

Abstract
Adult haematopoietic stem/progenitor cells (HSPCs) constitute the lifespan reserve for the generation of all the cellular lineages in the blood. Although massive progress in identifying the cluster of master genes controlling self-renewal and multipotency has been achieved in the past decade, some aspects of the physiology of HSPCs still need to be clarified. In particular, there is growing interest in the metabolic profile of HSPCs in view of their emerging role as determinants of cell fate. Indeed, stem cells and progenitors have distinct metabolic profiles, and the transition from stem to progenitor cell corresponds to a critical metabolic change, from glycolysis to oxidative phosphorylation. In this review, we summarize evidence, reported in the literature and provided by our group, highlighting the peculiar ability of HSPCs to adapt their mitochondrial oxidative/bioenergetic metabolism to survive in the hypoxic microenvironment of the endoblastic niche and to exploit redox signalling in controlling the balance between quiescence versus active cycling and differentiation. Especial prominence is given to the interplay between hypoxia inducible factor-1, globins and NADPH oxidases in managing the mitochondrial dioxygen-related metabolism and biogenesis in HSPCs under different ambient conditions. A mechanistic model is proposed whereby ‘mitochondrial differentiation’ is a prerequisite in uncommitted stem cells, paving the way for growth/differentiation factor-dependent processes. Advancing the understanding of stem cell metabolism will, hopefully, help to (i) improve efforts to maintain, expand and manipulate HSPCs ex vivo and realize their potential therapeutic benefits in regenerative medicine; (ii) reprogramme somatic cells to generate stem cells; and (iii) eliminate, selectively, malignant stem cells.
Linked Articles
This article is part of a themed section on Emerging Therapeutic Aspects in Oncology. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2013.169.issue-8
Keywords: haematopoietic stem/progenitor cells, mitochondria, oxidative phosphorylation, hypoxia inducible factor-1, NADPH oxidases, globins, redox signalling
Introduction
The mammalian blood system comprises an array of cell types, including erythrocytes, myeloid cells, megakaryocytes and platelets, lymphocytes, natural killer cells, dendritic cells and mast cells. As diverse as these cells are, they all originate from haematopoietic stem cells (HSCs), which are a limited pool of immature progenitors residing in the bone marrow (BM). A reservoir of long-term HSCs (LT-HSCs) lies at the very top of the cellular hierarchy. LT-HSCs guarantee a continuous supply of blood cells throughout an individual's lifetime due to their potential to self-renew (give rise to identical daughter cells) and differentiate. Pools of stem and progenitor cells with decreasing self-renewal potential are downstream of LT-HSCs: the short-term HSCs and multipotent progenitors, which retain full differentiation capacity, and the lineage-restricted progenitors.
These downstream progenitors are the real workhorses of the haematopoietic system, as they divide rapidly and generate a large number of differentiated progeny. In order to guarantee blood homeostasis, the system is tightly regulated but also highly resilient and is capable of modulating the production of specific progeny in response to bleeding, infections or environmental insults. This resilience is the result of two opposing forces – one sustaining HSC dormancy, or quiescence, which is critical to maintain a reservoir of stem cells, and the other activating proliferation and differentiation. Disrupting this balance can have a variety of pathological consequences, such as BM failure or haematological malignancy. Thus, it is important to understand the forces that regulate haematopoietic stem and progenitor cell (HSPC) function (Orkin and Zon, 2008).
Expression of cell surface markers on human haematopoietic cells has provided a method for purifying and characterizing the hierarchical organization of cell subsets with distinct biological functions. Although the identification of marker(s), which specifically individualize(s) the primitive multipotent HSC is still lacking and is the object of research and controversy (Schroeder, 2010), it is generally accepted that the CD34+/CD38−/lineage commitment marker (Lin)− is a phenotype signature consistent with most, if not all, of the haematopoietic activity in BM (Srour et al., 1991; Bhatia et al., 1997; Seita and Weissman, 2010). Successful engraftment of baboons with BM highly enriched for CD34+ cells led to the widespread use of CD34-enriched populations in human transplantation (Berenson et al., 1988). In general, patients transplanted with marrow or mobilized peripheral blood, enriched with CD34 cells [typically induced by granulocyte colony-stimulating factor (G-CSF) pretreatment of the donor] engraft rapidly and seem to have fewer transplant-related complications (Civin et al., 1996).
However, the HSPCs' CD34+/CD38−/Lin− probably constitutes a heterogeneous pool of cell subsets comprising active cycling and self-renewing stem cells and an array of proliferating (but not self-renewing) early progenitors and lineage (pre)-committed haematopoietic cells (Sakabe et al., 1997; Schroeder, 2010; Koutna et al., 2011). Importantly, the higher the expression of CD34 in a given HSPC subset is, the higher its efficacy both in an in vivo long-term re-populating assay (Krause et al., 1996) and in clinical BM transplantation outcome (Beksac and Preffer, 2012). This can be explained either as the occurrence of a specific stem cell subpopulation within the CD34high-HSPC pool or as a higher propensity of CD34high-HSPC to reverse their pre-commitment thereby recovering features of multipotent stem cells (Dooner et al., 2008; Knaän-Shanzer et al., 2008; Puca et al., 2012).
Mitochondrial and metabolic control of the HSPC fate
Transcriptome, epigenome and proteome studies are providing important insights into stem cell biology (Macarthur et al., 2009), indicating that specific metabolic properties are required to maintain self-renewal and the multipotent differentiation capacities of HSCs (Suda et al., 2011; Folmes et al., 2012). It is a common belief, recently supported by experimental evidence, that quiescent HSCs mainly rely on anaerobic glycolysis for energy production (Simsek et al., 2010; Takubo et al., 2013). This hypothesis is based on the notion that primitive HSCs reside in a specific BM tissue environment close to the endosteum where the limited oxygen supply makes this region hypoxic (this point will be discussed in more depth in the next paragraph) and therefore unsuited for mitochondrial oxidative phosphorylation (OXPHOS) (Eliasson and Jönsson, 2010). The OXPHOS pathway is made up of an inner mitochondrial membrane-located respiratory chain (RC), which transfers reducing equivalents along a set of respiratory complexes (I-IV) to O2. The free energy, as such made available, is converted to an electrochemical transmembrane potential, which drives the ATP synthesis, by the FoF1-ATP-synthase complex (Papa et al., 2012). The cytochrome c oxidase is the RC complex responsible for the final transfer of reducing equivalents to O2, which is converted into two H2O molecules. The estimated KM for O2 of cytochrome c oxidase is in the submicromolar range (Wilson et al., 1988; Scandurra and Gnaiger, 2010) (1.0 μM of dissolved O2 in water is in equilibrium with some 0.1% of pO2). This value is still much lower than that measured approximately in the BM (5–0.5% O2 ≅ 46–4.6 μM O2; Chow et al., 2001; Harrison et al., 2002; Parmar et al., 2007), indicating that the ambient O2 tension experienced in vivo by HSCs is not per se limiting for OXPHOS (Schroedl et al., 2002). Other factors must, therefore, contribute to the regulation of the activity of cytochrome c oxidase and, as a consequence, OXPHOS.
In a study by our group, the mitochondrial respiratory activity of human CD34+-HSPCs was measured for the first time, resulting in a cyanide-sensitive endogenous O2 consumption rate under resting conditions of about 125 pmol min−1 per 106 cells (Piccoli et al., 2005). This activity was depressed by oligomycin (inhibitor of the FoF1 ATP-synthase) and restored to values slightly higher than the resting respiration in the presence of the uncoupler FCCP. These results indicate that, although the respiratory activity of HSPCs is relatively low, their mitochondria are fully coupled to ATP synthesis and performing at almost the maximal respiratory capacity under resting conditions.
Staining of HSPCs with vital mitochondrion-tropic dyes reveals a heterogeneous pattern in the number and morphology of mitochondria. By double staining of human HSPCs for mitochondria and CD34, we were able to show that cells exhibiting the highest expression of CD34 displayed the lowest mitochondrial content (Piccoli et al., 2005). In a further study, the mitochondrial content in HSPCs proved to correlate inversely with an additional marker, CD133, which is diagnostic for more primitive HSPCs and lost during the early phase of commitment (Handgretinger et al., 2003; Piccoli et al., 2007a). A similar correlation was recently found in HSPCs isolated from the BM of mice stained with the stemness marker Sca1 (Piro et al., 2012). These results indicate that loss of primitive stemness is accompanied by a remodelling of the mitochondrial compartment. However, in another study, devoted to LT-HSCs, an increase in mitochondrial mass and mitochondrial membrane potential was found in cells with up-regulated CD34 (Mantel et al., 2010). While apparently inconsistent with our findings, these discrepancies may be explained by the fact that CD34, as a HSC marker, is poorly expressed in LT-HSCs. Therefore, a mitochondrial biogenesis is observed when HSCs lose their long-term re-populating ability, which is in agreement with the more general observation of a mitochondrial biogenesis during cell commitment and loss of multipotency. Consistent with this conclusion is the observed low retention of Rhodamine 123 in human HSCs with long-term self-renewal capacity within the CD34+CD38− Lin− population (Kim et al., 1998; McKenzie et al., 2007). Although Rhodamine 123 is commonly used to assess the efflux activity of plasma-membrane multi-drug transporters, it is also well established that, as a lipophilic cation, it specifically accumulates in active mitochondria. Moreover, very recently, it has been reported that cord blood-derived CD34+ haematopoietic cells with low levels of mitochondrial mass are enriched in haematopoietic re-populating stem cell function (Romero-Moya et al., 2013).
Theoretical models have been put forward inferring that the cell proliferation/differentiation may depend on the prevalence of a critical mitochondrial rather than a nuclear DNA ratio (von Wangenheim and Peterson, 1998). The energy needed to sustain proliferation/differentiation of quiescent stem cells requires bioenergetic adaptation. A four to five fold increase in ATP content of embryonic and adult stem cells has been found to follow their differentiation (Cho et al., 2006; Lonergan et al., 2006).
Using absolute real-time PCR, we estimated the mitochondrial DNA (mtDNA) and found it was 565 ± 100 copy number per HSPC (Piccoli et al., 2007a). If it is assumed that the average number of mitochondria per HSPC is 50 ± 20 (obtained by counting the organelles specifically loaded with a fluorescent probe using a confocal microscopy), the number of mtDNA molecules per mitochondria would be more than 13 (± 5). This number is relatively high when compared with that reported for typical differentiated mature cells [2–8 mtDNA per mitochondria (Robin and Wong, 1988)]. Interestingly, the expression of the factors controlling mtDNA replication [i.e. polymerase-gamma (POLG), mt-transcription factor A (MTFA)] has been found not to change significantly during the early phase of in vitro differentiation of HSCs (St John et al., 2005; Cho et al., 2006). This resembles that which occurs in the initial stages of mammalian embryogenesis, where regardless of the increase in mitochondrial mass, the total mitochondrial DNA in the 2 to 4/8 cell stage transition remains unchanged or even decreases (Pikó and Taylor, 1987; May-Panloup et al., 2005).
Based on the aforementioned observations, we hypothesized that the initial mitochondriogenesis, in pre-committed HSPCs consists primarily of mitochondriokinesis (Kuroiwa et al., 1994), with a re-distribution of pre-existing mtDNA among the newly formed organelles (Piccoli et al., 2007a). Such a ‘dilution’ of mtDNA may help to protect the cells from the risk of mutagenic events occurring during mtDNA replication, which may drive clonal selection of transformed haematopoietic precursors or premature haematopoietic ageing (Ralph et al., 2010; Norddahl et al., 2011). Once the mtDNA ‘density’ per mitochondria has reached a ‘normal’ value, the successive mitochondriogenesis would start synchronizing with mtDNA replication.
Accordingly, both in embryonic and in adult stem cells, enhanced mitochondrial mass was observed following in vitro differentiation. Interestingly, the newly formed mitochondria re-distribute from a perinuclear aggregation to a cytoplasmic diffused state (Handgretinger et al., 2003; Bavister, 2006; Cho et al., 2006; Nesti et al., 2007). The mitochondrial network is now well established as a dynamic entity resulting from the balance between mitochondrial fusion/fission, with the resulting organelle morphology seemingly dependent on the metabolic state of the cell (Westermann, 2010; Ferree and Shirihai, 2012). Moreover, the mitochondrial content of a cell is dependent on biogenetic rather than specific autophagic processes, also referred to as mitophagy (Novak, 2012). The latter process is necessary for the selective removal of damaged organelles. Elucidation of the mechanisms controlling mitochondrial dynamics is currently being determined for haematopoietic cell homeostasis. Conditional Atg7 (an autophagy-related protein)-deficient mice show loss of normal HSC function and die within weeks after this gene deletion (Mortensen et al., 2011). The HSCs populating these mice display an accumulation of mitochondria and reactive oxygen species (ROS), as well as increased proliferation and DNA damage. Atg7-deficient HSCs also fail to reconstitute the haematopoietic system of lethally irradiated mice. These results suggest a low amount of intracellular mitochondria content is needed in HSCs to preserve their stemness.
The notion that a different metabolic profile distinguishes quiescent from differentiating stem cells, with a shift from glycolysis to mitochondrial OXPHOS, is supported by recent experimental results (Simsek et al., 2010; Suda et al., 2011; Takubo et al., 2013). However, a key question to be answered is whether the bioenergetic shift is consequent to intrinsic nuclear reprogramming or is decided by extrinsic factors determining the fate of the stem cell. In the last few years, some important studies (briefly reviewed in the following) have contributed to the revelation that signal transduction pathways link the energy metabolism of the HSC to its self-renewing, proliferating and differentiating properties.
Mammalian target of rapamycin (mTOR), a serine/threonine PK has emerged as a key translational regulator of cellular metabolism in response to nutrient sensing (Wullschleger et al., 2006) and has been found to control mitochondrial oxidative function through an YY1-PGC-1α transcriptional complex (Cunningham et al., 2007). It has been reported that conditional deletion of TSC1, a negative controller of mTOR in HSCs, drives them from quiescence into rapid cycling, with increased mitochondrial biogenesis, respiratory activity and elevated levels of ROS (Chen et al., 2008a). Importantly, this deletion dramatically reduced both in vivo haematopoiesis and self-renewal of HSCs. In vivo treatment with an ROS antagonist or rapamycin (an inhibitor of mTOR) restored HSC numbers and functions. The detrimental effect of up-regulated ROS in metabolically active HSCs may explain the association between quiescence and the ‘stemness’ of HSCs.
The TSC-mTOR complex is part of the PI3K/Akt/mTOR pathway, which is activated when stem cells proliferate in response to growth factors, nutrients and high O2 (Yuan and Cantley, 2008). Constitutively active Akt signalling causes accelerated proliferation and depletion of HSCs (Kharas et al., 2010), whereas in Akt1/Akt2 double knockout mice, HSCs remain in a constantly quiescent state and cannot differentiate into multipotent progenitors (Juntilla et al., 2010). In contrast, deletion of Pten, a PIP3 phosphatase, induces constitutively activated PI3K/Akt signalling, resulting in an accelerated cell cycle and eventually leads to HSC exhaustion (Yilmaz et al., 2006; Zhang et al., 2006). These data indicate that mTOR is a critical modulator of stem cell maintenance and that two tumour suppressors, Pten and Tsc1, serve as important negative regulators of PI3K/mTOR signalling in HSCs.
Recently, a different signalling pathway linking Akt/mTOR to HSC metabolism has been described. The tumour suppressor liver kinase B1 (Lkb1) is a serine/threonine kinase that positively regulates the AMP-activated PK (AMPK), which, in turn, senses the AMP/ATP ratio (Hsu and Sabatini, 2008). AMPK phosphorylation inhibits mTOR, leading to reduced cell growth and proliferation. Thus, Lkb1/AMPK signalling regulates cell metabolism in response to changes in ATP availability. Three groups have independently identified an essential role for Lkb1 in HSC maintenance (Gan et al., 2010; Gurumurthy et al., 2010; Nakada et al., 2010). All three studies reported that conditional Lkb1 knockout mice show gradually impaired haematopoiesis, followed by transient increases in the number of haematopoietic progenitors and loss of reconstitution capacity after BM transplantation. Importantly, the typical Lkb1 null phenotype has dysfunctional mitochondria, a decreased mitochondrial membrane potential ΔΨm and ATP, and down-regulation of the PPARγ co-activator 1α (PGC-1α). These outcomes are partly dependent on AMPK but not on mTOR or oxidative stress-related mechanisms, indicating that the metabolic regulation of HSCs is mediated by a novel non-canonical mechanism involving Lkb1, such as PGC-1α-mediated modulation of mitochondrial function. Interestingly, in one of these studies (Gurumurthy et al., 2010), analysis of metabolic alterations in Lkb1-deficient haematopoietic cells resulted in significant alterations in lipid metabolism in the Lin− populations. Diminished mitochondrial function and elevated fatty acid levels are consistent with a model in which Lkb1 serves as a rheostat that sets the appropriate balance of anabolic and catabolic activities in haematopoietic cells.
