

Abstract
Cadmium (Cd) is a heavy metal that has received considerable concern environmentally and occupationally. Cd has a long biological half-life mainly due to its low rate of excretion from the body. Thus, prolonged exposure to Cd will cause toxic effect due to its accumulation over time in a variety of tissues, including kidneys, liver, central nervous system (CNS), and peripheral neuronal systems. Cd can be uptaken from the nasal mucosa or olfactory pathways into the peripheral and central neurons; for the latter, Cd can increase the blood brain barrier (BBB) permeability. However, mechanisms underlying Cd neurotoxicity remain not completely understood. Effect of Cd neurotransmitter, oxidative damage, interaction with other metals such as cobalt and zinc, estrogen-like, effect and epigenetic modification may all be the underlying mechanisms. Here, we review the in vitro and in vivo evidence of neurotoxic effects of Cd. The available finding indicates the neurotoxic effects of Cd that was associated with both biochemical changes of the cell and functional changes of central nervous system, suggesting that neurotoxic effects may play a role in the systemic toxic effects of the exposure to Cd, particularly the long-term exposure.
1. Introduction
Cadmium (Cd) is a toxic, nonessential transition metal and classified as a human carcinogen by the National Toxicology Program [1]. There are several sources of human exposure to Cd, including employment in primary metal industries, production of certain batteries, some electroplating processes (about 29% of year production), and consumption of tobacco products [2]. It is also of interest since the natural biogeochemical cycle of Cd has been overwhelmed. First, the concept of provisional tolerable weekly intake (PTWI) was established. The Joint FAO/WHO Expert Committee on Food Additives defines the PTWI for a chemical with no intended function as an estimate of the amount of the chemical that can be ingested weekly over a lifetime without appreciable health risk [3]. The first Cd PTWI was 400–500 μg per person per week. This level was based on a critical concentration of 200 mg Cd/g kidney cortex, attainable after a dietary Cd intake of 140–260 μg/d for over 50 years or 2000 mg over a lifetime. For several decades, the PTWI has been expressed more rationally in terms of the intake per kg body weight, and the value was constantly changed (Table 1). In 2010, the Consumer Product Safety Commission (CPSC) released a staff report recommending new guidance on Cd, that is, an acceptable daily intake level of 0.1 μg kg−1 body weight per day for chronic exposure [4]. In most studies, the half-life of Cd in humans is estimated at a range of 15 to 20 years [5]. Epidemiological and experimental studies have linked the occupational Cd exposure with lung cancer and other cancers such as the prostate, renal, liver, hematopoietic system, urinary bladder, pancreatic, testis, and stomach cancers [6–8]. Exposure to Cd also severely affects the function of the nervous system [9, 10], with symptoms including headache and vertigo, olfactory dysfunction, parkinsonian-like symptoms, slowing of vasomotor functioning, peripheral neuropathy, decreased equilibrium, decreased ability to concentrate, and learning disabilities [11–13]. The neurotoxicity of Cd in children was investigated in several studies in the 1970s and 1980s but has received little attention since then. In case-control studies in which the hair concentration of Cd in a clinically defined group was compared with that of a reference group, higher concentrations of hair Cd were reported in children with mental retardation [14] and learning difficulties or dyslexia [11, 15]. In cohort studies, Thatcher et al. reported that the concentration of Cd in hair was inversely related to adjust Intelligence Quotient (IQ) [16, 17]. Other investigators [18] also reported associations between hair Cd concentrations and children's performance on visual-motor tasks. These studies clearly indicate the association of increased total Cd concentration with mental retardation and reduced visual motor abilities. Effect of Cd neurotransmitter, oxidative damage, interaction with other metals such as cobalt and zinc, estrogen-like effect, and epigenetic modification may be the underlying mechanisms (Figure 1). However, the exact mechanism(s) through which Cd elicits its neurotoxic effects is still unresolved. In this review, therefore, we focus on recent evidence from experimental and epidemiological studies, showing that Cd exposure can induce its neurotoxin effects.
Table 1.
Summary of the “safe” level for human contacted of Cd.
| Organization/Authors | Year | Approach | Dose | Reference |
|---|---|---|---|---|
| FAO/WHO | 1988 | Intake | 400–500 μg/week | [3] |
| WHO | 1992 | Intake | 7 μg·kg−1 b.w./week or 1 μg·kg−1 b.w./day | [38] |
| FAO/WHO | 1993 | Intake | 7 μg·kg−1 b.w./week | [39] |
| Satarug et al. | 2000 | Intake | 30 μg/day | [40] |
| Nasreddine and Parent-Massin | 2002 | Intake | 10–30 μg/day | [41] |
| WHO | 2004 | Drinking-water | 3 μg/L | [42] |
| FAO/WHO | 2006 | Food | 0.4 mg/kg of the BMDL of R-Cd standard | [43] |
| ACGIH. | 2007 | Blood | 5 μg/L | [44] |
| EFSA | 2009 | Intake | 2.5 μg·kg−1 b.w./week | [45] |
| FAO/WHO | 2010 | Intake | 5.8 μg·kg−1 b.w./week | [46] |
| CPSC | 2010 | Intake | 0.1 μg·kg−1 b.w./day | [4] |
ACGIH: American conference of governmental industrial hygienists; CPSC: Consumer Product Safety Commission; FAO: Food and Agriculture Organization; WHO: World Health Organization; b.w.: body weight.
Figure 1.
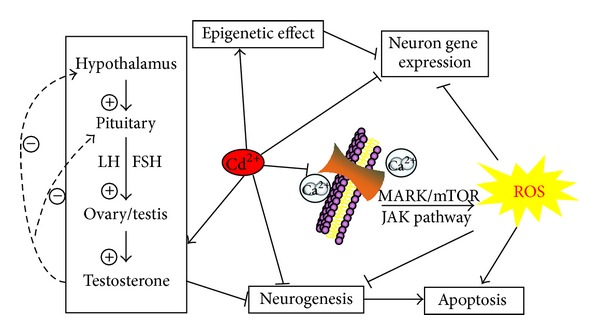
Mechanistic illustration of the neuronal toxicity of Cd. (1) Cd-induced neuron cell apoptosis and ROS (reactive oxygen species) are mediated through Ca2+-mitochondria signaling and Ca2+-membrane channels. (2) Cd impaired neurogenesis. (3) Cd accumulation in the brain leads to the altered gene expression and epigenetic effect. (4) Cd has estrogen-like effect, which can induce endocrine disruption by affecting the hypothalamic-pituitary-gonadal (HPG) axis in different aspects. Meanwhile, these potential mechanisms have possible interactions. The solid black arrows represent the stimulation, the solid black line segments indicate the inhibition, and the dotted lines represent the negative feedback control of the HPG axis.
2. The Absorption of Cd in the Nervous System and Distribution
The Cd exposure rate (based on meconium analysis) in infants was 8.5%, and the median concentration of the pollutants in the positive samples was 13.37 mg/mL. In most of these studies, the concentration of Cd in hair was measured. The CNS is especially vulnerable to damage during early neonatal development; Cd is able to readily pass to the fetus via the placenta and was detected in milk during lactation [19]. Cd can be uptaken from the nasal mucosa or olfactory pathways into the CNS; thus, the CNS is subjected to Cd toxicity [20–22]. Under normal conditions, Cd barely reaches the brain in adults due to the presence of the blood brain barrier (BBB); however, this structure is not fully developed in young animals [23]. The anatomical and physiological bases on which the choroids plexus becomes the target of xenobiotics have been examined. Cd tends to accumulate in the choroids plexus at concentrations much greater than those found in the cerebrospinal fluid (CSF) and elsewhere in brain tissues. A postmortem human study revealed that the Cd concentration in the choroids plexus was about 2-3 times higher than that found in the brain cortex [24]. As a general choroids plexus toxicant, Cd can directly damage the choroids plexus ultrastructure. Due to differences in the BBB integrity [25], Cd is thus more toxic to newborn and young rats than to adult rats. Cd can increase permeability of the BBB in rats [26] to penetrate and accumulate in the brain of developing and adult rats [27, 28], leading to brain intracellular accumulation, cellular dysfunction, and cerebral edema.
As a barrier, the choroidal epithelia are often the first and the most frequent ones to encounter metal insults from blood. Two mechanisms may aid resistance of choroids plexus to blood-borne toxicities. First, the choroids plexus contains abundant metal binding ligands that effectively sequester metal ions. Moreover, the concentration of cystine in the choroids plexus is 4-fold higher than that in the brain cortex [29]. Second, the choroids plexus owns an active defense system. The activities of superoxide dismutase and catalase are significantly higher in the choroids plexus than in cerebrum and cerebellum. Taken together, the choroids plexus likely forms the first line of defense against neurotoxicants. It must be kept in mind, however, that none of the cellular defense mechanisms would operate in an unlimited capacity. The pathophysiological changes can occur either as the threshold above which the protective capacity of the choroids plexus is exceeded or saturated, or as a direct result of barrier dysfunction. Thus, the need arises for a more comprehensive understanding of the detoxification mechanisms, such as antioxidant systems, induction of protective macromolecules (heat shock proteins, etc.), formation of specific metal inclusion bodies or binding proteins, and biotransformation reactions (methylation, conjugation, etc.) that operate in the choroids plexus.
