSF of rhesus monkeys infected intrathecally with Bb [154]. CCL2 is known to play a role in mediating nerve damage and demyelination of axons by causing an influx of monocytes and T cells in Wallerian degeneration [171] that may also possibly contribute to the axonal damage that affects patients with LNB of the PNS [172].
The cytokine IL-6 that has been reported in studies of LNB pathogenesis is known to be both helpful and harmful in the CNS [15, 173–176]. Dysregulated expression of IL-6 has been documented in several neurological disorders such as MS, acute transverse myelitis, AD, schizophrenia, epileptic seizures, and PD [177]. In addition, IL-6 has been shown to be involved in multiple physiological CNS processes such as neuron homeostasis, astrogliogenesis, and neuronal differentiation [178]. IL-6 is known to promote oligodendrocyte and neuronal survival in the presence of glutamate-mediated excitotoxicity in hippocampal slices [179] and promotes survival of oligodendrocytes in vitro [180]. It is possible that IL-6 could mediate both neuroprotection as well as neurodegeneration in inflammatory neurodegenerative diseases including LNB.
The chemokine IL-8, also seen to be elevated in the CSF of LNB patients [132, 179] and in rhesus microglia, astrocytes, and endothelial cells exposed to Bb [151–154], is associated with BBB dysfunction and plays a central role in recruitment of neutrophils and T cells into the CNS during bacterial meningitis [181–183]. IL-8 is known to induce the expression of proinflammatory proteases, the matrix metalloproteinases MMP-2 and MMP-9, and proapoptotic protein Bim (Bcl-2-interacting mediator of cell death) and cell death, in cultured neurons in 24 hours [184]. There are reports indicating the presence of a cytolytic phenotype of IFN-γ-producing cells from patients with LNB [185, 186], suggesting the possible involvement of cytotoxic cells in mediating the demyelination and axonal degeneration seen in LNB [113, 116, 121].
B. burgdorferi has also been shown to induce the late production of significant quantities of the anti-inflammatory cytokine IL-10 in murine microglia and astrocytes [187]. The delayed production of IL-10 suggests that a possible negative feedback loop to limit potentially damaging inflammation within the brain parenchyma during persistent infections may be operating in parallel with the harmful effects of proinflammatory mediators. The treatment of primary rhesus macaque microglia with the tetracycline analogs doxycycline and minocycline resulted in attenuated microglial proinflammatory mediator responses to Bb [188]. Doxycycline is used for the treatment of Lyme disease patients and has been shown to improve adverse clinical symptoms at a time when viable spirochetes can no longer be easily detected. Therefore, the dampening of microglial proinflammatory cascades to limit unchecked neuroinflammation and subsequent neuronal damage may be beneficial to LNB patients.
4. Cytokines and Chemokines in Human Immunodeficiency Virus Encephalitis
Infection with the human immunodeficiency virus-1 (HIV-1) and acquired immunodeficiency syndrome (AIDS) are a persistent health problem worldwide. HIV-1 seems to enter the brain very soon after peripheral infection and can induce severe and debilitating neurological problems that include behavioral abnormalities, motor dysfunction, and dementia. The neurological manifestations directly related to HIV are acute viral meningitis, chronic meningitis, HIV-associated dementia (HAD), vacuolar myelopathy, and involvement of the peripheral nervous system [189]. Infected peripheral immune-competent cells, in particular macrophages, appear to infiltrate the CNS and provoke a neuropathological response involving all cell types in the brain. HIV-1 encephalitis (HIVE), a common pathological manifestation of HAD, includes infiltration of macrophages into the brain where they become productively infected with the virus. This is accompanied by considerable cytokine and chemokine dysregulation in the brain that often culminates into the unique pathological features that characterize this syndrome. Once in the brain, HIV-1-infected blood-borne macrophages secrete proinflammatory cytokines such as TNF, IL-1β, and viral proteins such as HIV-1gp120 and Tat, which can affect neuronal function [190]. In the CNS, HIV-1 also incites activation of chemokine receptors, inflammatory mediators, extracellular matrix-degrading enzymes, and glutamate receptor-mediated excitotoxicity, all of which can initiate numerous downstream signaling pathways and disturb neuronal and glial function.
Lentiviruses are thought to enter the brain within circulating infected monocytes during immune surveillance. Numerous studies have been undertaken to determine the reasons underlying increased monocyte migration into the brain following lentiviral infection. HIV-infected leukocytes are primed for adhesion [191], having already shed L-selectin and increased expression of CD11b/CD18 compared with monocytes from healthy controls [192]. Therefore, it is possible that even marginal increases in the levels of chemokines expressed within the parenchyma would lead to increased migration of monocytes. Recent studies have shown that glial cells are stimulated to produce chemokines in response to inflammatory cytokines [193, 194] that are known to be secreted by simian immune-deficiency-virus- (SIV-)infected macrophages [195].
4.1. Astrocytes and Signaling in HIV Encephalitis
Astrocytes act to repel circulating immune cells through secretion of eotaxin [196], reinforcing the brain's immune-privileged status in conjunction with the selective physical properties of the BBB. Under normal conditions the brain allows only limited access by immune cells. Early in HIV infection the virus enters the brain through normal trafficking. This leads to a transient increase in BBB permeability and a localized immune response. As the disease progresses to encephalitis, the immune response is dramatically increased, marked by a loss of tight junction integrity, gliosis, and formation of multinucleated giant cells in the parenchyma.
Astrocytes are the primary cell type found in glia scar formation [197, 198], and secrete cytokines and chemokines to elicit increased trafficking of leukocytes into the brain [193, 199, 200]. Astrocytes may also provide a role for the resolution of inflammation by reducing the secretion of proinflammatory cytokines, and increasing anti-inflammatory processes [198, 201, 202]. Decreased BBB integrity early in SIV/HIV infection allows latently infected monocytes to enter the brain [203]. Circulating virus could induce brain microvessel endothelial cells (BMEC) to express CD106 diffusely [204, 205] leading to increased monocyte migration into brain, where they become productively infected. Astrocytes respond to these macrophages resulting in a wide-range of cellular changes referred to as astrogliosis.
4.2. Astrogliosis
On activation astrocytes undergo a morphological change, most notably an increase in ramification concomitant with upregulation of GFAP and thickened processes. Some astrocytes in the proximity of SIV lesions express peripherin, an alternative type III intermediate filament not normally expressed in brain [206]. Immunologically, astrocytes respond to HIV/SIV infection through increased production of inflammatory cytokines. As outlined above, the predominant inflammatory cell type in HIVE/SIVE is the monocyte-derived macrophage. The chemokines upregulated by astrocytes in HIVE/SIVE are largely specific to monocyte/macrophages [193, 207]. This suggests the possibility of a positive feedback system being initiated; a productively infected macrophage induces nearby astrocytes to up-regulate secretion of macrophage-specific chemokines, leading to lesion formation. The cytokine response of astrocytes includes a cornucopia of molecules including a variety of chemokines. It is intriguing that astrocytes will secrete a different “profile” of cytokines and chemokines in response to different classes of stimuli [208]. Below we discuss key cytokines and chemokines that are thought to play a role in SIVE/HIVE.
4.3. Microglia Activation and Cytokine Secretion
Microglia serve as a “first responder” to neuroinvasion by pathogens. An actin binding protein, AIF-1, is considered to be a microglial-specific marker within normal brain [209–211]. As such, AIF-1 is ideally suited to examining morphological changes in microglia. Ramified microglia sample their environment using long processes. These processes retract on activation, allowing the microglia to migrate to the source of infection [212]. Cultured microglia also have a ramified morphology until activated by, for example, SIV-infected macrophages [213].
Surprisingly, the presence of macrophages is more important to the microglial response rather than whether they are infected with virus or not. IL-6 and IL-8 are both induced to be secreted by microglia when coincubated with macrophages, highlighting their role as “first responders” to infiltrating innate immune cells such as monocyte/macrophages.
4.4. Expression and Secretion of Selected Cytokines
Productively infected macrophages in the encephalitic brain express TNF [195]. TNF-α receptors are present in the nonencephalitic brain [214], such that normal brains are primed to respond quickly to low levels of TNF. TNF induces increased chemokine production and secretion by astrocytes [215], and these chemokines induce monocyte migration preferentially over lymphocytes [193].
Vascular endothelial growth factor (VEGF) promotes proliferation of BMEC, resulting in reorganization of the cytoskeleton and tight junction proteins. This induces a decrease in BBB integrity, creating a permissive environment for monocyte migration, and also bidirectional leakage of proteins across the BBB. A possible mechanism for the VEGF pathway could be as follows: tat binds to the VEGF receptor [216], followed by the binding of the VEGF receptor to focal adhesion kinase [217], increases of which have been implicated in BBB disruption [218].
Other proinflammatory cytokines, including IFN-γ and IL-6, are upregulated in the encephalitic brain, with far-reaching effects in neuroinflammatory events [219]. The complement pathway is also known to be induced through IFN-γ and IL-6 signaling, resulting in propagation of inflammation in the area surrounding lesions. There are well-characterized neurotoxicity manifestations associated with HIV infection [220], including increased secretion of the neurotoxic IL-6 by glia in response to gp120 [221]. Therefore, rapid secretion of high levels of IL-6 by microglia would be anticipated to be a detrimental effect of SIV-infected macrophage infiltration into the brain [222].
4.5. Expression and Secretion of Selected Chemokines
Levels of the chemokine IL-8 has been recently demonstrated to be elevated in microglia in HIVE brain tissue [223], possibly in response to gp120 [224]. IL-8 have been shown to have neurotoxic effects and thus, plays a role in cognitive dysfunction associated with HIV [225]. Increased IL-8 expression observed in glial nodules may be largely due to a factor secreted by HIV-infected macrophages [213].
An early study of chemokine expression in brains of macaques infected with SIV showed increased CCL3, CCL5, CCL7, and CXCL10 [207], although no increase in CCL2, CCL8 (MCP-2), or CXCL8 was observed in this study. Other later studies have produced conflicting results. Penton-Rol used dexamethasone to stimulate cells to have increased CCL2 receptors before infecting with HIV 89.6 [226]. The Clements group at Johns Hopkins has shown increased CCL2 mRNA in brain extracts using a highly accelerated encephalitis model [227], although mRNA does not always equate with secreted protein. Additionally, the Berman group at Einstein College of Medicine has shown numerous effects of CCL2 on HIV-infected macrophages [200, 228]. CCL2 was among several chemokines in CSF that were not upregulated in one study using humans infected with HIV [229], although IP-10 was upregulated. In contrast, CCL2 was increased in pigtail macaques that develop encephalitis [230]. The precise cell types producing these chemokines were not identified in these studies. CCL2 mRNA was upregulated in cultured astrocytes, but remained at low levels compared to CCL7, suggesting a role for CCL7 in HIV-related encephalitis [193].
Even under noninflamed conditions, CCL7 is expressed in the brain [193, 207], which could contribute to basal levels of monocyte migration into the brain for “routine surveillance” [231]. That CCL7 is upregulated by astrocytes in response to cytokines present in encephalitic brains gives a potential role for controlling monocyte migration during encephalitis as well [193, 207]. In more recent studies, stimulation of astrocytes with TNF induced an increase in secretion of numerous cytokines [215]. Analyses of gene arrays of astrocytes treated with TNF showed that the only cytokine upregulated was CCL7. It is also possible that astrocytes provide a role for the resolution of inflammation through reduction in secretion of proinflammatory cytokines and increasing anti-inflammatory processes [198, 201, 202]. In the above study, polygonal astrocytes stimulated with TNF expressed higher levels of cytokines including VEGF than TNF-stimulated stellated astrocytes at the time points examined. Therefore, the order of stimulation of astrocytes is important in the subsequent secretion of cytokines [215].