The interplay between maintenance of the HSCs and mitochondria-dependent lipid metabolism has been further defined by a recent study describing a new pathway that encompasses the promyelocytic leukaemia (PML) tumour suppressor gene [previously found to control the homeostasis of HSCs (Ito et al., 2008)], PPARδ and fatty acid oxidation (FAO) (Ito et al., 2012). The PML protein is the essential component of subnuclear structures (nuclear bodies) implicated in a wide variety of processes, including post-translational modifications and regulation of transcription (Bernardi and Pandolfi, 2007). PPARδ is a member of the PPAR nuclear receptor superfamily of transcription factors that control nutrient sensing and the transcriptional regulation of metabolic pathways, especially fatty acid transport and FAO (Michalik and Wahli, 2006). In this study, it was found that conditional deletion of ppard (coding for PPARδ) or pharmacological inhibition of mitochondrial FAO induces loss of HSC maintenance, whereas treatment with PPARδ agonists improved HSC maintenance. Moreover, the maintenance defect of Pml−/− HSCs could be restored by activation of PPARδ, leading to haematopoietic recovery after BM transplantation with Pml−/− HSCs. These findings suggested that the altered functions of Pml−/− HSCs are at least, in part, due to dysfunctional PPARδ signalling and FAO.
Perhaps the most exciting aspect of this work was the discovery of a link between the PML–PPARδ–FAO pathway and the control of HSC divisions. HSCs have a number of division options, which include asymmetric divisions where an HSC gives rise to a daughter HSC and a daughter progenitor (which results in HSC maintenance), or symmetric divisions [which can result alternatively in two daughter HSCs (HSC expansion) or two daughter progenitors (stemness loss)]. Using a binary assay to detect surface markers and assess cell division in vitro, it was found that deletion of Ppard or Pml as well as inhibition of FAO resulted in the symmetric commitment of HSC daughter cells, whereas PPARδ activation increased asymmetric cell division. The mechanism linking mitochondrial FAO to asymmetric self-renewal remains to be elucidated.
An interesting point raised by the apparent role of mitochondria in controlling HSC division is the modality by which the organelles segregate in the daughter cells. Indeed, it has been suggested that asymmetric (instead of casual) segregation of active versus nascent mitochondria might reduce the risk of oxidative damage in one of the two daughter cells, preserving a more pristine genome in HSC during self-renewing asymmetric division (Mantel and Broxmeyer, 2008).
A recently reported study revealed another feature involved in the regulation of mitochondrial metabolism in HSCs. Genetic depletion of PTPMT1 (a PTEN-like mitochondrial phosphatase) in HSC inhibited haematopoiesis by blocking differentiation of these cells, whereas cycling of stem cell renewal was less affected (Yu et al., 2013). Further analyses demonstrated that PTPMT1 deficiency altered mitochondrial metabolism and that phosphatidylinositol phosphate substrates of PTPMT1 directly enhanced fatty acid-induced activation of mitochondrial uncoupling protein 2.
The apparent relevance of the described pathways exemplifies the complicated networks in the regulation of metabolism in HSCs. Indeed, on the one hand, the maintenance of the quiescent state in the G0 state of HSC seems to rely on a glycolysis-based metabolism and proliferating multipotent precursors with a different level of commitment to active aerobic OXPHOS. On the other hand, the role of mitochondria in cycling HSC appears to be to control the balance between self-renewing cell division and symmetrically committed cell division.
See Figure 1 for a schematic overview of the points reviewed in this paragraph.
Figure 1.
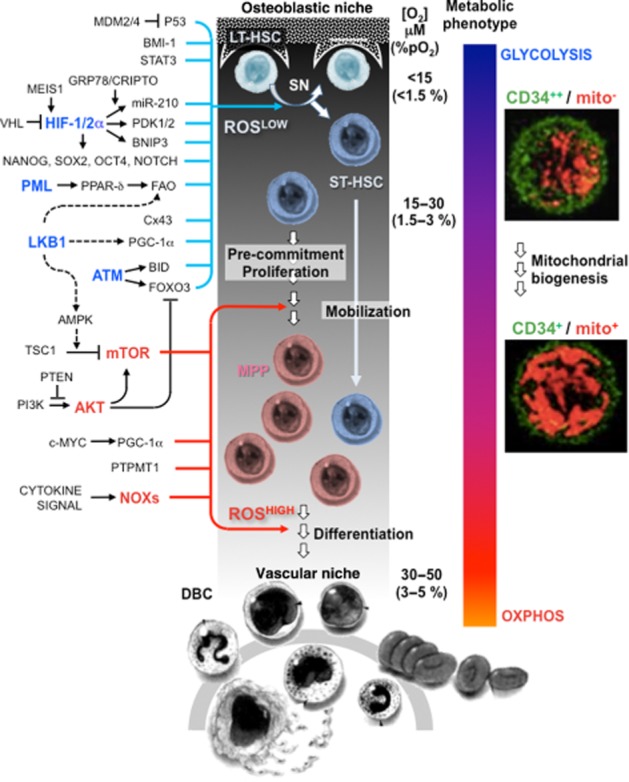
Redox signalling governing haematopoietic stem cell fate. Bone marrow stem cell endoblastic and vascular niches are schematically drawn on the left and bottom sides of the diagram respectively. The oxygen gradient, from the endoblastic niche to the sinusoid vessel, is also shown with the approximate values of concentration given in μM and % pO2 units (for conversion in other units, consider that 1.0 μM O2 = 0.77 mmHg = 5.77 kPa at 37°C). A quiescent long-term haematopoietic stem cell (LT-HSC) is elicited by niche-related factors to undergo asymmetric division. One of the two daughter cells remains in the niche, whereas the other short-term (ST)-HSC moves away from the hypoxic area and experiences a progressive increase in O2 concentration. The ST-HSC proliferate and convert to multipotent progenitor (MPP) cells that finally are committed to one or other of the fully differentiated blood cells (DBC). The progressive loss of the stemness profile is accompanied by a metabolic shift from glycolysis to oxidative phosphorylation. The two pictures on the right-hand side are confocal images of cells co-stained for the stemness marker CD34 and the mitochondrial compartment showing an increase in mitochondrial mass in the CD34low-HSPC. The increased number of functional mitochondria in the pre-commitment stage of the ST-HSC/MPP provides the cell with the energy-generating systems needed to cope with a proliferating/differentiating metabolism. On the left-hand side of the picture: schematic depictions of the main signalling pathways reported to control the quiescence/maintenance of the HSCs or to induce their proliferation/differentiation (the prime factors of the pathways are highlighted in blue and red respectively). All the pathways are linked directly or indirectly to the regulation of the cellular redox state with the low and high reactive oxygen species (ROSLOW and ROSHIGH) level priming the HSC maintenance and commitment respectively. See text for definitions of the abbreviations.
Redox signalling in HSPCs
ROS were once believed to be harmful by-products of dioxygen metabolism, but are now recognized as physiological messengers involved in controlling adaptive cell biology (Finkel, 2011). The term ROS encompasses many species, including superoxide (O2•−), hydroxyl radical (HO•) and hydrogen peroxide (H2O2). Other reactive species containing nitrogen (RNS) include NO• and peroxynitrite (ONOO−). The intracellular level of these redox active molecules depends on the balance between the generating versus the scavenging systems. According to a ‘window hypothesis’, a cell-type-dependent threshold level of such reactive species exists below which the altered oxido-reduction state of the cell is transduced in a physiological response.
Redox signalling has been shown to be involved in cell fate decision under the pressure of challenging conditions leading to activation of growth, proliferation, differentiation or cell cycle arrest, senescence and apoptosis (Sauer et al., 2001; Dröge, 2002; Finkel, 2003). The amount and duration of ROS are likely to provide a combination of conditions that result in apparently contradictory cell responses. So, a short-lived ROS production of low intensity is transduced in a signal leading to cell proliferation or differentiation, whereas the continuous generation of relatively large amounts of ROS elicits growth arrest and eventually cell death (Boonstra and Post, 2004; Burhans and Heintz, 2009; Chiu and Dawes, 2012). The specific chemical species involved and/or their cellular compartmentalization are also important elements in determining the signalling outcome.
ROS such as O2•− and H2O2 are generated from a number of sources including mitochondrial RC complexes, NADPH oxidases (NOXs), xanthine oxidase, cytochrome P450 and uncoupled NOS. Importantly, ROS generation is considered for some of these sources as an unwanted by-product of oxygen metabolism, as in the case of the mitochondrial RC, whereas for others, it is the intended product of the enzymatic reaction, as in the case of the NOXs. This distinction, however, may require more in-depth re-consideration. The superoxide anion O2•− represents the seminal ROS product wherefrom all the others derive. As O2•− is generated by a bimolecular reaction, it is generally assumed that the oxygen concentration has a significant effect on the total amount of ROS. However, this conviction is challenged by experimental evidence and needs further consideration. The O2•− reacts with NO to generate ONOO−, whereas it can be quickly converted to H2O2 by superoxide dismutases (SODs) such as Cu/Zn SOD (SOD1), MnSOD (SOD2) or extracellular SOD (SOD3). H2O2 is catalysed by catalase, glutathione peroxidases and the thioredoxin–peroxiredoxin system to water. Because H2O2 is relatively stable, membrane permeant and does not react with NO, it has been proposed to represent the main second messenger in physiological redox signalling.
Identifying the direct molecular target(s) of ROS in each cell type is important in understanding the cellular mechanism of redox regulation and is still a contentious issue. Signalling by ROS and RNS occurs through chemical reactions with specific atoms of target proteins; thus, recognition is at the sub-molecular, atomic level, and consequently has the potential for recognition of many ROS/RNS-specific receptors (Nathan, 2003). Nevertheless, with most of the redox-regulated proteins studied, the targeting of specific reactive groups within specific targeted proteins is evident. While in many of them, reactive cysteine residues function as redox sensors, others use a redox-sensitive metal iron (Baldwin and Benz, 2002; Ilbert et al., 2006).
The thiol-based ‘peroxide receptors’ or ‘sensors’ that react with H2O2 form a hierarchy in the oxidation of thiols in other proteins by interacting with them (Toledano et al., 2004). The model proposes that oxidation of most target proteins is not conducted directly, but rather mediated through the oxidation of a subset of very reactive thiols in sensor proteins, which, in turn, facilitate the oxidation of other target proteins through selective protein–protein interactions (Winterbourn and Hampton, 2008; Finkel, 2011). A redox signal is transmitted from the site of ROS generation through the sensor protein to its final destination, which may reside, as in the case of the transcription and replication factors, in the nucleus. Thus, different redox states within different subcellular compartments may present an additional constraint on the transmission of a redox signal.
In the past decade, specific interest and intense investigation have been addressed to the role of the redox status in controlling the balance between quiescence, self-renewal and differentiation of stem cells including HSCs and their function (Eliasson and Jönsson, 2010; Sardina et al., 2012; Urao and Ushio-Fukai, 2013). When isolated HSCs are stained with the ROS-sensitive fluorescent probe dichlorofluorescein (DCF), two subpopulations can be distinguished based on their basal ROS level, DCFhigh and DCFlow. Isolation and further characterization by serial transplantation of the two fractions reveals that the DCFlow HSC population retains a higher reconstitution ability (i.e. higher self-renewal potential) than the DCFhigh population (Jang and Sharkis, 2007). Interestingly, the DCFhigh fraction had higher myeloid differentiation capacity than the DCFlow cells, indicating that the DCFhigh fraction is ‘myeloid shifted,’ a characteristic of senescent HSCs. Treatment of the DCFhigh fraction with antioxidants, p38 MAPK or mTor inhibitors restored their colony-forming capacity in vitro. The authors proposed that the DCFlow fraction represents the low cycling quiescent HSC subset located in the hypoxic endoblastic niche. In a study on Drosophila haematopoietic progenitors, using genetic tools, it was shown that scavenging or moderately increasing ROS levels decreases or stimulates HSPC differentiation respectively (Owusu-Ansah and Banerjee, 2009). Thus, the effect of ROS on HSC function probably depends on its concentration and the exposure period.
Consistent with the importance of redox signalling in regulating division and driving differentiation of HSPC is the observation that a number of haematopoietic growth factors or cytokines stimulate signalling events, leading to cell growth (Sattler et al., 1999; Iiyama et al., 2006) or promote HSC mobilization into the circulation through the formation of H2O2 (Tesio et al., 2011). ROS-dependent HSC proliferation plays an important role in the early steps of haematopoietic reconstitution after HSC transplantation (Lewandowski et al., 2010).
The mechanism controlling the HSC redox status has been investigated using relevant gene-targeted mice. Conditional deletion of these genes in HSCs results in alterations in the cellular ROS balance towards a pro-oxidative state. This induces the HSCs to egress from quiescence, to lose self-renewal capacity, to accelerate proliferation/differentiation, to promote migration and to induce genome instability, thereby causing rapid depletion of the haematopoietic reserve (reviewed in Ushio-Fukai and Urao, 2009; Eliasson and Jönsson, 2010; Sardina et al., 2012; Urao and Ushio-Fukai, 2013). Importantly, treatment with an antioxidant in most cases restores the altered HSPC phenotype.
These studies have helped to identify the intertwined ROS-related regulatory network in HSCs comprising: the transcription factor FOXOs (Miyamoto et al., 2007; Tothova et al., 2007); p38 MAPK (Ito et al., 2006; Miyamoto et al., 2007); PI3K–Akt1/2 signalling (Juntilla et al., 2010; Kharas and Gritsman, 2010), PTEN (Yilmaz et al., 2006), TSC1/mTOR (Chen et al., 2008b), ATM-CDK-inhibitors p16Ink4a and p19Arf (Ito et al., 2004; 2006), ATM-BID (Maryanovich et al., 2012), the polycomb protein Bmi-1 (Lessard and Sauvageau, 2003; Park et al., 2003; Oguro et al., 2006; Liu et al., 2009), P53-Mdm2 (Abbas et al., 2010; 2011), the nuclear transcription factor Stat3 (Hankey, 2009; Mantel et al., 2012) and connexin-43, (Taniguchi Ishikawa et al., 2012). Of note, many of the transduction pathways that have been identified interact with each other, contributing by feed-forward and feedback mechanisms to fine-tune the redox homeostasis of HSPCs. Nevertheless, in some instances, it is unclear which ROS generating system is involved following targeted gene depletion. Although there are several sources of ROS, NADPH oxidases (NOXs) and mitochondria are the major ROS-generating systems in BM-derived HSPCs.
NADPH oxidase is a cell membrane bound di-haeme-containing enzyme that catalyses the transfer of electrons (one at a time) from NADPH to O2, leading to the formation of the superoxide radical anion O2•− (Cross and Segal, 2004). No other activity has been reported for NOX; thus, it is the only enzyme in the cell that ‘deliberately’ generates ROS. The best-characterized isoform NOX2 participates in the host-defence function in macrophagic blood cells by provoking the bactericidal ‘oxidative burst’. This reaction is tightly controlled and the catalytic oxidase subunit, which is usually silent, is promptly activated (within seconds) by the recruitment of a number of cytosolic regulatory subunits. This event is triggered by a signalling cascade, which starts from external stimuli and leads to the activation of the GTP-binding protein Rac1/2 and to phosphorylation of p47, both regulatory NOX2 subunits (Cross and Segal, 2004). In the last few years, NOX2 along with other evolutionary-related isoforms (NOX1, NOX3, NOX4, NOX5, DUOX1 and DUOX2) have been discovered in many other cell types not involved in host defence mechanisms (Cheng et al., 2001; Bokoch and knaus, 2003) and are thought to be involved in redox signalling (Sauer et al., 2000; 2001; Dröge, 2002; Bokoch and knaus, 2003; Finkel, 2003). These additional NOX2 isoforms rely on different subunit assembly patterns and appear to be differently regulated (Lambeth et al., 2007; Petry et al., 2010).