Cd is transported along the primary olfactory neurons to their terminations in the olfactory bulbs, thereby bypassing the intact BBB. The olfactory route could therefore be a likely way to reach the brain and should be taken into account for occupational risk assessments for this metal [30–32]. Occupational inhalation of Cd can be toxic to the olfactory sense [30]. Primary olfactory neurons are regularly replaced throughout the life span. Functional recovery and regeneration of olfactory neurons has been demonstrated in laboratory animals but not confirmed in humans [33]. Intranasal exposure to Cd has been related to olfactory dysfunction in humans and to nasal epithelial damage and altered odorant-guided behavior in rodent models. The pathophysiology underlying these deficits has been partly elucidated. Optical imaging revealed significant reductions in odorant-evoked release from the olfactory nerve at a Cd chloride dose two orders of magnitude less than that required to induce morphological changes in the nerve in the same animals, demonstrating that it is a more sensitive technique for assessing the consequences of intranasal neurotoxicant exposure. This approach is potentially useful in exploring the effects of any putative neurotoxicant that can be delivered intranasal [32].
3. The Influence of Cd to Central Nervous System (CNS)
3.1. The Morphology Change of CNS in Response to Cd
Cd-treated embryos developed a smaller head with unclear boundaries between the brain subdivisions, particularly in the mid-hindbrain region. Embryos display normal anterior to posterior regionalization; however, the commitment of neural progenitor cells was affected by Cd [34]. Cd has been shown to produce free radicals in the brain, which may potentially damage both neurons and oligodendrocytes (OLG). OLGs are the glial cells which myelinate axons in the CNS. An early study reported that Cd toxicity affected CNS white matter [35], and one laboratory demonstrated that OLGs are direct targets of this structure [36]. Experimental studies have shown that Cd can be a potent neurotoxicant for the peripheral nervous system. Moreover, Cd has a half-life of more than 15 years in humans. Elderly workers may be more susceptible to an increased Cd body burden and may develop a peripheral polyneuropathy (PNP) over time [37]. The primary olfactory neuron is the only sensory cell directly in contact with the environment and therefore potentially exposed to airborne toxicants; so olfactory damage may represent the early action of a toxicant on the nervous system; the primary olfactory neuron may represent the early target for airborne Cd toxic action.
In cortical neurons cell culture from fetal rats at 19 days of gestation, Cd modified the neuronal morphology after a 6 h treatment in a serum-free medium with 10 μM of Cd, whereas at 24 h it showed a great loss of neuronal integrity mainly evidenced by the almost complete disappearance of the axons. Lower concentrations of Cd than 1 μM could induce cell apoptosis and higher concentrations of Cd than 1 μM could induce necrotic cell death [9]. Cd affected the cell morphology; the morphological changes were mainly located in the neural extensions (axons and dendrites), which almost disappeared after 24 h of treatment with 1 μM of Cd in a serum-free medium [9, 47]. Such morphological changes induced by Cd have also been described in other cells [48]. Cd inhibited neurite outgrowth at concentrations that decreased viability in Neuroscreen-1 cells (NS-1) models, a subclone of PC12 cells, using high content screening. The previous studies have shown that cultured oligodendrocytes are directly damaged by Cd exposure, and continuous exposure (18–48 h) of OLPs to low micromolar concentrations (0.001–25 μM) of Cd significantly decreased mitochondrial metabolic activity, increased LDH leakage starting at 5 mM, and maximally activated caspase-3. These results suggest that Cd induces OLP cell death mainly by apoptosis, and at higher concentrations or with prolonged exposure to the heavy metal there is an increase in cytoplasmic membrane damage, an index of necrosis. More importantly, transient exposure to Cd is sufficient to damage OLPs and could in principle impair myelination in the neonate. Although the effects of Cd on neuronal cells in culture have been described in several papers, its effects on human central nervous system neurons in vivo have not yet been demonstrated. However, the recently described Cd-induced apoptosis of the motor neurons of the ventral horns in cultured explants from human fetal spinal cords (10-11 weeks gestational age) [49] suggests that the effects observed in vitro could actually occur also in vivo.
Increasing evidence has demonstrated that Cd is a possible etiological factor of neurodegenerative diseases, such as Alzheimer's disease (AD) and Parkinson's disease (PD) [50, 51]. Cerebral cortical neurons have been identified as targets of Cd-mediated toxicity and Cd-induced cell apoptosis [52, 53]. Apoptotic morphological changes induced by Cd in cerebral cortical neurons were assessed in some studies [53, 54].
In vivo study, the age and species of experimental animals could be effected the CNS results of damage. Perinatal exposure to Cd (50 ppm in drinking water) reduced the brain weights of pups and inhibited the activities of enzymes in nervous system, for example acetylcholine esterase, K+-ATPase, CNP (cyclic nucleotide phosphodiesterase), and 50-nucleotidase [55]. The Cd concentration in the choroids plexus was about 2-3 times higher than that found in the brain cortex. Cd-produced deterioration of the plexus structure can be characterized by the loss of microvilli, a rupture of the apical surface, and an increased number of blebs. Cellular debris present in the ventricular lumen may result from the breaking of the apical membrane. The epithelial cells display an abnormally high number of cytoplasmic vacuoles and lysosomes with condensed or irregular nuclei. As a general choroids plexus toxicant, Cd directly destroys the plexus ultrastructure. In both chronic (22 weeks) and acute (1–24 days) exposure models, the levels of Cd in the choroids plexus were high, while Cd in the CSF fell below the detection limit [29].
3.2. The Biochemical Changes of CNS in Response to Cd
The cholinergic system, with acetylcholine (ACh) as the neurotransmitter, is involved in cognitive processes, through the activation of metabotropic muscarinic and ionotropic nicotinic cholinergic receptors. The reaction responsible for the maintenance of levels of ACh is catalyzed by two cholinesterases (ChE): acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE). AChE is an important biomarker for several environmental contaminants in zebrafish [56, 57]. In addition, it is also known that this enzyme plays an important role in diseases with an increasing incidence in the elderly population, such as Alzheimer disease. Zebrafish (Danio rerio) is an emergent vertebrate model for studying several biological events, such as neurochemical alterations promoted by heavy metal toxicity. This teleost possesses only the gene for AChE, which is responsible for the whole ACh degradation, being the BuChE absent. The AChE gene has already been identified, cloned, and functionally detected in the zebrafish brain. The recently reported on the effects of long-term dietary-induced exposure to Cd on the AChE activity of adult rodents' brain regions. The authors studied the changes in the activities of AChE and Na+, K+-ATPase in the cerebral cortex, hippocampus, hypothalamus, cerebellum, and striatum of adult Wistar rats, following a 5-month (long-term) exposure to an experimental diet supplemented with low levels of Cd salt or with Cd-contaminated potato tubers. The authors also assessed the behavioral (cognitive-, motor-, and anxiety-related) outcomes following the above-mentioned treatment [58]. But the other research term queried that their study can be “regarded as a significant contribution to the field, as there is paucity of information on the AChE activity in brain regions following exposure to Cd” and “the experimental protocol used in the aforementioned study [58] is exceptionally well designed in order to simulate long-term dietary-induced exposure to Cd” [59].
Cd can affect the degree and balance of excitation inhibition in synaptic neurotransmission as well as the antioxidant levels in animal brain [28]. Cd increases the serotonin sensibility in the CNS [60]. Cd inhibits the release of acetylcholine, probably by interfering with calcium metabolism [61]. A remarkable reduction in the concentration of brain galactosylceramide (30%–43%) and 3′-sulphogalactosylceramide (24%–37%) at 21, 30, and 45 days of age was observed following pre- and postnatal exposure of Cd in comparison to the respective controls. The ontogenic profile of different brain phospholipids in the Cd-exposed group showed an increase in the levels of phosphatidylethanolamine at 21 (31%), 30 (25%) and 45 (19%) days of age; the phosphatidylcholine contents increased at day 21 (14%), followed by a significant decrease at 30 (14%) and 45 (19%) days compared to age-matched controls. Brain phosphatidylinositol, phosphatidylserine, and sphingomyelin did not show any alteration at early periods of exposure but decreased significantly following continued exposure by 34%, 45%, and 21%, respectively, at 45 days of age [62].
Numerous studies have shown that cadmium caused a decrease in depolarization-evoked exocytotic release of glutamate (as well as other neurotransmitters) from nerve terminals [73]. Cd2+ can stimulate [3H]-glutamate binding in human platelets. Cd2+ can increase lipid peroxidation levels and reactive oxygen species (ROS) measurement in platelets. Glutamatergic system may be used as a potential biomarker for neurotoxic action of Cd in humans [74].
3.3. The Central Activity Changes in Response to Cd
Cd may alter the stimulus properties of morphine in adult male rat model [75]. As shown in Table 2, perinatal Cd exposure has been shown to alter behaviors and reduce learning ability. In addition, high levels of Cd and lead in children's hair were associated with learning disabilities. Motor and perceptual abilities of children exposed to Cd in uteri were significantly affected [76]. Behavioral defects, neurochemical changes, and brain lesions were reported in experimental animals, while in humans acute Cd poisoning produced Parkinsonism symptoms [50]. Blood Cd in motor neuron disease (MND, with limited disability) was higher than controls [77, 78]. Plasma Cd levels in the sporadic motor neuron disease (SMND) group were significantly increased compared to controls [78]. A recent study using a fully automated, observer-independent procedure to study morphological alterations of the central nervous system demonstrated that myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) patients had an average decrease of the volume of the gray matter of about 8% compared to matched healthy controls [79]. The gray matter volume reduction was significantly associated with objective reduction of physical activity in ME/CFS patients. Deficits in learning and altered behaviors and activities were also observed in offspring exposed to Cd during gestational and/or lactational period. Cd induced neuronal death in cortical neurons through a combined mechanism of apoptosis and necrosis involving reactive oxygen species generation and lipid peroxidation [80]. With regard to the assessment of behavioral disorders, the cross-sectional study by Bao et al., from China, revealed a higher frequency of withdrawal, social problems, and attention problems associated with higher levels of cadmium in hair of children aged 7–16 years [71]. But another study did not find any significant association between cadmium exposure and ADHD [81].