5. Cytokines and Chemokines in Neuropathic Pain
Recent evidence suggests a strong correlation between inflammation following nerve damage and neuropathic pain [10, 23, 232]. Neuropathic pain is a complex syndrome resulting from many forms of peripheral nerve damage, for example, traumatic nerve injury, diabetes, infection, or drug-induced neuropathy, and immune and metabolic diseases [233]. Chronic pain can occur with peripheral nerve trauma and/or inflammation, autoimmune neuropathies and vasculitic neuropathies, or infection. Individuals who suffer from chronic pain experience prolonged pain at sites that may have been previously injured, yet are otherwise currently healthy. Chronic pain is associated with changes in neuroplasticity or changes in neural pathways, and synapses due to an erroneous reorganization of the nervous system, both peripherally and centrally. During the period of tissue damage, noxious stimuli and inflammation cause an elevation of nociceptive input from the periphery to the central nervous system. Prolonged nociception from the periphery elicits a neuroplastic response at the cortical level to change its somatotopic organization for the painful site, inducing central sensitization [234].
Peripheral nerves are the origin of almost all forms of neuropathic pain. Pain-responsive peripheral nerves reveal a remarkable degree of plasticity in both sensory neurons and spinal cord [10]. Immune processes can be directed against peripheral nerves, DRG, and dorsal roots resulting in pathological pain.
Immune activation near peripheral nerves may create increases in peripheral nerve excitability. Infectious agents as well as proinflammatory mediators produced by activated microglia can cause alterations in the blood-nerve barrier (BNB) as a result of chemoattractant molecules released at the site of the damaged peripheral nerve, which in turn recruit neutrophils and macrophages from the circulation into the nerve. Proinflammatory cytokines participate in this immune activation and orchestrate the early immune response. However, these inflammatory mediators can directly increase nerve excitability, damage myelin, and alter the permeability of the BNB leading to edema and further infiltration of immune cells. Schwann cells that ensheath peripheral nerves are macrophage-like and can present nonself-substances to T lymphocytes to further activate the immune cells. Schwann cells also participate in the removal of damaged myelin and cellular debris. Importantly, Schwann cells rapidly release the chemo-attractant CCL2 upon nerve damage that in turn recruits monocytes and T cells to the site of the nerve degeneration [10]. Proinflammatory cytokines have been repeatedly implicated in demyelination and degeneration of peripheral nerves, increases in sensory afferent excitability, and induction of neuropathic pain [23].
Inflammatory mediators elicited in the cells of the DRG and those produced by infiltrating immune cells and spinal microglial activation are key elements that mediate the signal transduction of the pain response [235]. Macrophages, lymphocytes, and satellite glial cells in the DRG and in the dorsal horn of the spinal cord participate in neuroimmune activation of glial cells, promoting the development of neuropathic pain. Since some chemokine receptors such as CCR2, CCR5, CXCR4, and CX3CR1 are located in primary afferent neurons or secondary neurons of the spinal dorsal horn [236], their chemokine ligands can potentially alter pain transmission. Peripheral administration of the chemokines CCL2, CCL3, CCL5, and CXCL12 has been shown to produce pain behaviors that are elicited by the activation of chemokine receptors in the DRG [236].
Importantly, CCL2 participates in pain regulation by directly interacting with sensory neurons and indirectly via peripheral leukocyte activation in the PNS [236, 237]. CCL2 has been shown to be elevated in primary sensory neurons after nerve injury, and CCR2 expression has been observed in both DRG neurons and activated Schwann cells in injured peripheral nerves [237]. Moreover, neuropathic pain induced by nerve injury is not elicited in CCR2 gene-deficient mice [236]. The addition of CCL2 to cultured DRG neurons triggered the release of calcitonin gene-related peptide (CGRP), a nociceptor neurotransmitter, from these cells, presumably as a result of increased neuronal excitation [238].
CCL3 has been found to be upregulated in activated Schwann cells and in infiltrating macrophages close to injured nerves. This chemokine participates in the development of neuropathic pain through its receptors CCR1 and CCR5, which are located in Schwann cells and macrophages [239]. Interestingly, there is an increase of fractalkine (CX3CL1), which is known to be both a pro- and anti-inflammatory molecule in injured nerves, and localization of its receptor CX3CR1 in recruited macrophages and DRG neurons. The activation of fractalkine-CX3CR1 has been shown to attenuate peripheral nerve injury-induced neuropathic pain [240].
The crosstalk between glial cells and neurons is important in the development of neuropathic pain [234]. Proinflammatory cytokines such as IL-1β, IL-6, and TNF produced by glial cells and neurons accelerate central pain sensitization, and inhibition of these cytokines in the CNS and PNS effectively reduces neuropathic pain [241]. Brain-derived neurotrophic factor (BDNF) derived from activated microglia potentiates the excitability of spinal neurons [242]. Microglial IL-18, a member of the IL-1 family, also plays a pivotal role in neuropathic pain [243]. IL-1β produced by macrophages and Schwann cells in injured nerves directly sensitizes nociceptors in primary afferent neurons [244]. IL-1 induces the release of substance P from DRG neurons [245] and neuropathic pain is reduced in IL-6 KO mice [246]. IL-6 can also contribute to pain by increasing the sensitivity of nerve endings [246]. IL-6 can enhance neuropathic pain in the dorsal horn by activating STAT3 signaling in glial cells after peripheral nerve injury. The STAT-3 pathway is a key mediator of signal transduction in neuropathic pain [247].
A recent study evaluated the role of axonal transport in neuroimmune communication following peripheral nerve injury, linking focal changes in Schwann cell activation and release of the proinflammatory cytokine TNF with subsequent activation and sensitization of ascending sensory neurons and glia that culminate in the neuropathic pain state. New data demonstrate that axonally transported (biotinylated) TNF-α activates and localizes with dorsal horn astrocytes within 96 hours after injection into the sciatic nerve and that glial GFAP-activation in these glial cells is diminished in TNF-α-receptor 1 KO mice [248].
IL-17 is an important regulator of immune responses and is involved in inducing and mediating proinflammatory reactions in a wide range of inflammatory and autoimmune diseases of the nervous system. Using IL-17 KO mice, it has been demonstrated that IL-17 contributes to neuroinflammatory responses and pain hypersensitivity following neuropathic injury [249]. Compared to wild-type, IL-17 KO mice displayed significantly decreased mechanical pain hypersensitivity as well as decreased infiltration of T cells and macrophages to the injured sciatic nerves, and the L3-L5 DRG, and decreased activation of microglia and astrocytes in the L3-L5 dorsal and ventral horns of the spinal cord. This work shows that IL-17 contributes to neuroinflammation and neuropathic pain following peripheral nerve injury and identifies IL-17 as a potential therapeutic target for treating neuropathic pain.
Recent studies have suggested that the C-C chemokine receptor (CCR)5 interacts with μ-opioid receptor and modifies a nociceptive reaction [250]. A study that examined effects of CCR5 deficiency on pain responses by employing CCR5 KO mice found that pain responses of CCR5 KO mice to chemical or inflammatory stimuli were milder than those of CCR5 wild-type mice [251]. Though the roles of proinflammatory cytokines and chemokines in neuropathic pain have been identified [252], the precise relationship between the chemokine-cytokine network and neuropathic pain is not yet well understood. Further studies are needed to understand the neuropathic regulatory mechanisms underlying neuroinflammation after nerve injury.
6. Conclusion
Cytokines and chemokines play an important role in mediating neuroinflammation and neurodegeneration in various kinds of inflammatory neurodegenerative diseases including bacterial meningitis, brain abscesses, Lyme neuroborreliosis, and HIV encephalitis described above. Interestingly, recent evidence suggests that peripheral and central neuroinflammation associated with cytokine-chemokine networks following nerve damage also play a central role in the pathogenesis of neuropathic pain. Although a link has been established between cytokines, chemokines, and neurodegeneration, their signaling mechanisms are complex and appear to involve a balance between promoting cell survival, apoptosis, and proinflammatory responses.
Although inflammation may induce beneficial effects such as pathogen clearance and phagocytosis of debris and apoptotic cells besides tissue repair processes, uncontrolled inflammation can result in detrimental outcomes via the production of neurotoxic factors that exacerbate neurodegenerative pathology. The factors that may disrupt this normal equilibrium remain largely unknown. Several cytokines and chemokines and their receptors orchestrate this immune response. Further, anti-inflammatory responses are regulated by proteins that inhibit signal transduction pathways, such as suppressor of cytokine signaling proteins, transcriptional repressors, and anti-inflammatory molecules that help control excessive inflammation. Chemokine recruitment of inflammatory cells to the sites of injury is instrumental in driving a secondary damage cascade.
Given the mounting evidence for their role in neurodegenerative disorders, cytokines and chemokines have received considerable attention as therapeutic targets. Considering that inflammation mediated by cytokines and chemokines is a common denominator in neurodegenerative diseases, targeting the correct timing of an immune response will be a pivotal factor in designing successful therapies. Further, it will be a challenge to design therapeutic agents that safely and effectively target only the detrimental mechanisms that contribute to disease pathogenesis, as cytokines and chemokines are vital for the normal functioning of the body. An understanding of the factors that dictate the switch from a protective to a deleterious inflammatory response will make possible interventions able to limit tissue damage. This type of intervention will also require a thorough understanding of the cell-subtype-specific action and cellular signaling pathways in CNS and PNS injury in order to design selective drug targets. Further investigations aimed at understanding chemokine-cytokine networks that are operative in the signal transduction of the pain response will prove beneficial in designing novel therapeutic strategies to alleviate neuropathic pain, a significant factor in neurodegenerative diseases.
Acknowledgments
The authors were supported by grant 51OD011104/P51RR000164 from the Office of Research Infrastructure Programs (ORIP) and the National Center for Research Resources of the National Institutes of Health. Geeta Ramesh and Mario T. Philipp were also supported by grant NS048952 (to Mario T. Philipp) from the National Institute of Neurologic Disorders and Stroke and Andrew G. MacLean by grant MH077544 from the National Institute of Mental Health.