By utilizing G-CSF-mobilized human CD34+-HSPCs, we were the first to provide functional evidence of O2 consumption attributable to NOX activity [i.e. ≅80 pmol min−1 per 106 cells (Piccoli et al., 2005)]. This was further confirmed at the transcriptional and translational level, revealing the presence of multiple isoforms of NADPH oxidase (i.e. NOX1, NOX2 and NOX4) (Piccoli et al., 2007b). Moreover, in a sequential study using high-resolution imaging of HSPC membranes, we immunodetected the presence of NOX in membrane ‘raft'-like microcompartments where the assembly/activation of the NOX components may be functionally integrated to create redox signalling platforms (Frassanito et al., 2008). The NOX activity in CD34+-HSPCs is constitutive and only slightly stimulated by PMA or G-CSF (Piccoli et al., 2005; 2007b). O2•− generated outside the cell can dismutate to form the freely diffusible H2O2. Consistently, we found that the extracellular isoform SOD3 was expressed in HSPCs (Piccoli et al., 2007b). H2O2 can enter into the cell and alter its redox status. In fact, we detected intracellular ROS by using the DCF probe and found that its level was largely (but not completely) decreased by specific inhibitors of NOX or by pretreatment of CD34+-HSPCs with externally added catalase or an inhibitor of the PI3K/Akt pathway (Piccoli et al., 2007b).
The catalytic properties of NOXs make them suited to function as oxygen sensors in the HSPC physiological environment. Indeed, the reported KM for O2 of NOXs is about 10–15 μM (Gabig and Babior, 1979; Chen et al., 2005), implying that their steep activity-slope falls in the range of the O2 concentration estimated in the BM from the endosteum to the sinusoidal vasculature (i.e. from <5 to 30–40 μM respectively). On this basis, we have proposed a model whereby the NOX4 works as a primary oxygen sensor in HSPCs (Piccoli et al., 2007b). NOX4 is, apparently, the only NOX isoform that does not require regulatory subunits and is constitutively active (Lambeth et al., 2007). Thus, the amount of NOX4-generated O2•− reflects the range of the environmental O2 experienced by the HSPCs. Furthermore, we proposed that intracellular H2O2 induces the activation of the other NOX isoforms by stimulation of their regulatory subunits. Indeed, most of the upstream factors, which activate NOX1/2, are themselves targets of redox control (Pendyala and Natarajan, 2010), thus placing NOX1/2 downstream and upstream of redox signalling. This, in turn, provides a positive feedback regulatory loop that gradually enhances intracellular ROS signalling upon displacement of the HSPCs from a hypoxic endoblastic function to a vasculature function activating/fostering therein proliferation/differentiation by HSPC reprogramming.
A number of reported findings, summarized in the following, support this model: (i) hypoxic conditions (<5% pO2) is a well-established setting for both quiescence maintenance and induction of HSPC from cord blood and BM in ex vivo cultures (Cipolleschi et al., 1993; Danet et al., 2003; Ivanovic et al., 2004; Hermitte et al., 2006; Shima et al., 2010); (ii) inhibitors of NOX preserve the progenitor features of HSPCs under normoxic conditions (Fan et al., 2007); (iii) activated Ras promoted both survival and growth factor-independent proliferation of CD34+-HSPCs through stimulation of NOX activity and ROS generation leading to up-regulation of the expression of D cyclins (Hole et al., 2010); (iv) NOX-related ROS are generated by haematopoietic cytokines and play key roles in the activation of receptor-mediated signalling and in the cell cycle progression (Iiyama et al., 2006; Woolley et al., 2012); and (v) ROS derived from NADPH oxidase play an important role in redox signalling linked to BM stem/progenitor cell post-ischaemic mobilization, homing and differentiation, thereby promoting neovascularization in hypoxic areas (Ushio-Fukai and Urao, 2009; Urao et al., 2012).
Moreover, NOX-mediated ROS signalling has been reported in the physiology of other stem cell populations, implying a more general role of NOXs in controlling stem cell biology. NOX-mediated ROS production promotes differentiation of (i) BM mesenchymal stem cells towards osteoclasts (Lee et al., 2005), neurons (Wang et al., 2007), adipocytes (Kanda et al., 2011); mouse embryonic stem (ES) cells towards endothelial cells (Sauer et al., 2005; Schmelter et al., 2006), smooth muscle cells (Xiao et al., 2009), phagocytic cells (Hannig et al., 2010) and cardiomyocytes (Sauer et al., 2000; Li et al., 2006; Buggisch et al., 2007). The ROS-mediated signalling appears to be time-dependent, being activated during the early stages of ES cell differentiation and then down-regulated during later stages. During the differentiation process, antioxidative genes are down-regulated (Saretzki et al., 2004), while NOXs 1/2/4 are up-regulated (Buggisch et al., 2007).
ROS are also produced from mitochondria as a consequence of aerobic metabolism (Murphy, 2009; Dröse and Brandt, 2012; Papa et al., 2012). The electron transport in mitochondrial RC starts with the extraction of electrons from NADH (complex I) or FADH2 (complex II) generated in the tricarboxylic acid cycle. Electrons are then transferred via ubiquinone to complex III and finally to complex IV. Electron leak during respiration leads to the formation of O2•− due to the incomplete reduction of O2 at complex I and complex III (Dröse and Brandt, 2012). O2•− formed at complex I is released into the mitochondrial matrix, whereas O2•− formed at complex III can be released either to the matrix or to the intermembrane space. Recent evidence that complex II may also be a site of ROS generation has been provided. Inhibition of complex III or IV and/or establishment of a large electrochemical gradient are all conditions that enhance electron leak by a mechanism known as ‘reverse electron flow’. Conversion of O2•− in H2O2 by outer mitochondrial membrane-associated or intra-mitochondrial SOD1 and SOD2, respectively, generates a relatively long-lasting membrane-permeant reactive species that, when escaped from the armoury of other antioxidant enzymes, can flux from the mitochondria to the cytoplasm. The role of mitochondria-related ROS-mediated signalling in the adaptive response to physiological stressors/inducers is recognized in a number of cell systems, including HSPCs as mentioned before (Hamanaka and Chandel, 2010; Rehman, 2010; Mantel et al., 2011; Rigoulet et al., 2011; Finkel, 2012; Lenaz, 2012; Maryanovich and Gross, 2012; Papa et al., 2012).
An intriguing aspect reported in various cell systems is the cross-talk between NOX and mitochondrial ROS (Daiber, 2010; Dikalov, 2011). It has been suggested that mitochondrial H2O2 regulates NOX activity. Conversely, NOX activation induces mitochondrial H2O2 formation by a number of different mechanisms. Given that H2O2 is a highly diffusible molecule, cross-talk between NOXs and mitochondrial ROS may represent a positive feed-forward mechanism that promotes sustained H2O2 production and activation of redox signalling. This may be part of a more general phenomenon described as ‘ROS-induced ROS release’ (Zorov et al., 2006; Zinkevich and Gutterman, 2011). Whether this regulatory mechanism is involved in stem and progenitor cell function is the subject of future investigation (Mantel et al., 2011). Considering the stability and membrane-permeant properties of H2O2, redox signalling in HSPCs can also be affected by H2O2 production from surrounding cells.
See Figure 1 for a schematic overview of the points reviewed in this paragraph.
HIF-1-mediated signalling in HSPC: beyond O2-sensing and adaptation to hypoxia
While a moderate increase in the ROS level has been shown to trigger a proliferative response and probably to provide conditions making HSPC more responsive to differentiation stimuli, in a low ROS level state, less active cells with preserved stemness properties have been identified (Jang and Sharkis, 2007). In this regard, it is not clear whether the ‘low ROS level’ regime is by itself functional to the maintenance of the undifferentiated state through a signal transduction pathway different from that attained under ‘high ROS level’ conditions.
The low ROS level displayed by the subset of more primitive HSPCs is thought to be a direct consequence of the hypoxic features of the endoblastic niche where this HSPC population resides (Chow et al., 2001; Parmar et al., 2007). This low-oxygen environment is not only tolerated by HSCs, but also appears to be essential for their function (Bradley et al., 1978; Katahira and Mizoguchi, 1987; Koller et al., 1992; Cipolleschi et al., 1993; LaIuppa et al., 1998; Danet et al., 2003; Kubota et al., 2008; Lo Celso et al., 2009; Eliasson and Jönsson, 2010).
The main experimental data supporting the localization of HSCs in a hypoxic BM niche are as follows: (i) administration of a perfusion tracer into mice showed that HSCs were relatively enriched in a low-perfusion cellular compartment in the BM (Parmar et al., 2007) and retained pimonidazole, a probe that selectively forms thiol-adducts under hypoxic conditions; (ii) administration of tirapazamine, a toxin selective for hypoxic cells, promoted loss of HSCs in vivo; and (iii) LT-HSCs proved to be pimonidazole+ in vivo (Simsek et al., 2010; Takubo et al., 2010).
Taken together, these findings support a model in which the hypoxic character of LT-HSCs is determined by their position within the BM. However, in contrast to the simple O2 gradient model for the BM hypoxic niche, two-dimensional observations of a segment of the BM suggest that a large fraction of the LT-HSCs reside adjacent to the vasculature (Kiel et al., 2005; Sugiyama et al., 2006). This would imply the existence of a different location for the HSCs defined as vascular, which, even considering the poor perfusion through the endosteal capillaries, is far from being a severe hypoxic environment. Thus, it is possible that subpopulations of HSCs exist that reside in different specific locations, in different cell cycle and/or metabolic states and that HSCs dynamically move within the BM acquiring distinct environment-dependent phenotypes (Lo Celso et al., 2009; Xie et al., 2009). The hypoxic endosteal niche would house quiescent non-cycling HSCs.
A key component for cellular and systemic responses to reduced O2 availability is the transcription factor hypoxia inducible factor-1 (HIF-1) (Semenza, 2009). HIF-1 is a heterodimeric protein consisting of an O2-regulated α subunit and a constitutively expressed β subunit (Wang and Semenza, 1995). Under normoxic conditions, HIF-1α is hydroxylated at two specific proline residues in the O2-dependent degradation domain (Kaelin and Ratcliffe, 2008) and at a specific asparagine residue in the C-terminal transactivation domain by three prolyl hydroxylases, PHD1–3 and the asparagine hydroxylase FIH-1, respectively; all require O2, Fe2+, 2-oxoglutarate and ascorbate for enzymatic activity (Epstein et al., 2001). Prolyl-hydroxylated HIF-1α is recognized by the von Hippel-Lindau (VHL) tumour suppressor protein, which recruits an ubiquitin-ligase complex, leading to ubiquitination and subsequent proteasomal degradation. Under hypoxic conditions, HIF-1α prolyl hydroxylation is suppressed, stabilizing the HIF-1α protein, which moves into the nucleus where it interacts with its cognate partner, HIF-1β (Kaelin and Ratcliffe, 2008). HIF-1α : HIF-1β heterodimers bind to hypoxia response elements (HREs) in numerous target genes and activate transcription. HIF-2α is an important isoform of HIF-1α, which displays a low level of redundancy. The two isoforms are believed to play a differential role under conditions of acute or chronic hypoxia (Koh and Powis, 2012).
Thus, HIF activity is regulated by an exceptionally tight, two-level mechanism in which HIF-1/2α stability is regulated through O2-dependent proline hydroxylation by PHDs (PHD-VHL-proteasome) and its transcriptional activity through the action of O2-dependent aspargine hydroxylation by FIH1. It has been reported that HIF hydroxylases are present in the cell in limited amounts and that the intracellular physiological concentration of O2 is below the apparent KM in this reaction (estimated to be around 230 and 90 μM for PHDs and FIH1 respectively (Hirsilä et al., 2003; Koivunen et al., 2004). Thus, hydroxylases have the capacity to respond to significant physiological changes in intracellular oxygen concentrations, linking them to the regulation of HIF. However, considering that the estimated range of O2 concentration from the sinusoidal vessels to the endoblastic niche is from 40–50 to 5 μM, the HIF hydroxylases are likely to work in a low-activity range with respect to their catalytic potential. Indeed, the activation of HIF is also controlled by a number of O2-independent mechanisms at the post-transcriptional and post-translational level (Koh et al., 2008). As discussed below, oxygen sensing by the hydroxylases may also proceed via an indirect process mediated through the generation of ROS in the mitochondria under hypoxic conditions.
HIF-1 activity promotes a switch from oxidative metabolism to glycolysis (reviewed in Semenza, 2011) through the transcriptional activation of genes encoding glucose transporters such as glucose transporter 1, glycolytic enzymes such as LDHA, and metabolic regulatory enzymes such as pyruvate dehydrogenase kinase 1 (PDK1) (Iyer et al., 1998; Suda et al., 2011). Importantly, HIF-1α positively controls the expression of the isoform cytochrome c oxidase 4-2, a subunit of the cytochrome c oxidase and of a mitochondrial protease Lon, which degrades the ‘normoxic’ cytochrome c oxidase 4-1(Fukuda et al., 2007). This change in the subunit composition of the cytochrome c oxidase optimizes the efficiency of respiration under hypoxia and reduces ROS production. Moreover, HIF-1 induces BNIP3, which triggers mitochondrial-selective autophagy (Zhang et al., 2008; Bellot et al., 2009) and the microRNA-210, which blocks the assembly of Fe/S clusters that are required for OXPHOS (Chan et al., 2009).
HIF-1 mRNA and protein are highly expressed in LT-HSCs (Simsek et al., 2010; Takubo et al., 2010). Conditionally HIF-1α-deleted HSCs lose their capacity for marrow reconstitution during serial BM transplantation, enter the cell cycle from G0 in a p16Ink4a/p19Arf-dependent manner, proliferate, and display reduced tolerance to stressors or aging (Takubo et al., 2010). Overstabilization of HIF-1α by biallelic loss of VHL induces cell cycle quiescence in HSCs and their progenitors but results in impaired transplantation capacity. In contrast, monoallelic loss of VHL induces cell cycle quiescence and improved BM engraftment during BM transplantation. These results indicate that there is an optimal HIF-1α protein level for HSC maintenance with an aberrant activation of its stabilization being detrimental to the HSC biology. Accordingly, long-term in vitro treatment of LT-HSCs with a PHD inhibitor, dimethyloxalylglycine, which also stabilizes HIF-1α, attenuates stem cell capacity during BM transplantation (Eliasson et al., 2010). In a recent study it was demonstrated that regulation of glycolysis by PDK functions as a metabolic checkpoint for cell cycle quiescence in HSCs, further supporting the role of a HIF-1 in conditioning the fate of HSC through metabolic control (Takubo et al., 2013). PDK overexpression in glycolysis-defective HSCs restores glycolysis, cell cycle quiescence and stem cell capacity, while loss of both Pdk2 and Pdk4 attenuates HSC quiescence, glycolysis and transplantation capacity. Moreover, treatment of HSCs with a PDK mimetic promotes their survival and transplantation capacity.
However, the glycolytic phenotype of LT-HSCs appears to be dependent on transcriptional activation of HIF-1α through the HSC-specific transcription factor MEIS1 (a homeobox protein) and not merely a product of a hypoxic condition (Simsek et al., 2010).
HIFs also interact with core stemness networks because HIF-2α can up-regulate OCT4 expression and modulate Wnt/β-catenin signalling, while silencing of HIF-2α and HIF-3α decreases the expression of transcription factors, NANOG, SOX2 and OCT4 (Covello et al., 2006; Forristal et al., 2010; Mazumdar et al., 2010). One of the heat shock proteins, GRP78, and its ligand, Cripto, regulate HSC quiescence and maintain HSCs in hypoxia as an intermediary of HIF-1α (Miharada et al., 2011). The Cripto promoter region has an HRE, onto which the stabilized HIF-1 complex binds. Moreover, it has been found that hypoxia synergizes with Notch to inhibit differentiation of myogenic and neural precursor cells. This effect requires the interaction between the transcriptionally active form of HIF-1α and the intracellular domain of Notch (Gustafsson et al., 2005).
