

Abstract
Background
Age-related macular degeneration (AMD) is the leading cause of blindness in the western world. To prevent what will certainly be a tremendous health and economic burden, effective therapeutics for AMD are urgently needed. To develop these agents in a timely fashion, the molecular pathways that cause disease progression must be elucidated.
Objective
To briefly describe the clinical features of AMD, and review the current understanding of the molecular basis of AMD.
Methods
A literature review.
Results
The discussion will primarily focus on the interplay of oxidative stress and complement dysregulation and the resulting chronic proinflammatory state thought to be central in AMD pathogenesis.
Conclusions
Oxidative stress and complement dysregulation play a substantive role in the development of AMD.
Keywords: Aging, Age-related macular degeneration, complement, innate immunity, oxidative stress, oxidation-specific epitopes
Introduction
Each year millions of individuals lose their central vision, compromising their ability to distinguish faces, read, and drive. They will add to the 15-20 million people in the US and more than 50 million worldwide who suffer from Age-Related Macular Degeneration (AMD) [1]. Since the principle risk factor for AMD is advanced age, the aging population will increase the health burden from this disease. A better understanding of the pathogenesis of AMD is crucially needed to develop effective therapeutics that prevent or reverse this disease.
The pathogenesis of AMD is influenced by complex crosstalk between various elements of the retinal microenvironment. It is a multifactorial, chronic, age-related disease with both environmental and genetic factors. The past decade has been highly fruitful at elucidating key molecular pathways in AMD. It is clear that oxidative stress resulting from cigarette smoking is strongly linked to AMD. Genetic studies have introduced the role of innate immune dysfunction as a pivotal factor in AMD. In this review, we will describe how oxidative stress and innate immune dysfunction contribute to the development of AMD. We present their roles from the perspective that both pathways are physiological, but with imbalance, they can play a pathological role.
Anatomical and Histological Considerations
The retina has the richest blood supply and greatest oxygen demand by weight of any organ in the body [2]. The retinal microenvironment is inherently pro-oxidative because it is exposed to high ambient oxygen (70-90 mmHg), photo-oxidative stress, and high metabolism [2]. The inner retina receives its blood from the retinal vasculature while the outer retina obtains its nourishment from the adjacent choriocapillaris. This dual circulation makes up the blood-retinal barrier, which provides relative immune privilege. The macula is the 6mm centermost area of the neurosensory retina that is responsible for high resolution vision (Fig. 1).
Figure 1.
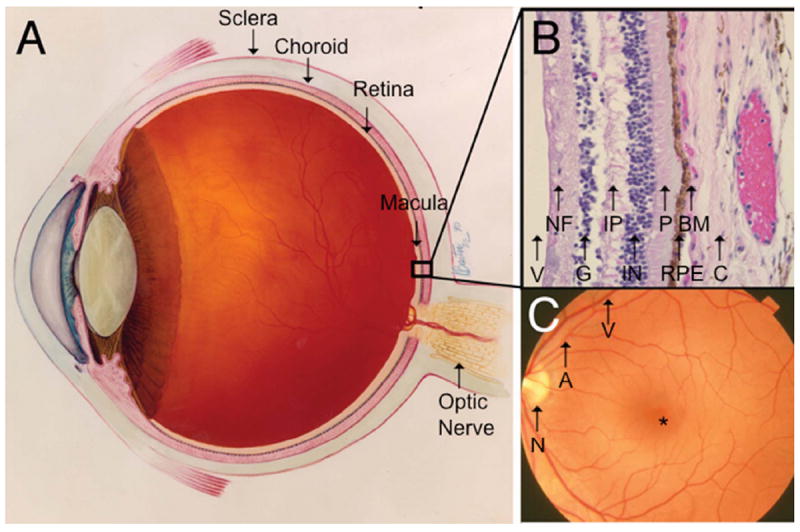
Ocular anatomy relevant to AMD. A. Cross-section of an eye showing the retina and neighboring anatomic structures. B. Histological cross-section of the retina moving inward along the path of incident light include the vitreous (V); Nerve fiber layer (NF) consisting of axons of the third neuron; Ganglion cell layer (G) consisting of the cell nuclei of the multipolar ganglion cells of the third neuron; Inner plexiform layer (IP) consisting of synapses between the axons of the second neuron and dendrites of the third neuron; Inner nuclear layer (IN) containing cell nuclei of the bipolar nerve cells on the second neuron, horizontal cells, and amacrine cells; Photoreceptors (P) consisting of rods and cones; Retinal pigment epithelium (RPE) consisting of a single cubic layer of heavily pigmented epithelial cells; Bruch’s membrane (BM) the matrix separating the RPE from the choroid; Choroid (C), the vasculature between the retina and sclera. C. Fundoscopic image showing the macula located between vascular arcades including the retinal artery (A) and vein (V) with the fovea (*) at the center, approximately 3 mm temporal to the optic nerve (N). Images courtesy of the National Eye Institute.
Following the path of incident light, the key players in AMD pathogenesis include the photoreceptors, the retinal pigment epithelium (RPE), Bruch’s membrane, and the choroid (Fig. 1). The RPE, a cuboidal epithelium, has multiple functions including photoreceptor outer segment phagocytosis, nutrient transport, cytokine secretion, and maintenance of the blood barrier. The RPE is attached to Bruch’s membrane, a pentalaminar, selectively permeable matrix that separates the RPE from the underlying choroid. The choroid is composed of the choriocapillaris, which nourishes the RPE and outer retina, and progressively larger vessels adjacent to the sclera (Fig. 1). The health of the overlying photoreceptors, which detect light, is dependent upon diffusion of oxygen and nutrient supply from the richly vascular choroid.
What is AMD?
AMD is a progressive, complex age-related disease. Early disease can go undetected because patients may be without appreciable vision loss [3]. Early “dry” AMD is characterized by whitish-yellow deposits coined drusen that develop in Bruch’s membrane (Fig. 2). Drusen can be categorized as either “hard” with distinct margins, or “soft” with indistinct edges [3]. While small hard drusen are typically not associated with disease, soft drusen larger than 125 μm in diameter are pathognomonic for AMD [3]. When large drusen coalesce, they can cause the RPE to separate from Bruch’s membrane, which can lead to RPE apoptosis, and when extensive, result in geographic atrophy (GA), an advanced form of AMD (Fig. 2). Accordingly, the risk for advanced AMD is directly related to the size of drusen and the degree of altered pigmentation in the RPE, which correlates with RPE dysfunction [3].