References
- 1.Vilcek J. The cytokines: an overview. In: Angus W, Thomson MTL, editors. The Cytokine Handbook. 4th edition. San Diego, Calif, USA: Academic Press; pp. 1–18. [Google Scholar]
- 2.Walz A, Peveri P, Aschauer H, Baggiolini M. Purification and amino acid sequencing of NAF, a novel neutrophil-activating factor produced by monocytes. Biochemical and Biophysical Research Communications. 1987;149(2):755–761. doi: 10.1016/0006-291x(87)90432-3. [DOI] [PubMed] [Google Scholar]
- 3.Yoshimura T, Matsushima K, Oppenheim JJ, Leonard EJ. Neutrophil chemotactic factor produced by lipopolysaccharide (LPS)-stimulated human blood mononuclear leukocytes: partial characterization and separation from interleukin 1 (IL 1) Journal of Immunology. 1987;139(3):788–793. [PubMed] [Google Scholar]
- 4.Moser B, Loetscher P. Lymphocyte traffic control by chemokines. Nature Immunology. 2001;2(2):123–128. doi: 10.1038/84219. [DOI] [PubMed] [Google Scholar]
- 5.Devi LA. G-protein-coupled receptor dimers in the lime light. Trends in Pharmacological Sciences. 2000;21(9):324–326. doi: 10.1016/s0165-6147(00)01519-4. [DOI] [PubMed] [Google Scholar]
- 6.Hickey WF. Leukocyte traffic in the central nervous system: the participants and their roles. Seminars in Immunology. 1999;11(2):125–137. doi: 10.1006/smim.1999.0168. [DOI] [PubMed] [Google Scholar]
- 7.Takeshita Y, Ransohoff RM. Inflammatory cell trafficking across the blood-brain barrier: chemokine regulation and in vitro models. Immunological Reviews. 2012;248:228–239. doi: 10.1111/j.1600-065X.2012.01127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Benveniste EN. Inflammatory cytokines within the central nervous system: sources, function, and mechanism of action. American Journal of Physiology. 1992;263(1):C1–C16. doi: 10.1152/ajpcell.1992.263.1.C1. [DOI] [PubMed] [Google Scholar]
- 9.Cámara-Lemarroy CR, Guzmán-de La Garza FJ, Fernández-Garza NE. Molecular inflammatory mediators in peripheral nerve degeneration and regeneration. NeuroImmunoModulation. 2010;17(5):314–324. doi: 10.1159/000292020. [DOI] [PubMed] [Google Scholar]
- 10.Watkins LR, Maier SF. Beyond neurons: evidence that immune and glial cells contribute to pathological pain states. Physiological Reviews. 2002;82(4):981–1011. doi: 10.1152/physrev.00011.2002. [DOI] [PubMed] [Google Scholar]
- 11.Nilupul PM, Ma HK, Arakawa S, et al. Inflammation following stroke. Journal of Clinical Neuroscience. 2006;13(1):1–8. doi: 10.1016/j.jocn.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 12.De Chiara G, marcocci ME, Sgarbanti R, et al. Infectious agents and neurodegeneration. Molecular Neurobiology. 2012 doi: 10.1007/s12035-012-8320-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vilhardt F. Microglia: phagocyte and glia cell. International Journal of Biochemistry and Cell Biology. 2005;37(1):17–21. doi: 10.1016/j.biocel.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 14.Gendelman HE. Neural immunity: friend or foe? Journal of NeuroVirology. 2002;8(6):474–479. doi: 10.1080/13550280290168631. [DOI] [PubMed] [Google Scholar]
- 15.Erta M, Quintana A, Hidalgo J. Interleukin-6 a major cytokine in the central nervous system. International Journal of Biological Sciences. 2012;8:1254–1266. doi: 10.7150/ijbs.4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ye L, Huang Y, Zhao L, et al. IL-1β and TNF-α induce neurotoxicity through glutamate production: a potential role for neuronal glutaminase. Journal of Neurochemistry. 2013;125(6):897–908. doi: 10.1111/jnc.12263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumar M, Verma S, Nerurkar VR. Pro-inflammatory cytokines derived from West Nile virus (WNV)-infected SK-N-SH cells mediate neuroinflammatory markers and neuronal death. Journal of Neuroinflammation. 2010;7, article 73 doi: 10.1186/1742-2094-7-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lazovic J, Basu A, Lin H-W, et al. Neuroinflammation and both cytotoxic and vasogenic edema are reduced in interleukin-1 type 1 receptor-deficient mice conferring neuroprotection. Stroke. 2005;36(10):2226–2231. doi: 10.1161/01.STR.0000182255.08162.6a. [DOI] [PubMed] [Google Scholar]
- 19.Takahashi JL, Giuliani F, Power C, Imai Y, Yong VW. Interleukin-1β promotes oligodendrocyte death through glutamate excitotoxicity. Annals of Neurology. 2003;53(5):588–595. doi: 10.1002/ana.10519. [DOI] [PubMed] [Google Scholar]
- 20.Sherwin C, Fern R. Acute lipopolysaccharide-mediated injury in neonatal white matter glia: role of TNF-α, IL-1β, and calcium. Journal of Immunology. 2005;175(1):155–161. doi: 10.4049/jimmunol.175.1.155. [DOI] [PubMed] [Google Scholar]
- 21.Abo-Ouf H, Hooper AW, White EJ, van Rensburg HJJ, Trigatti BL, Igdoura SA. Deletion of tumor necrosis factor–α ameliorates neurodegeneration in Sandhoff disease mice. Human Molecular Genetics. 2013:1–16. doi: 10.1093/hmg/ddt250. [DOI] [PubMed] [Google Scholar]
- 22.Lu MO, Duan RS, Quezada HC, et al. Aggravation of experimental autoimmune neuritis in TNF-α receptor 1 deficient mice. Journal of Neuroimmunology. 2007;186(1-2):19–26. doi: 10.1016/j.jneuroim.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 23.Chertoff M, Di Paolo N, Schoeneberg A, et al. Neuroprotective and neurodegenerative effects of the chronic expression of tumor necrosis factor α in the nigrostriatal dopaminergic circuit of adult mice. Experimental Neurology. 2011;227(2):237–251. doi: 10.1016/j.expneurol.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 24.Cartier L, Hartley O, Dubois-Dauphin M, Krause K-H. Chemokine receptors in the central nervous system: role in brain inflammation and neurodegenerative diseases. Brain Research Reviews. 2005;48(1):16–42. doi: 10.1016/j.brainresrev.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 25.Yang G, Meng Y, Li W, et al. Neuronal MCP-1 mediates microglia recruitment and neurodegeneration induced by the mild impairment of oxidative metabolism. Brain Pathology. 2011;21(3):279–297. doi: 10.1111/j.1750-3639.2010.00445.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eugenin EA, D’Aversa TG, Lopez L, Calderon TM, Berman JW. MCP-1 (CCL2) protects human neurons and astrocytes from NMDA or HIV-tat-induced apoptosis. Journal of Neurochemistry. 2003;85(5):1299–1311. doi: 10.1046/j.1471-4159.2003.01775.x. [DOI] [PubMed] [Google Scholar]
- 27.Thompson WL, Karpus WJ, Van Eldik LJ. MCP-1-deficient mice show reduced neuroinflammatory responses and increased peripheral inflammatory responses to peripheral endotoxin insult. Journal of Neuroinflammation. 2008;5, article 35 doi: 10.1186/1742-2094-5-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spittau B, Zhou X, Ming M, Krieglstein K. IL6 protects MN9D cells and midbrain dopaminergic neurons from MPP+-induced neurodegeneration. NeuroMolecular Medicine. 2012;4:317–327. doi: 10.1007/s12017-012-8189-7. [DOI] [PubMed] [Google Scholar]
- 29.Salmaggi A, Ciusani E, De Rossi M, et al. Expression and modulation of IFN-γ-inducible chemokines (IP-10, Mig, and I-TAC) in human brain endothelium and astrocytes: possible relevance for the immune invasion of the central nervous system and the pathogenesis of multiple sclerosis. Journal of Interferon and Cytokine Research. 2002;22(6):631–640. doi: 10.1089/10799900260100114. [DOI] [PubMed] [Google Scholar]
- 30.Cinque P, Bestetti A, Marenzi R, et al. Cerebrospinal fluid interferon-γ-inducible protein 10 (IP-10, CXCL10) in HIV-1 infection. Journal of Neuroimmunology. 2005;168(1-2):154–163. doi: 10.1016/j.jneuroim.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 31.Xia MQ, Bacskai BJ, Knowles RB, Qin SX, Hyman BT. Expression of the chemokine receptor CXCR3 on neurons and the elevated expression of its ligand IP-10 in reactive astrocytes: in vitro ERK1/2 activation and role in Alzheimer's disease. Journal of Neuroimmunology. 2000;108(1-2):227–235. doi: 10.1016/s0165-5728(00)00285-x. [DOI] [PubMed] [Google Scholar]
- 32.Sui Y, Stehno-Bittel L, Li S, et al. CXCL10-induced cell death in neurons: role of calcium dysregulation. European Journal of Neuroscience. 2006;23(4):957–964. doi: 10.1111/j.1460-9568.2006.04631.x. [DOI] [PubMed] [Google Scholar]
- 33.Mattison HA, Nie H, Gao H, Zhou H, Zhang JS. Suppressed pro-inflammatory response of microglia in CX3CR1 knockout mice. Journal of Neuroimmunology. 2013;257(1-2):110–115. doi: 10.1016/j.jneuroim.2013.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Semple BD, Bye N, Ziebell JM, Morganti-Kossmann MC. Deficiency of the chemokine receptor CXCR2 attenuates neutrophil infiltration and cortical damage following closed head injury. Neurobiology of Disease. 2010;40(2):394–403. doi: 10.1016/j.nbd.2010.06.015. [DOI] [PubMed] [Google Scholar]
- 35.De Paola M, Buanne P, Biordi L, Bertini R, Ghezzi P, Mennini T. Chemokine MIP-2/CXCL2, acting on CXCR2, induces motor neuron death in primary cultures. NeuroImmunoModulation. 2007;14(6):310–316. doi: 10.1159/000123834. [DOI] [PubMed] [Google Scholar]
- 36.Roy M, Richard J-F, Dumas A, Vallières L. CXCL1 can be regulated by IL-6 and promotes granulocyte adhesion to brain capillaries during bacterial toxin exposure and encephalomyelitis. Journal of Neuroinflammation. 2012;9, article 18 doi: 10.1186/1742-2094-9-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu Y, Murakami F. Chemokine CXCL12 and its receptors in the developing central nervous system: emerging themes and future perspectives. Developmental Neurobiology. 2012;72(10):1349–1362. doi: 10.1002/dneu.22041. [DOI] [PubMed] [Google Scholar]
- 38.Li M, Ransohoff RM. Multiple roles of chemokine CXCL12 in the central nervous system: a migration from immunology to neurobiology. Progress in Neurobiology. 2008;84(2):116–131. doi: 10.1016/j.pneurobio.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shimoji M, Pagan F, Healton EB, Mocchetti I. CXCR4 and CXCL12 expression is increased in the nigro-striatal system of Parkinson’s disease. Neurotoxicity Research. 2009;16(3):318–328. doi: 10.1007/s12640-009-9076-3. [DOI] [PubMed] [Google Scholar]
- 40.Savarin-Vuaillat C, Ransohoff RM. Chemokines and chemokine receptors in neurological disease: raise, retain, or reduce? Neurotherapeutics. 2007;4(4):590–601. doi: 10.1016/j.nurt.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Owens T, Babcock AA, Millward JM, Toft-Hansen H. Cytokine and chemokine inter-regulation in the inflamed or injured CNS. Brain Research Reviews. 2005;48(2):178–184. doi: 10.1016/j.brainresrev.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 42.Eskandari F, Webster JI, Sternberg EM. Neural immune pathways and their connection to inflammatory diseases. Arthritis Research and Therapy. 2003;5(6):251–265. doi: 10.1186/ar1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smith JA, Das A, Ray SK, Banik NL. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Research Bulletin. 2012;87(1):10–20. doi: 10.1016/j.brainresbull.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140(6):918–934. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fallon BA, Levin ES, Schweitzer PJ, Hardesty D. Inflammation and central nervous system Lyme disease. Neurobiology of Disease. 2010;37(3):534–541. doi: 10.1016/j.nbd.2009.11.016. [DOI] [PubMed] [Google Scholar]
