

Abstract
Background
Even though adverse event (AE) collection and official accounting are mandatory for clinical trials, there are limited detailed guidelines specifying how to summarize the event for reporting in a timely and expeditious manner. This manuscript details the AE and serious adverse event (SAE) reporting summary developed for a large multi-center National Institutes of Health (NIH) – sponsored clinical trial.
Purpose
To review and analyze the large volume of AE data reported by ten sites (806 SAEs and 19,034 AEs from August 2000 to May 2007) the Automated SAE Summary was developed. It was designed to ensure timeliness and clarity in the complex process of AE review and reporting.
Methods
The AE and SAE case report forms (CRFs) as well as the Automated SAE Summary were developed within a database management system developed by the Data Coordinating Center (DCC) which allowed for web-based data entry at the DCC and ten sites, and offered immediate overall and site-specific reports accessible by the DCC, site and NIH project staff.
Results
The Automated SAE Summary pulled data from multiple CRFs to create a succinct and informative summary and allowed for prompt and easy reporting to the regulatory agencies. The summary was adaptable to the needs of reviewers because of the availability of multiple search options.
Limitations
The advantages discussed in the manuscript include using the summary to identify trends quickly and facilitate the timely reporting of SAEs to the study monitoring entities; disadvantages include using ICD-9 Codes; monitoring open text fields for completeness and quality; and the process of completion of multiple CRFs for the same event.
Conclusions
The Automated SAE Summary was versatile in meeting the needs of multiple individuals. It can be reviewed for safety issues by the DCC, any regulatory agencies and local site Institutional Review Boards, as well as the Industry sponsor.
Keywords: Serious adverse events, automated, reporting, databases, clinical trials
Introduction
Submission and review of data regarding adverse events (AEs) and serious adverse events (SAEs) are an essential component of assuring the safety and protection of participants enrolled in clinical trials. AE data are reviewed on a regular basis by study safety committees, the Institutional Review Boards (IRB), and as appropriate by a Data Safety Monitoring Board (DSMB), the Food and Drug Administration (FDA), as well as any government and industry sponsors. Analysis of AE data is used to assess the risks associated with the product and or service that are being studied and to weigh it against any benefit that might be derived. Even though AE and SAE collection and official accounting are mandatory for clinical trials, there are limited detailed guidelines available that specify how to create a summary report of the event(s) in an expeditious manner to all of the different groups responsible for monitoring clinical trials. (1, 2) Prompt reporting of AEs and SAEs to the numerous interested parties, including regulatory agencies, as well as providing up to date information on each of the AEs are some of the challenges related to the maintaining of complete and accurate AE and SAE data. (3) This manuscript describes the use of a detailed SAE Summary developed for a large multi-center National Institutes of Health (NIH) – sponsored clinical trial.
Background
The Hepatitis C Antiviral Long-Term Treatment against Cirrhosis (HALT-C) study is a multi-center randomized clinical trial designed to determine whether long-term therapy with pegylated interferon in patients with chronic hepatitis C could reduce the risk of progression to cirrhosis, hepatic decompensation, hepatocellular carcinoma, or death. (4) There were 1,050 patients randomized to either treatment or control and then followed for three and a half years. Ten clinical centers, a central repository, a virology lab and a number of smaller ancillary studies laboratories participate in the trial. The data coordinating center (DCC) for the study at New England Research Institutes (NERI) provides project management, statistical services, data management, database design and development, and regulatory affairs services.
The trial is now in its tenth year. The treatment part of the trial ended on April 30, 2007. The study will continue to provide surveillance and follow-up to eligible patients for another 18 months through October 2009. Clinical outcomes), but not AEs, will continue to be reported and analyzed during the follow-up phase.
Definition of Adverse Events and Serious Adverse Events by FDA Guidelines
Currently, no universally accepted system exists for defining, documenting, and tracking AEs and SAEs that occur during biomedical research. (1) One widely accepted system is based on the FDA guidelines, Good Clinical Practice: Consolidated Guideline. This guideline defines “Good Clinical Practice” (GCP) and provides a unified standard for designing, conducting, recording, and reporting of AEs in trials that involve the participation of human subjects. (5) It defines an AE as any untoward medical occurrence in a study participant who has been administered a pharmaceutical product regardless of attribution of causality to that product. Thus, an AE can be any unfavorable and unintended event (including an abnormal laboratory finding), symptom, or disease temporally associated with the use of the product.