Figure 2.

A. Fundus photo of a right eye with non-neovascular (dry) AMD. Note the numerous large yellow drusen deposits. B. Fundus photo of a left eye with geographic atrophy characteristic of advanced AMD. Note area where RPE cells are absent (arrow). (c) Fundus photo of a right eye with neovascular or wet AMD. Note subretinal hemorrhage (arrow) adjacent to a choroidal neovascular membrane (arrowhead). Wet AMD image, courtesy of the National Eye Institute.
Advanced AMD can also present as “wet” AMD (Fig. 2), involving choroidal neovascularization (CNV), which represents approximately 20% of AMD cases. The aberrant expression of cytokines, such as vascular endothelial growth factor (VEGF), is a central driving factor in the development of the neovascularization. Fortunately, serial intravitreal injections of anti-VEGF antibodies can clinically control the neovascularization [4]. While antioxidant micronutrients can slow vision loss for intermediate AMD, unfortunately, there is no effective therapy for early, dry AMD. The impact of anti-VEGF therapeutics on wet AMD underscores the power of exploiting molecular targets. An understanding of the underlying molecular mechanisms that contribute to early AMD could enable the development of therapeutics for early AMD that prevent vision loss.
Epidemiological Evidence for AMD Risk
A variety of environmental influences have been linked with AMD risk. As the name suggests, advanced age is the strongest risk factor for AMD. Cigarette smoking is the second strongest risk factor, conferring more than twice the risk for developing AMD independent of known genetic risk factors. Tobacco smoke has been shown to be independently associated with AMD in persons with a 10 pack-year smoking history or those exposed to secondhand smoke. Using pooled data from three population-based cohort studies, the relative risk of the late AMD development is 2.4 among current smokers compared to non-smokers [5]. Cigarette smoke has been shown to increase RPE abnormalities in early disease, and is correlated with geographic atrophy, a late form of AMD characterized by RPE cell death [6].
Cigarette Smoke is an Exogenous Source of Oxidative Stress
With each puff of cigarette smoke, over 4700 chemical components including 1015 free radicals are inhaled. Chemical oxidants in cigarette smoke deplete tissues of ascorbic acid and protein sulfhydryl groups, causing the oxidation of DNA, lipids, and proteins [7]. In AMD, many of these oxidative changes result in the formation of malondialdehyde (MDA), 4-hydroxynonenal (4-HNE), carboxymethylpyrrole (CEP), and advanced glycation endproducts (AGEs) in the macula. Coupled with basic research indicating an oxidative etiology in AMD [8], such findings support oxidation as a key mechanism in AMD pathogenesis.
Cardinal features of AMD develop in animals exposed to smoke [27]. The RPE of mice exposed to cigarette smoke for 6 months developed an increase in oxidatively damaged DNA, profound loss of basal infoldings, increased intracellular membranous vacuoles, and increased apoptosis compared to the RPE of mice raised in air [27]. These changes are similar to that seen in RPE cells overlying drusen in AMD. A companion study demonstrated increased RPE mitochondrial DNA damage, decreased factor H (CFH) and complement activation with increased C3, C5, and C5b-9 complexes within Bruch’s membrane in smoke treated mice [28]. Mice given a high fat diet and smoke developed prominent sub-RPE deposits and Bruch’s membrane thickening [29]. Taken together, a smoke-induced mouse model of AMD is an effective tool for studying AMD pathogenesis as well as validating new therapeutic targets.
Genetics of AMD
Genetic variants associated with AMD risk [30] include genes that encode proteins involved in oxidative stress and complement pathway modulation (Table 1). The single nucleotide polymorphism (SNP) rs1061170 in CFH, as well as polymorphisms in C2, CFB, C3, and CFI confer risk for AMD. Genome-wide association studies (GWAS) have identified the chromosome 10q26 susceptibility locus that contains the rs10490924 variant. Within this region is the LOC387715 gene, which codes for the ARMS2 (age-related maculopathy susceptibility 2) protein whose function remains an area of active inquiry. Also within this region is the rs11200638 SNP which codes for the promoter of HTRA1, a serine protease that is associated with wet AMD [25]. GWAS studies have also uncovered genes variants involved in lipid metabolism such as ABCA1, LIPC, CETP, and LPL.
Table 1.
Selected polymorphisms that confer AMD risk.