Table 2.
Literature review of Cd neurotoxicity in humans and rats.
| Year | Study design | Age group | E/C (n) | Exposure to Cd | Expose pathways | Effects | Reference |
|---|---|---|---|---|---|---|---|
| 1961 | Cross-sectional | Male worker | 106E/84C | — | Occupational exposure | Anosmia | [63] |
|
| |||||||
| 1977 | Cross- sectional | Children | 31E/22C | CdH | Daily life | Neurological disorders, such as learning disabilities and hyperactivity | [11] |
|
| |||||||
| 1981 | Cross-sectional | Children | 73E/44C | CdH | Daily life | Dyslexic, learning disorder | [15] |
|
| |||||||
| 1981 | Cross-sectional | Workers | 49E | CdU | Occupational exposure | Polyneuropathy | [64] |
|
| |||||||
| 1982 | Cross-sectional | Children | 149 | CdH | Daily life | Effect on verbal I.Q. | [16] |
|
| |||||||
| 1985 | Case-control | Young men | 40 | CdH | Daily life | Behavioural difficulty | [65] |
|
| |||||||
| 1985 | Cross-sectional | Children | 69 | CdH | Daily life | Nonadaptive classroom behavior, affected behavioral development visuomotor skills ↓ | [18] |
|
| |||||||
| 1989 | Cross-sectional | Male workers | 31E | CdU | Occupational exposure | ↓ Attention, memory, and psychomotor speed | [66] |
|
| |||||||
| 1992 | Cross-sectional | Worker | 38E | CdU | Occupational exposure | 90% Headache; 42% dizzy spells 21% weakness; 16% brain atrophy | [67] |
|
| |||||||
| 1992 | Cross-sectional | Worker | 55E/16C | CdU | Occupational exposure | Hyposmia | [68] |
|
| |||||||
| 1997 | Case report | Old man | 1 | Multiple organ failure | Occupational exposure, acute | Parkinsonism | [50] |
|
| |||||||
| 1999 | Cross-sectional | Worker | 13E/19C | CdU | Occupational exposure | Polyneuropathy | [37] |
|
| |||||||
| 2000 | Cross-sectional | Adult worker | 42E/47C | CdU | Occupational exposure | ↓ Motor speed, attention, memory ↑ equilibrium, PNP, and concentration complaints | [69] |
|
| |||||||
| 2006 | Case report | Adult workers | 1 | CdU | Inhale the fumes | Peripheral neuropathy | [70] |
|
| |||||||
| 2009 | Cross-sectional | Children | 549 | CdH | Daily life | Withdrawal, social problems and attention problems associated | [71] |
|
| |||||||
| 2012 | Wistar rats | Male | 20E/20C | Intratracheal instillation | Experiment exposure | Dose- and time-dependent shift from slower to faster waves | [72] |
E: exposed subjects, C: control subjects; CdU: urinary cadmium concentration; CdH: concentration of cadmium in hair; I.Q.: Intelligence Quotient.
In animal study, the role of the nanosized Cd in the causation of nervous system damages and shows the possibility of modeling human neurotoxic damage in rats [72].
4. The Mechanism by Which Cd Affects CNS
4.1. Cd and Oxidative Damage
Cd-induced injury in the cerebral microvessels is thought to be associated with oxidative stress. Following in vivo Cd exposure, there was an early increase followed by a later decrease in microvessel enzymes involved in cellular redox reactions, such as superoxide dismutase, glutathione peroxidase, and catalase. Thus, a depletion of microvessel antioxidant defense systems and a resultant increase in lipid peroxidation (LPO) may provoke microvessel damage [82]. Upon exposure, Cd has been shown to induce heavy metal-binding proteins such as metallothionein (MT) in various organs. MT is a low-molecular-weight protein, 6,500 Da with high cysteine content and high metal affinity, which plays a major role in the kinetics and metabolism of Cd. Four isoforms have been identified, namely, MT-I to MT-IV. Metallothionein-3 (MT-III) is specifically expressed in the brain; however, it is downregulated, and thus deficient in Alzheimer's disease [83]. The choroids plexus also expresses MT proteins. Nishimura et al. observed a strong MT immunostaining in ependymal cells and choroids plexus epithelium in younger rats (1–3 weeks old) poisoned with Cd. Thus, the sequestration of Cd by MT may partly contribute to the high accumulation of Cd in the choroids plexus [84]. Cd significantly increases the levels of lipid peroxidation in parietal cortex, striatum, and cerebellum as compared to a control group, and dexamethasone (Dx) treatment prevented the increase in LPO levels associated to Cd exposure, probably through the increase in MT content [85]. So MT as a protective mechanism against Cd-induced neurotoxicity is suggested.
In addition, mitochondria play a role in stress responses and can produce ROS when damaged. Mitochondria are indeed a major source of ROS. The enzyme COX can serve as an indicator of mitochondrial function. This is because COX dysfunction increases ROS, reduces energy stores, and impairs energy metabolism [86]. Accumulating evidences indicate that Cd-induced neuronal toxicity is due to induction of ROS, which leads to oxidative stress [54, 87]. Recently, Cd induced ROS generation in a time- and concentration-dependent manner in PC12 and SH-SY5Y cells [54], which causes apoptosis of neuronal cells via activation of MAPKs (mitogen-activated protein kinases) and mTOR (mammalian target of rapamycin) signaling pathways [54, 87, 88].
4.2. Interaction and/or Impact of Other Metals Including Calcium, Zinc, and Cobalt on Cd Neurotoxicity
Cerebral cortical neurons have been identified as targets of Cd-mediated toxicity [52]. And Cd-induced cerebral cortical neurons apoptosis occurs through Ca2+-mitochondria signaling [52, 53]. Cd disrupts intracellular free calcium ([Ca(2+)] i) homeostasis, leading to apoptosis in a variety of cells including primary murine neurons. Calcium is a ubiquitous intracellular ion which acts as a signaling mediator in numerous cellular processes including cell proliferation, differentiation, and survival/death. Few studies have focused on the interaction between Cd and Ca-binding molecules, such as calmodulin.
Cd may block the influx of Ca2+ through membrane channels into the nerve terminal following the action potential; these decreases in calcium influx caused by Cd would be associated with an altered transmitter release [89]. Of the receptor agonists tested partially inhibited by Cd2+ [90]. Cd ions are often used to block high-threshold Ca2+ currents [91] but may also block low-threshold Ca2+ currents [92]. When 100 μM Cd was added to the external solution, a minor fraction, 8.6% ± 8.9%, of the low-threshold current at −40 mV (holding potential −90 mV) was blocked [93]. Further studies are needed to define which molecular effects are indeed elicited by Cd-calmodulin and/or whether Cd2+ binds to other regulators of the Ca2+-induced signaling pathways.
In a new study, Cd-induced apoptosis is associated with calcium-induced massive production of ROS, dissipation of mitochondrial membrane potential (ΔΨm), cleavage of caspase-9, caspase-3, and PARP. And the results demonstrate that Cd-induced apoptosis is mediated by calcium signaling pathway, and calcium-mediated apoptosis occurs through the mitochondria-caspase signaling pathway [53].
Although Cd is not accumulated in significant quantities into the brain following exposure, it disturbs the metabolism of Cu and Zn. Because zinc (Zn) and Cd are cations of similar size and charge, and Cd has been shown to inhibit Zn uptake in a variety of systems, Cd is using transport systems that normally function to regulate Zn levels in brain. Metal analysis in the brain showed a reduction in zinc and copper levels at 15 and 21 days of age in Cd-exposed animals. It may be concluded that an early exposure of Cd may produce alteration in the development of different lipids, which may produce CNS dysfunctions with a possibility of being manifested even in later life [62]. The most well studied metallothioneins are isoforms of metallothioneins I and II that are expressed in almost all mammalian tissues. Metallothionein III is expressed in brain and is rich in zinc. Since the blood-brain barrier keeps Cd outside the CNS, reported neurotoxic effects of Cd during development are likely to be secondary to an interference of Cd with Zn-metabolism and not a direct effect of Cd on brain cells. It is therefore of importance to investigate whether neurotoxicity induced by Cd is related to mechanisms involving MT III in brain [5].
Very few articles study neurotoxin effects about the effect of cobalt (Co2+). This study addition of inorganic Ca2+ channel blockers (Cd2+ or Co2+) was used to demonstrate the Ca2+ dependence of Ca2+-activated k+ currents [94]. Cd2+ and Co2+ were the nonspecific calcium channel antagonists in the study [95, 96]. Co2+ did not modify significantly the ATP-evoked current [97]. Application of high potassium media to growth cones inhibited neurite outgrowth, an effect that was blocked by 2 mM cobalt or 100 μM Cd, suggesting that Ca2+ influx via voltage-gated channels contributes to glutamate-induced regulation of neuritis outgrowth [98].