Studies that use investigational new drugs must request authorization by the FDA for their use in clinical studies and utilize the investigational new drug (IND) application. Specific regulations govern the submission process for obtaining an IND and also specify the procedures and requirements for the initial and ongoing review of the IND by the FDA. Reporting requirements for AEs that are related to the investigational product use, unexpected and serious in nature, are all defined in these regulations. The FDA considers an AE to be serious if it: results in death; is life-threatening; requires inpatient hospitalization or prolongation of existing hospitalization; results in persistent or significant disability/incapacity; results in a congenital anomaly/birth defect; or requires medical intervention to prevent any of these outcomes. (6)
Definition of Adverse Events and Serious Adverse Events in the Study Protocol
Based on International Conference on Harmonization (ICH) guidelines (5), the HALT-C study protocol defined an AE as any adverse change from the study participant's baseline condition prior to initiation of any drug therapy associated with the study. An intercurrent illness, which might occur during the entire course of the trial, even if not thought to be related to the study treatment, was included. The decision to report all AEs was based on the overlap between some of the symptoms of the disease (fatigue, nausea, vomiting) and AEs related to treatment with interferon. In addition, some of the hematological AEs of treatment with interferon (anemia, neutropenia, and thrombocytopenia) also occur frequently in patients with hepatitis, who are not undergoing treatment. Since the study was conducted under an FDA IND, the FDA definition of seriousness was used. If an AE was considered serious, then additional information was obtained including attribution of relatedness to treatment by the clinical site investigator. SAEs which were reported as unexpected and related to treatment, even remotely, required expedited reporting to the FDA after analysis by the industry sponsor (see below).
Serious Adverse Events versus Clinical Outcomes
Even though clinical outcomes (study endpoints) were serious in nature, they were not reported as SAEs but were collected and reported to the DSMB. Clinical outcomes were defined as events expected as a result of progressive liver disease: 1) death from any cause, 2) development of hepatic decompensation [variceal hemorrhage, ascites, spontaneous bacterial peritonitis, hepatic encephalopathy], 3) hepatocellular carcinoma and 4) a Child-Turcotte-Pugh score of 7 or higher on two consecutive study visits. All possible clinical outcomes were reviewed with source documentation by an Outcome Review Board of 2–3 physicians. These events were identified as separate from (S)AEs. Distinguishing SAEs from clinical outcomes was a challenge. Per protocol, an event defined as a clinical outcome was not an SAE, but if the patient was hospitalized for the event, by protocol definition it could have been an SAE. The one exception was the event of a death, which was defined as both an SAE and a clinical outcome.
Creating a Case Report Form for Serious Adverse Events
As a standard of practice, the DCC has developed a uniform standard for the design of case report forms (CRFs) across the study as well as for the process of data entry and revision. At the time this reporting system was set up (1999) and throughout the trial, all CRFs, including the SAE forms, were completed on paper and filed in a notebook for each participant. All data filled out on the paper form were entered. When any correction was made, previous information was crossed out, initialed and dated on the paper form. The new information was written beside the crossed out answer, and then was entered in the Data Management System (DMS). For each paper CRF, there was always only one corresponding electronic form in the DMS. Any data stored in a CRF and entered in the database also were available electronically and could be summarized in the form of a report.
The SAE CRF developed by the DCC deviated from the CRF standard design because it served a dual purpose: to collect data for the study database and to monitor all SAEs for safety and possible expedited review. The CRF was purposely designed to simulate the drug sponsor's AE report, thus facilitating searches of the sponsor's database for previous similar events. If criteria for expedited reporting to the FDA were met, the Safety Officer at the Industry Sponsor was responsible for preparing a safety packet containing a MedWatch form, an “Analysis of Similar Events,” and a letter to the FDA describing the SAE. In turn, the DCC released these safety packets to the study investigators. Therefore, some basic medical information was added to the paper CRF because the Safety Officer did not have access to the study database. The Industry sponsor also required that any new or additional information about an SAE be recorded on a new paper form.
Therefore, the paper CRF needed to satisfy the industry sponsor's template to facilitate ease in searching their database for possible expedited review, but also needed to be entered and updated in the DMS in order to create the Automated SAE Summary. This CRF was different from any other CRF in the study in two ways: 1) only partial information could be entered and 2) each time there was an update to the information, a new paper CRF was to be used. This dual system caused problems addressed in the Advantages and Limitations section.