| Gene | Loci | Function | Odds Ratio | References |
|---|---|---|---|---|
|
| ||||
| C2/CFB (Complement 2/Complement factor B) | 6p21 | Regulator of complement activation | Het=0.32–0.40 | Gold et al Nat Genet. 2006 [9] |
|
| ||||
| C3 (Complement 3) | 19p13 | Innate immunity (classical pathway component, alternative complement pathway activator) | Homo=1.93–3.91 | Yates et al. NEJM. 2007 [10] |
|
| ||||
| CFH (Complement factor H) | 1q32 | Regulator of alternative complement pathway | Homo=6.32 | Klein et al. Science. 2005 [11] |
| Edwards et al. Science. 2005 [12] | ||||
| Haines et al. Science. 2005; [13] | ||||
| Hageman et al. PNAS. 2005 [14] | ||||
|
| ||||
| CFHR1/CFHR3 (Complement factor H-related 1, 3) | 1q31- q32 | Unknown, possible overlapping function with CFH | Homo=0.29 | Spencer et al. Hum Mol Genet. 2008 [15] |
|
| ||||
| CFI (Complement Factor I) | 4q25 | C3b/C4b inactivator | Homo=0.28 | Kondo, N, Eur J Hum Genet 2010 [16] |
|
| ||||
| CX3CR1 (Chemokine [C-X3-C motif] receptor 1) | 3p21 | Inflammation (chemokine receptor) | Homo=3.57 | Tuo et al. FASEB J. 2004 [17] |
|
| ||||
| ARMS2/LOC3877 15 (Age-related maculopathy susceptibility 2) | 10q26 | Unknown; Localized to mitochondrial outer membrane | Homo=8.59 | Rivera et al. Hum Mol Genet. 2005 [18] |
|
| ||||
| HTRA1 (High temperature requirement A1) | 10q26 | Serine protease | Homo=6.92, 7.46 | Dewan et al. Science. 2006 [19] |
| Yang et al. Science. 2006 [20] | ||||
|
| ||||
| A4917G | mitDNA | Unknown; T haplogroup polymorphism | Homo=2.16 | Canter et al. PLoS One. 2008 [21] |
|
| ||||
| SOD2 (Superoxide dismutase 2) | 6q25 | Mitochondrial matrix enzyme that scavenges oxygen radicals | Homo=1.79 | Kondo et al. Mol Vis. 2009 [22] |
|
| ||||
| ABCA4 (ATP-binding cassette, sub-family A, member 4) | 1p22 | Transport of N-retinylidene-PE across photorecptor outer segment disc membrane | Het=5.0 | Allikmets et al. Am J Hum Genet. 2000 [23] |
|
| ||||
| APOE (Apolipoprotein E) | 19q13 | Cholesterol and lipid transport | Homo=1.046 | Thakkinstian et al Am J Epidemiol. 2006 [24] |
|
| ||||
| CETP (Cholesteryl ester transfer protein) | 16q21 | Transfer of cholesteryl esters between lipoproteins | Het=2.69 Homo=8.21 | Rivera A. Hum Mol Genet. 2005 [18] |
|
| ||||
| LPL (Lipoprotein Lipase) | 8p21 | Hydrolization of triglycerides in lipoproteins | Homo=0.82 | Neale et al. PNAS. 2010 [25] |
|
| ||||
| LIPC (Lipase C) | 15q21-q23 | Hepatic triglyceride lipase | Homo=0.39 | Yu et al. Invest Ophthalmol Vis Sci. 2011 [26] |
|
| ||||
| ERCC6 (Excision- repair cross-complementing, group 6) | 10q11 | DNA repair | Homo=1.6 | Tuo J. Proc Natl Acad Sci U S A. 2006 Jun 13;103(24):9256-61. [17] |
Het, heterozygous; Homo, homozygous.
Genes related to oxidative stress have also been associated with AMD susceptibility (Table 1). The mitochondrial DNA polymorphism A4917G, as well as mitochondrial superoxide dismutase 2 (SOD2), confers risk for AMD. Similarly, the polymorphism in the LOC387715 locus is associated with a higher risk for AMD in smokers than either smoking or the polymorphism alone.
Mitochondrial Damage is an Endogenous Source of Oxidative Stress
As mitochondrial DNA is inherently prone to oxidative damage, it is not surprising that mitochondrial DNA recovered from macular RPE of AMD patients has more damage than that of age-matched controls [31]. Mitochondrial defects observed in the RPE of AMD eyes include DNA mutations, impaired structural integrity, and defective mitochondrial function. Damaged mitochondria can lead to diminished energy production and imbalance of pro- and anti-apoptotic signals, leading to cell death [31]. Mitochondrial damage leads to an increase in reactive oxygen species (ROS) production, which may damage other key cellular elements. As mentioned above, SOD2 is the isoform that is located in the mitochondria. Since global SOD2 deficient mice die soon after birth, Justilien et al. used a ribozyme that targets SOD2 packaged in an adeno-associated virus for subretinal injection to knockdown SOD2 in the RPE [31]. Local SOD2 knockdown induced oxidative damage to proteins, RPE vacuolization and degeneration, Bruch’s membrane thickening, and shortened, disorganized photoreceptor outer and inner segments. As with AMD patients, SOD2 knockdown eyes had increased autofluorescence and elevated lipofuscin. The photoreceptor outer nuclear layer became progressively thinned from apoptotic cell death that was coincident with loss of electroretinographic signaling. Because RPE degeneration preceded photoreceptor damage and retinal dysfunction, the authors concluded that RPE is the primary cell type initiating disease.
The Central Role of the RPE and its Antioxidant System
The RPE plays a central and arguably primary role because RPE dysfunction and atrophy precede the advanced stages of AMD (GA and CNV). The literature is replete with examples of the RPE responding to numerous stimuli to orchestrate its own fate and that of the other members of the retinal microenvironment. RNA transcriptome analysis of in vitro RPE cells [32] and human AMD donor eyes [32] have demonstrated a common response to and insults from pathological stressors. Interestingly, significant interindividual variation in RPE transcript expression in AMD donor eyes is seen, further supporting the redundant number of molecular mechanisms leading to an AMD phenotype [33]. In addition, genome-wide stress-response transciptome and proteome assays have begun to elucidate the molecular changes in retina with age, and in response to AMD-associated stresses on whole-genome RPE gene expression [34]. Many RPE-elaborated cytokines released from oxidative stress have been identified in human and experimental AMD specimens [31]. Under physiologic conditions, this robust antioxidant system can neutralize the ROS that result from stressors such as cigarette smoke or light exposure. The generation of ROS has been thought to be a major factor in aging and disease. However, it is clear that ROS participate in a number of physiological processes that are essential to the normal functioning of cells. Redox signaling can act as an important regulatory element in both physiological and pathological processes. Physiological ROS can be generated from several sources [35]. In a pathologic state, in which the balance is tipped and the stressor overwhelms the antioxidant response, ROS persist and can induce the formation of oxidation specific epitopes (OSEs), which will be discussed below.