4.3. Cd and Neurogenesis
There is evidence that relates arsenic and manganese exposure with neurodevelopmental problems in children, but there is little information on cadmium exposure. Cd-induced neurotoxicity might be caused by impaired neurogenesis, resulting in markedly reduced neuronal differentiation and axonogenesis, leading to neuronal cell death [99]. The present study compares the sensitivity of human (ReN CX) and mouse (mCNS) neuroprogenitor cell lines to chemicals using a multiplex assay for proliferation and apoptosis, endpoints that are critical for neural development. Cells were exposed to 0.001–100 μM concentrations of Cd to affect proliferation and/or apoptosis. Cd decreased proliferation by at least 50% of control in either the ReN CX or mCNS cells. Compared to control, Cd decreased cell viability (ATP levels) by at least 50% in the ReN CX cells, while Cd decreased viability by at least 50% in the mCNS cells. Based on these results, BrdU is an appropriate marker for assessing chemical effects on proliferation, and human cells are more sensitive than mouse cells for this endpoint [100]. In the mammalian brain, the complex molecular pathways underlying neurogenesis provide a variety of possible targets that might be impacted by Cd exposure and identifying which pathways are disrupted is difficult.
Gender-related differences in susceptibility to chemical exposure to neurotoxicants have not received sufficient attention. There is abundant available information on the gender-specific health effects of mercury and lead, and exposure to lead seems to affect boys more than girls [101]. The internal cadmium dose is generally higher in women than in men, due to a higher gastrointestinal absorption at low iron stores. This was probably one major reason why Itai-itai disease was mainly a woman's disease. Yet, data are sparse regarding the risk for women relative to men to develop cadmium-induced kidney damage in populations exposed to low levels of cadmium [102]. Information regarding gender differences in susceptibility of cadmium is still too scarce to draw any definite conclusion. More research is highly warranted about this matter. Environmental epidemiological studies should be designed to quantify differential gender-based exposures and outcomes, and this may provide new insights into prevention strategies [101].
4.4. Cd and Gene Expression
Cd accumulation prior to and at birth, however, might cause irreversible or lasting changes in the brain, which in turn leads to the altered gene expression [103, 104]. The expression of several proneuronal genes including ngn1 in cell clusters, zash1a in the developing optic tectum, and zash1b in the telencephalon and tectum. Cd-treated embryos also have fewer differentiated neurons and glia in the facial sensory ganglia as indicated by decreased zn-12 expression. Also, a lower transcription level of neurogenic genes, ngn1 and neuroD, is observed in neurons [34]. Cd-induced neurotoxicity can be caused by impaired neurogenesis, resulting in markedly reduced neuronal differentiation and axonogenesis.
By contrast, caspase 3 and p53 were altered by environmental chemicals in mouse, but not in human cells. Therefore, these markers are not appropriate to assess the ability of environmental chemicals to induce apoptosis in the ReN CX cells [100]. Among the brain-specific genes, neurogranin (RC3) and myelin basic protein (MBP) are regulated by serum thyroid hormones (THs) [105]. Since Cd exposure affects THs, Cd exposure will alter expression of these genes in the brain, and furthermore, those effects might be enhanced by a hypothyroid state. In conclusion, Cd combined with MMI decreased the RC mRNA expression in the brain of female offspring. The reduced expression of RC3 mRNA may explain the effect of Cd on brain function. Perinatal Cd exposure disrupted the reproductive function of offspring indicated by the reduced expression of ERs and PgR mRNA, which was not considered to be estrogenic action.
Cd-induced apoptosis in the neuronal cells has a time- and concentration-dependent manner. Cd induces apoptosis of neuronal cells by activation of JNK, Erk1/2, and mTOR signaling network. These findings support the notion that inhibitors of these pathways may be exploited for prevention of Cd-induced Parkinson's disease, Alzheimer's disease, and other neurodegenerative disorders [88].
4.5. Estrogen-Like Effect (Hormones Regulating Shaft)
Cd can modify hormone levels by affecting the hypothalamic-pituitary-testicular axis in different aspects, not only via its effects on Leydig cells. Cd affected the circadian pattern release of noradrenaline, a regulator of hypothalamus hormone secretion, which resulted in changes in the daily pattern of plasma testosterone and LH levels [106]. In addition, plasma levels of pituitary hormones (e.g., LH, FSH, prolactin, ACTH) were also modified after Cd exposure [107]. Nevertheless, it remains to be investigated if Cd acts as an endocrine modulator by interacting with ERs or ARs in the testis and/or Leydig cells. The study of the hypothalamic-pituitary-gonadal (HPG) axis in animals exposed to the metal is of great interest since the levels of Cd in air, water, soil, and foods have increased by several folds in many parts of the world as a result of emissions from industrial activities. Cd accumulation increased in the hypothalamus and testes in all the Cd-treated animals, whereas the accumulation of Cd in the pituitary was found only in postpubertal rats. These data suggest that Cd exerts age-dependent effects on the hypothalamic-pituitary-testicular (HPT) axis function, and a disruption of the regulatory mechanisms of the HPG axis emerges [108]. Previous studies have shown that the heavy metal Cd mimics the effects of estradiol in estrogen-responsive breast cancer cell lines. Cd activates ER-alpha through an interaction with the hormone-binding domain of the receptor [109]. Cd globally effects HPT axis function by acting at the three levels analyzed and that an interaction between Cd exposure and age emerge [110]. The effects of Cd on sGnRH and rtERa gene expression in the brain in rainbow trout model, and these genes are strongly involved in reproduction [111]. But one study show that the effect of Cd on the ability of medaka gonads to produce steroids without the presence of physiological signals from brain, pituitary or circulating blood was examined in vitro [112]. Plasma levels of luteinizing hormone (LH) were not modified by Cd in both age groups, but follicle stimulating hormone (FSH) levels decreased in postpubertal rats, and was not altered in pubertal rats. Plasma levels of testosterone increased in pubertal rats but decreased in postpubertal rats [108].
In summary, it is important to note that the endocrine disruption induced by Cd is likely to be multi-factorial, mediated via its effects on Leydig cells and/or the hypothalamic-pituitary-gonadal axis.
4.6. Epigenetic Effect
Cd binds DNA in a weak fashion, indicating this is not a primary mode of action. Because DNA sequence is static, genetic susceptibility from DNA sequence variation cannot explain the mechanisms by which prenatal or early childhood metal exposures impact cognition and behavior later in life. Cd may well act as an epigenetic or indirectly genotoxic carcinogen since it is, in general, poorly mutagenic [6, 113]. There is growing evidence that exposure to toxicants in early life may cause later health effects. Children of women exposed to Cd during pregnancy display lower motor and perceptual abilities. High Cd body burden in children is also related to impaired intelligence and lowered school achievement. One possible mechanistic pathway for this phenomenon, which has yet to be fully explored in humans, is epigenetics. Epigenetics is the study of heritable changes in gene expression that occur without changes in DNA sequence. Such changes can have influences as profound as those exerted by mutations but, unlike mutations, are reversible and responsive to environmental influences. DNA methylation is the best studied of the epigenetic processes that regulate gene silencing. DNA methylation results in the addition of a methyl group to the 5′position of the cytosine ring in the context of CpG dinucleotides (or island) to form 5-methylcytosine (5-MeC) [114]. Several studies have demonstrated that DNA methylation was facilitated by long-term exposure to Cd [115, 116]. Oxidative stress may be a unifying process to explain these findings across different metals. Metals are known to increase reactive oxygen species production in a catalytic fashion via redox cycling. Oxidative DNA damage can interfere with the ability of methyltransferases to interact with DNA [117], thus resulting in a generalized hypomethylation of cytosine residues at CpG sites [118]. Cd-induced alterations in methylation metabolism could initiate a cascade of events including gene-specific DNA hypo- or hyper-methylation [119], resulting in aberrant gene expression and also in diminished glutathione activity leaving cells more vulnerable to oxidative stress. Although the results of these epigenetic changes on neurodevelopment have remained unexplored, given the clear importance of DNA methylation to processes of neurodevelopment, the metal-induced disruption of DNA methylation clearly deserves further study. Neurotoxicity is a common health endpoint for excess Cd exposure. Finally, there is intriguing evidence that epigenetic phenomena may underlie observed effects of fetal or early life exposure and late onset of disease. In addition, Cd inhibited DNA methyltransferases in a manner that was noncompetitive with respect to the DNA substrate. This finding is suggestive of interference in enzyme DNA interaction, possibly through an interaction of Cd with the methyltransferase DNA binding domain [115]. Failure of DNA methylation systems in the brain leads to clinical syndromes such as mental retardation and autistic-like behaviors [120]. Animal studies increasingly demonstrate that environmental factors can alter DNA methylation patterns and that these changes correlate with animal behavior [121]. Further research in Cd should include the role of epigenetics in determining long-term and late-onset health effects from metal exposure.
5. Summary
Cd plays a critical role in neurobiology; a growing number of clinical investigations have pointed to Cd intoxication as a possible etiological factor of neurodegenerative diseases, including Parkinson's disease, Alzheimer's disease, and Huntington's disease [50, 122, 123]. Many individuals in Europe and Asian already exceed these exposure levels, and the margin is very narrow for large groups. The question remains open about the impact of long term low level exposure to Cd on vulnerable subgroups of the general population (foetuses, pregnant women, young children, and elderly people) living in historically polluted areas where nonferrous industries are or were in operation. The neurotoxic effects of Cd were complex associated with both biochemical changes of the cell and functional changes of central nervous system, suggesting that neurotoxic effects may play a role in the systemic toxic effects of the exposure to Cd, particularly the long-term exposure. So the mechanism of Cd neurotoxicity should be enhancing, and measures should be taken to reduce cadmium exposure in the general population in order to minimize the risk of adverse health effects. The recent report on toxicity testing in the 21st century by the National Research Council of the National Academy of Sciences recommends the use of cell lines of human origin. The future of environmental research on Cd should include the role of neurotoxic in determining the long-term and late-onset health effects following Cd exposure.