- 46.Asbury AK, Arnason BG, Adams RD. The inflammatory lesion in idiopathic polyneuritis. Its role in pathogenesis. Medicine. 1969;48(3):173–215. doi: 10.1097/00005792-196905000-00001. [DOI] [PubMed] [Google Scholar]
- 47.Halperin JJ. Lyme disease and the peripheral nervous system. Muscle and Nerve. 2003;28(2):133–143. doi: 10.1002/mus.10337. [DOI] [PubMed] [Google Scholar]
- 48.Bogliun G, Beghi E. Incidence and clinical features of acute inflammatory polyradiculoneuropathy in Lombardy, Italy, 1996. Acta Neurologica Scandinavica. 2004;110(2):100–106. doi: 10.1111/j.1600-0404.2004.00272.x. [DOI] [PubMed] [Google Scholar]
- 49.Kiguchi N, Kobayashi Y, Kishioka S. Chemokines and cytokines in neuroinflammation leading to neuropathic pain. Current Opinion in Pharmacology. 2012;12(1):55–61. doi: 10.1016/j.coph.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 50.Scheld WM, Koedel U, Nathan B, Pfister H-W. Pathophysiology of bacterial meningitis: mechanism(s) of neuronal injury. Journal of Infectious Diseases. 2002;186(2):S225–S233. doi: 10.1086/344939. [DOI] [PubMed] [Google Scholar]
- 51.Nau R, Brück W. Neuronal injury in bacterial meningitis: mechanisms and implications for therapy. Trends in Neurosciences. 2002;25(1):38–45. doi: 10.1016/s0166-2236(00)02024-5. [DOI] [PubMed] [Google Scholar]
- 52.Zysk G, Brück W, Huitinga I, et al. Elimination of blood-derived macrophages inhibits the release of interleukin-1 and the entry of leukocytes into the cerebrospinal fluid in experimental pneumococcal meningitis. Journal of Neuroimmunology. 1997;73(1-2):77–80. doi: 10.1016/s0165-5728(96)00173-7. [DOI] [PubMed] [Google Scholar]
- 53.Täuber MG, Moser B. Cytokines and chemokines in meningeal inflammation: biology and clinical implications. Clinical Infectious Diseases. 1999;28(1):1–12. doi: 10.1086/515079. [DOI] [PubMed] [Google Scholar]
- 54.Trostdorf F, Brück W, Schmitz-Salue M, et al. Reduction of meningeal macrophages does not decrease migration of granulocytes into the CSF and brain parenchyma in experimental pneumococcal meningitis. Journal of Neuroimmunology. 1999;99(2):205–210. doi: 10.1016/s0165-5728(99)00121-6. [DOI] [PubMed] [Google Scholar]
- 55.Pfister H-W, Scheld WM. Brain injury in bacterial meningitis: therapeutic implications. Current Opinion in Neurology. 1997;10(3):254–259. doi: 10.1097/00019052-199706000-00015. [DOI] [PubMed] [Google Scholar]
- 56.Koedel U, Scheld WM, Pfister H-W. Pathogenesis and pathophysiology of pneumococcal meningitis. Lancet Infectious Diseases. 2002;2(12):721–736. doi: 10.1016/s1473-3099(02)00450-4. [DOI] [PubMed] [Google Scholar]
- 57.Nau R, Brück W. Neuronal injury in bacterial meningitis: mechanisms and implications for therapy. Trends in Neurosciences. 2002;25(1):38–45. doi: 10.1016/s0166-2236(00)02024-5. [DOI] [PubMed] [Google Scholar]
- 58.Hanisch UK, Prinz M, Angstwurm K, et al. The protein tyrosine kinase inhibitor AG126 prevents the massive microglial cytokine induction by pneumococcal cell walls. European Journal of Immunology. 2001;31(7):2104–2115. doi: 10.1002/1521-4141(200107)31:7<2104::aid-immu2104>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 59.Rock RB, Gekker G, Hu S, et al. Role of microglia in central nervous system infections. Clinical Microbiology Reviews. 2004;17(4):942–964. doi: 10.1128/CMR.17.4.942-964.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Braun JS, Novak R, Murray PJ, et al. Apoptosis-inducing factor mediates microglial and neuronal apoptosis caused by pneumococcus. Journal of Infectious Diseases. 2001;184(10):1300–1309. doi: 10.1086/324013. [DOI] [PubMed] [Google Scholar]
- 61.Gerber J, Nau R. Mechanisms of injury in bacterial meningitis. Current Opinion in Neurology. 2010;23(3):312–318. doi: 10.1097/WCO.0b013e32833950dd. [DOI] [PubMed] [Google Scholar]
- 62.Fassbender K, Schminke U, Ries S, et al. Endothelial-derived adhesion molecules in bacterial meningitis: association to cytokine release and intrathecal leukocyte-recruitment. Journal of Neuroimmunology. 1997;74(1-2):130–144. doi: 10.1016/s0165-5728(96)00214-7. [DOI] [PubMed] [Google Scholar]
- 63.Täuber MG, Moser B. Cytokines and chemokines in meningeal inflammation: biology and clinical implications. The Journal of Infectious Diseases. 1985;151(3):528–534. doi: 10.1086/515079. [DOI] [PubMed] [Google Scholar]
- 64.Saukkonen K, Sande S, Cioffe C, et al. The role of cytokines in the generation of inflammation and tissue damage in experimental gram-positive meningitis. Journal of Experimental Medicine. 1990;171(2):439–448. doi: 10.1084/jem.171.2.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gerber J, Raivich G, Wellmer A, et al. A mouse model of Streptococcus pneumoniae meningitis mimicking several features of human disease. Acta Neuropathologica. 2001;101(5):499–508. doi: 10.1007/s004010000326. [DOI] [PubMed] [Google Scholar]
- 66.Grandgirard D, Steiner O, Täuber MG, Leib SL. An infant mouse model of brain damage in pneumococcal meningitis. Acta Neuropathologica. 2007;114(6):609–617. doi: 10.1007/s00401-007-0304-8. [DOI] [PubMed] [Google Scholar]
- 67.Saladino RA, Stack AM, Fleisher GR, et al. Development of a model of low-inoculum Streptococcus pneumoniae intrapulmonary infection in infant rats. Infection and Immunity. 1997;65(11):4701–4704. doi: 10.1128/iai.65.11.4701-4704.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Grandgirard D, Burri M, Agyeman P, Leib SL. Adjunctive daptomycin attenuates brain damage and hearing loss more efficiently than rifampin in infant rat pneumococcal meningitis. Antimicrob Agents Chemother. 2012;56(8):4289–4295. doi: 10.1128/AAC.00674-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gerber J, Böttcher T, Hahn M, Siemer A, Bunkowski S, Nau R. Increased mortality and spatial memory deficits in TNF-α-deficient mice in ceftriaxone-treated experimental pneumococcal meningitis. Neurobiology of Disease. 2004;16(1):133–138. doi: 10.1016/j.nbd.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 70.Sharief MK, Ciardi M, Thompson EJ. Blood-brain barrier damage in patients with bacterial meningitis: association with tumor necrosis factor-α but not interleukin-1β . Journal of Infectious Diseases. 1992;166(2):350–358. doi: 10.1093/infdis/166.2.350. [DOI] [PubMed] [Google Scholar]
- 71.Zwijnenburg PJG, Van der Poll T, Florquin S, Roord JJ, Van Furth AM. Interleukin-10 negatively regulates local cytokine and chemokine production but does not influence antibacterial host defense during murine pneumococcal meningitis. Infection and Immunity. 2003;71(4):2276–2279. doi: 10.1128/IAI.71.4.2276-2279.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zwijnenburg PJG, Van der Poll T, Florquin S, Roord JJ, Van Furth AM. Interleukin-10 negatively regulates local cytokine and chemokine production but does not influence antibacterial host defense during murine pneumococcal meningitis. Infection and Immunity. 2003;71(4):2276–2279. doi: 10.1128/IAI.71.4.2276-2279.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Orihuela CJ, Fillon S, Smith-Sielicki SH, et al. Cell wall-mediated neuronal damage in early sepsis. Infection and Immunity. 2006;74(7):3783–3789. doi: 10.1128/IAI.00022-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Koedel U, Bernatowicz A, Frei K, Fontana A, Pfister H-W. Systemically (but not intrathecally) administered IL-10 attenuates pathophysiologic alterations in experimental pneumococcal meningitis. Journal of Immunology. 1996;157(11):5185–5191. [PubMed] [Google Scholar]
- 75.Malipiero U, Koedel U, Pfister W, Fontana A. Bacterial meningitis: the role of transforming growth factor-beta in innate immunity and secondary brain damage. Neurodegenerative Diseases. 2007;4(1):43–50. doi: 10.1159/000100358. [DOI] [PubMed] [Google Scholar]
- 76.Ebert S, Phillips DJ, Jenzewski P, Nau R, O’Connor AE, Michel U. Activin A concentrations in human cerebrospinal fluid are age-dependent and elevated in meningitis. Journal of the Neurological Sciences. 2006;250(1-2):50–57. doi: 10.1016/j.jns.2006.06.026. [DOI] [PubMed] [Google Scholar]
- 77.Wilms H, Schwark T, Brandenburg L-O, et al. Regulation of activin A synthesis in microglial cells: pathophysiological implications for bacterial meningitis. Journal of Neuroscience Research. 2010;88(1):16–23. doi: 10.1002/jnr.22185. [DOI] [PubMed] [Google Scholar]
- 78.Kastenbauer S, Angele B, Sporer B, Pfister H-W, Koedel U. Patterns of protein expression in infectious meningitis: a cerebrospinal fluid protein array analysis. Journal of Neuroimmunology. 2005;164(1-2):134–139. doi: 10.1016/j.jneuroim.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 79.Spanaus K-S, Nadal D, Pfister H-W, et al. C-X-C and C-C chemokines are expressed in the cerebrospinal fluid in bacterial meningitis and mediate chemotactic activity on peripheral blood-derived polymorphonuclear and mononuclear cells in vitro. Journal of Immunology. 1997;158(4):1956–1964. [PubMed] [Google Scholar]
- 80.Zwijnenburg PJG, De Bie HMA, Roord JJ, Van Der Poll T, Van Furth AM. Chemotactic activity of CXCL5 in cerebrospinal fluid of children with bacterial meningitis. Journal of Neuroimmunology. 2003;145(1-2):148–153. doi: 10.1016/j.jneuroim.2003.09.013. [DOI] [PubMed] [Google Scholar]
- 81.Holub M, Beran O, Dzupová O, et al. Cortisol levels in cerebrospinal fluid correlate with severity and bacterial origin of meningitis. Critical Care. 2007;11(2):p. R41. doi: 10.1186/cc5729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Klein M, Paul R, Angele B, Popp B, Pfister H-W, Koedel U. Protein expression pattern in experimental pneumococcal meningitis. Microbes and Infection. 2006;8(4):974–983. doi: 10.1016/j.micinf.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 83.Stergaard C, Yieng-Kow RV, Larsen CG, et al. Treatment with a monoclonal antibody to IL-8 attenuates the pleocytosis in experimental pneumococcal meningitis in rabbits when given intravenously, but not intracisternally. Clinical and Experimental Immunology. 2000;122(2):207–211. doi: 10.1046/j.1365-2249.2000.01357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Prinz M, Kann O, Draheim HJ, et al. Microglial activation by components of gram-positive and -negative bacteria: distinct and common routes to the induction of ion channels and cytokines. Journal of Neuropathology and Experimental Neurology. 1999;58(10):1078–1089. doi: 10.1097/00005072-199910000-00006. [DOI] [PubMed] [Google Scholar]