Electronic Data Capture
Electronic data capture of a large number of variables that will be sorted and organized in different reports is preferable to using older handwritten methods and/or importing data files. Although this trial used the DCC's Advanced Data Entry and Protocol Tracking System (ADEPT), any DMS has the possibility of creating a similar summary report. The ADEPT DMS is user-friendly and specifically designed to support reliable and secure data entry for research purposes. (7) The system allows for web-based data entry at the DCC and ten sites and offers immediate overall and site-specific reports accessible by DCC staff, site staff and NIH project staff. Reports available are on enrollment, forms status, follow-up schedules, patient summaries, and adverse events.
Reporting the Occurrence of Adverse Events
The reporting of AEs and SAEs commenced at the first Screening Eligibility visit in August 2000. When an event occurred, an AE CRF was completed on paper, entered in the DMS, and the data instantaneously became available at the DCC. (Figure 1) Events marked as serious automatically created an ”expectancy” for an SAE CRF in the DMS, which prompted the site to complete a follow-up report on the SAE. Initial data entry or editing of the SAE CRF triggered an email informing the DCC that the SAE was added or edited, enabling the DCC to review the SAE Summary immediately. Any incomplete fields requiring follow-up were flagged by the system for inclusion in queries.
Figure 1.

Flow chart of the process of adverse events reporting
Serious Adverse Event Description and Guidelines for Writing the Summary
Two different fields for an SAE description were created on the CRF: an ongoing summary (Summary 1) for ongoing events requiring follow-up, and a final summary (Summary 2), for resolved events. Summary 1 provided an overview of the event in a manner similar to a hospital progress note, giving the date and recent summary of events. Often this section included multiple updates until the event was finally resolved. The section allowed for up to 3,000 characters (including punctuation and spaces) to be entered. Summary 2 was completed when the final status of the SAE was known. If the event were continuing or unknown, a special value of −9 (Missing) was entered. The CRF also included a brief (one sentence) description of the SAE and an ICD-9 code (International Statistical Classification of Diseases and Related Health Problems). (8) These ICD-9 codes were used to tabulate SAEs by body system.
Monitoring Accuracy of Adverse Event Reporting and Updates
To ensure accuracy and safety of data, every SAE CRF was faxed to the Project Director at the DCC for 100% data check. The SAE CRF was also reviewed for coding, medical facts, dates, pertinent drug information, and safety reporting for possible expedited review. The Project Director ensured that the data reported on the SAE CRF were consistent with the information reported for the patient to date. Sites were queried regarding any inconsistencies. If follow-up or additional information were needed, the sites were requested to complete a follow-up SAE CRF.
In addition, the medical records of randomly selected patients were reviewed and monitored for possible AEs and SAEs during on-site visits. Any AE described in the medical record was monitored for a corresponding AE in the DMS. Each AE marked as serious was monitored to ensure there was a corresponding SAE CRF completed and entered. Simultaneously, AE and SAE CRFs were checked for accuracy of coding and event description. (9, 10)
Monitoring of the final SAE summary included checking for completeness of relevant drug and medical history. The DSMB developed guidelines over the course of the trial for specific information needed in the SAE summaries. For example, all cardiac and/or pulmonary SAEs (i.e., syncope, shortness of breath, chest pain, etc.) required hemoglobin results from baseline and time of SAE; infections required white blood count and the absolute neutrophil count results from baseline and time of SAE ; “bleeding” SAEs required the platelet count from baseline and time of SAE; cellulitis SAEs required the site/location, especially in relation to the injection site; depression SAEs required any previous history of depression or other mental conditions. A history of cardiac disease or diabetes or other significant medical history as well as relevant treatment information and date the study drug was reduced or stopped also were included. The written summaries were screened carefully for any potential Health Insurance Portability and Accountability Act (HIPAA) violations: specific physician or institutional names were not used.(11) Up to 3,000 characters (including punctuation and spaces) were available for data entry.
Purpose of the Automated SAE Summary
At the commencement of the trial reporting of SAEs to the DSMB, the NIH Project Scientist the FDA and the Industry sponsor, as necessary was cumbersome. Information was typed, cut and pasted from the database into an Excel file. Every month the Excel file was updated and re-sorted by most current SAE and/or current update. This method was time-consuming and prone to error and required separate versions to be produced, depending on the needs of the separate monitoring entities.