In addition to the low oxygen availability, in normoxic conditions HIF-1α is also responsive to a variety of stimuli, including various growth factors, cytokines and hormones. It has been found that many of these non-hypoxic stimuli use ROS in the regulation of HIF-1α (Kietzmann and Görlach, 2005; Pouysségur and Mechta-Grigoriou, 2006). The notion of redox control/modulation of HIF-1α activity has been supported, in the last two decades, by a large body of evidence, although the sources and mechanisms of ROS generation are still controversial. H2O2 and NO donors were found to stabilize HIF, and genetic and pharmacological interventions that affect ROS generation have been shown to affect the accumulation of HIF-1α (Brüne and Zhou, 2003; Kietzmann and Görlach, 2005). In various studies, NOX1-2 isoforms have been suggested as important sources of ROS in the regulation of both HIF-1 and -2α (Görlach et al., 2001; Kim et al., 2002; Goyal et al., 2004; Maranchie and Zhan, 2005; Bonello et al., 2007).
Our group provided evidence that G-CSF-mobilized CD34+- and CD133+-HSPCs stabilized a cytoplasmic form of HIF-1α under normoxic conditions (Piccoli et al., 2007c). It was shown that HIF-1α stabilization correlated with the down-regulation of pVHL and was positively controlled by NADPH-oxidase-dependent production of ROS. Conversely, normoxic stabilization of HSPCs was not observed in BM-resident HSPCs, thus indicating a specific O2-independent post-transcriptional control of HIF in mobilized HSPCs. Normoxic stabilization of the HIF-2α was also recently assessed in the same cell phenotype (unpublished data). This finding led us to propose that HIF functions as a mediator of HSPCs recruitment to injured ischaemic tissues while preserving the maintenance of the undifferentiated state. Accordingly, hypoxic tissues, like cardiac peri-ischaemic areas, promote HSC mobilization by HIF-1-mediated release of stromal-derived factor 1, which acts at the level of BM, recruiting undifferentiated HSCs into the circulating blood (Ceradini et al., 2004). Following a chemotactic gradient, HSCs can reach the hypoxic tissue and provide it with a source of endothelial precursors that could help in the re-vascularization process, as recently suggested by a study showing that endothelial cells are an intrinsic component of adult myeloid lineage differentiation (Bailey et al., 2006). Given the interchangeable features shared by chemokines and growth factors (Vandervelde et al., 2005), it is possible that G-CSF conditioning might mimic the physiological BM-mobilization of HSCs (Powell et al., 2005) under crisis conditions. This mechanism in circulating HSPCs might also support the re-population of exhausted BM niches in physiological conditions, as well as in clinical practice during BM reconstitution/transplantation.
The main mechanism suggested for the ROS-mediated activation of HIF is the redox modification of the catalytic site of PHD1-3 and FIH1. As described earlier, both these types of hydroxylases are dioxygenases that require Fe2+ and are affected by changes in the levels of this co-factor. Fe2+, which is oxidized during an occasional decoupled pathway in the hydroxylation reaction, requires ascorbate to be regenerated. Hence, the accumulation of ROS would inactivate PHDs and FIH1 and, consequently, stabilize HIF-1 as a consequence of direct oxidation of Fe2+ or depletion of antioxidants (Gerald et al., 2004; Pouysségur and Mechta-Grigoriou, 2006).
A subject of intense investigation and debate is the role of ROS-mediated signalling as an important, if not essential, factor in controlling the HIF-1 activity even under conditions of limited O2 availability. Although counterintuitive, a large body of accumulated evidence shows that under hypoxia, cells generate ROS at levels that, depending on the conditions, can be even higher than that observed under normoxia (Chandel et al., 1998). Using genetically manipulated cells lacking either SOD1/2 or GPX1/catalase, it was found that hypoxic stabilization of HIF-1α occurs specifically with the latter, thus suggesting that H2O2 is the ROS likely to be involved in this process (Brunelle et al., 2005).
A series of studies supports a role for mitochondrial RC in the generation of the ROS involved in the hypoxic response. It was shown that HIF-1α activation was impeded in ρ0 cells, which lack functional mitochondrial DNA, and in cells treated with inhibitors of the mitochondrial RC complexes. Further in-depth studies demonstrated that in the electron transport chain, complex III is an important source of hypoxic ROS (Brunelle et al., 2005; Guzy et al., 2005). Pharmacological and genetic evidence suggests that the ubiquinone (Q) cycle of complex III is the main site of ROS generation linked to hypoxic stabilization of the HIF-1α protein (Chandel et al., 1998; 2000; Brunelle et al., 2005; Guzy et al., 2005; Mansfield et al., 2005). Complex III can release O2 into the mitochondrial intermembrane space and subsequently into the cytosol (Muller et al., 2004). In accord with these observations, it was found, using cytochrome b mutant cybrids, that when hypoxic ROS production was up-regulated, HIF-1α was stabilized. Moreover, the mitochondrial-targeted antioxidant Mito-Q inhibited the ROS-mediated activation of the HIF pathway in hypoxia (Bell et al., 2007). On the other hand, several other reports have challenged these data, demonstrating that in ρ0 cell lines, HIF-1α was stabilized under severe hypoxic conditions (Doege et al., 2005).
To note, under severe hypoxia (anoxia), ROS production is impaired by the absence of O2 but nevertheless HIF-1 is activated (Schroedl et al., 2002). Importantly, cells that reside under hypoxia do not undergo cell death, whereas a sustained period of anoxia causes cell death. Thus, reduction of O2 depletion under hypoxia might be a way to prevent the creation of harmful anoxic conditions. As the RC is the primary consumer of intracellular O2, the HIF-mediated inhibition of the mitochondrial metabolism prevents the depletion of O2, maintaining it at acceptable levels of hypoxia. The failure to stabilize HIF-1α under hypoxia in cells with impaired mitochondrial function was interpreted as being the result of reduced mitochondrial respiration and intracellular O2 redistribution. According to this proposal, known as the oxygen redistribution model, this led to an increase in O2 availability and, consequently, the relief of hypoxia, rather than reduced ROS production by the electron transfer chain (Hagen et al., 2003). However, the two proposed mechanistic models are not necessarily reciprocally excluding and can both contribute an explanation of the role of functional mitochondria in the hypoxic induction of HIF.
See Figures 1 and 2 for a schematic overview of the points reviewed in this paragraph.
Figure 2.
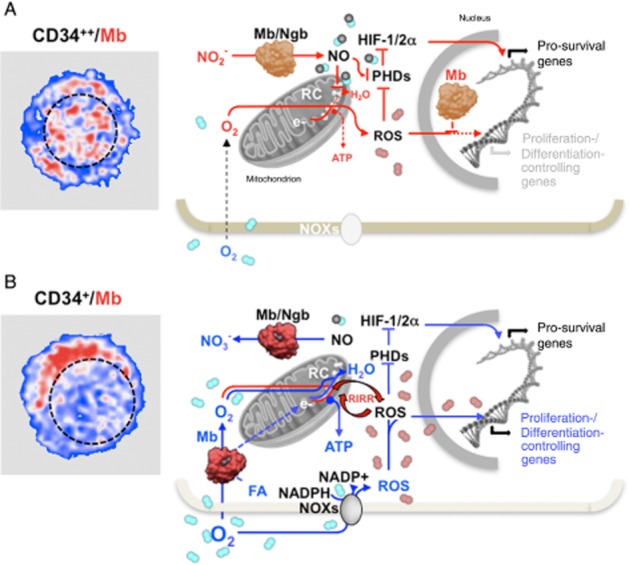
Schematic view of the suggested functional role of globins under hypoxic and normoxic conditions in quiescent and early committed HSPCs. The pictures on the left are artificial colour images of CD34++ (A) and CD34+ (B) in human HSPCs immunostained with a myoglobin (Mb)-Ab. The Mb-related fluorescence signal is shown in red and the nuclear compartment outlined with a dashed black line. The schemes on the right illustrate the suggested functions of Mb and neuroglobin (Ngb) in the context of hypoxic (A) and normoxic (B) conditions in early and late HSPCs respectively. The prevailing de-oxygenated and oxygenated state of the globins is highlighted with a different colour tone in (A) and (B). It is shown that under hypoxia (A) the de-oxygenated form of Mb/Ngb function as a nitrite reductase generating NO. This inhibits the mitochondrial respiratory chain (RC) and the oxidative phosphorylation promoting electron leak to O2 with formation of ROS, which, along with NO, inhibit the prolyl-hydroxylases (PHDs) thereby stabilizing the hypoxia inducible factors (HIF-1/2α). The transcriptional activity of HIF induces the expression of a set of pro-survival genes. The nuclear localization of Mb under this condition prevents ROS from scavenging the redox-linked activation of NFs/genes involved in the proliferation/differentiation of HSPCs or oxidative damage of DNA. Under normoxic conditions (B) the oxygenated form of the globins converts NO in nitrate unleashing the NO inhibition of the RC. This restores mitochondrial O2 consumption and ATP production by oxidative phosphorylation. Under these conditions the globins might favour the delivery of O2 and/or fatty acids (FA) to the mitochondria. It is further shown that the normoxic condition promotes the activity of the NADPH oxidases (NOXs) with the formation of ROS. The ROS level is suggested to be eventually increased by means of a ROS-induced ROS release (RIRR) mechanism involving the mitochondrial RC. The consequent enhanced pro-oxidative state of the cell functions as a signal inducing the HSPC to differentiate. The protective role of the nuclear Mb under this condition is removed by its cytoplasmic re-localization. Depending on the prevailing conditions, set by extrinsic signals, ROS can also activate under normoxic conditions HIF thereby delaying differentiation of HSPCs if these are mobilized to restore exhausted niches or act at hypoxia-injured tissues. See text for further considerations.
O2-binding/releasing globins: a new entry in HSPC physiology
A further adaptive response to low O2 tension found in different cell types is the induced expression of O2-binding/releasing globins comprising myoglobin (Mb) and Hb and the recently discovered cytoglobin (Cygb) and neuroglobin (Ngb). We have obtained functional and molecular evidence that human HSPCs express Mb at a level comparable with that of a muscle-derived cell line. A search for other globins revealed the significant expression of Ngb but not of Cygb. Moreover, confocal microscopy immune-detection strikingly demonstrated nuclear localization of Mb in cell subsets expressing a high level of CD34, whereas Ngb was homogeneously distributed in the cytoplasm of all the HSPCs (unpublished data).
The dogma that Mb is exclusively present in cardiac or skeletal muscle of vertebrates has been recently challenged (Fraser et al., 2006; Roesner et al., 2008) and, notably, Mb expression has been found in a number of non-myogenic human tumours as well as in cancer-derived cell lines (Flonta et al., 2009; Kristiansen et al., 2010; Gorr et al., 2011; Oleksiewicz et al., 2011). Ngb is almost exclusively expressed in the nervous system and in the retina (Burmester et al., 2000; Reuss et al., 2002; Schmidt et al., 2003). However, in non-small cell lung cancer, simultaneous expression of Mb and Ngb was recently reported (Oleksiewicz et al., 2011).
From the differential O2-sensitive optical spectra reported in our study, it was possible to estimate an overall content of ≍0.01 nmol haemes per 106 HSPC corresponding to ≍180 ng globin per 106 HSPC. This value is significantly higher than the upper range reported in tumour samples and cancer-derived cell lines (Flonta et al., 2009; Kristiansen et al., 2010). As the cellular volume of CD34+-HSPC is about 400 μm3 (Sharma et al., 2008), an intracellular concentration of ≍25 μM can be estimated. This value is about one order of magnitude lower than that in muscular cells. However, considering the relatively large nucleus/cytoplasm volume ratio in HSPCs, the cytoplasmic concentration of the globins might be significant.
In myocytes, Mb is widely accepted to function as temporary ‘store’ for O2, able to buffer short phases of exercise-induced increases in O2 flux during which it supplies the gas to the mitochondria (Ordway and Garry, 2004). More controversial, is the role of Mb-mediated facilitated diffusion of O2 within muscle cells (Wittenberg, 1970; Jürgens et al., 1994). In addition to these canonical activities, Mb has been shown to participate in the NO homeostasis in muscle through either scavenging (Brunori and Gibson, 2001; Flögel et al., 2001) or producing the NO molecule (Hendgen-Cotta et al., 2008). Further possible functions of Mb in muscle include synthesis of peroxides (Khan et al., 1998), scavenging of ROS (Flögel et al., 2004) and binding of fatty acids (Sriram et al., 2008). Concerning Ngb, its exact physiological role is still debatable. Several proposed functions of Ngb overlap with those of Mb (Burmester and Hankeln, 2009). Ngb was also found to co-localize with mitochondria (Yu et al., 2012). Moreover, Ngb might be involved in a signal transduction pathway, for example, by inhibiting the dissociation of GDP from G protein α or might be part of a redox process that is, for example, instrumental in preventing apoptosis via reduction of cytochrome c.
The transcript analysis for Mb expression in HSPCs revealed, in addition to the standard mRNA expressed in muscle, the prevailing occurrence of an alternative transcript recently described in breast cancer cells (Kristiansen et al., 2010). In this alternative Mb mRNA, a distal HRE from the transcription start accounts for the HIF-1/2α-dependent up-regulation of Mb in hypoxic cancer cells (Kristiansen et al., 2011). In the case of Ngb, albeit its promoter apparently lacks the conventional HREs, nevertheless, a number of studies have clearly established that hypoxia and HIF-1 are necessary for the up-regulation of Ngb expression in the nervous system (Haines et al., 2012a, b). Accordingly, we found that the expression of Mb and Ngb are dependent on hypoxia and the stabilization of both HIF-1α and-2α in HSPCs (Piccoli et al., 2007c and unpublished data).
Normal and cancer stem cells share, to a certain extent, similar properties, such as the propensity to survive in hypoxic milieu, a predisposition towards a glycolysis-based metabolism, the ability to maintain a long-lasting quiescent undifferentiated state and to undergo self-renewing divisions (Reya et al., 2001; Li et al., 2006). Therefore, studies aimed at elucidating the role played by members of the globin family in cancer cell biology might provide clues to rationalize the functional role of Mb and Ngb in HSPC. In this context, results from a study wherein Mb-negative human lung carcinoma cells were engineered by lentiviral transfection to ectopically express mouse Mb are noteworthy (Galluzzo et al., 2009). The experimental Mb-expressing tumours displayed reduced or no hypoxia, minimal HIF-1α levels, decreased vessel density, a more differentiated cancer cell phenotype and a largely suppressed local and distal metastatic spreading. Although these beneficial outcomes in Mb-overexpressing tumours might result primarily from the reduction in tumour hypoxia, nevertheless, the relatively high levels of forced ectopic expression of Mb are not comparable with the low, picomolar, concentrations of Mb detected in cancer cells. Intriguingly, knock down of Mb in breast cancer cell lines resulted in an unexpected increase in O2 uptake and elevated activities of mitochondrial enzymes during hypoxia and silencing of Mb transcription attenuated proliferation rates and the motility of cancer cells under both hypoxic and fully oxygenated conditions (Kristiansen et al., 2011). To note, Cygb, another Mb-related globin detected in lung and breast cancer cells, was shown to exert a tumour-suppressor effect as its silencing in Cygb-positive cells resulted in increased colony formation, whereas induced Cygb expression in Cygb-negative cells reduced colony formation. All together, the evidence available in the literature converge to define a role for Mb as tumour suppressor, as well as for other members of the globin family (Shivapurkar et al., 2008), but seemingly not directly linked to the binding and transport of O2.