- 85.Banerjee A, van Sorge NM, Sheen TR, Uchiyama S, Mitchell TJ, Doran KS. Activation of brain endothelium by pneumococcal neuraminidase NanA promotes bacterial internalization. Cellular Microbiology. 2010;12(11):1576–1588. doi: 10.1111/j.1462-5822.2010.01490.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mook-Kanamori BB, Geldhoff M, van der Poll T, van de Beek D. Pathogenesis and pathophysiology of pneumococcal meningitis. Clinical Microbiology Reviews. 2011;24(3):557–591. doi: 10.1128/CMR.00008-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mathisen GE, Johnson JP. Brain abscess. Clinical Infectious Diseases. 1997;4:763–779. doi: 10.1086/515541. [DOI] [PubMed] [Google Scholar]
- 88.Prasad KN, Mishra AM, Gupta D, Husain N, Husain M, Gupta RK. Analysis of microbial etiology and mortality in patients with brain abscess. Journal of Infection. 2006;53(4):221–227. doi: 10.1016/j.jinf.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 89.Townsend GC, Scheld WM. Infections of the central nervous system. Advances in Internal Medicine. 1998;43:403–447. [PubMed] [Google Scholar]
- 90.Kielian T, Bearden ED, Baldwin AC, Esen N. IL-1 and TNF-α play a pivotal role in the host immune response in a mouse model of Staphylococcus aureus-induced experimental brain abscess. Journal of Neuropathology and Experimental Neurology. 2004;63(4):381–396. doi: 10.1093/jnen/63.4.381. [DOI] [PubMed] [Google Scholar]
- 91.Baldwin AC, Kielian T. Persistent immune activation associated with a mouse model of Staphylococcus aureus-induced experimental brain abscess. Journal of Neuroimmunology. 2004;151(1-2):24–32. doi: 10.1016/j.jneuroim.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 92.Kielian T, Barry B, Hickey WF. CXC chemokine receptor-2 ligands are required for neutrophil-mediated host defense in experimental brain abscesses. Journal of Immunology. 2001;166(7):4634–4643. doi: 10.4049/jimmunol.166.7.4634. [DOI] [PubMed] [Google Scholar]
- 93.Kielian T, Phulwani NK, Esen N, et al. MyD88-dependent signals are essential for the host immune response in experimental brain abscess. Journal of Immunology. 2007;178(7):4528–4537. doi: 10.4049/jimmunol.178.7.4528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kielian T, Mayes P, Kielian M. Characterization of microglial responses to Staphylococcus aureus: effects on cytokine, costimulatory molecule, and Toll-like receptor expression. Journal of Neuroimmunology. 2002;130(1-2):86–99. doi: 10.1016/s0165-5728(02)00216-3. [DOI] [PubMed] [Google Scholar]
- 95.Kielian T, Esen N, Bearden ED. Toll-like receptor 2 (TLR2) is pivotal for recognition of S. aureus peptidoglycan but not intact bacteria by microglia. GLIA. 2005;49(4):567–576. doi: 10.1002/glia.20144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gurley C, Nichols J, Liu S, Phulwani NK, Esen N, Kielian T. Microglia and astrocyte activation by Toll-like receptor ligands: modulation by PPAR-γ agonists. PPAR Research. 2008;2008:15 pages. doi: 10.1155/2008/453120.453120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Esen N, Kielian T. Central role for MyD88 in the responses of microglia to pathogen-associated molecular patterns. Journal of Immunology. 2006;176(11):6802–6811. doi: 10.4049/jimmunol.176.11.6802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rath TJ, Hughes M, Arabi M, Shah GV. Imaging of cerebritis, encephalitis, and brain abscess. Neuroimaging Clinics of North America. 2012;4:585–607. doi: 10.1016/j.nic.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 99.Flaris NA, Hickey WF. Development and characterization of an experimental model of brain abscess in the rat. American Journal of Pathology. 1992;141(6):1299–1307. [PMC free article] [PubMed] [Google Scholar]
- 100.Kielian T, Hickey WF. Proinflammatory cytokine, chemokine, and cellular adhesion molecule expression during the acute phase of experimental brain abscess development. American Journal of Pathology. 2000;157(2):647–658. doi: 10.1016/S0002-9440(10)64575-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Esen N, Wagoner G, Philips N. Evaluation of capsular and acapsular strains of S. aureus in an experimental brain abscess model. Journal of Neuroimmunology. 2010;218(1-2):83–93. doi: 10.1016/j.jneuroim.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kielian T. Immunopathogenesis of brain abscess. Journal of Neuroinflammation. 2004;1(1, article 16) doi: 10.1186/1742-2094-1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cassateua MA. Neutrophil-derived proteins: selling cytokines by the pound. Advances in Immunology. 1999;(73):369–509. doi: 10.1016/s0065-2776(08)60791-9. [DOI] [PubMed] [Google Scholar]
- 104.Lo WD, Chen R, Boue DR, Stokes BT. Effect of neutrophil depletion in acute cerebritis. Brain Research. 1998;802(1-2):175–183. doi: 10.1016/s0006-8993(98)00600-3. [DOI] [PubMed] [Google Scholar]
- 105.Kielian T, Esen N, Liu S, et al. Minocycline modulates neuroinflammation independently of its antimicrobial activity in Staphylococcus aureus-induced brain abscess. American Journal of Pathology. 2007;171(4):1199–1214. doi: 10.2353/ajpath.2007.070231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Burgdorfer W, Barbour AG, Hayes SF. Lyme disease—a tick-borne spirochetosis? Science. 1982;216(4552):1317–1319. doi: 10.1126/science.7043737. [DOI] [PubMed] [Google Scholar]
- 107.Rupprecht TA, Koedel U, Fingerle V, Pfister H-W. The pathogenesis of Lyme neuroborreliosis: from infection to inflammation. Molecular Medicine. 2008;14(3-4):205–212. doi: 10.2119/2007-00091.Rupprecht. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Halperin JJ. Nervous system Lyme disease. Journal of the Royal College of Physicians of Edinburgh. 2010;40(3):248–255. doi: 10.4997/JRCPE.2010.314. [DOI] [PubMed] [Google Scholar]
- 109.Fallon BA, Levin ES, Schweitzer PJ, Hardesty D. Inflammation and central nervous system Lyme disease. Neurobiology of Disease. 2010;37(3):534–541. doi: 10.1016/j.nbd.2009.11.016. [DOI] [PubMed] [Google Scholar]
- 110.Elamin M, Monaghan T, Mulllins G, et al. The clinical spectrum of Lyme neuroborreliosis. Irish Medical Journal. 2010;103(2):46–49. [PubMed] [Google Scholar]
- 111.Lana-Peixoto MA. Multiple sclerosis and positive Lyme serology. Arquivos de Neuro-Psiquiatria. 1994;52(4):566–571. doi: 10.1590/s0004-282x1994000400019. [DOI] [PubMed] [Google Scholar]
- 112.Brinar VV, Habek M. Rare infections mimicking MS. Clinical Neurology and Neurosurgery. 2010;112(7):625–628. doi: 10.1016/j.clineuro.2010.04.011. [DOI] [PubMed] [Google Scholar]
- 113.Oksi J, Kalimo H, Marttila RJ, et al. Inflammatory brain changes in Lyme borreliosis A report on three patients and review of literature. Brain. 1996;119(6):2143–2154. doi: 10.1093/brain/119.6.2143. [DOI] [PubMed] [Google Scholar]
- 114.Keil R, Baron R, Kaiser R, Deuschl G. Vasculitic course of neuroborreliosis with thalamic infarction. Nervenarzt. 1997;68(4):339–341. doi: 10.1007/s001150050133. [DOI] [PubMed] [Google Scholar]
- 115.Topakian R, Stieglbauer K, Nussbaumer K, Aichner FT. Cerebral vasculitis and stroke in Lyme neuroborreliosis: two case reports and review of current knowledge. Cerebrovascular Diseases. 2008;26(5):455–461. doi: 10.1159/000155982. [DOI] [PubMed] [Google Scholar]
- 116.Kohler J. Lyme borreliosis: a case of transverse myelitis with syrinx cavity. Neurology. 1989;39(11):1553–1554. doi: 10.1212/wnl.39.11.1553-a. [DOI] [PubMed] [Google Scholar]
- 117.Benach JL, Cameron DJ, Donnenberg MS, Luft BJ, Dattwyler RJ. Borrelia burgdorferi in the central nervous system. Journal of the American Medical Association. 1992;268(7):872–873. [Google Scholar]
- 118.Duray PH, Steere AC. Clinical pathologic correlations of Lyme disease by stage. Annals of the New York Academy of Sciences. 1988;539:65–79. doi: 10.1111/j.1749-6632.1988.tb31839.x. [DOI] [PubMed] [Google Scholar]
- 119.Blanc F, Ballonzoli L, Marcel C, De Martino S, Jaulhac B, de Seze J. Lyme optic neuritis. Journal of the Neurological Sciences. 2010;295(1-2):117–119. doi: 10.1016/j.jns.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 120.Durovska J, Bazovska S, Pancak J, Zaborska M, Derdakova M, Traubner P. Infection with B. burgdorferi s.l., and the CNS demyelinating disease. A case report. Neuroendocrinology Letters. 2011;32(4):411–414. [PubMed] [Google Scholar]
- 121.Bigi S, Aebi C, Nauer C, Bigler S, Steinlin M. Acute transverse myelitis in Lyme neuroborreliosis. Infection. 2010;38(5):413–416. doi: 10.1007/s15010-010-0028-x. [DOI] [PubMed] [Google Scholar]
- 122.Vallat JM, Hugon J, Lubeau M. Tick-bite meningoradiculoneuritis: clinical, electrophysiologic, and histologic findings in 10 cases. Neurology. 1987;37(5):749–753. doi: 10.1212/wnl.37.5.749. [DOI] [PubMed] [Google Scholar]
- 123.Logigian EL. Peripheral nervous system Lyme borreliosis . Seminars in Neurology. 1997;17(1):25–28. doi: 10.1055/s-2008-1040909. [DOI] [PubMed] [Google Scholar]
- 124.Meurs L, Labeye D, Declercq I, Piéret F, Gille M. Acute transverse myelitis as a main manifestation of early stage II neuroborreliosis in two patients. European Neurology. 2004;52(3):186–188. doi: 10.1159/000081864. [DOI] [PubMed] [Google Scholar]
- 125.Koc F, Bozdemir H, Pekoz T, Aksu HS, Ozcan S, Kurdak H. Lyme disease presenting as subacute transverse myelitis. Acta Neurologica Belgica. 2009;109(4):326–329. [PubMed] [Google Scholar]
- 126.Halperin J, Luft BJ, Volkman DJ, Dattwyler RJ. Lyme neuroborreliosis: peripheral nervous system manifestations. Brain. 1990;113(4):1207–1221. doi: 10.1093/brain/113.4.1207. [DOI] [PubMed] [Google Scholar]
- 127.Halperin JJ. Neuroborreliosis. American Journal of Medicine. 1995;98(4):52S–56S. doi: 10.1016/s0002-9343(99)80044-8. [DOI] [PubMed] [Google Scholar]
- 128.Sellati TJ, Burns MJ, Ficazzola MA, Furie MB. Borrelia burgdorferi upregulates expression of adhesion molecules on endothelial cells and promotes transendothelial migration of neutrophils in vitro. Infection and Immunity. 1995;63(11):4439–4447. doi: 10.1128/iai.63.11.4439-4447.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Garcia-Monco JC, Fernandez Villar B, Benach JL. Lyme borreliosis: neurological features. Neurologia. 1990;5(9):315–322. [PubMed] [Google Scholar]
- 130.Meurers B, Kohlhepp W, Gold R, Rohrbach E, Mertens HG. Histopathological findings in the central and peripheral nervous systems in neuroborreliosis. A report of three cases. Journal of Neurology. 1990;237(2):113–116. doi: 10.1007/BF00314674. [DOI] [PubMed] [Google Scholar]
- 131.Duray PH. Histopathology of clinical phases of human Lyme disease. Rheumatic Disease Clinics of North America. 1989;15(4):691–710. [PubMed] [Google Scholar]