Conflict of Interests
The authors have no conflict of interests to declare.
Acknowledgment
This paper was supported by National Natural Science Foundation of China (NSFC, no. 81260310).
Abbreviations
- 5-MeC:
5-Methylcytosine
- ACGIH:
American conference of governmental industrial hygienists
- Ach:
Acetylcholine
- AChE:
Acetylcholinesterase
- AD:
Alzheimer's disease
- ADHD:
Attention deficit hyperactivity disorder
- BBB:
Blood brain barrier
- BuChE:
Butyrylcholinesterase
- B.W.:
Body weight
- Cd:
Cadmium
- ChE:
Cholinesterases
- CFS:
Chronic fatigue syndrome
- CNP:
Cyclic nucleotide phosphodiesterase
- CPSC:
Consumer product safety commission
- CNS:
Central nervous system
- CSF:
Cerebrospinal fluid
- Dx:
Dexamethasone
- FAO:
Food and Agriculture Organization
- FSH:
Follicle stimulating hormone
- HPG:
Hypothalamus-pituitary-gonadal axis
- HPT:
Hypothalamic-pituitary-testicular
- Im:
Intramuscular
- IQ:
Intelligence quotient
- LDH:
Lactate dehydrogenase
- LH:
Luteinizing hormone
- LPO:
Lipid peroxidation
- MAPKs:
Mitogen-activated protein kinases
- mTOR:
Mammalian target of rapamycin
- MBP:
Myelin basic protein
- Mcns:
Mouse cortical neural stem cells
- ME:
Myalgic encephalomyelitis
- MND:
Motor neuron disease
- MT:
Metallothionein
- NS-1:
Neuroscreen-1 cells
- OLP:
Oligodendrocyte progenitors
- PNP:
Polyneuropathy
- PTWI:
Provisional tolerable weekly intake
- ReN CX:
Human ReNcell CX
- Sc:
Subcutaneous
- SMND:
Sporadic motor neuron disease
- THs:
Thyroid hormones
- WHO:
World Health Organization
- Zn:
Zinc.
References
- 1.ATSDR. Guidance Manual for the Joint Toxic Action of Chemical Mixtures. 2004, http://www.atsdr.cdc.gov/interactionprofiles/ipga.html. [DOI] [PubMed]
- 2.Beryllium, cadmium, mercury, and exposures in the glass manufacturing industry. Working Group views and expert opinions. IARC Monographs on the Evaluation of Carcinogenic Risks To Humans. 1993;58:1–415. [PMC free article] [PubMed] [Google Scholar]
- 3.FAO/WHO. WHO Technical Report Series No. 776. Geneva, Switzerland: WHO; 1988. Evaluation of certain food additives and contaminants (Thirty-third report of the Joint FAO/WHO Expert Committee on Food Additives) [PubMed] [Google Scholar]
- 4.Mead MN. Cadmium confusion do consumers need protection? Environmental Health Perspectives. 2010;118(12):A528–A534. doi: 10.1289/ehp.118-a528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jin T, Lu J, Nordberg M. Toxicokinetics and biochemistry of cadmium with special emphasis on the role of metallothionein. NeuroToxicology. 1998;19(4-5):529–536. [PubMed] [Google Scholar]
- 6.Waalkes MP. Cadmium carcinogenesis in review. Journal of Inorganic Biochemistry. 2000;79(1–4):241–244. doi: 10.1016/s0162-0134(00)00009-x. [DOI] [PubMed] [Google Scholar]
- 7.Pesch B, Haerting J, Ranft U, Klimpel A, Oelschlägel B, Schill W. Occupational risk factors for renal cell carcinoma: agent-specific results from a case-control study in Germany. MURC Study Group. Multicenter urothelial and renal cancer study. International Journal of Epidemiology. 2000;29(6):1014–1024. doi: 10.1093/ije/29.6.1014. [DOI] [PubMed] [Google Scholar]
- 8.Joseph P, Muchnok TK, Klishis ML, et al. Cadmium-induced cell transformation and tumorigenesis are associated with transcriptional activation of c-fos, c-jun, and c-myc proto-oncogenes: role of cellular calcium and reactive oxygen species. Toxicological Sciences. 2001;61(2):295–303. doi: 10.1093/toxsci/61.2.295. [DOI] [PubMed] [Google Scholar]
- 9.López E, Figueroa S, Oset-Gasque MJ, González MP. Apoptosis and necrosis: two distinct events induced by cadmium in cortical neurons in culture. British Journal of Pharmacology. 2003;138(5):901–911. doi: 10.1038/sj.bjp.0705111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cao Y, Chen A, Radcliffe J, et al. Postnatal cadmium exposure, neurodevelopment, and blood pressure in children at 2, 5, and 7 years of age. Environmental Health Perspectives. 2009;117(10):1580–1586. doi: 10.1289/ehp.0900765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pihl RO, Parkes M. Hair element content in learning disabled children. Science. 1977;198(4313):204–206. doi: 10.1126/science.905825. [DOI] [PubMed] [Google Scholar]
- 12.Kim SD, Moon CK, Eun S-Y, Ryu PD, Jo SA. Identification of ASK1, MKK4, JNK, c-Jun, and caspase-3 as a signaling cascade involved in cadmium-induced neuronal cell apoptosis. Biochemical and Biophysical Research Communications. 2005;328(1):326–334. doi: 10.1016/j.bbrc.2004.11.173. [DOI] [PubMed] [Google Scholar]
- 13.Monroe RK, Halvorsen SW. Cadmium blocks receptor-mediated Jak/STAT signaling in neurons by oxidative stress. Free Radical Biology and Medicine. 2006;41(3):493–502. doi: 10.1016/j.freeradbiomed.2006.04.023. [DOI] [PubMed] [Google Scholar]
- 14.Marlowe M, Errera J, Jacobs J. Increased lead and cadmium burdens among mentally retarded children and children with borderline intelligence. American Journal of Mental Deficiency. 1983;87(5):477–483. [PubMed] [Google Scholar]
- 15.Capel ID, Pinnock MH, Dorrell HM. Comparison of concentrations of some trace, bulk, and toxic metals in the hair of normal and dyslexic children. Clinical Chemistry. 1981;27(6):879–881. [PubMed] [Google Scholar]
- 16.Thatcher RW, Lester ML, McAlaster R, Horst R. Effects of low levels of cadmium and lead on cognitive functioning in children. Archives of Environmental Health. 1982;37(3):159–166. doi: 10.1080/00039896.1982.10667557. [DOI] [PubMed] [Google Scholar]
- 17.Thatcher RW, McAlaster R, Lester ML. Evoked potentials related to hair cadmium and lead in children. Annals of the New York Academy of Sciences. 1984;425:384–390. doi: 10.1111/j.1749-6632.1984.tb23560.x. [DOI] [PubMed] [Google Scholar]
- 18.Marlowe M, Cossairt A, Moon C. Main and interaction effects of metallic toxins on classroom behavior. Journal of Abnormal Child Psychology. 1985;13(2):185–198. doi: 10.1007/BF00910641. [DOI] [PubMed] [Google Scholar]
- 19.Korpela H, Loueniva R, Yrjanheikki E, Kauppila A. Lead and cadmium concentrations in maternal and umbilical cord blood, amniotic fluid, placenta, and amniotic membranes. American Journal of Obstetrics and Gynecology. 1986;155(5):1086–1089. doi: 10.1016/0002-9378(86)90356-x. [DOI] [PubMed] [Google Scholar]
- 20.Lafuente A, Esquifino AI. Cadmium effects on hypothalamic activity and pituitary hormone secretion in the male. Toxicology Letters. 1999;110(3):209–218. doi: 10.1016/s0378-4274(99)00159-9. [DOI] [PubMed] [Google Scholar]
- 21.Esquifino AI, Márquez N, Alvarez-Demanuel E, Lafuente A. Effects of chronic alternating cadmium exposure on the episodic secretion of prolactin in male rats. Journal of Trace Elements in Medicine and Biology. 1999;12(4):205–210. doi: 10.1016/S0946-672X(99)80059-5. [DOI] [PubMed] [Google Scholar]
- 22.Lafuente A, Márquez N, Piquero S, Esquifino AI. Cadmium affects the episodic luteinizing hormone secretion in male rats: possible age-dependent effects. Toxicology Letters. 1999;104(1-2):27–33. doi: 10.1016/s0378-4274(98)00349-x. [DOI] [PubMed] [Google Scholar]
- 23.Pal R, Nath R, Gill KD. Influence of ethanol on cadmium accumulation and its impact on lipid peroxidation and membrane bound functional enzymes (Na+, K+-ATPase and acetylcholinesterase) in various regions of adult rat brain. Neurochemistry International. 1993;23(5):451–458. doi: 10.1016/0197-0186(93)90129-s. [DOI] [PubMed] [Google Scholar]
- 24.Manton WI, Cook JD. High accuracy (stable isotope dilution) measurements of lead in serum and cerebrospinal fluid. British Journal of Industrial Medicine. 1984;41(3):313–319. doi: 10.1136/oem.41.3.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Antonio MT, Corredor L, Leret ML. Study of the activity of several brain enzymes like markers of the neurotoxicity induced by perinatal exposure to lead and/or cadmium. Toxicology Letters. 2003;143(3):331–340. doi: 10.1016/s0378-4274(03)00194-2. [DOI] [PubMed] [Google Scholar]
- 26.Shukla GS, Chandra SV. Concurrent exposure to lead, manganese, and cadmium and their distribution to various brain regions, liver, kidney, and testis of growing rats. Archives of Environmental Contamination and Toxicology. 1987;16(3):303–310. doi: 10.1007/BF01054947. [DOI] [PubMed] [Google Scholar]