The macula and in particular, the RPE are protected from oxidative stress through signaling pathways. The most powerful of these transcription systems is the Nrf2 (nuclear factor erythroid-2 related factor 2), a basic leucine zipper transcription factor [31]. Nrf2 regulates a comprehensive, coordinated transcriptional program that maintains cellular redox homeostasis and protects the cell from oxidative injury [31]. Nrf2 is normally sequestered in the cytosol by interacting with Kelch-like ECH-associated protein 1 (Keap1). Keap1 also functions as a substrate adaptor protein for the Cul3-dependent E3 ubiquitin ligase complex, which maintains steady-state levels of Nrf2 via proteolysis by the ubiquitin-proteasome pathway [36]. In the absence of stress, Keap1 constitutively suppresses Nrf2 signaling by degrading Nrf2 and preventing nuclear Nrf2 translocation, which results in a low baseline expression of antioxidant genes [37]. However, upon exposure to ROS, Keap1 undergoes a conformational change when its multiple cysteine residues interact with ROS, which releases Nrf2 and inhibits Keap1-mediated proteasomal Nrf2 degradation. The released Nrf2 then translocates to the nucleus, where it dimerizes with Maf proteins, and binds to the antioxidant response element (ARE) in the promoters of its target genes to initiate transcription [38]. The Nrf2 signaling response regulates both an early acute phase through actions of the “direct” enzymes, such as catalase or SOD, which neutralize H2O2 and superoxide, respectively, a chronic phase through maintenance of cellular glutathione and thioredoxin, and xenobiotic metabolism enzymes that produce reducing equivalents, such as NADPH quinine oxidoreductase (NQO-1) [39]. When ROS depletes cellular glutathione, cells can die from oxidatively mediated apoptosis. Importantly, Nrf2 signaling plays an essential antioxidant role in the RPE [39].
In elderly persons who smoke, Nrf2 signaling is impaired. Suzuki et al. showed that compared with young smokers whose Nrf2 level was independent of smoking status, smoking reduced Nrf2 mRNA in the macrophages of elderly smokers [40]. This study also demonstrated an inverse relation between Nrf2 mRNA levels and oxidized glutathione and carbonylated albumin levels. In pulmonary aspirates from patients with emphysema, Nrf2 signaling is reduced at both the transcription and protein levels, and as a result, antioxidant and glutathione levels are reduced relative to aged-matched controls [40].
Impaired Nrf2 signaling from smoking has been corroborated in animal models. When exposed to tobacco smoke, mice have impaired Nrf2 signaling when measured by changes in Nrf2 and target genes transcription levels in alveolar macrophages [40]. Nrf2 deficient mice exposed to cigarette smoke develop emphysema while wild-type (WT) mice remain unaffected [41]. In these mice, if Keap1 is impaired or Nrf2 is upregulated, the emphysematous changes can be prevented [40].
In the presence of oxidative stress, the innate immune response can be amplified, thereby converting it from a protective to a pathologic response. The Nrf2 signaling system can act to buffer oxidative stress and thus the innate immune response including complement-mediated inflammation. In the lipopolysaccharide (LPS) induced septic shock model, Nrf2 deficient mice, compared to WT mice, had increased innate immune activity, inflammation, and death [37]. Nrf2 signaling showed a similar protective effect in a model of ocular inflammation [42]. After intraperitoneal LPS injection, the retinas of Nrf2 deficient mice had increased ROS and elevated expression of a number of cytokines compared to WT controls [42]. After LPS challenge, the retinas of WT mice demonstrated increased expression of the Nrf2-responsive antioxidant enzymes (NQO-1 and glutamate-cysteine ligase, modifier subunit (GCLM)) compared to Nrf2 deficient mice [42]. Leukocyte adhesion to the vascular endothelium is a key event that precedes leukocyte invasion into tissue. LPS treatment also increased the number of leukocytes adherent to the retinal vascular endothelium after LPS treatment in Nrf2 deficient and WT mice alike. If pretreated with 2-Cyano-3,12-dioxooleana-1,9-dien-28-imidazolide (CDDO-Im), a small molecule Nrf2 activator, the retinas of WT mice had increased antioxidant gene expression, reduced inflammatory mediator expression, and reduced leukocyte adherence to retinal vasculature. As expected, CDDO-Im did not confer a similar protective effect in Nrf2 deficient mice.
In addition to acute inflammation, deficient Nrf2 signaling can create a chronic proinflammatory environment, a known susceptibility factor for AMD. Nrf2 deficient mice have a prolonged inflammatory response [43], increased oxidative damage, and complement C3 deposition in several organs including the brain [44] compared to WT. Conversely, Nrf2 activation prevents inflammation mediated by oxidative stress. For example, in a model of liver necrosis, Nrf2 activation by triterpenoids prevented late phase pro-inflammatory gene expression, thereby halting the inflammatory amplification loop [39]. Recently, Zhao et al showed several features of AMD in 12 month old Nrf2 deficient mice that included the accumulation of lipofuscin and membranous vacuoles within the RPE, and drusen-like deposits along with the accumulation of inflammatory proteins in Bruch’s membrane. Taken together, it appears that upregulating the Nrf2 pathway can dually provide direct antioxidant effect and reduce the accompanying pro-inflammatory response during conditions like AMD.
Oxidation Specific Epitopes are a Link to Chronic Oxidative Stress and Innate Immune Activation
Inadequately neutralized ROS can cause oxidize lipids, such as the peroxidation of polyunsaturated fatty acids. Resulting lipid hydroperoxides, in the presence of iron and other transition metals, can give rise to esterified and unesterified aldehydes. These lipid peroxidation products can modify proteins, lipids, and DNA. These oxidized self structures have been coined “oxidation specific epitopes” (OSEs) [45]. Unlike ROS, which are short lived, OSEs can produce a chronic inflammatory nidus if not neutralized or removed. For example, apoptotic bodies are highly enriched with OSEs. These OSEs are thought to act as immunogenic danger-associated molecular pattern molecules (DAMPS), tags that enable the body to identify and clear biological waste. A delicate balance exists between a protective clearance response and pathologic autoinflammatory response to DAMPS/OSEs. In the pathologic state, ROS are inadequately neutralized, thus forming abundant OSEs that overwhelm innate immune clearance. Not surprisingly, OSEs have been identified in numerous diseases in which oxidative stress and inflammation are thought to be key factors, including Alzheimer’s disease, artherosclerosis, diabetes, muscular dystrophy, rheumatoid arthritis, and lupus [45].