- 132.Weller M, Stevens A, Sommer N, Wietholter H, Dichgans J. Cerebrospinal fluid interleukins, immunoglobulins, and fibronectin in neuroborreliosis. Archives of Neurology. 1991;48(8):837–841. doi: 10.1001/archneur.1991.00530200079022. [DOI] [PubMed] [Google Scholar]
- 133.Grusell M, Widhe M, Ekerfelt C. Increased expression of the Th1-inducing cytokines interleukin-12 and interleukin-18 in cerebrospinal fluid but not in sera from patients with Lyme neuroborreliosis. Journal of Neuroimmunology. 2002;131(1-2):173–178. doi: 10.1016/s0165-5728(02)00255-2. [DOI] [PubMed] [Google Scholar]
- 134.Widhe M, Jarefors S, Ekerfelt C, et al. Borrelia-specific interferon-γ and interleukin-4 secretion in cerebrospinal fluid and blood during Lyme borreliosis in humans: association with clinical outcome. Journal of Infectious Diseases. 2004;189(10):1881–1891. doi: 10.1086/382893. [DOI] [PubMed] [Google Scholar]
- 135.Widhe M, Skogman BH, Jarefors S, et al. Up-regulation of Borrelia-specific IL-4- and IFN-γ-secreting cells in cerebrospinal fluid from children with Lyme neuroborreliosis. International Immunology. 2005;17(10):1283–1291. doi: 10.1093/intimm/dxh304. [DOI] [PubMed] [Google Scholar]
- 136.Grygorczuk S, Zajkowska J, Swierzbińska R, Pancewicz S, Kondrusik M, Hermanowska-Szpakowicz T. Concentration of interferon-inducible T cell chemoattractant and monocyte chemotactic protein-1 in serum and cerebrospinal fluid of patients with Lyme borreliosis . Roczniki Akademii Medycznej w Białymstoku. 2005;50:173–178. [PubMed] [Google Scholar]
- 137.Rupprecht TA, Koedel U, Muhlberger B, Wilske B, Fontana A, Pfister H-W. CXCL11 is involved in leucocyte recruitment to the central nervous system in neuroborreliosis. Journal of Neurology. 2005;252(7):820–823. doi: 10.1007/s00415-005-0752-9. [DOI] [PubMed] [Google Scholar]
- 138.Rupprecht TA, Pfister H-W, Angele B, Kastenbauer S, Wilske B, Koedel U. The chemokine CXCL13 (BLC): a putative diagnostic marker for neuroborreliosis. Neurology. 2005;65(3):448–450. doi: 10.1212/01.wnl.0000171349.06645.79. [DOI] [PubMed] [Google Scholar]
- 139.Ljøstad U, Mygland Å. CSF B—Lymphocyte chemoattractant (CXCL13) in the early diagnosis of acute Lyme neuroborreliosis. Journal of Neurology. 2008;255(5):732–737. doi: 10.1007/s00415-008-0785-y. [DOI] [PubMed] [Google Scholar]
- 140.Kowarik MC, Cepok S, Sellner J, et al. CXCL13 is the major determinant for B cell recruitment to the CSF during neuroinflammation. Journal of Neuroinflammation. 2012;9:p. 93. doi: 10.1186/1742-2094-9-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Rupprecht T, Koedel U, Angele B, Fingerle V, Pfister H-W. Cytokine CXCL13—a possible early CSF marker for neuroborreliosis. Nervenarzt. 2006;77(4):470–473. doi: 10.1007/s00115-005-2021-7. [DOI] [PubMed] [Google Scholar]
- 142.Cadavid D. The mammalian host response to borrelia infection. Wiener Klinische Wochenschrift. 2006;118(21-22):653–658. doi: 10.1007/s00508-006-0692-0. [DOI] [PubMed] [Google Scholar]
- 143.Halperin JJ, Heyes MP. Neuroactive kynurenines in Lyme borreliosis . Neurology. 1992;42(1):43–50. doi: 10.1212/wnl.42.1.43. [DOI] [PubMed] [Google Scholar]
- 144.Petit TL. The neurobiology of learning and memory: elucidation of the mechanisms of cognitive dysfunction. NeuroToxicology. 1988;9(3):413–428. [PubMed] [Google Scholar]
- 145.Chaparro-Huerta V, Rivera-Cervantes MC, Flores-Soto ME, Gómez-Pinedo U, Beas-Zárate C. Proinflammatory cytokines and apoptosis following glutamate-induced excitotoxicity mediated by p38 MAPK in the hippocampus of neonatal rats. Journal of Neuroimmunology. 2005;165(1-2):53–62. doi: 10.1016/j.jneuroim.2005.04.025. [DOI] [PubMed] [Google Scholar]
- 146.Philipp MT, Aydintug MK, Bohm RP, Jr., et al. Early and early disseminated phases of Lyme disease in the rhesus monkey: a model for infection in humans. Infection and Immunity. 1993;61(7):3047–3059. doi: 10.1128/iai.61.7.3047-3059.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Pachner AR, Delaney E, O’Neill T, Major E. Inoculation of nonhuman primates with the N40 strain of Borrelia burgdorferi leads to a model of Lyme neuroborreliosis faithful to the human disease. Neurology. 1995;45(1):165–172. doi: 10.1212/wnl.45.1.165. [DOI] [PubMed] [Google Scholar]
- 148.Norgard MV, Arndt LL, Akins DR, Curetty LL, Harrich DA, Radolf JD. Activation of human monocytic cells by Treponema pallidum and Borrelia burgdorferi lipoproteins and synthetic lipopeptides proceeds via a pathway distinct from that of lipopolysaccharide but involves the transcriptional activator NF-κB. Infection and Immunity. 1996;64(9):3845–3852. doi: 10.1128/iai.64.9.3845-3852.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ramesh G, Alvarez AL, Roberts ED, et al. Pathogenesis of Lyme neuroborreliosis: Borrelia burgdorferi lipoproteins induce both proliferation and apoptosis in rhesus monkey astrocytes. European Journal of Immunology. 2003;33(9):2539–2550. doi: 10.1002/eji.200323872. [DOI] [PubMed] [Google Scholar]
- 150.Ramesh G, Philipp MT. Pathogenesis of Lyme neuroborreliosis: mitogen-activated protein kinases Erk1, Erk2, and p38 in the response of astrocytes to Borrelia burgdorferi lipoproteins. Neuroscience Letters. 2005;384(1-2):112–116. doi: 10.1016/j.neulet.2005.04.069. [DOI] [PubMed] [Google Scholar]
- 151.Bernardino ALF, Myers TA, Alvarez X, Hasegawa A, Philipp MT. Toll-like receptors: insights into their possible role in the pathogenesis of Lyme neuroborreliosis. Infection and Immunity. 2008;76(10):4385–4395. doi: 10.1128/IAI.00394-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Myers TA, Kaushal D, Philipp MT. Microglia are mediators of Borrelia burgdorferi-induced apoptosis in SH-SY5Y neuronal cells. PLoS Pathogens. 2009;5(11) doi: 10.1371/journal.ppat.1000659.e1000659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ramesh G, Borda JT, Dufour J, et al. Interaction of the Lyme disease spirochete Borrelia burgdorferi with brain parenchyma elicits inflammatory mediators from glial cells as well as glial and neuronal apoptosis. American Journal of Pathology. 2008;173(5):1415–1427. doi: 10.2353/ajpath.2008.080483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ramesh G, Borda JT, Gill A, et al. Possible role of glial cells in the onset and progression of Lyme neuroborreliosis. Journal of Neuroinflammation. 2009;6, article 23 doi: 10.1186/1742-2094-6-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Pachner AR, Amemiya K, Delaney E, O’Neill T, Hughes CAN, Zhang W-F. Interleukin-6 is expressed at high levels in the CNS in Lyme neuroborreliosis. Neurology. 1997;49(1):147–152. doi: 10.1212/wnl.49.1.147. [DOI] [PubMed] [Google Scholar]
- 156.Narayan K, Dail D, Li L, et al. The nervous system as ectopic germinal center: CXCL13 and IgG in Lyme neuroborreliosis. Annals of Neurology. 2005;57(6):813–823. doi: 10.1002/ana.20486. [DOI] [PubMed] [Google Scholar]
- 157.Pachner AR, Steiner I. Lyme neuroborreliosis: infection, immunity, and inflammation. Lancet Neurology. 2007;6(6):544–552. doi: 10.1016/S1474-4422(07)70128-X. [DOI] [PubMed] [Google Scholar]
- 158.Wei XH, Na XD, Liao GJ, et al. The up-regulation of IL-6 in DRG and spinal horn contributes to neuopathic pain following L5 ventral horn transection. Experimental Neurology. 2013;241:159–168. doi: 10.1016/j.expneurol.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 159.Ramesh G, Benge S, Pahar B, Philipp MT. A possible role for inflammation in mediating apoptosis of oligodendrocytes as induced by the Lyme disease spirochete Borrelia burgdorferi . Journal of Neuroinflammation. 2012;2012:p. 72. doi: 10.1186/1742-2094-9-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Grütter MG. Caspases: key players in programmed cell death. Current Opinion in Structural Biology. 2000;10(6):649–655. doi: 10.1016/s0959-440x(00)00146-9. [DOI] [PubMed] [Google Scholar]
- 161.Bø L, Vedeler CA, Nyland HI, Trapp BD, Mørk SJ. Subpial demyelination in the cerebral cortex of multiple sclerosis patients. Journal of Neuropathology and Experimental Neurology. 2003;62(7):723–732. doi: 10.1093/jnen/62.7.723. [DOI] [PubMed] [Google Scholar]
- 162.Bradl M, Lassmann H. Oligodendrocytes: biology and pathology. Acta Neuropathologica. 2010;119(1):37–53. doi: 10.1007/s00401-009-0601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Hisahara S, Okano H, Miura M. Caspase-mediated oligodendrocyte cell death in the pathogenesis of autoimmune demyelination. Neuroscience Research. 2003;46(4):387–397. doi: 10.1016/s0168-0102(03)00127-5. [DOI] [PubMed] [Google Scholar]
- 164.Stadelmann C, Wegner C, Brück W. Inflammation, demyelination, and degeneration—recent insights from MS pathology. Biochimica et Biophysica Acta. 2011;1812(2):275–282. doi: 10.1016/j.bbadis.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 165.Conductier G, Blondeau N, Guyon A, Nahon J-L, Rovère C. The role of monocyte chemoattractant protein MCP1/CCL2 in neuroinflammatory diseases. Journal of Neuroimmunology. 2010;224(1-2):93–100. doi: 10.1016/j.jneuroim.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 166.Gerard C, Rollins BJ. Chemokines and disease. Nature Immunology. 2001;2(2):108–115. doi: 10.1038/84209. [DOI] [PubMed] [Google Scholar]
- 167.Rollins BJ. Monocyte chemoattractant protein 1: a potential regulator of monocyte recruitment in inflammatory disease. Molecular Medicine Today. 1996;2(5):198–204. doi: 10.1016/1357-4310(96)88772-7. [DOI] [PubMed] [Google Scholar]
- 168.Simpson JE, Newcombe J, Cuzner ML, Woodroofe MN. Expression of monocyte chemoattractant protein-1 and other β- chemokines by resident glia and inflammatory cells in multiple sclerosis lesions. Journal of Neuroimmunology. 1998;84(2):238–249. doi: 10.1016/s0165-5728(97)00208-7. [DOI] [PubMed] [Google Scholar]
- 169.Ransohoff RM, Hamilton TA, Tani M, et al. Astrocyte expression of mRNA encoding cytokines IP-10 and JE/MCP-1 in experimental autoimmune encephalomyelitis. FASEB Journal. 1993;7(6):592–600. doi: 10.1096/fasebj.7.6.8472896. [DOI] [PubMed] [Google Scholar]
- 170.Hinojosa AE, Garcia-Bueno B, Leza JC, Madrigal JLM. CCL2/MCP-1 modulation of microglial activation and proliferation. Journal of Neuroinflammation. 2011;8, article 77 doi: 10.1186/1742-2094-8-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Perrin FE, Lacroix S, Avilés-Trigueros M, David S. Involvement of moncyte chemo-attractant protein-1, macrophage inflammatory protein-1 alpha and interleukin-1 beta in Wallerian degeneration. Brain. 2005;128, part 4:854–866. doi: 10.1093/brain/awh407. [DOI] [PubMed] [Google Scholar]
- 172.Kindstrand E, Nilsson BY, Hovmark A, et al. Polyneuropathy in late Lyme borreliosis—a clinical, neurophysiological and morphological description. Acta Neurologica Scandinavica. 2000;101(1):47–52. doi: 10.1034/j.1600-0404.2000.00009.x. [DOI] [PubMed] [Google Scholar]