- 27.Gonçalves JF, Fiorenza AM, Spanevello RM, et al. N-acetylcysteine prevents memory deficits, the decrease in acetylcholinesterase activity and oxidative stress in rats exposed to cadmium. Chemico-Biological Interactions. 2010;186(1):53–60. doi: 10.1016/j.cbi.2010.04.011. [DOI] [PubMed] [Google Scholar]
- 28.Mendez-Armenta M, Rios C. Cadmium neurotoxicity. Environmental Toxicology and Pharmacology. 2007;23(3):350–358. doi: 10.1016/j.etap.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 29.Zheng W, Perry DF, Nelson DL, Aposhian HV. Choroid plexus protects cerebrospinal fluid against toxic metals. FASEB Journal. 1991;5(8):2188–2193. doi: 10.1096/fasebj.5.8.1850706. [DOI] [PubMed] [Google Scholar]
- 30.Tjalve H, Henriksson J. Uptake of metals in the brain via olfactory pathways. NeuroToxicology. 1999;20(2-3):181–195. [PubMed] [Google Scholar]
- 31.Bondier J-R, Michel G, Propper A, Badot P-M. Harmful effects of cadmium on olfactory system in mice. Inhalation Toxicology. 2008;20(13):1169–1177. doi: 10.1080/08958370802207292. [DOI] [PubMed] [Google Scholar]
- 32.Czarnecki LA, Moberly AH, Rubinstein T, Turkel DJ, Pottackal J, McGann JP. In vivo visualization of olfactory pathophysiology induced by intranasal cadmium instillation in mice. NeuroToxicology. 2011;32(4):441–449. doi: 10.1016/j.neuro.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mascagni P, Consonni D, Bregante G, Chiappino G, Toffoletto F. Olfactory function in workers exposed to moderate airborne cadmium levels. NeuroToxicology. 2003;24(4-5):717–724. doi: 10.1016/S0161-813X(03)00024-X. [DOI] [PubMed] [Google Scholar]
- 34.Chow ESH, Hui MNY, Lin CC, Cheng SH. Cadmium inhibits neurogenesis in zebrafish embryonic brain development. Aquatic Toxicology. 2008;87(3):157–169. doi: 10.1016/j.aquatox.2008.01.019. [DOI] [PubMed] [Google Scholar]
- 35.Fern R, Black JA, Ransom BR, Waxman SG. Cd2+-induced injury in CNS white matter. Journal of Neurophysiology. 1996;76(5):3264–3273. doi: 10.1152/jn.1996.76.5.3264. [DOI] [PubMed] [Google Scholar]
- 36.Almazan G, Liu H-N, Khorchid A, Sundararajan S, Martinez-Bermudez AK, Chemtob S. Exposure of developing oligodendrocytes to cadmium causes HSP72 induction, free radical generation, reduction in glutathione levels, and cell death. Free Radical Biology and Medicine. 2000;29(9):858–869. doi: 10.1016/s0891-5849(00)00384-1. [DOI] [PubMed] [Google Scholar]
- 37.Viaene MK, Roels HA, Leenders J, et al. Cadmium: a possible etiological factor in peripheral polyneuropathy. NeuroToxicology. 1999;20(1):7–16. [PubMed] [Google Scholar]
- 38.WHO. Cadmium-Environmental Aspects (Environmental Health Criteria 135) Geneva, Switzerland: . WHO; 1992. [Google Scholar]
- 39.FAO/WHO. WHO Technical Report Series No. 837. Geneva, Switzerland: WHO; 1993. Toxicology evaluation of certain food additives and contaminants. [Google Scholar]
- 40.Satarug S, Haswell-Elkins MR, Moore MR. Safe levels of cadmium intake to prevent renal toxicity in human subjects. British Journal of Nutrition. 2000;84(6):791–802. [PubMed] [Google Scholar]
- 41.Nasreddine L, Parent-Massin D. Food contamination by metals and pesticides in the European Union. Should we worry? Toxicology Letters. 2002;127(1–3):29–41. doi: 10.1016/s0378-4274(01)00480-5. [DOI] [PubMed] [Google Scholar]
- 42.WHO. Guidelines For Drinking-Water Quality. Geneva, Switzerland, 3rd edition, 2004, http://www.who.int/water_sanitation_health/dwq/gdwq3/en/ [Google Scholar]
- 43.FAO/WHO. Codex Alimentarius Commission. Proceedings of the 29th Session; July 2006; Geneva, Switzerland. International Conference Centre; [Google Scholar]
- 44.ACGIH. Cadmium. Threshold limit values for chemical substances and physical agents and biological exposure indices. Proceedings of the American Conference of Governmental Industrial Hygienists; 2007; Cincinnati, OH, USA. p. p. 17. [Google Scholar]
- 45.EFSA. Scientific Opinion of the Panel on Contaminants in the Food Chain on a Request From the European Commission on cadmium in Food. 2009. [Google Scholar]
- 46.FAO/WHO. Geneva, Switzerland: WHO; 2010. The Summary report of the 73rd JECFA meeting. [Google Scholar]
- 47.Yoshida S. Re-evaluation of acute neurotoxic effects of Cd2+ on mesencephalic trigeminal neurons of the adult rat. Brain Research. 2001;892(1):102–110. doi: 10.1016/s0006-8993(00)03240-6. [DOI] [PubMed] [Google Scholar]
- 48.Bucio L, Souza V, Albores A, et al. Cadmium and mercury toxicity in a human fetal hepatic cell line (WRL-68 cells) Toxicology. 1995;102(3):285–299. doi: 10.1016/0300-483x(95)03095-w. [DOI] [PubMed] [Google Scholar]
- 49.Sarchielli E, Pacini S, Morucci G, et al. Cadmium induces alterations in the human spinal cord morphogenesis. BioMetals. 201;25(1):63–74. doi: 10.1007/s10534-011-9483-9. [DOI] [PubMed] [Google Scholar]
- 50.Okuda B, Iwamoto Y, Tachibana H, Sugita M, et al. Parkinsonism after acute cadmium poisoning. Clinical Neurology and Neurosurgery. 1997;99(4):263–265. doi: 10.1016/s0303-8467(97)00090-5. [DOI] [PubMed] [Google Scholar]
- 51.Jiang L-F, Yao T-M, Zhu Z-L, Wang C, Ji L-N. Impacts of Cd(II) on the conformation and self-aggregation of Alzheimer’s tau fragment corresponding to the third repeat of microtubule-binding domain. Biochimica et Biophysica Acta. 2007;1774(11):1414–1421. doi: 10.1016/j.bbapap.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 52.Xu B, Chen S, Luo Y, et al. Calcium signaling is involved in cadmium-induced neuronal apoptosis via induction of reactive oxygen species and activation of MAPK/mTOR network. PLoS One. 2011;6(4) doi: 10.1371/journal.pone.0019052.e19052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yuan Y, Jiang C-Y, Xu XH, et al. Cadmium-induced apoptosis in primary rat cerebral cortical neurons culture is mediated by a calcium signaling pathway. PLoS One. 2013;8(5) doi: 10.1371/journal.pone.0064330.e64330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen L, Liu L, Huang S. Cadmium activates the mitogen-activated protein kinase (MAPK) pathway via induction of reactive oxygen species and inhibition of protein phosphatases 2A and 5. Free Radical Biology and Medicine. 2008;45(7):1035–1044. doi: 10.1016/j.freeradbiomed.2008.07.011. [DOI] [PubMed] [Google Scholar]
- 55.Gupta A, Chandra SV. Gestational cadmium exposure and brain development: a biochemical study. Industrial Health. 1991;29(2):65–71. doi: 10.2486/indhealth.29.65. [DOI] [PubMed] [Google Scholar]
- 56.Rico EP, Rosemberg DB, Senger MR, et al. Methanol alters ecto-nucleotidases and acetylcholinesterase in zebrafish brain. Neurotoxicology and Teratology. 2006;28(4):489–496. doi: 10.1016/j.ntt.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 57.Senger MR, Rosemberg DB, Rico EP, et al. In vitro effect of zinc and cadmium on acetylcholinesterase and ectonucleotidase activities in zebrafish (Danio rerio) brain. Toxicology in Vitro. 2006;20(6):954–958. doi: 10.1016/j.tiv.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 58.Goncalves JF, Nicoloso FT, da Costa P, et al. Behavior and brain enzymatic changes after long-term intoxication with cadmium salt or contaminated potatoes. Food and Chemical Toxicology. 2012;50(10):3709–3718. doi: 10.1016/j.fct.2012.07.016. [DOI] [PubMed] [Google Scholar]
- 59.Zarros A, Kalopita K, Tsakiris S, Baillie GS. Can acetylcholinesterase activity be considered as a reliable biomarker for the assessment of cadmium-induced neurotoxicity? Food and Chemical Toxicology. 2013;56:406–410. doi: 10.1016/j.fct.2013.02.048. [DOI] [PubMed] [Google Scholar]
- 60.Herba E, Pojda-Wilczek D, Pojda SM, Plech A, Brus R. The effect of serotonin on flash visual evoked potential in the rat prenatally exposed to cadmium. Klinika Oczna. 2001;103(2-3):81–84. [PubMed] [Google Scholar]