The Automated SAE Summary was developed by the DCC to review and analyze the large volume of AE data reported by sites (806 SAEs and 19,034 AEs from August 2000 to May 2007). It ensured timeliness and clarity in the complex process of AE review and reporting. (Figure 1) The user-friendly interface and searching were helpful for browsing, updating and reviewing SAEs. It was easily adaptable to the needs of all reviewers because of the availability of multiple search options. At the DCC, AEs were reviewed for coding errors and inconsistencies, and for completeness of the description of SAEs before submission to the DSMB Chairperson and NIH Project Scientist (monthly), DSMB (semi-annually) and the FDA (annually). DCC data managers and clinical site study coordinators used the Automated SAE Summary for data cleaning purposes.
Automated SAE Summary Workflow
The Automated SAE Summary data could be sorted by the following specific criteria: patient ID, all clinical centers or a specific clinical center, study phase, relationship to study medication, event outcome (continuing or resolved) and specific time period. Once the sorting criteria were chosen, the SAE Summary pulled data from multiple sources and created a half - page document for each SAE, outlining treatment group, age and sex of participant, study week, dose of medication at time of the event, reason the event meets SAE definitions, outcome of the event, the relationship of the event to study drug, and a concise summary of the event. (Figure 2) The SAE summary was reviewed by the DSMB, the FDA and the NIH Project Scientist. Queries from the DSMB could be addressed by the DCC within 24 hours. The SAE summary could be amended easily to provide additional information in response to specific DSMB questions. Each clinical site could only view its own SAE summaries and these reports could be used for preparing site specific IRB submissions. The DCC and NIH project scientist could view SAE summaries from all the sites, thus enabling them to see trends in SAE reporting by site, outcome, relationship to treatment and or specific event.
Figure 2.
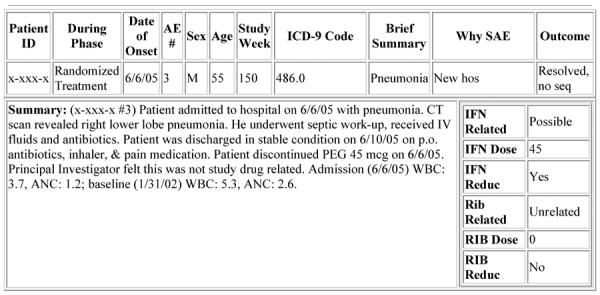
Automated SAE Summary Output
If an SAE was not yet resolved, the ongoing summary (Summary 1) detailed the event as it stood and was shown in the SAE Summary. Once the SAE was resolved, the final summary (Summary 2) was chosen for viewing in the SAE Summary. This final summary was particularly useful in reporting SAEs to the FDA, DSMB, and the NIH Project Scientist because it gave significant medical history as well as the completed summary of the event.
Advantages and Limitations
Advantages of the Automated Reporting system included ability to monitor trends in SAEs, ensure quality of data and the tracking of completeness of the reporting. Limitations were in the use of the ICD-9 coding system and having a dual case report form. Using an open text field had some advantages and some limitations.
Monitoring Trends
The Automated SAE Summary facilitated DCC's monitoring of SAEs for accuracy as well as for trends. For example, during the randomized phase of the trial, the DSMB received monthly SAE Summaries and noted a possible trend with infections and cardiac events. They, therefore, requested the DCC to specifically track infections in order to ensure patient safety. All infections were closely monitored and sites were asked to report absolute neutrophil and white blood cell counts for possible relatedness to study drug.
Quality of Data
Sites were required to fax the completed SAE CRF to the DCC and the Industry Safety Officer, and to enter the form. Having a paper copy of the SAE was very useful for ensuring correct form completion. The Project Director monitored every paper copy and the most recent update to an existing SAE against what was entered for accuracy as well as clinical content (medical history, lab values, stop date of study drug). Any queries to sites included checking the source documentation. Having the paper form to check was helpful in ensuring a succinct and thorough report. Paperless SAE reporting would be more difficult.
Tracking Completeness of Serious Adverse Events
Since mandatory SAE reporting was required within 48 hours of the site's knowledge of the event, some pertinent information was often missing on initial report.(12) The cause, the complete course of treatment or the outcome may have been unknown. It may have taken weeks until a study coordinator was able to obtain medical records from an outside hospital to complete all information required. When the Automated SAE Summary was run, the most up-to-date information was displayed in the output. If the CRF were still missing important information, the study coordinators were able to put a special code (−9) and an explanation. This missing code value created a query in the DMS. The DCC data managers were able to easily send queries to the sites about these specific data fields. Every time an SAE CRF was added or updated, an automatic email listing the patient ID, initials and the type of change was sent to the DCC. The CRF was reviewed on the same day for accuracy and completeness. If additional information were needed, the site was notified.