- 173.Merrill JE, Benveniste EN. Cytokines in inflammatory brain lesions: helpful and harmful. Trends in Neurosciences. 1996;19(8):331–338. doi: 10.1016/0166-2236(96)10047-3. [DOI] [PubMed] [Google Scholar]
- 174.Rothwell NJ, Strijbos PJLM. Cytokines in neurodegeneration and repair. International Journal of Developmental Neuroscience. 1995;13(3-4):179–185. doi: 10.1016/0736-5748(95)00018-c. [DOI] [PubMed] [Google Scholar]
- 175.Raivich G, Jones LL, Werner A, Blüthmann H, Doetschmann T, Kreutzberg GW. Molecular signals for glial activation: pro- and anti-inflammatory cytokines in the injured brain. Acta Neurochirurgica, Supplement. 1999;1999(73):21–30. doi: 10.1007/978-3-7091-6391-7_4. [DOI] [PubMed] [Google Scholar]
- 176.Minghetti L. Role of inflammation in neurodegenerative diseases. Current Opinion in Neurology. 2005;18(3):315–321. doi: 10.1097/01.wco.0000169752.54191.97. [DOI] [PubMed] [Google Scholar]
- 177.Schmitz T, Chew L-J. Cytokines and myelination in the central nervous system. The Scientific World Journal. 2008;8:1119–1147. doi: 10.1100/tsw.2008.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Spooren A, Kolmus K, Laureys G, et al. Interleukin-6, a mental cytokine. Brain Research Reviews. 2011;67(1-2):157–183. doi: 10.1016/j.brainresrev.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 179.Pizzi M, Sarnico I, Boroni F, et al. Prevention of neuron and oligodendrocyte degeneration by interleukin-6 (IL-6) and IL-6 receptor/IL-6 fusion protein in organotypic hippocampal slices. Molecular and Cellular Neuroscience. 2004;25(2):301–311. doi: 10.1016/j.mcn.2003.10.022. [DOI] [PubMed] [Google Scholar]
- 180.Barres BA, Schmid R, Sendnter M, Raff MC. Multiple extracellular signals are required for long-term oligodendrocyte survival. Development. 1993;118(1):283–295. doi: 10.1242/dev.118.1.283. [DOI] [PubMed] [Google Scholar]
- 181.Grygorczuk S, Pancewicz S, Zajkowska J, Kondrusik M, Świerzbińska R, Hermanowska-Szpakowicz T. Concentrations of macrophage inflammatory proteins MIP-1α and MIP-1β and interleukin 8 (Il-8) in Lyme borreliosis . Infection. 2004;32(6):350–355. doi: 10.1007/s15010-004-3110-4. [DOI] [PubMed] [Google Scholar]
- 182.Kossmann T, Stahel PF, Lenzlinger PM, et al. Interleukin-8 released into the cerebrospinal fluid after brain injury is associated with blood-brain barrier dysfunction and nerve growth factor production. Journal of Cerebral Blood Flow and Metabolism. 1997;17(3):280–289. doi: 10.1097/00004647-199703000-00005. [DOI] [PubMed] [Google Scholar]
- 183.Dumont RA, Car BD, Voitenok NN, et al. Systemic neutralization of interleukin-8 markedly reduces neutrophilic pleocytosis during experimental lipopolysaccharide-induced meningitis in rabbits. Infection and Immunity. 2000;68(10):5756–5763. doi: 10.1128/iai.68.10.5756-5763.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Thirumangalakudi L, Yin L, Rao HV, Grammas P. IL-8 induces expression of matrix metalloproteinases, cell cycle and pro-apoptotic proteins, and cell death in cultured neurons. Journal of Alzheimer’s Disease. 2007;11(3):305–311. doi: 10.3233/jad-2007-11307. [DOI] [PubMed] [Google Scholar]
- 185.Nordberg M, Forsberg P, Johansson A, et al. Cytotoxic mechanisms may play a role in the local immune response in the central nervous system in neuroborreliosis. Journal of Neuroimmunology. 2011;232(1-2):186–193. doi: 10.1016/j.jneuroim.2010.09.028. [DOI] [PubMed] [Google Scholar]
- 186.Ekerfelt C, Jarefors S, Tynngård N, et al. Phenotypes indicating cytolytic properties of Borrelia-specific interferon-γ secreting cells in chronic Lyme neuroborreliosis. Journal of Neuroimmunology. 2003;145(1-2):115–126. doi: 10.1016/j.jneuroim.2003.08.037. [DOI] [PubMed] [Google Scholar]
- 187.Rasley A, Tranguch SL, Rati DM, Marriott I. Murine glia express the immunosuppressive cytokine, interleukin-10, following exposure to Borrelia burgdorferi or Neisseria meningitidis. GLIA. 2006;53(6):583–592. doi: 10.1002/glia.20314. [DOI] [PubMed] [Google Scholar]
- 188.Bernardino ALF, Kaushal D, Philipp MT. The antibiotics doxycycline and minocycline inhibit the inflammatory responses to the Lyme disease spirochete borrelia burgdorferi . Journal of Infectious Diseases. 2009;199(9):1379–1388. doi: 10.1086/597807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.McArthur JC, Brew BJ, Nath A. Neurological complications of HIV infection. Lancet Neurology. 2005;4(9):543–555. doi: 10.1016/S1474-4422(05)70165-4. [DOI] [PubMed] [Google Scholar]
- 190.Brabers NACH, Nottet HSLM. Role of the pro-inflammatory cytokines TNF-α and IL-1β in HIV-associated dementia. European Journal of Clinical Investigation. 2006;36(7):447–458. doi: 10.1111/j.1365-2362.2006.01657.x. [DOI] [PubMed] [Google Scholar]
- 191.Hallett MB, Lloyds D. Neutrophil priming: the cellular signals that say “amber” but not “green”. Immunology Today. 1995;16(6):264–268. doi: 10.1016/0167-5699(95)80178-2. [DOI] [PubMed] [Google Scholar]
- 192.Elbim C, Pillet S, Prevost MH, et al. Redox and activation status of monocytes from human immunodeficiency virus-infected patients: relationship with viral load. Journal of Virology. 1999;73(6):4561–4566. doi: 10.1128/jvi.73.6.4561-4566.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Renner NA, Ivey NS, Redmann RK, Lackner AA, MacLean AG. MCP-3/CCL7 production by astrocytes: implications for SIV neuroinvasion and AIDS encephalitis. Journal of NeuroVirology. 2011;17(2):146–152. doi: 10.1007/s13365-010-0017-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Thompson WL, Van Eldik LJ. Inflammatory cytokines stimulate the chemokines CCL2/MCP-1 and CCL7/MCP-7 through NFκB and MAPK dependent pathways in rat astrocytes. Brain Research. 2009;1287:47–57. doi: 10.1016/j.brainres.2009.06.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Orandle MS, MacLean AG, Sasseville VG, Alvarez X, Lackner AA. Enhanced expression of proinflammatory cytokines in the central nervous system is associated with neuroinvasion by simian immunodeficiency virus and the development of encephalitis. Journal of Virology. 2002;76(11):5797–5802. doi: 10.1128/JVI.76.11.5797-5802.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Cardona AE, Gonzalez PA, Teale JM. CC chemokines mediate leukocyte trafficking into the central nervous system during murine neurocysticercosis: role of γδ T cells in amplification of the host immune response. Infection and Immunity. 2003;71(5):2634–2642. doi: 10.1128/IAI.71.5.2634-2642.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Voskuhl RR, Peterson RS, Song B, et al. Reactive astrocytes form scar-like perivascular barriers to leukocytes during adaptive immune inflammation of the CNS. Journal of Neuroscience. 2009;29(37):11511–11522. doi: 10.1523/JNEUROSCI.1514-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Kielian T. Immunopathogenesis of brain abscess. Journal of Neuroinflammation. 2004;1(1, article 16) doi: 10.1186/1742-2094-1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Cota M, Kleinschmidt A, Ceccherini-Silberstein F, et al. Upregulated expression of interleukin-8, RANTES and chemokine receptors in human astrocytic cells infected with HIV-1. Journal of NeuroVirology. 2000;6(1):75–83. doi: 10.3109/13550280009006384. [DOI] [PubMed] [Google Scholar]
- 200.Eugenin EA, Osiecki K, Lopez L, Goldstein H, Calderon TM, Berman JW. CCL2/monocyte chemoattractant protein-1 mediates enhanced transmigration of human immunodeficiency virus (HIV)-infected leukocytes across the blood-brain barrier: a potential mechanism of HIV-CNS invasion and NeuroAIDS. Journal of Neuroscience. 2006;26(4):1098–1106. doi: 10.1523/JNEUROSCI.3863-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Hauwel M, Furon E, Canova C, Griffiths M, Neal J, Gasque P. Innate (inherent) control of brain infection, brain inflammation and brain repair: the role of microglia, astrocytes, “protective” glial stem cells and stromal ependymal cells. Brain Research Reviews. 2005;48(2):220–233. doi: 10.1016/j.brainresrev.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 202.Park EJ, Park SY, Joe E-H, Jou I. 15d-PGJ2 and rosiglitazone suppress Janus kinase-STAT inflammatory signaling through induction of suppressor of cytokine signaling 1 (SOCS1) and SOCS3 in glia. Journal of Biological Chemistry. 2003;278(17):14747–14752. doi: 10.1074/jbc.M210819200. [DOI] [PubMed] [Google Scholar]
- 203.Fischer-Smith T, Rappaport J. Evolving paradigms in the pathogenesis of HIV-1-associated dementia. Expert Reviews in Molecular Medicine. 2005;7(27):1–26. doi: 10.1017/S1462399405010239. [DOI] [PubMed] [Google Scholar]
- 204.Sasseville VG, Lane JH, Walsh D, Ringler DJ, Lackner AA. VCAM-1 expression and leukocyte trafficking to the CNS occur early in infection with pathogenic isolates of SIV. Journal of Medical Primatology. 1995;24(3):123–131. doi: 10.1111/j.1600-0684.1995.tb00157.x. [DOI] [PubMed] [Google Scholar]
- 205.Sasseville VG, Newman WA, Lackner AA, et al. Elevated vascular cell adhesion molecule-1 in AIDS encephalitis induced by simian immunodeficiency virus. American Journal of Pathology. 1992;141(5):1021–1030. [PMC free article] [PubMed] [Google Scholar]
- 206.Mathew JS, Westmoreland SV, Alvarez X, et al. Expression of peripherin in the brain of macaques (Macaca mulatta and Macaca fascicularis) occurs in astrocytes rather than neurones and is associated with encephalitis. Neuropathology and Applied Neurobiology. 2001;27(6):434–443. doi: 10.1046/j.1365-2990.2001.00361.x. [DOI] [PubMed] [Google Scholar]
- 207.Sasseville VG, Smith MM, Mackay CR, et al. Chemokine expression in simian immunodeficiency virus-induced AIDS encephalitis. American Journal of Pathology. 1996;149(5):1459–1467. [PMC free article] [PubMed] [Google Scholar]
- 208.McKimmie CS, Graham GJ. Astrocytes modulate the chemokine network in a pathogen-specific manner. Biochemical and Biophysical Research Communications. 2010;394(4):1006–1011. doi: 10.1016/j.bbrc.2010.03.111. [DOI] [PubMed] [Google Scholar]
- 209.Sopper S, Demuth M, Stahl-Hennig C, et al. The effect of simian immunodeficiency virus infection in vitro and in vivo on the cytokine production of isolated microglia and peripheral macrophages from Rhesus monkey. Virology. 1996;220(2):320–329. doi: 10.1006/viro.1996.0320. [DOI] [PubMed] [Google Scholar]
- 210.Köhler C. Allograft inflammatory factor-1/Ionized calcium-binding adapter molecule 1 is specifically expressed by most subpopulations of macrophages and spermatids in testis. Cell and Tissue Research. 2007;330(2):291–302. doi: 10.1007/s00441-007-0474-7. [DOI] [PubMed] [Google Scholar]
- 211.Ohsawa K, Imai Y, Sasaki Y, Kohsaka S. Microglia/macrophage-specific protein Iba1 binds to fimbrin and enhances its actin-bundling activity. Journal of Neurochemistry. 2004;88(4):844–856. doi: 10.1046/j.1471-4159.2003.02213.x. [DOI] [PubMed] [Google Scholar]