- 61.Desi I, Nagymajtenyi L, Schulz H. Behavioural and neurotoxicological changes caused by cadmium treatment of rats during development. Journal of Applied Toxicology. 1998;18(1):63–70. doi: 10.1002/(sici)1099-1263(199801/02)18:1<63::aid-jat475>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 62.Gupta A, Shukla GS. Ontogenic profile of brain lipids following perinatal exposure to cadmium. Journal of Applied Toxicology. 1996;16(3):227–233. doi: 10.1002/(SICI)1099-1263(199605)16:3<227::AID-JAT337>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 63.Adams RG, Crabtree N. Anosmia in alkaline battery workers. British Journal of Industrial Medicine. 1961;18:216–221. doi: 10.1136/oem.18.3.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.MNusioł A, Szyrocka-Szwed K, Wojczuk J, Kudybka E. Evaluation of the neurological status and EEG findings in workers chronically exposed to cadmium. Wiadomosci Lekarskie. 1981;34(19):1615–1620. [PubMed] [Google Scholar]
- 65.Struempler RE, Larson GE, Rimland B. Hair mineral analysis and disruptive behavior in clinically normal young men. Journal of Learning Disabilities. 1985;18(10):609–612. doi: 10.1177/002221948501801009. [DOI] [PubMed] [Google Scholar]
- 66.Hart RP, Rose CS, Hamer RM. Neuropsychological effects of occupational exposure to cadmium. Journal of Clinical and Experimental Neuropsychology. 1989;11(6):933–943. doi: 10.1080/01688638908400946. [DOI] [PubMed] [Google Scholar]
- 67.Bar-Sela S, Levy M, Westin JB, Laster R, Richter ED. Medical findings in nickel-cadmium battery workers. Israel Journal of Medical Sciences. 1992;28(8-9):578–583. [PubMed] [Google Scholar]
- 68.Rose CS, Heywood PG, Costanzo RM. Olfactory impairment after chronic occupational cadmium exposure. Journal of Occupational Medicine. 1992;34(6):600–605. [PubMed] [Google Scholar]
- 69.Viaene MK, Masschelein R, Leenders J, Swerts LJVC, de Groof M, Roels HA. Neurobehavioural effects of occupational exposure to cadmium: a cross sectional epidemiological study. Occupational and Environmental Medicine. 2000;57(1):19–27. doi: 10.1136/oem.57.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sethi PK, Khandelwal D. Cadmium exposure: health hazards of silver cottage industry in developing countries. Journal of Medical Toxicology. 2006;2(1):14–15. doi: 10.1007/BF03161007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bao Q-S, Lu C-Y, Song H, et al. Behavioural development of school-aged children who live around a multi-metal sulphide mine in Guangdong province, China: a cross-sectional study. BMC Public Health. 2009;9(article 217) doi: 10.1186/1471-2458-9-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Papp A, Oszlánczi G, Horváth E, et al. Consequences of subacute intratracheal exposure of rats to cadmium oxide nanoparticles: electrophysiological and toxicological effects. Toxicology & Industrial Health. 2012;28(10):933–941. doi: 10.1177/0748233711430973. [DOI] [PubMed] [Google Scholar]
- 73.Borisova T, Krisanova N, Sivko R, et al. Presynaptic malfunction: the neurotoxic effects of cadmium and lead on the proton gradient of synaptic vesicles and glutamate transport. Neurochemistry International. 2011;59(2):272–279. doi: 10.1016/j.neuint.2011.05.014. [DOI] [PubMed] [Google Scholar]
- 74.Borges VC, Santos FW, Rocha JBT, Nogueira CW. Heavy metals modulate glutamatergic system in human platelets. Neurochemical Research. 2007;32(6):953–958. doi: 10.1007/s11064-006-9231-7. [DOI] [PubMed] [Google Scholar]
- 75.Nation JR, Miller DK, Bratton GR. Dietary cadmium exposure alters characteristics of training, substitution, and tolerance when morphine is used as a discriminative stimulus. NeuroToxicology. 2000;21(4):553–567. [PubMed] [Google Scholar]
- 76.Bonithon-Kopp C, Huel G, Grasmick C. Effects of pregnancy on the inter-individual variations in blood levels of lead, cadmium and mercury. Biological Research in Pregnancy and Perinatology. 1986;7(1):37–42. [PubMed] [Google Scholar]
- 77.Vinceti M, Guidetti D, Bergomi M, et al. Lead, cadmium, and selenium in the blood of patients with sporadic amyotrophic lateral sclerosis. Italian Journal of Neurological Sciences. 1997;18(2):87–92. doi: 10.1007/BF01999568. [DOI] [PubMed] [Google Scholar]
- 78.Pamphlett R, McQuilty R, Zarkos K. Blood levels of toxic and essential metals in motor neuron disease. NeuroToxicology. 2001;22(3):401–410. doi: 10.1016/s0161-813x(01)00029-8. [DOI] [PubMed] [Google Scholar]
- 79.de Lange FP, Kalkman JS, Bleijenberg G, Hagoort P, Van Der Meer JWM, Toni I. Gray matter volume reduction in the chronic fatigue syndrome. NeuroImage. 2005;26(3):777–781. doi: 10.1016/j.neuroimage.2005.02.037. [DOI] [PubMed] [Google Scholar]
- 80.López E, Arce C, Oset-Gasque MJ, Cañadas S, González MP. Cadmium induces reactive oxygen species generation and lipid peroxidation in cortical neurons in culture. Free Radical Biology and Medicine. 2006;40(6):940–951. doi: 10.1016/j.freeradbiomed.2005.10.062. [DOI] [PubMed] [Google Scholar]
- 81.Yousef S, Adem A, Zoubeidi T, Kosanovic M, Mabrouk AA, Eapen V. Attention deficit hyperactivity disorder and environmental toxic metal exposure in the United Arab Emirates. Journal of Tropical Pediatrics. 2011;57(6):457–460. doi: 10.1093/tropej/fmq121.fmq121 [DOI] [PubMed] [Google Scholar]
- 82.Shukla A, Shukla GS, Srimal RC. Cadmium-induced alterations in blood—brain barrier permeability and its possible correlation with decreased microvessel antioxidant potential in rat. Human and Experimental Toxicology. 1996;15(5):400–405. doi: 10.1177/096032719601500507. [DOI] [PubMed] [Google Scholar]
- 83.Uchida Y, Takio K, Titani K, Ihara Y, Tomonaga M. The growth inhibitory factor that is deficient in the Alzheimer’s disease brain is a 68 amino acid metallothionein-like protein. Neuron. 1991;7(2):337–347. doi: 10.1016/0896-6273(91)90272-2. [DOI] [PubMed] [Google Scholar]
- 84.Nishimura N, Nishimura H, Ghaffar A, Tohyama C. Localization of metallothionein in the brain of rat and mouse. Journal of Histochemistry and Cytochemistry. 1992;40(2):309–315. doi: 10.1177/40.2.1552172. [DOI] [PubMed] [Google Scholar]
- 85.Méndez-Armenta M, Villeda-Hernández J, Barroso-Moguel R, Nava-Ruíz C, Jiménez-Capdeville ME, Ríos C. Brain regional lipid peroxidation and metallothionein levels of developing rats exposed to cadmium and dexamethasone. Toxicology Letters. 2003;144(2):151–157. doi: 10.1016/s0378-4274(03)00199-1. [DOI] [PubMed] [Google Scholar]
- 86.Onyango IG, Bennett JP, Jr., Tuttle JB. Endogenous oxidative stress in sporadic Alzheimer’s disease neuronal cybrids reduces viability by increasing apoptosis through pro-death signaling pathways and is mimicked by oxidant exposure of control cybrids. Neurobiology of Disease. 2005;19(1-2):312–322. doi: 10.1016/j.nbd.2005.01.026. [DOI] [PubMed] [Google Scholar]
- 87.Chen L, Xu B, Liu L, et al. Cadmium induction of reactive oxygen species activates the mTOR pathway, leading to neuronal cell death. Free Radical Biology and Medicine. 2011;50(5):624–632. doi: 10.1016/j.freeradbiomed.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chen L, Liu L, Luo Y, Huang S. MAPK and mTOR pathways are involved in cadmium-induced neuronal apoptosis. Journal of Neurochemistry. 2008;105(1):251–261. doi: 10.1111/j.1471-4159.2007.05133.x. [DOI] [PubMed] [Google Scholar]
- 89.Antonio MT, Corpas I, Leret ML. Neurochemical changes in newborn rat’s brain after gestational cadmium and lead exposure. Toxicology Letters. 1999;104(1-2):1–9. doi: 10.1016/s0378-4274(98)00125-8. [DOI] [PubMed] [Google Scholar]
- 90.Hayat S, Wigley CB, Robbins J. Intracellular calcium handling in rat olfactory ensheathing cells and its role in axonal regeneration. Molecular and Cellular Neuroscience. 2003;22(2):259–270. doi: 10.1016/s1044-7431(03)00051-4. [DOI] [PubMed] [Google Scholar]
- 91.Fox AP, Nowycky MC, Tsien RW. Kinetic and pharmacological properties distinguishing three types of calcium currents in chick sensory neurones. Journal of Physiology. 1987;394:149–172. doi: 10.1113/jphysiol.1987.sp016864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Huguenard JR. Low-threshold calcium currents in central nervous system neurons. Annual Review of Physiology. 1996;58:329–348. doi: 10.1146/annurev.ph.58.030196.001553. [DOI] [PubMed] [Google Scholar]