OSEs activate the innate immune response through interaction with pattern recognition receptors (PRRs). Each PRR, which is germ line encoded, is capable of binding highly conserved motifs on numerous pathogens, and provides a first line defense against invading pathogens. In addition to pathogens, PRRs bind self-epitopes such as OSEs. OSEs can be endocytosed after binding to PRRs on natural killer cells, monocytes/macrophages, and dendritic cells. When macrophages, dendritic cells, and other antigen-presenting cells have PRR bound, they can interact with T cells, thereby converting innate to adaptive immunity. When in fluid-phase, PRRs can bind and neutralize OSEs and promote their removal. Under physiologic conditions, phagocytic cells can clear OSEs and other DAMPs without causing collateral damage to the neighboring healthy tissue. In the pathologic state, PRR-mediated signaling cascades can cause elevated cytokine and chemokine release, which induces pathologic inflammation, and in certain instances, autoimmunity.
MDA is an OSE that accumulates in numerous oxidative stress related diseases. CFH, traditionally considered a major regulator of the alternative complement pathway, was recently found to be a PRR that specifically binds MDA [8]. In pull down experiments with MDA covered beads, CFH was the predominant protein bound in pooled plasma from mice lacking immunoglobulins. CFH in human plasma also selectively bound MDA. The study demonstrated MDA-CFH specificity because other OSEs including oxidized phosphocholine–BSA, CEP–BSA, and 4-hydroxynonenal-BSA did not bind to CFH to any measurable extent [8].
Of the 20 globular short consensus repeats (SCRs) of CFH [46], only the SCR7 or SCR20 fragments bound MDA. These domains are clustering points of various disease-related mutations including the 402H variant in SCR7 that confers risk for AMD. Plasma from patients with the 402H CFH variant have impaired MDA binding [46]. Irrespective of total plasma CFH levels, MDA binding is further decreased in persons homozygous for the 402H CFH variant compared to heterozygotes. CFH also bound MDA decorated apoptotic debris resulting in the generation of iC3b inactivation fragments. The 402H CFH variant had impaired C3b inactivation when incubated with MDA apoptotic particles. Since iC3b opsonins promote the clearance of apoptotic cells without generating inflammation [47], the 402H CFH variant predisposes to a proinflammatory state during the clearance of apoptotic cells. Thus, during RPE cell apoptosis in early AMD, the risk 402H CFH variant could potentially promote tissue-injuring inflammation while apoptotic RPE debris are being cleared.
CFH blocked the uptake of MDA-modified proteins by macrophages in vitro and prevented Il-8 induction by RPE cells in vitro and in vivo. Mice challenged with intravitreal injections of MDA had increased Il-8 production in the RPE/choroid, compared to mice receiving injections of MDA first bound and neutralized by CFH. These novel results suggest that therapeutic targets which modulate PRR OSEs such as CFH mimetics, may prove useful in treating AMD and other chronic inflammatory diseases.
Another area of active inquiry in the AMD field is the relation of inflammasome activation to AMD development. Inflammasomes are multiprotein complexes composed of three components including the pattern recognition receptor NOD-like receptor family, pyrin domain containing 3 (NLRP3), the adaptor protein apoptosis-associated speck-like protein containing a CARD (ASC), and pro-caspase-1. When activated by DAMPs, inflammatory cells such as macrophages, orchestrate assembly of the inflammasome [48]. The inflammasome may have different influence depending upon the stage of AMD. In a paradigm shifting study, Doyle et al. [48] found that inflammasome generated inflammation may be protective in AMD. The study demonstrated that drusen isolated from patients with AMD and drusen components complement C1q and the OSE CEP, activate the NLRP3 inflammasome [48]. When immunized with CEP, mice develop AMD-like lesions, including drusen accumulation [49]. In these mice, NLRP3 and caspase-1 were upregulated in the macrophages near drusen [48], suggesting direct inflammasome activation by drusen components. NLRP3 inflammasomes, once activated, increase the conversion of pro-interleukin-1β (IL-1β) and pro-IL-18 into active IL-1β and IL-18, respectively, well as activating other inflammatory cytokines [48]. In a laser-induced CNV mouse model of wet AMD, a lack of NLRP3 or IL-18 exacerbated CNV formation [48]. Accordingly, the study supports a protective role of the NLRP3 inflammasome and IL-18 in preventing neovascular AMD. Current efforts in the field are directed towards determining if knocking out other inflammasome components (such as ASC and caspase-1) will similarly worsen CNV. Conversely, others are exploring whether inflammasome activation or increased IL-18 can suppress CNV.
On the other hand, the Ambati lab reported that toxic accumulation of abnormal RNA products in patients and mice deficient in DICER1 can modulate NLRP3 inflammasome activation and AMD development. They found that inhibiting the inflammasome prevented RPE degeneration and GA. They suggested that efforts to control the inflammasome, just as in early infection, may prevent AMD development [50].
Putting it together: The Interplay of Metabolic Oxidative Stress and Innate Immune Dysfunction in AMD Pathogenesis
Fig. 3 summarizes a proposed mechanism for how metabolic oxidative stress and innate immune hyperactivity play a role in early AMD development. In this model, no component is inherently pathologic or protective. Instead, each has a physiologic role, and in cases of imbalance, a pathologic role. In the physiologic state, low levels of ROS provide oxidant signaling that maintains healthy cellular function. When stressors such as chronic light exposure or cigarette smoking overwhelm the antioxidant system of the retina, ROS become pathological by introducing a cascade of tissue injuring events related to oxidative and inflammatory damage. With decreased Nrf2 signaling seen with aging, inadequately neutralized ROS create OSEs. OSEs activate the innate immune response when they interact with PRRs. Membrane bound PRRs facilitate phagocytosis of OSEs while fluid-phase PRRs may bind and neutralize OSEs by signaling their removal by immune cells. When this system is overwhelmed by an overabundance or chronic exposure of OSEs, the inflammation can convert from a physiologic to pathologic response. In this pathologic state, the combination of inadequately neutralized oxidative stress and inflammation induce both RPE cell injury and death. The photoreceptors, which rely on the RPE for their health, become dysfunctional, resulting in vision loss. Oxidative modification, such as the formation of AGEs, a collection of OSEs, promotes the retention of other OSEs that may prevent their neutralization or clearance by PRRs. These changes can magnify the inflammatory response and the retention of inflammatory debris or cellular remnants that ultimately contribute to drusen formation.