- 212.Tassi M, Calvente R, Marín-Teva JL, et al. Behavior of in vitro cultured ameboid microglial cells migrating on Müller cell end-feet in the quail embryo retina. GLIA. 2006;54(5):376–393. doi: 10.1002/glia.20393. [DOI] [PubMed] [Google Scholar]
- 213.Renner NA, Sansing HA, Morici LA, Inglis FM, Lackner AA, Maclean AG. Microglia activation by SIV-infected macrophages: alterations in morphology and cytokine secretion. Journal For Neurovirology. 2012;18(3):213–221. doi: 10.1007/s13365-012-0100-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Shaw KTY, Greig NH. Chemokine receptor mRNA expression at the in vitro blood-brain barrier during HIV infection. NeuroReport. 1999;10(4):53–56. doi: 10.1097/00001756-199901180-00010. [DOI] [PubMed] [Google Scholar]
- 215.Renner NA, Sansing HA, Inglis FM, et al. Transient acidification and subsequent proinflammatory cytokine stimulation of astrocytes induce distinct activation phenotypes. Journal of Cellular Physiology. 2013;228(6):1284–1294. doi: 10.1002/jcp.24283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Nyagol J, De Falco G, Lazzi S, et al. HIV-1 Tat mimetic of VEGF correlates with increased microvessels density in AIDS-related diffuse large B-cell and Burkitt lymphomas. Journal of Hematopathology. 2008;1(1):3–10. doi: 10.1007/s12308-008-0002-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Garces CA, Kurenova EV, Golubovskaya VM, Cance WG. Vascular endothelial growth factor receptor-3 and focal adhesion kinase bind and suppress apoptosis in breast cancer cells. Cancer Research. 2006;66(3):1446–1454. doi: 10.1158/0008-5472.CAN-05-1661. [DOI] [PubMed] [Google Scholar]
- 218.Ivey NS, Renner NA, Moroney T, et al. Association of FAK activation with lentivirus-induced disruption of blood-brain barrier tight junction-associated ZO-1 protein organization. Journal for Neurovirology. 2009;10:1–12. doi: 10.1080/13550280902998413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Roberts ES, Burudi EME, Flynn C, et al. Acute SIV infection of the brain leads to upregulation of IL6 and interferon-regulated genes: expression patterns throughout disease progression and impact on neuroAIDS. Journal of Neuroimmunology. 2004;157(1-2):81–92. doi: 10.1016/j.jneuroim.2004.08.030. [DOI] [PubMed] [Google Scholar]
- 220.Williams KC, Hickey WF. Central nervous system damage, monocytes and macrophages, and neurological disorders in AIDS. Annual Review of Neuroscience. 2002;25:537–562. doi: 10.1146/annurev.neuro.25.112701.142822. [DOI] [PubMed] [Google Scholar]
- 221.Shah A, Verma AS, Patel KH, et al. HIV-1 gp120 induces expression of IL-6 through a nuclear factor-kappa B-dependent mechanism: suppression by gp120 specific small interfering RNA. PLoS ONE. 2011;6(6) doi: 10.1371/journal.pone.0021261.e21261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Sugama S, Takenouchi T, Cho BP, Joh TH, Hashimoto M, Kitani H. Possible roles of microglial cells for neurotoxicity in clinical neurodegenerative diseases and experimental animal models. Inflammation and Allergy. 2009;8(4):277–284. doi: 10.2174/187152809789352249. [DOI] [PubMed] [Google Scholar]
- 223.D’Aversa TG, Eugenin EA, Berman JW. CD40-CD40 ligand interactions in human microglia induce CXCL8 (interleukin-8) secretion by a mechanism dependent on activation of ERK1/2 and nuclear translocation of nuclear factor-κB (NFκB) and activator protein-1 (AP-1) Journal of Neuroscience Research. 2008;86(3):630–639. doi: 10.1002/jnr.21525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Shah A, Kumar A. HIV-1 gp120-mediated increases in IL-8 production in astrocytes are mediated through the NF-κB pathway and can be silenced by gp120-specific siRNA. Journal of Neuroinflammation. 2010;7, article 96 doi: 10.1186/1742-2094-7-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Xiong H, Boyle J, Winkelbauer M, et al. Inhibition of long-term potentiation by interleukin-8: implications for human immunodeficiency virus-1-associated dementia. Journal of Neuroscience Research. 2003;71(4):600–607. doi: 10.1002/jnr.10503. [DOI] [PubMed] [Google Scholar]
- 226.Penton-Rol G, Cota M, Polentarutti N, et al. Up-regulation of CCR2 chemokine receptor expression and increased susceptibility to the multitropic HIV strain 89.6 in monocytes exposed, to glucocorticoid hormones. Journal of Immunology. 1999;163(6):3524–3529. [PubMed] [Google Scholar]
- 227.Witwer KW, Gama L, Li M, et al. Coordinated regulation of SIV replication and immune responses in the CNS. PLoS ONE. 2009;4(12):p. e8129. doi: 10.1371/journal.pone.0008129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Eugenin EA, D’Aversa TG, Lopez L, Calderon TM, Berman JW. MCP-1 (CCL2) protects human neurons and astrocytes from NMDA or HIV-tat-induced apoptosis. Journal of Neurochemistry. 2003;85(5):1299–1311. doi: 10.1046/j.1471-4159.2003.01775.x. [DOI] [PubMed] [Google Scholar]
- 229.Kolb SA, Sporer B, Lahrtz F, Koedel U, Pfister H-W, Fontana A. Identification of a T cell chemotactic factor in the cerebrospinal fluid of HIV-1-infected individuals as interferon-γ inducible protein 10. Journal of Neuroimmunology. 1999;93(1-2):172–181. doi: 10.1016/s0165-5728(98)00223-9. [DOI] [PubMed] [Google Scholar]
- 230.Mankowski JL, Queen SE, Clements JE, Zink MC. Cerebrospinal fluid markers that predict SIV CNS disease. Journal of Neuroimmunology. 2004;157(1-2):66–70. doi: 10.1016/j.jneuroim.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 231.Williams KC, Hickey WF. Traffic of hematogenous cells through the central nervous system. Current Topics in Microbiology and Immunology. 1995;202:221–245. doi: 10.1007/978-3-642-79657-9_15. [DOI] [PubMed] [Google Scholar]
- 232.Scholz J, Woolf CJ. The neuropathic pain triad: neurons, immune cells and glia. Nature Neuroscience. 2007;10(11):1361–1368. doi: 10.1038/nn1992. [DOI] [PubMed] [Google Scholar]
- 233.Dubový P. Wallerian degeneration and peripheral nerve conditions for both axonal regeneration and neuropathic pain induction. Annals of Anatomy. 2011;193(4):267–275. doi: 10.1016/j.aanat.2011.02.011. [DOI] [PubMed] [Google Scholar]
- 234.Seifert F, Maihöfner C. Functional and structural imaging of pain-induced neuroplasticity. Current Opinion in Anaesthesiology. 2011;24(5):515–523. doi: 10.1097/ACO.0b013e32834a1079. [DOI] [PubMed] [Google Scholar]
- 235.Abbadie C. Chemokines, chemokine receptors and pain. Trends in Immunology. 2005;26(10):529–534. doi: 10.1016/j.it.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 236.Abbadie C, Lindia JA, Cumiskey AM, et al. Impaired neuropathic pain responses in mice lacking the chemokine receptor CCR2. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(13):7947–7952. doi: 10.1073/pnas.1331358100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.White FA, Bhangoo SK, Miller RJ. Chemokines: integrators of pain and inflammation. Nature Reviews Drug Discovery. 2005;4(10):834–844. doi: 10.1038/nrd1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.White FA, Feldman P, Miller RJ. Chemokine signaling and the management of neuropathic pain. Molecular Interventions. 2009;9(4):188–195. doi: 10.1124/mi.9.4.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Ren K, Dubner R. Interactions between the immune and nervous systems in pain. Nature Medicine. 2010;16(11):1267–1276. doi: 10.1038/nm.2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Holmes FE, Arnott N, Vanderplank P, et al. Intra-neural administration of fractalkine attenuates neuropathic pain-related behaviour. Journal of Neurochemistry. 2008;106(2):640–649. doi: 10.1111/j.1471-4159.2008.05419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Moalem G, Tracey DJ. Immune and inflammatory mechanisms in neuropathic pain. Brain Research Reviews. 2006;51(2):240–264. doi: 10.1016/j.brainresrev.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 242.Coull JAM, Beggs S, Boudreau D, et al. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438(7070):1017–1021. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- 243.Miyoshi K, Obata K, Kondo T, Okamura H, Noguchi K. Interleukin-18-mediated microglia/astrocyte interaction in the spinal cord enhances neuropathic pain processing after nerve injury. Journal of Neuroscience. 2008;28(48):12775–12787. doi: 10.1523/JNEUROSCI.3512-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Binshtok AM, Wang H, Zimmermann K, et al. Nociceptors are interleukin-1β sensors. Journal of Neuroscience. 2008;28(52):14062–14073. doi: 10.1523/JNEUROSCI.3795-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Skoff AM, Zhao C, Adler JE. Interleukin-1α regulates substance P expression and release in adult sensory neurons. Experimental Neurology. 2009;217(2):395–400. doi: 10.1016/j.expneurol.2009.03.022. [DOI] [PubMed] [Google Scholar]
- 246.Xu X-J, Hao J-X, Andell-Jonsson S, Poli V, Bartfai T, Wiesenfeld-Hallin Z. Nociceptive responses in interleukin-6-deficient mice to peripheral inflammation and peripheral nerve section. Cytokine. 1997;9(12):1028–1033. doi: 10.1006/cyto.1997.0243. [DOI] [PubMed] [Google Scholar]
- 247.Dominguez E, Mauborgne A, Mallet J, Desclaux M, Pohl M. SOCS3-mediated blockade of JAK/STAT3 signaling pathway reveals its major contribution to spinal cord neuroinflammation and mechanical allodynia after peripheral nerve injury. Journal of Neuroscience. 2010;30(16):5754–5766. doi: 10.1523/JNEUROSCI.5007-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Myers RR, Shubayev VI. The ology of neuropathy: an integrative review of the role of neuroinflammation and TNF-α axonal transport in neuropathic pain. Journal of the Peripheral Nervous System. 2011;16(4):277–286. doi: 10.1111/j.1529-8027.2011.00362.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Kim CF, Moalem-Taylor G. Interleukin-17 contributes to neuroinflammation and neuropathic pain following peripheral nerve injury in mice. Journal of Pain. 2011;12(3):370–383. doi: 10.1016/j.jpain.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 250.Nũnéz S, Lee J-S, Zhang Y, Bai G, Ro JY. Role of peripheral μ-opioid receptors in inflammatory orofacial muscle pain. Neuroscience. 2007;146(3):1346–1354. doi: 10.1016/j.neuroscience.2007.02.024. [DOI] [PubMed] [Google Scholar]
- 251.Lee YK, Choi DY, Jung YY, et al. Decreased pain responses of C-C chemokine receptor 5 knockout mice to chemical or inflammatory stimuli. Neuropharmacology. 2013;67:57–65. doi: 10.1016/j.neuropharm.2012.10.030. [DOI] [PubMed] [Google Scholar]
- 252.Kiguchi N, Kobayashi Y, Kishioka S. Chemokines and cytokines in neuroinflammation leading to neuropathic pain. Current Opinion in Pharmacology. 2012;12(1):55–61. doi: 10.1016/j.coph.2011.10.007. [DOI] [PubMed] [Google Scholar]