- 93.Sundgren-Andersson AK, Johansson S. Calcium spikes and calcium currents in neurons from the medial preoptic nucleus of rat. Brain Research. 1998;783(2):194–209. doi: 10.1016/s0006-8993(97)01342-5. [DOI] [PubMed] [Google Scholar]
- 94.Aoki T, Baraban SC. Properties of a calcium-activated K+ current on interneurons in the developing rat hippocampus. Journal of Neurophysiology. 2000;83(6):3453–3461. doi: 10.1152/jn.2000.83.6.3453. [DOI] [PubMed] [Google Scholar]
- 95.Liu X, Zhou J-L, Chung K, Chung JM. Ion channels associated with the ectopic discharges generated after segmental spinal nerve injury in the rat. Brain Research. 2001;900(1):119–127. doi: 10.1016/s0006-8993(01)02274-0. [DOI] [PubMed] [Google Scholar]
- 96.Hsieh Y-C, Hsu E-L, Chow Y-S, Kou R. Effects of calcium channel antagonists on the corpora allata of adult male loreyi leafworm Mythimna loreyi: Juvenile hormone acids release and intracellular calcium level. Archives of Insect Biochemistry and Physiology. 2001;48(2):89–99. doi: 10.1002/arch.1061. [DOI] [PubMed] [Google Scholar]
- 97.Acuña-Castillo C, Morales B, Huidobro-Toro JP. Zinc and copper modulate differentially the P2X4 receptor. Journal of Neurochemistry. 2000;74(4):1529–1537. doi: 10.1046/j.1471-4159.2000.0741529.x. [DOI] [PubMed] [Google Scholar]
- 98.Ryan SK, Shotts LR, Hong S-K, et al. Glutamate regulates neurite outgrowth of cultured descending brain neurons from larval lamprey. Developmental Neurobiology. 2007;67(2):173–188. doi: 10.1002/dneu.20335. [DOI] [PubMed] [Google Scholar]
- 99.Son J, Lee S-E, Park B-S, et al. Biomarker discovery and proteomic evaluation of cadmium toxicity on a collembolan species, Paronychiurus kimi (Lee) Proteomics. 2011;11(11):2294–2307. doi: 10.1002/pmic.200900690. [DOI] [PubMed] [Google Scholar]
- 100.Culbreth ME, Harrill JA, Freudenrich TM, Mundy WR, Shafer TJ. Comparison of chemical-induced changes in proliferation and apoptosis in human and mouse neuroprogenitor cells. Neurotoxicology. 2012;33(6):1499–1510. doi: 10.1016/j.neuro.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 101.Llop S, Lopez-Espinosa MJ, Rebagliato M, Ballester F. Gender differences in the neurotoxicity of metals in children. Toxicology. 2013 doi: 10.1016/j.tox.2013.04.015. [DOI] [PubMed] [Google Scholar]
- 102.Vahter M, Åkesson A, Lidén C, Ceccatelli S, Berglund M. Gender differences in the disposition and toxicity of metals. Environmental Research. 2007;104(1):85–95. doi: 10.1016/j.envres.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 103.Ishitobi H, Mori K, Yoshida K, Watanabe C. Effects of perinatal exposure to low-dose cadmium on thyroid hormone-related and sex hormone receptor gene expressions in brain of offspring. NeuroToxicology. 2007;28(4):790–797. doi: 10.1016/j.neuro.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 104.Ishitobi H, Watanabe C. Effects of low-dose perinatal cadmium exposure on tissue zinc and copper concentrations in neonatal mice and on the reproductive development of female offspring. Toxicology Letters. 2005;159(1):38–46. doi: 10.1016/j.toxlet.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 105.Farsetti A, Mitsuhashi T, Desvergne B, Robbins J, Nikodem VM. Molecular basis of thyroid hormone regulation of myelin basic protein gene expression in rodent brain. Journal of Biological Chemistry. 1991;266(34):23226–23232. [PubMed] [Google Scholar]
- 106.Lafuente A, González-Carracedo A, Romero A, Cano P, Esquifino AI. Cadmium exposure differentially modifies the circadian patterns of norepinephrine at the median eminence and plasma LH, FSH and testosterone levels. Toxicology Letters. 2004;146(2):175–182. doi: 10.1016/j.toxlet.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 107.Lafuente A, Cano P, Esquifino AI. Are cadmium effects on plasma gonadotropins, prolactin, ACTH, GH and TSH levels, dose-dependent? BioMetals. 2003;16(2):243–250. doi: 10.1023/a:1020658128413. [DOI] [PubMed] [Google Scholar]
- 108.Lafuente A, Márquez N, Pérez-Lorenzo M, Pazo D, Esquifino AI. Pubertal and postpubertal cadmium exposure differentially affects the hypothalamic-pituitary-testicular axis function in the rat. Food and Chemical Toxicology. 2000;38(10):913–923. doi: 10.1016/s0278-6915(00)00077-6. [DOI] [PubMed] [Google Scholar]
- 109.Stoica A, Katzenellenbogen BS, Martin MB. Activation of estrogen receptor-α by the heavy metal cadmium. Molecular Endocrinology. 2000;14(4):545–553. doi: 10.1210/mend.14.4.0441. [DOI] [PubMed] [Google Scholar]
- 110.Lafuente A, Márquez N, Pérez-Lorenzo M, Pazo D, Esquifino AI. Cadmium effects on hypothalamic-pituitary-testicular axis in male rats. Experimental Biology and Medicine. 2001;226(6):605–611. doi: 10.1177/153537020122600615. [DOI] [PubMed] [Google Scholar]
- 111.Vetillard A, Bailhache T. Cadmium: an endocrine disrupter that affects gene expression in the liver and brain of juvenile rainbow trout. Biology of Reproduction. 2005;72(1):119–126. doi: 10.1095/biolreprod.104.029520. [DOI] [PubMed] [Google Scholar]
- 112.Tilton SC, Foran CM, Benson WH. Effects of cadmium on the reproductive axis of Japanese medaka (Oryzias latipes) Comparative Biochemistry and Physiology C. 2003;136(3):265–276. doi: 10.1016/j.cca.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 113.Koyama H, Kitoh H, Satoh M, Tohyama C. Low dose exposure to cadmium and its health effects (1). Genotoxicity and carcinogenicity. Japanese Journal of Hygiene. 2002;57(3):547–555. doi: 10.1265/jjh.57.547. [DOI] [PubMed] [Google Scholar]
- 114.Miller OJ, Schnedl W, Allen J, Erlanger BF. 5 Methylcytosine localised in mammalian constitutive heterochromatin. Nature. 1974;251(5476):636–637. doi: 10.1038/251636a0. [DOI] [PubMed] [Google Scholar]
- 115.Takiguchi M, Achanzar WE, Qu W, Li G, Waalkes MP. Effects of cadmium on DNA-(Cytosine-5) methyltransferase activity and DNA methylation status during cadmium-induced cellular transformation. Experimental Cell Research. 2003;286(2):355–365. doi: 10.1016/s0014-4827(03)00062-4. [DOI] [PubMed] [Google Scholar]
- 116.Benbrahim-Tallaa L, Waterland RA, Dill AL, Webber MM, Waalkes MP. Tumor suppressor gene inactivation during cadmium-induced malignant transformation of human prostate cells correlates with overexpression of de Novo DNA methyltransferase. Environmental Health Perspectives. 2007;115(10):1454–1459. doi: 10.1289/ehp.10207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Valinluck V, Tsai H-H, Rogstad DK, Burdzy A, Bird A, Sowers LC. Oxidative damage to methyl-CpG sequences inhibits the binding of the methyl-CpG binding domain (MBD) of methyl-CpG binding protein 2 (MeCP2) Nucleic Acids Research. 2004;32(14):4100–4108. doi: 10.1093/nar/gkh739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Turk PW, Laayoun A, Smith SS, Weitzman SA. DNA adduct 8-hydroxyl-2′-deoxyguanosine (8-hydroxyguanine) affects function of human DNA methyltransferase. Carcinogenesis. 1995;16(5):1253–1255. doi: 10.1093/carcin/16.5.1253. [DOI] [PubMed] [Google Scholar]
- 119.Wang B, Li Y, Tan Y, et al. Low-dose Cd induces hepatic gene hypermethylation, along with the persistent reduction of cell death and increase of cell proliferation in rats and mice. PLoS One. 2012;7(3) doi: 10.1371/journal.pone.0033853.e33853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Shahbazian MD, Zoghbi HY. Rett syndrome and MeCP2: linking epigenetics and neuronal function. American Journal of Human Genetics. 2002;71(6):1259–1272. doi: 10.1086/345360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Schwartz BS, Lee B-K, Bandeen-Roche K, et al. Occupational lead exposure and longitudinal decline in neurobehavioral test scores. Epidemiology. 2005;16(1):106–113. doi: 10.1097/01.ede.0000147109.62324.51. [DOI] [PubMed] [Google Scholar]
- 122.Johnson S. Gradual micronutrient accumulation and depletion in Alzheimer’s disease. Medical Hypotheses. 2001;56(6):595–597. doi: 10.1054/mehy.2000.1301. [DOI] [PubMed] [Google Scholar]
- 123.Panayi AE, Spyrou NM, Iversen BS, White MA, Part P. Determination of cadmium and zinc in Alzheimer’s brain tissue using Inductively Coupled Plasma Mass Spectrometry. Journal of the Neurological Sciences. 2002;195(1):1–10. doi: 10.1016/s0022-510x(01)00672-4. [DOI] [PubMed] [Google Scholar]