As discussed in the previous paragraph, the notion that hypoxia is a condition preventing oxidative stress is challenged by the repeatedly reported observation that under hypoxia cells produce more ROS than under normal O2 tension and that the source of such ‘hypoxic’ ROS is the mitochondrial RC complex III. However, the physiological mechanism of ROS production by the hypoxic RC still needs to be defined. Indeed, the low KM for O2 of the cytochrome c oxidase sets the environmental O2 concentration that effectively would limit the RC function to values below 1 μM (i.e. <0.1% pO2), which is unlikely to be reached in the BM endoblastic niche. Moreover, even if such a severe hypoxic condition occurred, the low concentration of O2 would limit ROS production, which is, in any case, a second-order reaction with single electron donors. Therefore, other mechanisms might contribute to inhibit cytochrome c oxidase and, at the same time, to account for a significant generation of ROS. Among those, the reversible NO inhibition of cytochrome c oxidase has been shown to be physiologically relevant in shifting ATP production from OXPHOS to glycolysis (Sarti et al., 2012a,b,c) as well as in eliciting signalling events, including the diversion of O2 to non-respiratory substrates and the generation of ROS (Erusalimsky and Moncada, 2007).
Combining these caveats and stimulated by our finding of globins in HSPCs, we would like to propose a model whereby Mb/Ngb provides, under hypoxic conditions, a source of NO by their hypoxia-dependent nitrite-reductase activity (Figure 2). NO is a potent competitive inhibitor of cytochrome c oxidase, especially under conditions of limited O2 availability (Sarti et al., 2012a). Thus, inhibition of the terminal part of the mitochondrial RC would elicit ROS formation at the complex III site under non-severe hypoxia conditions. Moreover, NO has also reported to be an inhibitor of complex I (Brown and Borutaite, 2004). Of note, nitrite-reductase activity has also been recently demonstrated for cytochrome c oxidase itself with production and auto-inhibition of NO (Ball et al., 2012). However, the low amount of cytochrome c oxidase in HSPC as compared with other cell types would be inadequate to guarantee an optimal generation of NO.
The process described above would contribute to the hypoxic stabilization of HIF-1/2α by the mechanism explained earlier, involving ROS- and NO-mediated inactivation of PDHs and FIH-1. The beneficial up-regulation of HIF-1, which, in addition to affecting HSPC metabolism, preserves the HSPC undifferentiated state, needs, however, to be reconciled with the known effect that redox signalling has on the cell cycle. Indeed, the main adaptive response of many cell types to a mild oxidative insult is activation of the cell cycle, enhanced proliferative activity and eventually differentiation (Noble et al., 2005; Burhans and Heintz, 2009; Finkel, 2011; Antico Arciuch et al., 2012; Chiu and Dawes, 2012). This would be detrimental in HSCPs if maintenance of the stemness profile is required. However, it is important to recall that redox signalling operates differentially in distinct intracellular compartments and oxidative modifications of nuclear located transcription factors are emerging as an important mechanism controlling gene expression (Go and Jones, 2010; Lukosz et al., 2010).
In this context, our finding that Mb specifically accumulates in the nucleus of CD34high-HSPC might be relevant to the cellular programme of multipotency maintenance. Conceivably, the Mb accumulated in the nucleus is not related to O2-storage/-facilitated diffusion, but rather suggests its involvement in the scavenging of reactive species to prevent nitro-oxidative modification of DNA or of redox-sensitive transcription of genes participating in the differentiation process (Ogasawara and Zhang, 2009; Owusu-Ansah and Banerjee, 2009; Brigelius-Flohé and Flohé, 2011; Kobayashi and Suda, 2012; Sardina et al., 2012). Interestingly, it has been shown that ROS-mediated oxidation of sensitive cysteine residues in PMLs controls their degradation and nuclear body biogenesis in acute promyelocytic leukaemia and BM progenitor cells (Jeanne et al., 2010). Moreover, Mb might interact directly with stem cell-specific nuclear transcription complexes. A recently reported MS-based analysis revealed a complex interatomic network of Mb with partners known to have antioxidant and anti-apoptotic functions, as well as with a number of nuclear proteins involved in DNA duplication/transcription/repair, chromatin stabilization, ribosomal assembly and nuclear import (Haines et al., 2012a, b). Importantly, many Mb-interactants overlapped with those of Ngb and were modified by hypoxia. Therefore, the nuclear localization of Mb that we observed in CD34high-HSPC might serve as a compartmentalized antioxidant buffer or as NF interactants to prevent unwanted activation of specialization genes, thereby preserving quiescence.
An alternative but consistent scenario could be that Ngb acts as a generator of cytoplasmic NO and Mb as a nuclear antioxidant or protein interactant. This would account for the occurrence of two different globins, which act in concert to fine-tune the level and intracellular compartmentalization of reactive species.
Once HSPCs move from the hypoxic osteoblastic niche to a more oxygenated milieu, for example, the vascular niche, this is perceived as an activating signal. Indeed, there is accumulating evidence to support the notion that, even in the absence of specific biochemical signals, changes in environmental physical and chemical parameters may cause the stem cell to egress from its quiescence. Changes in the intracellular redox state seemingly represent a signalling system that anticipates the growth/differentiation factor-mediated commitment and/or makes the stem cell more responsive to specific proliferative/differentiation stimuli. Assuming that this pertains to the CD34low-HSPC subtype, the re-localization of Mb thereof from the nucleus to the cytoplasm might be a way to enable reactive species to signal, at the nuclear level, proliferative/differentiation stimuli.
Given the kinetic features discussed previously, NOXs are well-suited to sense and signal by ROS production the change in O2 concentration when HSPCs move from the periosteal to the vascular niche. This is consistent with the documented primary role of NOX-generated ROS in fostering proliferation/differentiation of HSPCs. However, the contribution of mitochondria to ROS-production cannot be ruled out and the possibility that a ‘ROS-induced ROS release’ process and/or the shift towards a more active oxidative metabolism exists cannot be ruled out.
It should be noted that the nitrite-reductase activity of globins requires that they are in their deoxygenated form; therefore, under normoxia, a very low, if any, Mb/Ngb-mediated NO production is expected. Instead, both Mb and Ngb function under normoxia as dioxygenases, converting enhanced NOS-mediated NO production to nitrate (Sarti et al., 2012a), thereby unleashing the NO-dependent inhibition of the mitochondrial respiratory activity. This would result in stimulation of oxidative phosphorylation and related ATP production adapting the HSPC bioenergetic metabolism to cope with the larger energy demand required for proliferation/differentiation. Under this more bioenergetically active condition, the cytoplasmic localization of Mb and Ngb might also serve to smooth the intracellular O2 gradients nearby the peri-mitochondrial space and/or to transfer fatty acids.
Conclusions
Taken together, the growing body of accumulating evidence convincingly indicates that a complex interaction between ROS-mediated signalling and metabolic profile determines the fate of a cell. Stem cells and specifically HSCs are not an exception and exploit redox signalling to optimize the delicate balance between quiescence, self-renewal and commitment. What makes HSCs (and progenitors) different is that they exploit ROS to control both the maintenance of a quiescent condition and activation of self-renewal followed by proliferation and differentiation. This apparent paradox can be explained by the following: (i) the oxidizable targets may have a different sensitivity to ROS, so that a low ROS level would activate pathways suited to promote cell survival by a glycolysis-based metabolism, whereas higher ROS levels would activate different pathways leading to mitochondrial biogenesis and to more oxidative metabolism; (ii) the chemical properties of diverse reactive species (i.e. O2•−, H2O2 and OH•) are different and affect distinct targets; (iii) the source, stability and localization of ROS can contribute to the compartmentalization of the redox signalling; (iv) the interplay with other reactive species such as NO may add a further level of complexity and robustness of the signal transduction responsiveness; and (v) the cross-talk between upstream and downstream ROS-involving signal transduction pathways may generate negative or positive feedback loops resulting in different adaptive outcomes.
It is important to realize that redox signalling alone may not be sufficient to orchestrate the complex programme that regulates the fate of a stem cell, but rather may set the pre-commitment conditions allowing stem/progenitor cells to be more or less responsive to growth/differentiation factors. In this context, the control of ‘mitochondrial differentiation’ appears to be a checkpoint step that precedes and influences the stem cell fate.
Importantly, all the above-mentioned pathways are capable of being manipulated by specific pharmacological agonists or antagonists, paving the way to regulate the metabolic signature of HSPCs for future clinical applications.
Acknowledgments
This work is dedicated to the memory of Antonio Tabilio who enthusiastically inspired the Foggia group to this research issue. Supporting grants from Italian Ministry of University and Research (PRIN-2008FJJHKM_001) to N. C and Italian Association against Leukemia-Lymphoma and Myeloma (AIL-Foggia) to C. P. are acknowledged.
Glossary
- AMPK
AMP-activated PK
- BM
bone marrow
- FAO
fatty acid oxidation
- HIF-1
hypoxia inducible factor-1
- HRE
hypoxia response elements
- HSC
haematopoietic stem cell
- HSPC
haematopoietic stem/progenitor cell
- Lkb1
liver kinase B1
- LT-HSC
long-term HSC
- OXPHOS
mitochondrial oxidative phosphorylation
- NOX
NADPH oxidase
- PGC-1α
PPARγ co-activator 1α
- PML
promyelocytic leukaemia
- RC
respiratory chain
Conflict of interest
The authors have no conflicting financial interests.
References
- Abbas HA, Maccio DR, Coskun S, Jackson JG, Hazen AL, Sills TM, et al. Mdm2 is required for survival of hematopoietic stem cells/progenitors via dampening of ROS-induced p53 activity. Cell Stem Cell. 2010;7:606–617. doi: 10.1016/j.stem.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbas HA, Pant V, Lozano G. The ups and downs of p53 regulation in hematopoietic stem cells. Cell Cycle. 2011;10:3257–3262. doi: 10.4161/cc.10.19.17721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antico Arciuch VG, Elguero ME, Poderoso JJ, Carreras MC. Mitochondrial regulation of cell cycle and proliferation. Antioxid Redox Signal. 2012;16:1150–1180. doi: 10.1089/ars.2011.4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey AS, Willenbring H, Jiang S, Anderson DA, Schroeder DA, Wong MH. Myeloid lineage progenitors give rise to vascular endothelium. Proc Natl Acad Sci U S A. 2006;103:13156–13161. doi: 10.1073/pnas.0604203103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin MA, Benz CC. Redox control of zinc finger proteins. Methods Enzymol. 2002;353:54–69. doi: 10.1016/s0076-6879(02)53036-6. [DOI] [PubMed] [Google Scholar]
- Ball KA, Nelson AW, Foster DG, Poyton RO. Nitric oxide produced by cytochrome c oxidase helps stabilize HIF-1α in hypoxic mammalian cells. Biochem Biophys Res Commun. 2012;420:727–732. doi: 10.1016/j.bbrc.2012.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bavister BD. The mitochondrial contribution to stem cell biology. Reprod Fertil Dev. 2006;18:829–838. doi: 10.1071/rd06111. [DOI] [PubMed] [Google Scholar]
- Beksac M, Preffer F. Is it time to revisit our current hematopoietic progenitor cell quantification methods in the clinic? Bone Marrow Transplant. 2012;47:1391–1396. doi: 10.1038/bmt.2011.240. [DOI] [PubMed] [Google Scholar]
- Bell EL, Klimova TA, Eisenbart J, Moraes CT, Murphy MP, Budinger GR, et al. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J Cell Biol. 2007;177:1029–1036. doi: 10.1083/jcb.200609074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouysségur J, et al. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol. 2009;29:2570–2581. doi: 10.1128/MCB.00166-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berenson RJ, Andrews RG, Bensinger WI, Kalamasz D, Knitter G, Buckner CD, et al. Antigen CD34+ marrow cells engraft lethally irradiated baboons. J Clin Invest. 1988;81:951–955. doi: 10.1172/JCI113409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi R, Pandolfi PP. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat Rev Mol Cell Biol. 2007;8:1006–1016. doi: 10.1038/nrm2277. [DOI] [PubMed] [Google Scholar]
- Bhatia M, Wang JC, Kapp U, Bonnet D, Dick JE. Purification of primitive human hematopoietic cells capable of repopulating immune-deficient mice. Proc Natl Acad Sci U S A. 1997;94:5320–5325. doi: 10.1073/pnas.94.10.5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokoch GM, knaus UG. NADPH oxidases: not just for leukocytes anymore! Trends Biochem Sci. 2003;28:502–508. doi: 10.1016/S0968-0004(03)00194-4. [DOI] [PubMed] [Google Scholar]
- Bonello S, Zähringer C, BelAiba RS, Djordjevic T, Hess J, Michiels C, et al. Reactive oxygen species activate the HIF-1alpha promoter via a functional NFkappaB site. Arterioscler Thromb Vasc Biol. 2007;27:755–761. doi: 10.1161/01.ATV.0000258979.92828.bc. [DOI] [PubMed] [Google Scholar]
- Boonstra J, Post JA. Molecular events associated with reactive oxygen species and cell cycle progression in mammalian cells. Gene. 2004;337:1–13. doi: 10.1016/j.gene.2004.04.032. [DOI] [PubMed] [Google Scholar]
- Bradley TR, Hodgson GS, Rosendaal M. The effect of oxygen tension on haemopoietic and fibroblast cell proliferation in vitro. J Cell Physiol. 1978;97:517–522. doi: 10.1002/jcp.1040970327. [DOI] [PubMed] [Google Scholar]
- Brigelius-Flohé R, Flohé L. Basic principles and emerging concepts in the redox control of transcription factors. Antioxid Redox Signal. 2011;15:2335–2381. doi: 10.1089/ars.2010.3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown GC, Borutaite V. Inhibition of mitochondrial respiratory complex I by nitric oxide, peroxynitrite and S-nitrosothiols. Biochim Biophys Acta. 2004;1658:44–49. doi: 10.1016/j.bbabio.2004.03.016. [DOI] [PubMed] [Google Scholar]
- Brüne B, Zhou J. The role of nitric oxide (NO) in stability regulation of hypoxia inducible factor-1alpha (HIF-1alpha) Curr Med Chem. 2003;10:845–855. doi: 10.2174/0929867033457746. [DOI] [PubMed] [Google Scholar]