Figure 3.

Model of oxidative stress and resulting protective or pathologic immune response. Through the antioxidant systems including the Nrf2 signaling system and the innate immune system’s activation by oxidation specific epitopes (OSEs) the retinal microenvironment can prevent itself from oxidative damage. When overwhelmed by exogenous stressors such as cigarette smoke, chronic inflammation may result leading to a pathologic immune response.
Future directions
The line between physiologic and pathological ROS and complement activation in the retina beckons to be understood. The current understanding of how Nrf2 signaling protects the retina from oxidative stress is also in its infancy. Future efforts aimed at bolstering the cell’s inherent defense mechanisms against oxidative stress, such as enhancing Nrf2 signaling, are attractive therapeutic targets for AMD. Such a strategy is also intended to reduce oxidative stress before it triggers pathologic innate immune activation. In a similar vein, exploiting PRRs that neutralize OSEs could dampen chronic oxidative stress or complement activation. One of the most exciting PRR targets, clinically relevant in AMD, is CFH, which acts to neutralize the ubiquitous OSE MDA. As the PRR OSE story is also still in its infancy, it is likely that other PRR therapeutic targets will be uncovered. Central to these strategies is a sound understanding of how oxidative stress and complement activation trigger AMD. With this knowledge, it is likely that new therapeutics will soon emerge as a much needed treatment for dry AMD.
Acknowledgments
Funding: NEI EY14005 (JTH), EY019904 (JTH), Thome Foundation (JTH), Beckman Foundation Initiative for Macular Research, Research to Prevent Blindness Senior Scientist Award (JTH), Unrestricted grant from Research to Prevent Blindness to the Wilmer Eye Institute, NIH P30EY001765 core grant, Robert Bond Welch Professorship (JTH), and a gift from the Merlau family and Aleda Wright.
References
- 1.Chakravarthy U, Wong TY, Fletcher A, Piault E, Evans C, Zlateva G, Buggage R, Pleil A, Mitchell P. Clinical risk factors for age-related macular degeneration: A systematic review and meta-analysis. BMC Ophthalmol. 10:31. doi: 10.1186/1471-2415-10-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Warburg O. The chemical constitution of respiration ferment. Science. 1928;68:437–443. doi: 10.1126/science.68.1767.437. [DOI] [PubMed] [Google Scholar]
- 3.Bird AC, Bressler NM, Bressler SB, Chisholm IH, Coscas G, Davis MD, de Jong PT, Klaver CC, Klein BE, Klein R, et al. An international classification and grading system for age-related maculopathy and age-related macular degeneration. The international arm epidemiological study group. Surv Ophthalmol. 1995;39:367–374. doi: 10.1016/s0039-6257(05)80092-x. [DOI] [PubMed] [Google Scholar]
- 4.Zampros I, Praidou A, Brazitikos P, Ekonomidis P, Androudi S. Antivascular endothelial growth factor agents for neovascular age-related macular degeneration. J Ophthalmol. 2012:319728. doi: 10.1155/2012/319728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tomany SC, Wang JJ, Van Leeuwen R, Klein R, Mitchell P, Vingerling JR, Klein BE, Smith W, De Jong PT. Risk factors for incident age-related macular degeneration: Pooled findings from 3 continents. Ophthalmology. 2004;111:1280–1287. doi: 10.1016/j.ophtha.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 6.The age-related eye disease study system for classifying age-related macular degeneration from stereoscopic color fundus photographs: The age-related eye disease study report number 6. Am J Ophthalmol. 2001;132:668–681. doi: 10.1016/s0002-9394(01)01218-1. [DOI] [PubMed] [Google Scholar]
- 7.Lykkesfeldt J, Christen S, Wallock LM, Chang HH, Jacob RA, Ames BN. Ascorbate is depleted by smoking and repleted by moderate supplementation: A study in male smokers and nonsmokers with matched dietary antioxidant intakes. Am J Clin Nutr. 2000;71:530–536. doi: 10.1093/ajcn/71.2.530. [DOI] [PubMed] [Google Scholar]
- 8.Weismann D, Hartvigsen K, Lauer N, Bennett KL, Scholl HP, Charbel Issa P, Cano M, Brandstatter H, Tsimikas S, Skerka C, Superti-Furga G, Handa JT, Zipfel PF, Witztum JL, Binder CJ. Complement factor h binds malondialdehyde epitopes and protects from oxidative stress. Nature. 478:76–81. doi: 10.1038/nature10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gold B, Merriam JE, Zernant J, Hancox LS, Taiber AJ, Gehrs K, Cramer K, Neel J, Bergeron J, Barile GR, Smith RT, Hageman GS, Dean M, Allikmets R. Variation in factor b (bf) and complement component 2 (c2) genes is associated with age-related macular degeneration. Nat Genet. 2006;38:458–462. doi: 10.1038/ng1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yates JR, Sepp T, Matharu BK, Khan JC, Thurlby DA, Shahid H, Clayton DG, Hayward C, Morgan J, Wright AF, Armbrecht AM, Dhillon B, Deary IJ, Redmond E, Bird AC, Moore AT. Complement c3 variant and the risk of age-related macular degeneration. N Engl J Med. 2007;357:553–561. doi: 10.1056/NEJMoa072618. [DOI] [PubMed] [Google Scholar]