ICD-9 Codes
To identify AEs, ICD-9 Codes were used that were cross checked with the brief description of the event. The ICD-9 Coding system was chosen in 1999 when the protocol was developed. This system was the one most available and familiar to the protocol developers at that time, but proved difficult to use. ICD-9 codes are best used for diagnoses, with knowledge of the disease etiology. Coding symptoms, therefore, such as confusion, malaise, tingling in extremities was difficult. To overcome these difficulties, the DCC came up with specific codes for all conditions and symptoms that could not be coded with the existing ICD-9 codes. The MedDRA® (Medical Dictionary for Regulatory Activities) (13) and SNOMED CT® (Systematized Nomenclature of Medicine-Clinical Terms) (14) systems are now readily available and should be considered for future trials. The CTCAE (Common Terminology Criteria for Adverse Events) (NCI, CTEP) system is the standard dictionary for reporting AEs in oncology clinical trials (15) and, therefore, not used in HALT-C. Each of these systems has pros and cons and only a detailed evaluation and comparison of the content, structure, access, ease of use and support, although often times not straightforward (15), can provide a meaningful conclusion as to which of the three may best accommodate the needs of a specific study.
Dual Case Report Form Completion
Per the industry sponsor's standard operating procedures, each time a SAE was updated, a new paper CRF was completed and the new information entered. With most events there were multiple SAE paper CRFs for the same participant with the same event number, but only one CRF in the DMS. The DMS kept a log of all updates that were entered and the date that they were entered. However, the task of keeping track of the most recent updates on paper and monitoring for accuracy in the DMS was time consuming and confusing. Site coordinators and data managers were given multiple training sessions on how to complete the paper CRF and how to data enter. A better choice would have been to maintain the standard paper and data entering system of the CRF as for all other forms in the study.
Open Text Field
The summary of the SAE was written by the coordinators in an open text field. Although the summary enabled physicians who were unfamiliar with the subject to get a quick snapshot of the event, it proved the most difficult data to collect. The majority of summaries were accurate, but often too lengthy or missing important medical history information. The DSMB requested specific medical information on these Automated SAE Summaries. Instructions were provided to all sites on appropriate summary content. However, since SAE CRFs were not completed every day or week, repeat training was required and careful monitoring of forms was intense. Some of the challenges included: a large number of sites and site staff (10 sites, with 2–3 coordinators at each site), staff turnover, difference in professional training among coordinators, and writing skills. The Project Director at the DCC monitored and suggested edits to the sites for these summaries.
Conclusions
The Automated SAE Summary was versatile in meeting the needs of monitoring entities. It could be reviewed for safety issues by the DCC Project Director, the FDA, the DSMB, the NIH Project Scientist and local site Institutional Review Boards as well as the Industry sponsor. The SAE Summary output could be quickly and accurately modified in response to the needs and concerns of the monitors.
One of the biggest challenges in the process of maintaining complete SAE data was the actual updating of the SAEs and tracking completion of said updates. (12) The Automated SAE Summary facilitated intense monitoring and tracking of updates of all SAEs. Instead of writing lengthy emails, the SAE Summary could be viewed and/or downloaded at the clinical site as well as the DCC, making it easy to query any inconsistencies for data cleaning. It also facilitated the entering of more accurate and timely data by clearing identifying new data that needed to be collected in response to requests for additional data from the monitoring entities.
Recording of adverse events can become burdensome for site staff. To decrease this burden, user-friendly systems need to be in place to facilitate efficient and accurate AE documentation. Suggestions from lessons learned in the HALT-C Trial would include: using an adverse event CRF consistent with the rest of the study CRFs, providing a concise outline of desired clinical information requested (if there is a free text field), and coding AEs with the most up-to date and easy to use system available.
The Automated SAE Summary was easy and quick to use. Inconsistencies in data were monitored, sites queried and data cleaned in an appropriate timeframe. A paperless system for monitoring AEs and SAEs would be a challenge because data summarized in a report are only as good as the information which is entered. Certainly, in this clinical trial, having a paper system as back up for monitoring and accurate tracking was beneficial. It remains a question whether SAE reporting is ready to go entirely paperless.