- Brunelle JK, Bell EL, Quesada NM, Vercauteren K, Tiranti V, Zeviani M, et al. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab. 2005;1:409–414. doi: 10.1016/j.cmet.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Brunori M, Gibson QH. Cavities and packing defects in the structural dynamics of myoglobin. EMBO Rep. 2001;2:674–679. doi: 10.1093/embo-reports/kve159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buggisch M, Ateghang B, Ruhe C, Strobel C, Lange S, Wartenberg M. Stimulation of ES-cell-derived cardiomyogenesis and neonatal cardiac cell proliferation by reactive oxygen species and NADPH oxidase. J Cell Sci. 2007;120:885–894. doi: 10.1242/jcs.03386. [DOI] [PubMed] [Google Scholar]
- Burhans WC, Heintz NH. The cell cycle is a redox cycle: linking phase-specific targets to cell fate. Free Radic Biol Med. 2009;7:1282–1293. doi: 10.1016/j.freeradbiomed.2009.05.026. [DOI] [PubMed] [Google Scholar]
- Burmester T, Hankeln TJ. What is the function of neuroglobin? Exp Biol. 2009;212:1423–1428. doi: 10.1242/jeb.000729. [DOI] [PubMed] [Google Scholar]
- Burmester T, Weich B, Reinhardt S, Hankeln T. A vertebrate globin expressed in the brain. Nature. 2000;407:520–523. doi: 10.1038/35035093. [DOI] [PubMed] [Google Scholar]
- Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, Bastidas N, Kleinman ME, et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med. 2004;10:858–864. doi: 10.1038/nm1075. [DOI] [PubMed] [Google Scholar]
- Chan SY, Zhang YY, Hemann C, Mahoney CE, Zweier JL, Loscalzo J. MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab. 2009;10:273–284. doi: 10.1016/j.cmet.2009.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci U S A. 1998;95:11715–11720. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, et al. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem. 2000;275:25130–25138. doi: 10.1074/jbc.M001914200. [DOI] [PubMed] [Google Scholar]
- Chen C, Liu Y, Liu R, Ikenoue T, Guan KL, Liu Y, et al. TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J Exp Med. 2008a;205:2397–2408. doi: 10.1084/jem.20081297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CT, Shih YR, Kuo TK, Lee OK, Wei YH. Coordinated changes of mitochondrial biogenesis and antioxidant enzymes during osteogenic differentiation of human mesenchymal stem cells. Stem Cells. 2008b;26:960–968. doi: 10.1634/stemcells.2007-0509. [DOI] [PubMed] [Google Scholar]
- Chen Y, Gill PS, Welch WJ. Oxygen availability limits renal NADPH-dependent superoxide production. Am J Physiol Renal Physiol. 2005;289:F749–F753. doi: 10.1152/ajprenal.00115.2005. [DOI] [PubMed] [Google Scholar]
- Cheng G, Cao Z, Xu X, van Meir EG, Lambeth JD. Homologs of gp91phox: cloning and tissue expression of Nox3, Nox4, and Nox5. Gene. 2001;269:131–140. doi: 10.1016/s0378-1119(01)00449-8. [DOI] [PubMed] [Google Scholar]
- Chiu J, Dawes IW. Redox control of cell proliferation. Trends Cell Biol. 2012;22:592–601. doi: 10.1016/j.tcb.2012.08.002. [DOI] [PubMed] [Google Scholar]
- Cho YM, Kwon S, Pak YK, Seol HW, Choi YM, Park do J, et al. Dynamic changes in mitochondrial biogenesis and antioxidant enzymes during the spontaneous differentiation of human embryonic stem cells. Biochem Biophys Res Commun. 2006;348:1472–1478. doi: 10.1016/j.bbrc.2006.08.020. [DOI] [PubMed] [Google Scholar]
- Chow DC, Wenning LA, Miller WM, Papoutsakis ET. Modeling pO(2) distributions in the bone marrow hematopoietic compartment. II. Modified Kroghian models. Biophys J. 2001;81:685–696. doi: 10.1016/S0006-3495(01)75733-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolleschi MG, Dello Sbarba P, Olivotto M. The role of hypoxia in the maintenance of hematopoietic stem cells. Blood. 1993;82:2031–2037. [PubMed] [Google Scholar]
- Civin CI, Trischmann T, Kadan NS, Davis J, Noga S, Cohen K, et al. Highly purified CD34-positive cells reconstitute hematopoiesis. J Clin Oncol. 1996;14:2224–2233. doi: 10.1200/JCO.1996.14.8.2224. [DOI] [PubMed] [Google Scholar]
- Covello KL, Kehler J, Yu H, Gordan JD, Arsham AM, Hu CJ, et al. HIF-2alpha regulates Oct-4: effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 2006;20:557–570. doi: 10.1101/gad.1399906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross AR, Segal AW. The NADPH oxidase of professional phagocytes – prototype of the NOX electron transport chain systems. Biochim Biophys Acta. 2004;1657:1–22. doi: 10.1016/j.bbabio.2004.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature. 2007;450:736–740. doi: 10.1038/nature06322. [DOI] [PubMed] [Google Scholar]
- Daiber A. Redox signaling (cross-talk) from and to mitochondria involves mitochondrial pores and reactive oxygen species. Biochim Biophys Acta. 2010;1797:897–906. doi: 10.1016/j.bbabio.2010.01.032. [DOI] [PubMed] [Google Scholar]
- Danet GH, Pan Y, Luongo JL, Bonnet DA, Simon MC. Expansion of human SCID-repopulating cells under hypoxic conditions. J Clin Invest. 2003;112:126–135. doi: 10.1172/JCI17669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikalov S. Cross talk between mitochondria and NADPH oxidases. Free Radic Biol Med. 2011;51:1289–1301. doi: 10.1016/j.freeradbiomed.2011.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doege K, Heine S, Jensen I, Jelkmann W, Metzen E. Inhibition of mitochondrial respiration elevates oxygen concentration but leaves regulation of hypoxia-inducible factor (HIF) intact. Blood. 2005;106:2311–2317. doi: 10.1182/blood-2005-03-1138. [DOI] [PubMed] [Google Scholar]
- Dooner GJ, Colvin GA, Dooner MS, Johnson KW, Quesenberry PJ. Gene expression fluctuations in murine hematopoietic stem cells with cell cycle progression. J Cell Physiol. 2008;214:786–795. doi: 10.1002/jcp.21273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dröge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- Dröse S, Brandt U. Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Adv Exp Med Biol. 2012;748:145–169. doi: 10.1007/978-1-4614-3573-0_6. [DOI] [PubMed] [Google Scholar]
- Eliasson P, Jönsson JI. The hematopoietic stem cell niche: low in oxygen but a nice place to be. J Cell Physiol. 2010;222:17–22. doi: 10.1002/jcp.21908. [DOI] [PubMed] [Google Scholar]
- Eliasson P, Rehn M, Hammar P, Larsson P, Sirenko O, Flippin LA, et al. Hypoxia mediates low cell-cycle activity and increases the proportion of long-term-reconstituting hematopoietic stem cells during in vitro culture. Exp Hematol. 2010;38:301–310. doi: 10.1016/j.exphem.2010.01.005. [DOI] [PubMed] [Google Scholar]
- Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O'Rourke J, Mole DR, et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107:43–54. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- Erusalimsky JD, Moncada S. Nitric oxide and mitochondrial signaling: from physiology to pathophysiology. Arterioscler Thromb Vasc Biol. 2007;27:2524–2531. doi: 10.1161/ATVBAHA.107.151167. [DOI] [PubMed] [Google Scholar]
- Fan J, Cai H, Tan WS. Role of the plasma membrane ROS-generating NADPH oxidase in CD34+ progenitor cells preservation by hypoxia. J Biotechnol. 2007;130:455–462. doi: 10.1016/j.jbiotec.2007.05.023. [DOI] [PubMed] [Google Scholar]
- Ferree A, Shirihai O. Mitochondrial dynamics: the intersection of form and function. Adv Exp Med Biol. 2012;748:13–40. doi: 10.1007/978-1-4614-3573-0_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T. Oxidant signals and oxidative stress. Curr Opin Cell Biol. 2003;15:247–254. doi: 10.1016/s0955-0674(03)00002-4. [DOI] [PubMed] [Google Scholar]
- Finkel T. Signal transduction by reactive oxygen species. J Cell Biol. 2011;194:7–15. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T. Signal transduction by mitochondrial oxidants. J Biol Chem. 2012;287:4434–4440. doi: 10.1074/jbc.R111.271999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flögel U, Merx MW, Godecke A, Decking UK, Schrader J. Myoglobin: a scavenger of bioactive NO. Proc Natl Acad Sci U S A. 2001;98:735–740. doi: 10.1073/pnas.011460298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flögel U, Gödecke A, Klotz LO, Schrader J. Role of myoglobin in the antioxidant defense of the heart. FASEB J. 2004;18:1156–1158. doi: 10.1096/fj.03-1382fje. [DOI] [PubMed] [Google Scholar]
- Flonta SE, Arena S, Pisacane A, Michieli P, Bardelli A. Expression and functional regulation of myoglobin in epithelial cancers. Am J Pathol. 2009;175:201–206. doi: 10.2353/ajpath.2009.081124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folmes CD, Dzeja PP, Nelson TJ, Terzic A. Metabolic plasticity in stem cell homeostasis and differentiation. Cell Stem Cell. 2012;11:596–606. doi: 10.1016/j.stem.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forristal CE, Wright KL, Hanley NA, Oreffo RO, Houghton FD. Hypoxia inducible factors regulate pluripotency and proliferation in human embryonic stem cells cultured at reduced oxygen tensions. Reproduction. 2010;139:85–97. doi: 10.1530/REP-09-0300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser J, de Mello LV, Ward D, Rees HH, Williams DR, Fang Y, et al. Hypoxia-inducible myoglobin expression in nonmuscle tissues. Proc Natl Acad Sci U S A. 2006;103:2977–2981. doi: 10.1073/pnas.0508270103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frassanito MC, Piccoli C, Capozzi V, Boffoli D, Tabilio A, Capitanio N. Topological organization of NADPH-oxidase in haematopoietic stem cell membrane: preliminary study by fluorescence near-field optical microscopy. J Microsc. 2008;229:517–524. doi: 10.1111/j.1365-2818.2008.01937.x. [DOI] [PubMed] [Google Scholar]
- Fukuda R, Zhang H, Kim JW, Shimoda L, Dang CV, Semenza GL. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007;129:111–122. doi: 10.1016/j.cell.2007.01.047. [DOI] [PubMed] [Google Scholar]
- Gabig TG, Babior BM. The O2(-)-forming oxidase responsible for the respiratory burst in human neutrophils. Properties of the solubilized enzyme. J Biol Chem. 1979;254:9070–9074. [PubMed] [Google Scholar]
- Galluzzo M, Pennacchietti S, Rosano S, Comoglio PM, Michieli P. Prevention of hypoxia by myoglobin expression in human tumor cells promotes differentiation and inhibits metastasis. J Clin Invest. 2009;119:865–875. doi: 10.1172/JCI36579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan B, Hu J, Jiang S, Liu Y, Sahin E, Zhuang L, et al. Lkb1 regulates quiescence and metabolic homeostasis of haematopoietic stem cells. Nature. 2010;468:701–704. doi: 10.1038/nature09595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerald D, Berra E, Frapart YM, Chan DA, Giaccia AJ, Mansuy D, et al. JunD reduces tumor angiogenesis by protecting cells from oxidative stress. Cell. 2004;118:781–794. doi: 10.1016/j.cell.2004.08.025. [DOI] [PubMed] [Google Scholar]
- Go YM, Jones DP. Redox control systems in the nucleus: mechanisms and functions. Antioxid Redox Signal. 2010;13:489–509. doi: 10.1089/ars.2009.3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Görlach A, Diebold I, Schini-Kerth VB, Berchner-Pfannschmidt U, Roth U, Brandes RP, et al. Thrombin activates the hypoxia-inducible factor-1 signaling pathway in vascular smooth muscle cells: role of the p22(phox)-containing NADPH oxidase. Circ Res. 2001;89:47–54. doi: 10.1161/hh1301.092678. [DOI] [PubMed] [Google Scholar]
- Gorr TA, Wichmann D, Pilarsky C, Theurillat JP, Fabrizius A, Laufs T, et al. Old proteins – new locations: myoglobin, haemoglobin, neuroglobin and cytoglobin in solid tumours and cancer cells. Acta Physiol (Oxf) 2011;202:563–581. doi: 10.1111/j.1748-1716.2010.02205.x. [DOI] [PubMed] [Google Scholar]
- Goyal P, Weissmann N, Grimminger F, Hegel C, Bader L, Rose F. Upregulation of NAD(P)H oxidase 1 in hypoxia activates hypoxia-inducible factor 1 via increase in reactive oxygen species. Free Radic Biol Med. 2004;36:1279–1288. doi: 10.1016/j.freeradbiomed.2004.02.071. [DOI] [PubMed] [Google Scholar]
- Gurumurthy S, Xie SZ, Alagesan B, Kim J, Yusuf RZ, Saez B, et al. The Lkb1 metabolic sensor maintains haematopoietic stem cell survival. Nature. 2010;468:659–663. doi: 10.1038/nature09572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson MV, Zheng X, Pereira T, Gradin K, Jin S, Lundkvist J, et al. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev Cell. 2005;9:617–628. doi: 10.1016/j.devcel.2005.09.010. [DOI] [PubMed] [Google Scholar]
- Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, et al. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005;1:401–408. doi: 10.1016/j.cmet.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Hagen T, Taylor CT, Lam F, Moncada S. Redistribution of intracellular oxygen in hypoxia by nitric oxide: effect on HIF1alpha. Science. 2003;302:1975–1978. doi: 10.1126/science.1088805. [DOI] [PubMed] [Google Scholar]
- Haines B, Demaria M, Mao X, Xie L, Campisi J, Jin K, et al. Hypoxia-inducible factor-1 and neuroglobin expression. Neurosci Lett. 2012a;514:137–140. doi: 10.1016/j.neulet.2012.01.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haines BA, Davis DA, Zykovich A, Peng B, Rao R, Mooney SD, et al. Comparative protein interactomics of neuroglobin and myoglobin. J Neurochem. 2012b;123:192–198. doi: 10.1111/j.1471-4159.2012.07881.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamanaka RB, Chandel NS. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem Sci. 2010;35:505–513. doi: 10.1016/j.tibs.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handgretinger R, Gordon PR, Leimig T, Chen X, Buhring HJ, Niethammer D, et al. Biology and plasticity of CD133+ hematopoietic stem cells. Ann N Y Acad Sci. 2003;996:141–151. doi: 10.1111/j.1749-6632.2003.tb03242.x. [DOI] [PubMed] [Google Scholar]
- Hankey PA. Regulation of hematopoietic cell development and function by Stat3. Front Biosci. 2009;14:5273–5290. doi: 10.2741/3597. [DOI] [PubMed] [Google Scholar]
- Hannig M, Figulla HR, Sauer H, Wartenberg M. Control of leucocyte differentiation from embryonic stem cells upon vasculogenesis and confrontation with tumour tissue. J Cell Mol Med. 2010;14:303–312. doi: 10.1111/j.1582-4934.2008.00424.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison JS, Rameshwar P, Chang V, Bandari P. Oxygen saturation in the bone marrow of healthy volunteers. Blood. 2002;99:394. doi: 10.1182/blood.v99.1.394. [DOI] [PubMed] [Google Scholar]
- Hendgen-Cotta UB, Merx MW, Shiva S, Schmitz J, Becher S, Klare JP, et al. Nitrite reductase activity of myoglobin regulates respiration and cellular viability in myocardial ischemia-reperfusion injury. Proc Natl Acad Sci U S A. 2008;105:10256–10261. doi: 10.1073/pnas.0801336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermitte F, Brunet de la Grange P, Belloc F, Praloran V, Ivanovic Z. Very low O2 concentration (0.1%) favors G0 return of dividing CD34+ cells. Stem Cells. 2006;24:65–73. doi: 10.1634/stemcells.2004-0351. [DOI] [PubMed] [Google Scholar]
- Hirsilä M, Koivunen P, Günzler V, Kivirikko KI, Myllyharju J. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. J Biol Chem. 2003;278:30772–30780. doi: 10.1074/jbc.M304982200. [DOI] [PubMed] [Google Scholar]
- Hole PS, Pearn L, Tonks AJ, James PE, Burnett AK, Darley RL, et al. Ras-induced reactive oxygen species promote growth factor-independent proliferation in human CD34+ hematopoietic progenitor cells. Blood. 2010;115:1238–1246. doi: 10.1182/blood-2009-06-222869. [DOI] [PubMed] [Google Scholar]