- 11.Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, Henning AK, SanGiovanni JP, Mane SM, Mayne ST, Bracken MB, Ferris FL, Ott J, Barnstable C, Hoh J. Complement factor h polymorphism in age-related macular degeneration. Science. 2005;308:385–389. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Edwards AO, Ritter R, 3rd, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor h polymorphism and age-related macular degeneration. Science. 2005;308:421–424. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- 13.Haines JL, Hauser MA, Schmidt S, Scott WK, Olson LM, Gallins P, Spencer KL, Kwan SY, Noureddine M, Gilbert JR, Schnetz-Boutaud N, Agarwal A, Postel EA, Pericak-Vance MA. Complement factor h variant increases the risk of age-related macular degeneration. Science. 2005;308:419–421. doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- 14.Hageman GS, Anderson DH, Johnson LV, Hancox LS, Taiber AJ, Hardisty LI, Hageman JL, Stockman HA, Borchardt JD, Gehrs KM, Smith RJ, Silvestri G, Russell SR, Klaver CC, Barbazetto I, Chang S, Yannuzzi LA, Barile GR, Merriam JC, Smith RT, Olsh AK, Bergeron J, Zernant J, Merriam JE, Gold B, Dean M, Allikmets R. A common haplotype in the complement regulatory gene factor h (hf1/cfh) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U S A. 2005;102:7227–7232. doi: 10.1073/pnas.0501536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spencer KL, Olson LM, Anderson BM, Schnetz-Boutaud N, Scott WK, Gallins P, Agarwal A, Postel EA, Pericak-Vance MA, Haines JL. C3 r102g polymorphism increases risk of age-related macular degeneration. Hum Mol Genet. 2008;17:1821–1824. doi: 10.1093/hmg/ddn075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kondo N, Bessho H, Honda S, Negi A. Additional evidence to support the role of a common variant near the complement factor i gene in susceptibility to age-related macular degeneration. Eur J Hum Genet. 18:634–635. doi: 10.1038/ejhg.2009.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tuo J, Smith BC, Bojanowski CM, Meleth AD, Gery I, Csaky KG, Chew EY, Chan CC. The involvement of sequence variation and expression of cx3cr1 in the pathogenesis of age-related macular degeneration. FASEB J. 2004;18:1297–1299. doi: 10.1096/fj.04-1862fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rivera A, Fisher SA, Fritsche LG, Keilhauer CN, Lichtner P, Meitinger T, Weber BH. Hypothetical loc387715 is a second major susceptibility gene for age-related macular degeneration, contributing independently of complement factor h to disease risk. Hum Mol Genet. 2005;14:3227–3236. doi: 10.1093/hmg/ddi353. [DOI] [PubMed] [Google Scholar]
- 19.Dewan A, Liu M, Hartman S, Zhang SS, Liu DT, Zhao C, Tam PO, Chan WM, Lam DS, Snyder M, Barnstable C, Pang CP, Hoh J. Htra1 promoter polymorphism in wet age-related macular degeneration. Science. 2006;314:989–992. doi: 10.1126/science.1133807. [DOI] [PubMed] [Google Scholar]
- 20.Yang Z, Camp NJ, Sun H, Tong Z, Gibbs D, Cameron DJ, Chen H, Zhao Y, Pearson E, Li X, Chien J, Dewan A, Harmon J, Bernstein PS, Shridhar V, Zabriskie NA, Hoh J, Howes K, Zhang K. A variant of the htra1 gene increases susceptibility to age-related macular degeneration. Science. 2006;314:992–993. doi: 10.1126/science.1133811. [DOI] [PubMed] [Google Scholar]
- 21.Canter JA, Olson LM, Spencer K, Schnetz-Boutaud N, Anderson B, Hauser MA, Schmidt S, Postel EA, Agarwal A, Pericak-Vance MA, Sternberg P, Jr, Haines JL. Mitochondrial DNA polymorphism a4917g is independently associated with age-related macular degeneration. PLoS One. 2008;3:e2091. doi: 10.1371/journal.pone.0002091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kondo N, Bessho H, Honda S, Negi A. Sod2 gene polymorphisms in neovascular age-related macular degeneration and polypoidal choroidal vasculopathy. Mol Vis. 2009;15:1819–1826. [PMC free article] [PubMed] [Google Scholar]
- 23.Allikmets R. Further evidence for an association of abcr alleles with age-related macular degeneration. The international abcr screening consortium. Am J Hum Genet. 2000;67:487–491. doi: 10.1086/303018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thakkinstian A, Bowe S, McEvoy M, Smith W, Attia J. Association between apolipoprotein e polymorphisms and age-related macular degeneration: A huge review and meta-analysis. Am J Epidemiol. 2006;164:813–822. doi: 10.1093/aje/kwj279. [DOI] [PubMed] [Google Scholar]
- 25.Neale BM, Fagerness J, Reynolds R, Sobrin L, Parker M, Raychaudhuri S, Tan PL, Oh EC, Merriam JE, Souied E, Bernstein PS, Li B, Frederick JM, Zhang K, Brantley MA, Jr, Lee AY, Zack DJ, Campochiaro B, Campochiaro P, Ripke S, Smith RT, Barile GR, Katsanis N, Allikmets R, Daly MJ, Seddon JM. Genome-wide association study of advanced age-related macular degeneration identifies a role of the hepatic lipase gene (lipc) Proc Natl Acad Sci U S A. 107:7395–7400. doi: 10.1073/pnas.0912019107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu Y, Reynolds R, Fagerness J, Rosner B, Daly MJ, Seddon JM. Association of variants in the lipc and abca1 genes with intermediate and large drusen and advanced age-related macular degeneration. Invest Ophthalmol Vis Sci. 52:4663–4670. doi: 10.1167/iovs.10-7070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fujihara M, Nagai N, Sussan TE, Biswal S, Handa JT. Chronic cigarette smoke causes oxidative damage and apoptosis to retinal pigmented epithelial cells in mice. PLoS One. 2008;3:e3119. doi: 10.1371/journal.pone.0003119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang AL, Lukas TJ, Yuan M, Du N, Handa JT, Neufeld AH. Changes in retinal pigment epithelium related to cigarette smoke: Possible relevance to smoking as a risk factor for age-related macular degeneration. PLoS One. 2009;4:e5304. doi: 10.1371/journal.pone.0005304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Espinosa-Heidmann DG, Suner IJ, Catanuto P, Hernandez EP, Marin-Castano ME, Cousins SW. Cigarette smoke-related oxidants and the development of sub-rpe deposits in an experimental animal model of dry amd. Invest Ophthalmol Vis Sci. 2006;47:729–737. doi: 10.1167/iovs.05-0719. [DOI] [PubMed] [Google Scholar]