Acknowledgments
In addition to the authors of this manuscript, the following individuals were instrumental in the planning, conduct and/or care of patients enrolled in this study at each of the participating institutions as follows:
University of Massachusetts Medical Center, Worcester, MA: (Contract N01-DK-9-2326) Gyongyi Szabo, MD, Donna Giansiracusa, RN
University of Connecticut Health Center, Farmington, CT: (Grant M01RR-06192) Herbert L. Bonkovsky, MD
Saint Louis University School of Medicine, St Louis, MO: (Contract N01-DK-9-2324) Adrian M. Di Bisceglie, MD, Bruce Bacon, MD, Brent Neuschwander-Tetri, MD
Massachusetts General Hospital, Boston, MA: (Contract N01-DK-9-2319, Grant M01RR-01066) Jules L. Dienstag, MD, Raymond T. Chung, MD, Andrea E. Reid, MD
University of Colorado School of Medicine, Denver, CO: (Contract N01-DK-9-2327, Grant M01RR-00051) Gregory T. Everson, MD, Carol McKinley, RN, Brenda Easley, RN
University of California - Irvine, Irvine, CA: (Contract N01-DK-9-2320, Grant M01RR-00827) Timothy R. Morgan, MD, John C. Hoefs, MD,
University of Texas Southwestern Medical Center, Dallas, TX: (Contract N01-DK-9-2321, Grant M01RR-00633) William M. Lee, MD, Nicole Crowder, LVN, Rivka Elbein, RN, BSN
University of Southern California, Los Angeles, CA: (Contract N01-DK-9-2325, Grant M01RR-00043) Karen L. Lindsay, MD, MMM, Carol B. Jones, RN
University of Michigan Medical Center, Ann Arbor, MI: (Contract N01-DK-9-2323, Grant M01RR-00042) Anna S.F. Lok, MD, Robert J. Fontana, MD
Virginia Commonwealth University Health System, Richmond, VA: (Contract N01-DK-9-2322, Grant M01RR-00065) Mitchell L. Shiffman, MD, Richard K. Sterling, MD, Paula Smith, RN
Liver Diseases Branch, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD: Marc G. Ghany, MD, T. Jake Liang, MD, Elenita Rivera, RN, Vanessa Haynes-Williams, RN
National Institute of Diabetes and Digestive and Kidney Diseases, Division of Digestive Diseases and Nutrition, Bethesda, MD: James E. Everhart, MD, Leonard B. Seeff, MD, Jay H. Hoofnagle, MD
University of Washington, Seattle, WA: (Contract N01-DK-9-2318), Chihiro Morishima, MD, David R. Gretch, MD, Minjun Chung, BS, ASCP
New England Research Institutes, Watertown, MA: (Contract N01-DK-9-2328) Dinh Tran, Linda J. Massey, Kristin K. Snow, ScD
Armed Forces Institute of Pathology, Washington, DC: Zachary D. Goodman, MD
Data and Safety Monitoring Board Members: (Chair) Gary L. Davis, MD, Guadalupe Garcia-Tsao, MD, Michael Kutner, PhD, Stanley M. Lemon, MD, Robert P. Perrillo, MD
Financial support: This study was supported by the National Institute of Diabetes & Digestive & Kidney Diseases (contract numbers are listed in the Acknowledgements). Additional support was provided by the National Institute of Allergy and Infectious Diseases, the National Cancer Institute, the National Center for Minority Health and Health Disparities, the National Center for Research Resources (grant numbers are listed below), and National Institute on Alcohol Abuse and Alcoholism grant K24 AA13736. Additional funding to conduct this study was supplied by Hoffmann-La Roche, Inc., through a Cooperative Research and Development Agreement (CRADA) with the National Institutes of Health.
Authors with no financial relationships with Hoffmann-La Roche, Inc., to disclose are: M.C. Bell, P.R. Robuck, E.C. Wright, M.S. Mihova, C. Hofmann, J.L. De Santo, S.L. Milstein, P.A. Richtmyer, J. L. Shelton, D.L. King, C.J. Park, W.A. Molchen, Y.J. Park, and M. Kelley. Financial relationships of the authors with Hoffmann-La Roche, Inc., are as follows: M. Cormier was on the speaker's bureau and received research support.