- 30.Deangelis MM, Silveira AC, Carr EA, Kim IK. Genetics of age-related macular degeneration: Current concepts, future directions. Semin Ophthalmol. 26:77–93. doi: 10.3109/08820538.2011.577129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kanda A, Chen W, Othman M, Branham KE, Brooks M, Khanna R, He S, Lyons R, Abecasis GR, Swaroop A. A variant of mitochondrial protein loc387715/arms2, not htra1, is strongly associated with age-related macular degeneration. Proc Natl Acad Sci U S A. 2007;104:16227–16232. doi: 10.1073/pnas.0703933104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Strunnikova NV, Maminishkis A, Barb JJ, Wang F, Zhi C, Sergeev Y, Chen W, Edwards AO, Stambolian D, Abecasis G, Swaroop A, Munson PJ, Miller SS. Transcriptome analysis and molecular signature of human retinal pigment epithelium. Hum Mol Genet. 19:2468–2486. doi: 10.1093/hmg/ddq129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Booij JC, ten Brink JB, Swagemakers SM, Verkerk AJ, Essing AH, van der Spek PJ, Bergen AA. A new strategy to identify and annotate human rpe-specific gene expression. PLoS One. 5:e9341. doi: 10.1371/journal.pone.0009341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Glenn JV, Mahaffy H, Dasari S, Oliver M, Chen M, Boulton ME, Xu H, Curry WJ, Stitt AW. Proteomic profiling of human retinal pigment epithelium exposed to an advanced glycation-modified substrate. Graefes Arch Clin Exp Ophthalmol. 250:349–359. doi: 10.1007/s00417-011-1856-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Finkel T. Signal transduction by reactive oxygen species. J Cell Biol. 194:7–15. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu X, Kensler T. Nrf2 as a target for cancer chemoprevention. Mutat Res. 2005;591:93–102. doi: 10.1016/j.mrfmmm.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 37.Thimmulappa RK, Lee H, Rangasamy T, Reddy SP, Yamamoto M, Kensler TW, Biswal S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest. 2006;116:984–995. doi: 10.1172/JCI25790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dinkova-Kostova AT, Holtzclaw WD, Kensler TW. The role of keap1 in cellular protective responses. Chem Res Toxicol. 2005;18:1779–1791. doi: 10.1021/tx050217c. [DOI] [PubMed] [Google Scholar]
- 39.Osburn WO, Yates MS, Dolan PD, Chen S, Liby KT, Sporn MB, Taguchi K, Yamamoto M, Kensler TW. Genetic or pharmacologic amplification of nrf2 signaling inhibits acute inflammatory liver injury in mice. Toxicol Sci. 2008;104:218–227. doi: 10.1093/toxsci/kfn079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Suzuki M, Betsuyaku T, Ito Y, Nagai K, Nasuhara Y, Kaga K, Kondo S, Nishimura M. Down-regulated nf-e2-related factor 2 in pulmonary macrophages of aged smokers and patients with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2008;39:673–682. doi: 10.1165/rcmb.2007-0424OC. [DOI] [PubMed] [Google Scholar]
- 41.Rangasamy T, Cho CY, Thimmulappa RK, Zhen L, Srisuma SS, Kensler TW, Yamamoto M, Petrache I, Tuder RM, Biswal S. Genetic ablation of nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J Clin Invest. 2004;114:1248–1259. doi: 10.1172/JCI21146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nagai N, Thimmulappa RK, Cano M, Fujihara M, Izumi-Nagai K, Kong X, Sporn MB, Kensler TW, Biswal S, Handa JT. Nrf2 is a critical modulator of the innate immune response in a model of uveitis. Free Radic Biol Med. 2009;47:300–306. doi: 10.1016/j.freeradbiomed.2009.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Braun S, Hanselmann C, Gassmann MG, auf dem Keller U, Born-Berclaz C, Chan K, Kan YW, Werner S. Nrf2 transcription factor, a novel target of keratinocyte growth factor action which regulates gene expression and inflammation in the healing skin wound. Mol Cell Biol. 2002;22:5492–5505. doi: 10.1128/MCB.22.15.5492-5505.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li J, Stein TD, Johnson JA. Genetic dissection of systemic autoimmune disease in nrf2-deficient mice. Physiol Genomics. 2004;18:261–272. doi: 10.1152/physiolgenomics.00209.2003. [DOI] [PubMed] [Google Scholar]
- 45.Chou MY, Hartvigsen K, Hansen LF, Fogelstrand L, Shaw PX, Boullier A, Binder CJ, Witztum JL. Oxidation-specific epitopes are important targets of innate immunity. J Intern Med. 2008;263:479–488. doi: 10.1111/j.1365-2796.2008.01968.x. [DOI] [PubMed] [Google Scholar]
- 46.Jozsi M, Zipfel PF. Factor h family proteins and human diseases. Trends Immunol. 2008;29:380–387. doi: 10.1016/j.it.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 47.Amarilyo G, Verbovetski I, Atallah M, Grau A, Wiser G, Gil O, Ben-Neriah Y, Mevorach D. Ic3b-opsonized apoptotic cells mediate a distinct anti-inflammatory response and transcriptional nf-kappab-dependent blockade. Eur J Immunol. 40:699–709. doi: 10.1002/eji.200838951. [DOI] [PubMed] [Google Scholar]
- 48.Doyle SL, Campbell M, Ozaki E, Salomon RG, Mori A, Kenna PF, Farrar GJ, Kiang AS, Humphries MM, Lavelle EC, O’Neill LA, Hollyfield JG, Humphries P. Nlrp3 has a protective role in age-related macular degeneration through the induction of il-18 by drusen components. Nat Med. 18:791–798. doi: 10.1038/nm.2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hollyfield JG, Bonilha VL, Rayborn ME, Yang X, Shadrach KG, Lu L, Ufret RL, Salomon RG, Perez VL. Oxidative damage-induced inflammation initiates age-related macular degeneration. Nat Med. 2008;14:194–198. doi: 10.1038/nm1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428–435. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]