List of Abbreviations
- ADEPT
Advanced Data Entry and Protocol Tracking System
- AE
Adverse Event
- CRF
Case Report Form
- CTP
Child-Turcotte-Pugh
- DCC
Data Coordinating Center
- DMS
Data Management System
- DSMB
Data Safety Monitoring Board
- FDA
Food and Drug Administration
- GCP
Good Clinical Practice
- HALT-C
Hepatitis C Antiviral Long-term Treatment against Cirrhosis
- HIPAA
Health Insurance Portability and Accountability Act
- ICD-9 Codes
International Statistical Classification of Diseases and Related Health Problems
- ICH
International Conference on Harmonization
- IND
Investigational New Drug
- IRB
Institutional Review Board
- MedDRA®
Medical Dictionary for Regulatory Activities
- NERI
New England Research Institutes
- NIH
National Institutes of Health
- SAE
Serious Adverse Event
- SNOMED CT®
Systematized Nomenclature of Medicine-Clinical Terms
Footnotes
This is publication number 31 from the HALT-C Trial Group.
References
- 1.Mitchell R, Shah M, Ahmad S, Rogers AS, Ellenberg JH. A unified web-based query and notification system (QNS) for subject management, adverse events, regulatory, and IRB components of clinical trials. Clin Trials. 2005;2(1):61–71. doi: 10.1191/1740774505cn68oa. [DOI] [PubMed] [Google Scholar]
- 2.Silverman DI, Cirullo L, DeMartinis NA, Damato K, DeMeo M, Fernandez GA, et al. Systematic identification and classification of adverse events in human research. Contemp Clin Trials. 2006;27(3):295–303. doi: 10.1016/j.cct.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 3.Wisniewski SR, Eng H, Meloro L, Gatt R, Ritz L, Stegman D, et al. Web-based communications and management of a multi-center clinical trial: the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) project. Clin Trials. 2004;1(4):387–98. doi: 10.1191/1740774504cn035oa. [DOI] [PubMed] [Google Scholar]
- 4.Lee WM, Dienstag JL, Lindsay KL, Lok AS, Bonkovsky HL, Shiffman ML, et al. Evolution of the HALT-C Trial: pegylated interferon as maintenance therapy for chronic hepatitis C in previous interferon nonresponders. Control Clin Trials. 2004;25(5):472–92. doi: 10.1016/j.cct.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 5.International Conference on Harmonization (ICH) Guideline for Industry Clinical Safety Data Management: Definitions and Standards for Expedited Reporting. [Google Scholar]
- 6.FDA 21 CFR Part 312 Investigational New Drug Application (IND)
- 7.Clinical Data Management . New England Research Institutes, Inc; [Last accessed 02/19/2009]. ;Available from: http://www.neriscience.com/web/MultiPiecePage.asp_Q_PageID_E_13_A_PageName_E_Datamanagement. [Google Scholar]
- 8. [Last accessed 02/19/2009];Online ICD-9 Codes. ; Available from: http://icd9cm.chrisendres.com/icd9cm/
- 9.Jinjuvadia K, Kwan W, Fontana RJ. Searching for a needle in a haystack: use of ICD-9-CM codes in drug-induced liver injury. Am J Gastroenterol. 2007;102(11):2437–43. doi: 10.1111/j.1572-0241.2007.01456.x. [DOI] [PubMed] [Google Scholar]
- 10.Reuben A. Needles in haystacks. Am J Gastroenterol. 2007;102(11):2444–6. doi: 10.1111/j.1572-0241.2007.01450.x. [DOI] [PubMed] [Google Scholar]
- 11.Health Insurance Portability and Accountability Act . Fed Regist. 2000. Standards for Privacy of Individually Identifiable Health Information - Rules and Regulations; p. 250. [Google Scholar]
- 12.Trotti A, Colevas AD, Setser A, Basch E. Patient-reported outcomes and the evolution of adverse event reporting in oncology. J Clin Oncol. 2007;25(32):5121–7. doi: 10.1200/JCO.2007.12.4784. [DOI] [PubMed] [Google Scholar]
- 13.MedDRA® . The International Federation of Pharmaceutical Manufacturers and Associations (IFPMA) [DOI] [PubMed] [Google Scholar]
- 14.Snomed . International Health Terminology Standards Development Organisation (IHTSDO) [Google Scholar]
- 15.Richesson RL, Fung KW, Krischer JP. Heterogeneous but “standard” coding systems for adverse events: Issues in achieving interoperability between apples and oranges. Contemp Clin Trials. 2008;29(5):635–45. doi: 10.1016/j.cct.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]