- Hsu PP, Sabatini DM. Cancer cell metabolism: Warburg and beyond. Cell. 2008;134:703–707. doi: 10.1016/j.cell.2008.08.021. [DOI] [PubMed] [Google Scholar]
- Iiyama M, Kakihana K, Kurosu T, Miura O. Reactive oxygen species generated by hematopoietic cytokines play roles in activation of receptor-mediated signaling and in cell cycle progression. Cell Signal. 2006;18:174–182. doi: 10.1016/j.cellsig.2005.04.002. [DOI] [PubMed] [Google Scholar]
- Ilbert M, Graf PC, Jakob U. Zinc center as redox switch – new function for an old motif. Antioxid Redox Signal. 2006;8:835–846. doi: 10.1089/ars.2006.8.835. [DOI] [PubMed] [Google Scholar]
- Ito K, Hirao A, Arai F, Matsuoka S, Takubo K, Hamaguchi I, et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature. 2004;431:997–1002. doi: 10.1038/nature02989. [DOI] [PubMed] [Google Scholar]
- Ito K, Hirao A, Arai F, Takubo K, Matsuoka S, Miyamoto K, et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat Med. 2006;12:446–451. doi: 10.1038/nm1388. [DOI] [PubMed] [Google Scholar]
- Ito K, Bernardi R, Morotti A, Matsuoka S, Saglio G, Ikeda Y, et al. PML targeting eradicates quiescent leukaemia-initiating cells. Nature. 2008;453:1072–1078. doi: 10.1038/nature07016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Carracedo A, Weiss D, Arai F, Ala U, Avigan DE, et al. A PML–PPAR-δ pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat Med. 2012;18:1350–1358. doi: 10.1038/nm.2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanovic Z, Hermitte F, Brunet de la Grange P, Dazey B, Belloc F, Lacombe F, et al. Simultaneous maintenance of human cord blood SCID-repopulating cells and expansion of committed progenitors at low O2 concentration (3%) Stem Cells. 2004;22:716–724. doi: 10.1634/stemcells.22-5-716. [DOI] [PubMed] [Google Scholar]
- Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998;12:149–162. doi: 10.1101/gad.12.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang YY, Sharkis SJ. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood. 2007;110:3056–3063. doi: 10.1182/blood-2007-05-087759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeanne M, Lallemand-Breitenbach V, Ferhi O, Koken M, Le Bras M, Duffort S, et al. PML/RARA oxidation and arsenic binding initiate the antileukemia response of As2O3. Cancer Cell. 2010;18:88–98. doi: 10.1016/j.ccr.2010.06.003. [DOI] [PubMed] [Google Scholar]
- Juntilla MM, Patil VD, Calamito M, Joshi RP, Birnbaum MJ, Koretzky GA. AKT1 and AKT2 maintain hematopoietic stem cell function by regulating reactive oxygen species. Blood. 2010;115:4030–4038. doi: 10.1182/blood-2009-09-241000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jürgens KD, Peters T, Gros G. Diffusivity of myoglobin in intact skeletal muscle cells. Proc Natl Acad Sci U S A. 1994;91:3829–3833. doi: 10.1073/pnas.91.9.3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin WG, Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- Kanda Y, Hinata T, Kang SW, Watanabe Y. Reactive oxygen species mediate adipocyte differentiation in mesenchymal stem cells. Life Sci. 2011;89:250–258. doi: 10.1016/j.lfs.2011.06.007. [DOI] [PubMed] [Google Scholar]
- Katahira J, Mizoguchi H. Improvement of culture conditions for human megakaryocytic and pluripotent progenitor cells by low oxygen tension. Int J Cell Cloning. 1987;5:412–420. doi: 10.1002/stem.5530050506. [DOI] [PubMed] [Google Scholar]
- Khan KK, Mondal MS, Padhy L, Mitra S. The role of distal histidine in peroxidase activity of myoglobin – transient-kinetics study of the reaction of H2O2 with wild-type and distal-histidine-mutanted recombinant human myoglobin. Eur J Biochem. 1998;257:547–555. doi: 10.1046/j.1432-1327.1998.2570547.x. [DOI] [PubMed] [Google Scholar]
- Kharas MG, Gritsman K. Akt: a double-edged sword for hematopoietic stem cells. Cell Cycle. 2010;9:1223–1224. doi: 10.4161/cc.9.7.11362. [DOI] [PubMed] [Google Scholar]
- Kharas MG, Okabe R, Ganis JJ, Gozo M, Khandan T, Paktinat M, et al. Constitutively active AKT depletes hematopoietic stem cells and induces leukemia in mice. Blood. 2010;115:1406–1415. doi: 10.1182/blood-2009-06-229443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiel MJ, Iwashita T, Yilmaz OH, Morrison SJ. Spatial differences in hematopoiesis but not in stem cells indicate a lack of regional patterning in definitive hematopoietic stem cells. Dev Biol. 2005;283:29–39. doi: 10.1016/j.ydbio.2005.03.037. [DOI] [PubMed] [Google Scholar]
- Kietzmann T, Görlach A. Reactive oxygen species in the control of hypoxia-inducible factor-mediated gene expression. Semin Cell Dev Biol. 2005;16:474–486. doi: 10.1016/j.semcdb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Kim CH, Cho YS, Chun YS, Park JW, Kim MS. Early expression of myocardial HIF-1alpha in response to mechanical stresses: regulation by stretch-activated channels and the phosphatidylinositol 3-kinase signaling pathway. Circ Res. 2002;90:E25–E33. doi: 10.1161/hh0202.104923. [DOI] [PubMed] [Google Scholar]
- Kim M, Cooper DD, Hayes SF, Spangrude GJ. Rhodamine-123 staining in hematopoietic stem cells of young mice indicates mitochondrial activation rather than dye efflux. Blood. 1998;9:4106–4117. [PubMed] [Google Scholar]
- Knaän-Shanzer S, van der Velde-van Dijke I, van de Watering MJ, de Leeuw PJ, Valerio D, van Bekkum DW, et al. Phenotypic and functional reversal within the early human hematopoietic compartment. Stem Cells. 2008;26:3210–3217. doi: 10.1634/stemcells.2007-0117. [DOI] [PubMed] [Google Scholar]
- Kobayashi CI, Suda T. Regulation of reactive oxygen species in stem cells and cancer stem cells. J Cell Physiol. 2012;227:421–430. doi: 10.1002/jcp.22764. [DOI] [PubMed] [Google Scholar]
- Koh MY, Powis G. Passing the baton: the HIF switch. Trends Biochem Sci. 2012;37:364–372. doi: 10.1016/j.tibs.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh MY, Darnay BG, Powis G. Hypoxia-associated factor, a novel E3-ubiquitin ligase, binds and ubiquitinates hypoxia-inducible factor 1alpha, leading to its oxygen-independent degradation. Mol Cell Biol. 2008;28:7081–7095. doi: 10.1128/MCB.00773-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koivunen P, Hirsilä M, Günzler V, Kivirikko KI, Myllyharju J. Catalytic properties of the asparaginyl hydroxylase (FIH) in the oxygen sensing pathway are distinct from those of its prolyl 4-hydroxylases. J Biol Chem. 2004;279:9899–9904. doi: 10.1074/jbc.M312254200. [DOI] [PubMed] [Google Scholar]
- Koller MR, Bender JG, Miller WM, Papoutsakis ET. Reduced oxygen tension increases hematopoiesis in long-term culture of human stem and progenitor cells from cord blood and bone marrow. Exp Hematol. 1992;20:264–270. [PubMed] [Google Scholar]
- Koutna I, Peterkova M, Simara P, Stejskal S, Tesarova L, Kozubek M. Proliferation and differentiation potential of CD133+ and CD34+ populations from the bone marrow and mobilized peripheral blood. Ann Hematol. 2011;90:127–137. doi: 10.1007/s00277-010-1058-2. [DOI] [PubMed] [Google Scholar]
- Krause DS, Fackler MJ, Civin CI, May WS. CD34: structure, biology, and clinical utility. Blood. 1996;87:1–13. [PubMed] [Google Scholar]
- Kristiansen G, Rose M, Geisler C, Fritzsche FR, Gerhardt J, Lüke C, et al. Endogenous myoglobin in human breast cancer is a hallmark of luminal cancer phenotype. Br J Cancer. 2010;102:1736–1745. doi: 10.1038/sj.bjc.6605702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristiansen G, Hu J, Wichmann D, Stiehl DP, Rose M, Gerhardt J, et al. Endogenous myoglobin in breast cancer is hypoxia-inducible by alternative transcription and functions to impair mitochondrial activity: a role in tumor suppression? J Biol Chem. 2011;286:43417–43428. doi: 10.1074/jbc.M111.227553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota Y, Takubo K, Suda T. Bone marrow long label-retaining cells reside in the sinusoidal hypoxic niche. Biochem Biophys Res Commun. 2008;366:335–339. doi: 10.1016/j.bbrc.2007.11.086. [DOI] [PubMed] [Google Scholar]
- Kuroiwa T, Ohta T, Kuroiwa H, Shigeyuki K. Molecular and cellular mechanisms of mitochondrial nuclear division and mitochondriokinesis. Microsc Res Tech. 1994;27:220–232. doi: 10.1002/jemt.1070270304. [DOI] [PubMed] [Google Scholar]
- LaIuppa JA, Papoutsakis ET, Miller WM. Oxygen tension alters the effects of cytokines on the megakaryocyte, erythrocyte, and granulocyte lineages. Exp Hematol. 1998;26:835–843. [PubMed] [Google Scholar]
- Lambeth JD, Kawahara T, Diebold B. Regulation of Nox and Duox enzymatic activity and expression. Free Radic Biol Med. 2007;43:319–331. doi: 10.1016/j.freeradbiomed.2007.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee NK, Choi YG, Baik JY, Han SY, Jeong DW, Bae YS, et al. A crucial role for reactive oxygen species in RANKL-induced osteoclast differentiation. Blood. 2005;106:852–859. doi: 10.1182/blood-2004-09-3662. [DOI] [PubMed] [Google Scholar]
- Lenaz G. Mitochondria and reactive oxygen species. Which role in physiology and pathology? Adv Exp Med Biol. 2012;942:93–136. doi: 10.1007/978-94-007-2869-1_5. [DOI] [PubMed] [Google Scholar]
- Lessard J, Sauvageau G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature. 2003;423:255–260. doi: 10.1038/nature01572. [DOI] [PubMed] [Google Scholar]
- Lewandowski D, Barroca V, Ducongé F, Bayer J, Van Nhieu JT, Pestourie C, et al. In vivo cellular imaging pinpoints the role of reactive oxygen species in the early steps of adult hematopoietic reconstitution. Blood. 2010;115:443–452. doi: 10.1182/blood-2009-05-222711. [DOI] [PubMed] [Google Scholar]
- Li J, Stouffs M, Serrander L, Banfi B, Bettiol E, Charnay Y, et al. The NADPH oxidase NOX4 drives cardiac differentiation: role in regulating cardiac transcription factors and MAP kinase activation. Mol Biol Cell. 2006;17:3978–3988. doi: 10.1091/mbc.E05-06-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Cao L, Chen J, Song S, Lee IH, Quijano C, et al. Bmi1 regulates mitochondrial function and the DNA damage response pathway. Nature. 2009;459:387–392. doi: 10.1038/nature08040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Celso C, Fleming HE, Wu JW, Zhao CX, Miake-Lye S, Fujisaki J, et al. Live-animal tracking of individual haematopoietic stem/progenitor cells in their niche. Nature. 2009;457:92–96. doi: 10.1038/nature07434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonergan T, Brenner C, Bavister B. Differentiation-related changes in mitochondrial properties as indicators of stem cell competence. J Cell Physiol. 2006;208:149–153. doi: 10.1002/jcp.20641. [DOI] [PubMed] [Google Scholar]
- Lukosz M, Jakob S, Büchner N, Zschauer TC, Altschmied J, Haendeler J. Nuclear redox signaling. Antioxid Redox Signal. 2010;12:713–742. doi: 10.1089/ars.2009.2609. [DOI] [PubMed] [Google Scholar]
- Macarthur BD, Ma'ayan A, Lemischka IR. Systems biology of stem cell fate and cellular reprogramming. Nat Rev Mol Cell Biol. 2009;10:672–681. doi: 10.1038/nrm2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie JL, Takenaka K, Gan OI, Doedens M, Dick JE. Low rhodamine 123 retention identifies long-term human hematopoietic stem cells within the Lin-CD34+CD38- population. Blood. 2007;109:543–545. doi: 10.1182/blood-2006-06-030270. [DOI] [PubMed] [Google Scholar]
- Mansfield KD, Guzy RD, Pan Y, Young RM, Cash TP, Schumacker PT, et al. Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-alpha activation. Cell Metab. 2005;1:393–399. doi: 10.1016/j.cmet.2005.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantel C, Broxmeyer HE. Sirtuin 1, stem cells, aging, and stem cell aging. Curr Opin Hematol. 2008;15:326–331. doi: 10.1097/MOH.0b013e3283043819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantel C, Messina-Graham S, Broxmeyer HE. Upregulation of nascent mitochondrial biogenesis in mouse hematopoietic stem cells parallels upregulation of CD34 and loss of pluripotency: a potential strategy for reducing oxidative risk in stem cells. Cell Cycle. 2010;9:2008–2017. doi: 10.4161/cc.9.10.11733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantel C, Messina-Graham SV, Broxmeyer HE. Superoxide flashes, reactive oxygen species, and the mitochondrial permeability transition pore: potential implications for hematopoietic stem cell function. Curr Opin Hematol. 2011;18:208–213. doi: 10.1097/MOH.0b013e3283475ffe. [DOI] [PubMed] [Google Scholar]
- Mantel C, Messina-Graham S, Moh A, Cooper S, Hangoc G, Fu XY, et al. Mouse hematopoietic cell-targeted STAT3 deletion: stem/progenitor cell defects, mitochondrial dysfunction, ROS overproduction, and a rapid aging-like phenotype. Blood. 2012;120:2589–2599. doi: 10.1182/blood-2012-01-404004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maranchie JK, Zhan Y. Nox4 is critical for hypoxia-inducible factor 2-alpha transcriptional activity in von Hippel-Lindau-deficient renal cell carcinoma. Cancer Res. 2005;65:9190–9193. doi: 10.1158/0008-5472.CAN-05-2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maryanovich M, Gross A. A ROS rheostat for cell fate regulation. Trends Cell Biol. 2012;23:128–134. doi: 10.1016/j.tcb.2012.09.007. [DOI] [PubMed] [Google Scholar]
- Maryanovich M, Oberkovitz G, Niv H, Vorobiyov L, Zaltsman Y, Brenner O, et al. The ATM-BID pathway regulates quiescence and survival of haematopoietic stem cells. Nat Cell Biol. 2012;14:535–541. doi: 10.1038/ncb2468. [DOI] [PubMed] [Google Scholar]
- May-Panloup P, Vignon X, Chrétien MF, Heyman Y, Tamassia M, Malthièry Y, et al. Increase of mitochondrial DNA content and transcripts in early bovine embryogenesis associated with upregulation of mtTFA and NRF1 transcription factors. Reprod Biol Endocrinol. 2005;3:65. doi: 10.1186/1477-7827-3-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazumdar J, O'Brien WT, Johnson RS, LaManna JC, Chavez JC, Klein PS, et al. O2 regulates stem cells through Wnt/β-catenin signalling. Nat Cell Biol. 2010;12:1007–1013. doi: 10.1038/ncb2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalik L, Wahli WJ. Involvement of PPAR nuclear receptors in tissue injury and wound repair. J Clin Invest. 2006;116:598–606. doi: 10.1172/JCI27958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miharada K, Karlsson G, Rehn M, Rörby E, Siva K, Cammenga J, et al. Cripto regulates hematopoietic stem cells as a hypoxic-niche-related factor through cell surface receptor GRP78. Cell Stem Cell. 2011;9:330–344. doi: 10.1016/j.stem.2011.07.016. [DOI] [PubMed] [Google Scholar]
- Miyamoto K, Araki KY, Naka K, Arai F, Takubo K, Yamazaki S, et al. Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell. 2007;1:101–112. doi: 10.1016/j.stem.2007.02.001. [DOI] [PubMed] [Google Scholar]
- Mortensen M, Soilleux EJ, Djordjevic G, Tripp R, Lutteropp M, Sadighi-Akha E, et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J Exp Med. 2011;208:455–467. doi: 10.1084/jem.20101145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller FL, Liu Y, Van Remmen H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J Biol Chem. 2004;279:49064–49073. doi: 10.1074/jbc.M407715200. [DOI] [PubMed] [Google Scholar]
- Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakada D, Saunders TL, Morrison SJ. Lkb1 regulates cell cycle and energy metabolism in haematopoietic stem cells. Nature. 2010;468:653–658. doi: 10.1038/nature09571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan CJ. Specificity of a third kind: reactive oxygen and nitrogen intermediates in cell signaling. J Clin Invest. 2003;111:769–778. doi: 10.1172/JCI18174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesti C, Pasquali L, Vaglini F, Siciliano G, Murri L. The role of mitochondria in stem cell biology. Biosci Rep. 2007;27:165–171. doi: 10.1007/s10540-007-9044-1. [DOI] [PubMed] [Google Scholar]
- Noble M, Mayer-Pröschel M, Pröschel C. Redox regulation of precursor cell function: insights and paradoxes. Antioxid Redox Signal. 2005;7:1456–1467. doi: 10.1089/ars.2005.7.1456. [DOI] [PubMed] [Google Scholar]
- Norddahl GL, Pronk CJ, Wahlestedt M, Sten G, Nygren JM, Ugale A, et al. Accumulating mitochondrial DNA mutations drive premature hematopoietic aging phenotypes distinct from physiological stem cell aging. Cell Stem Cell. 2011;8:499–510. doi: 10.1016/j.stem.2011.03.009. [DOI] [PubMed] [Google Scholar]
- Novak I. Mitophagy: a complex mechanism of mitochondrial removal. Antioxid Redox Signal. 2012;17:794–802. doi: 10.1089/ars.2011.4407. [DOI] [PubMed] [Google Scholar]
- Ogasawara MA, Zhang H. Redox regulation and its emerging roles in stem cells and stem-like cancer cells. Antioxid Redox Signal. 2009;11:1107–1122. doi: 10.1089/ars.2008.2308. [DOI] [PubMed] [Google Scholar]
- Oguro H, Iwama A, Morita Y, Kamijo T, van Lohuizen M, Nakauchi H. Differential impact of Ink4a and Arf on hematopoietic stem cells and their bone marrow microenvironment in Bmi1-deficient mice. J Exp Med. 2006;203:2247–2253. doi: 10.1084/jem.20052477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleksiewicz U, Daskoulidou N, Liloglou T, Tasopoulou K, Bryan J, Gosney JR, et al. Neuroglobin and myoglobin in non-small cell lung cancer: expression, regulation and prognosis. Lung Cancer. 2011;74:411–418. doi: 10.1016/j.lungcan.2011.05.001. [DOI] [PubMed] [Google Scholar]
- Ordway GA, Garry DJ. Myoglobin: an essential hemoprotein in striated muscle. J Exp Biol. 2004;207:3441–3446. doi: 10.1242/jeb.01172. [DOI] [PubMed] [Google Scholar]
- Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 2008;132:631–644. doi: 10.1016/j.cell.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owusu-Ansah E, Banerjee U. Reactive oxygen species prime Drosophila haematopoietic progenitors for differentiation. Nature. 2009;461:537–541. doi: 10.1038/nature08313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papa S, Martino PL, Capitanio G, Gaballo A, De Rasmo D, Signorile A, et al. The oxidative phosphorylation system in mammalian mitochondria. Adv Exp Med Biol. 2012;942:3–37. doi: 10.1007/978-94-007-2869-1_1. [DOI] [PubMed] [Google Scholar]
- Park IK, Qian D, Kiel M, Becker MW, Pihalja M, Weissman IL, et al. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 2003;423:302–305. doi: 10.1038/nature01587. [DOI] [PubMed] [Google Scholar]
- Parmar K, Mauch P, Vergilio JA, Sackstein R, Down JD. Distribution of hematopoietic stem cells in the bone marrow according to regional hypoxia. Proc Natl Acad Sci U S A. 2007;104:5431–5436. doi: 10.1073/pnas.0701152104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendyala S, Natarajan V. Redox regulation of Nox proteins. Respir Physiol Neurobiol. 2010;174:265–271. doi: 10.1016/j.resp.2010.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petry A, Weitnauer M, Görlach A. Receptor activation of NADPH oxidases. Antioxid Redox Signal. 2010;13:467–487. doi: 10.1089/ars.2009.3026. [DOI] [PubMed] [Google Scholar]
- Piccoli C, Ria R, Scrima R, Cela O, D'Aprile A, Boffoli D, et al. Characterization of mitochondrial and extra-mitochondrial oxygen consuming reactions in human hematopoietic stem cells. Novel evidence of the occurrence of NAD(P)H oxidase activity. J Biol Chem. 2005;280:26467–26476. doi: 10.1074/jbc.M500047200. [DOI] [PubMed] [Google Scholar]
- Piccoli C, D'Aprile A, Scrima R, Ripoli M, Boffoli D, Tabilio A, et al. Role of reactive oxygen species as signal molecules in the pre-commitment phase of adult stem cells. Ital J Biochem. 2007a;56:295–301. [PubMed] [Google Scholar]
- Piccoli C, D'Aprile A, Ripoli M, Scrima R, Lecce L, Boffoli D, et al. Bone-marrow derived hematopoietic stem/progenitor cells express multiple isoforms of NADPH oxidase and produce constitutively reactive oxygen species. Biochem Biophys Res Commun. 2007b;353:965–972. doi: 10.1016/j.bbrc.2006.12.148. [DOI] [PubMed] [Google Scholar]
- Piccoli C, D'Aprile A, Ripoli M, Scrima R, Boffoli D, Tabilio A, et al. The hypoxia-inducible factor is stabilized in circulating hematopoietic stem cells under normoxic conditions. FEBS Lett. 2007c;581:3111–3119. doi: 10.1016/j.febslet.2007.05.077. [DOI] [PubMed] [Google Scholar]
- Pikó L, Taylor KD. Amounts of mitochondrial DNA and abundance of some mitochondrial gene transcripts in early mouse embryos. Dev Biol. 1987;123:364–374. doi: 10.1016/0012-1606(87)90395-2. [DOI] [PubMed] [Google Scholar]
- Piro D, Piccoli C, Guerra L, Sassone F, D'Aprile A, Favia M, et al. Hematopoietic stem/progenitor cells express functional mitochondrial energy-dependent cystic fibrosis transmembrane conductance regulator. Stem Cells Dev. 2012;21:634–646. doi: 10.1089/scd.2011.0041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouysségur J, Mechta-Grigoriou F. Redox regulation of the hypoxia-inducible factor. Biol Chem. 2006;387:1337–1346. doi: 10.1515/BC.2006.167. [DOI] [PubMed] [Google Scholar]
- Powell TM, Paul JD, Hill JM, Thompson M, Benjamin M, Rodrigo M, et al. Granulocyte colony-stimulating factor mobilizes functional endothelial progenitor cells in patients with coronary artery disease. Arterioscler Thromb Vasc Biol. 2005;25:296–301. doi: 10.1161/01.ATV.0000151690.43777.e4. [DOI] [PubMed] [Google Scholar]
- Puca A, Russo G, Romano G, Giordano A. Chaotic dynamic stabilities and instabilities of hematopoietic stem cell growth plasticity. J Cell Physiol. 2012;213:672–678. doi: 10.1002/jcp.21181. [DOI] [PubMed] [Google Scholar]
- Ralph SJ, Rodríguez-Enríquez S, Neuzil J, Saavedra E, Moreno-Sánchez R. The causes of cancer revisited: ‘mitochondrial malignancy’ and ROS-induced oncogenic transformation – why mitochondria are targets for cancer therapy. Mol Aspects Med. 2010;31:145–170. doi: 10.1016/j.mam.2010.02.008. [DOI] [PubMed] [Google Scholar]
- Rehman JJ. Empowering self-renewal and differentiation: the role of mitochondria in stem cells. Mol Med (Berl) 2010;88:981–986. doi: 10.1007/s00109-010-0678-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuss S, Saaler-Reinhardt S, Weich B, Wystub S, Reuss MH, Burmester T, et al. Expression analysis of neuroglobin mRNA in rodent tissues. Neuroscience. 2002;115:645–656. doi: 10.1016/s0306-4522(02)00536-5. [DOI] [PubMed] [Google Scholar]
- Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- Rigoulet M, Yoboue ED, Devin A. Mitochondrial ROS generation and its regulation: mechanisms involved in H(2)O(2) signaling. Antioxid Redox Signal. 2011;14:459–468. doi: 10.1089/ars.2010.3363. [DOI] [PubMed] [Google Scholar]
- Robin ED, Wong R. Mitochondrial DNA molecules and virtual number of mitochondria per cell in mammalian cells. J Cell Physiol. 1988;136:507–513. doi: 10.1002/jcp.1041360316. [DOI] [PubMed] [Google Scholar]
- Roesner A, Mitz SA, Hankeln T, Burmester T. Globins and hypoxia adaptation in the goldfish, Carassius auratus. FEBS J. 2008;275:3633–3643. doi: 10.1111/j.1742-4658.2008.06508.x. [DOI] [PubMed] [Google Scholar]
- Romero-Moya D, Bueno C, Montes R, Navarro-Montero O, Iborra FJ, López LC, et al. Cord blood-derived CD34+ hematopoietic cells with low levels of mitochondrial mass are enriched in hematopoietic repopulating stem cell function. Haematologica. 2013 doi: 10.3324/haematol.2012.079244. doi: 10.3324/haematol.2012.079244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakabe H, Ohmizono Y, Tanimukai S, Kimura T, Mori KJ, Abe T, et al. Functional differences between subpopulations of mobilized peripheral blood-derived CD34+ cells expressing different levels of HLA-DR, CD33, CD38 and c-kit antigens. Stem Cells. 1997;15:73–81. doi: 10.1002/stem.150073. [DOI] [PubMed] [Google Scholar]
- Sardina JL, López-Ruano G, Sánchez-Sánchez B, Llanillo M, Hernández-Hernández A. Reactive oxygen species: are they important for haematopoiesis? Crit Rev Oncol Hematol. 2012;81:257–274. doi: 10.1016/j.critrevonc.2011.03.005. [DOI] [PubMed] [Google Scholar]
- Saretzki G, Armstrong L, Leake A, Lako M, von Zglinicki T. Stress defense in murine embryonic stem cells is superior to that of various differentiated murine cells. Stem Cells. 2004;22:962–971. doi: 10.1634/stemcells.22-6-962. [DOI] [PubMed] [Google Scholar]
- Sarti P, Forte E, Giuffrè A, Mastronicola D, Magnifico MC, Arese M. The chemical interplay between nitric oxide and mitochondrial cytochrome c oxidase: reactions, effectors and pathophysiology. Int J Cell Biol. 2012a doi: 10.1155/2012/571067. doi: 10.1155/2012/571067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarti P, Forte E, Mastronicola D, Giuffrè A, Arese M. Cytochrome c oxidase and nitric oxide in action: molecular mechanisms and pathophysiological implications. Biochim Biophys Acta. 2012b;1817:610–619. doi: 10.1016/j.bbabio.2011.09.002. [DOI] [PubMed] [Google Scholar]
- Sarti P, Arese M, Forte E, Giuffrè A, Mastronicola D. Mitochondria and nitric oxide: chemistry and pathophysiology. Adv Exp Med Biol. 2012c;942:75–92. doi: 10.1007/978-94-007-2869-1_4. [DOI] [PubMed] [Google Scholar]
- Sattler M, Winkler T, Verma S, Byrne CH, Shrikhande G, Salgia R, et al. Hematopoietic growth factors signal through the formation of reactive oxygen species. Blood. 1999;93:2928–2935. [PubMed] [Google Scholar]
- Sauer H, Rahimi G, Hescheler J, Wartenberg M. Role of reactive oxygen species and phosphatidylinositol 3-kinase in cardiomyocyte differentiation of embryonic stem cells. FEBS Lett. 2000;476:218–223. doi: 10.1016/s0014-5793(00)01747-6. [DOI] [PubMed] [Google Scholar]
- Sauer H, Wartenberg M, Hescheler J. Reactive oxygen species as intracellular messengers during cell growth and differentiation. Cell Physiol Biochem. 2001;11:173–186. doi: 10.1159/000047804. [DOI] [PubMed] [Google Scholar]
- Sauer H, Bekhite MM, Hescheler J, Wartenberg M. Redox control of angiogenic factors and CD31-positive vessel-like structures in mouse embryonic stem cells after direct current electrical field stimulation. Exp Cell Res. 2005;304:380–390. doi: 10.1016/j.yexcr.2004.11.026. [DOI] [PubMed] [Google Scholar]
- Scandurra FM, Gnaiger E. Cell respiration under hypoxia: facts and artefacts in mitochondrial oxygen kinetics. Adv Exp Med Biol. 2010;662:7–25. doi: 10.1007/978-1-4419-1241-1_2. [DOI] [PubMed] [Google Scholar]
- Schmelter M, Ateghang B, Helmig S, Wartenberg M, Sauer H. Embryonic stem cells utilize reactive oxygen species as transducers of mechanical strain-induced cardiovascular differentiation. FASEB J. 2006;20:1182–1184. doi: 10.1096/fj.05-4723fje. [DOI] [PubMed] [Google Scholar]
- Schmidt M, Giessl A, Laufs T, Hankeln T, Wolfrum U, Burmester T. How does the eye breathe? Evidence for neuroglobin-mediated oxygen supply in the mammalian retina. J Biol Chem. 2003;278:1932–1935. doi: 10.1074/jbc.M209909200. [DOI] [PubMed] [Google Scholar]
- Schroeder T. Hematopoietic stem cell heterogeneity: subtypes, not unpredictable behavior. Cell Stem Cell. 2010;6:203–207. doi: 10.1016/j.stem.2010.02.006. [DOI] [PubMed] [Google Scholar]
- Schroedl C, McClintock DS, Budinger GR, Chandel NS. Hypoxic but not anoxic stabilization of HIF-1alpha requires mitochondrial reactive oxygen species. Am J Physiol Lung Cell Mol Physiol. 2002;283:L922–L923. doi: 10.1152/ajplung.00014.2002. [DOI] [PubMed] [Google Scholar]
- Seita J, Weissman IL. Hematopoietic stem cell: self-renewal versus differentiation. Wiley Interdiscip Rev Syst Biol Med. 2010;2:640–653. doi: 10.1002/wsbm.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiology (Bethesda) 2009;24:97–106. doi: 10.1152/physiol.00045.2008. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Regulation of metabolism by hypoxia-inducible factor 1. Cold Spring Harb Symp Quant Biol. 2011;76:347–353. doi: 10.1101/sqb.2011.76.010678. [DOI] [PubMed] [Google Scholar]
- Sharma S, Cabana R, Shariatmadar S, Krishan A. Cellular volume and marker expression in human peripheral blood apheresis stem cells. Cytometry A. 2008;73:160–167. doi: 10.1002/cyto.a.20524. [DOI] [PubMed] [Google Scholar]
- Shima H, Takubo K, Tago N, Iwasaki H, Arai F, Takahashi T, et al. Acquisition of G0 state by CD34-positive cord blood cells after bone marrow transplantation. Exp Hematol. 2010;38:1231–1240. doi: 10.1016/j.exphem.2010.08.004. [DOI] [PubMed] [Google Scholar]
- Shivapurkar N, Stastny V, Okumura N, Girard L, Xie Y, Prinsen C, et al. Cytoglobin, the newest member of the globin family, functions as a tumor suppressor gene. Cancer Res. 2008;68:7448–7456. doi: 10.1158/0008-5472.CAN-08-0565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simsek T, Kocabas F, Zheng J, Deberardinis RJ, Mahmoud AI, Olson EN, et al. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell. 2010;7:380–390. doi: 10.1016/j.stem.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriram R, Kreutzer U, Shih L, Jue T. Interaction of fatty acid with myoglobin. FEBS Lett. 2008;582:3643–3649. doi: 10.1016/j.febslet.2008.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srour EF, Brandt JE, Briddell RA, Leemhuis T, van Besien K, Hoffman R. Human CD34+ HLA-DR- bone marrow cells contain progenitor cells capable of self-renewal, multilineage differentiation, and long-term in vitro hematopoiesis. Blood Cells. 1991;17:287–295. [PubMed] [Google Scholar]
- St John JC, Ramalho-Santos J, Gray HL, Petrosko P, Rawe VY, Navara CS, et al. The expression of mitochondrial DNA transcription factors during early cardiomyocyte in vitro differentiation from human embryonic stem cells. Cloning Stem Cells. 2005;7:141–153. doi: 10.1089/clo.2005.7.141. [DOI] [PubMed] [Google Scholar]
- Suda T, Takubo K, Semenza GL. Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell. 2011;9:298–310. doi: 10.1016/j.stem.2011.09.010. [DOI] [PubMed] [Google Scholar]
- Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25:977–988. doi: 10.1016/j.immuni.2006.10.016. [DOI] [PubMed] [Google Scholar]
- Takubo K, Goda N, Yamada W, Iriuchishima H, Ikeda E, Kubota Y, et al. Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell. 2010;7:391–402. doi: 10.1016/j.stem.2010.06.020. [DOI] [PubMed] [Google Scholar]
- Takubo K, Nagamatsu G, Kobayashi CI, Nakamura-Ishizu A, Kobayashi H, Ikeda E, et al. Regulation of glycolysis by pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell. 2013;12:49–61. doi: 10.1016/j.stem.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi Ishikawa E, Gonzalez-Nieto D, Ghiaur G, Dunn SK, Ficker AM, Murali B, et al. Connexin-43 prevents hematopoietic stem cell senescence through transfer of reactive oxygen species to bone marrow stromal cells. Proc Natl Acad Sci U S A. 2012;109:9071–9076. doi: 10.1073/pnas.1120358109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesio M, Golan K, Corso S, Giordano S, Schajnovitz A, Vagima Y, et al. Enhanced c-Met activity promotes G-CSF-induced mobilization of hematopoietic progenitor cells via ROS signaling. Blood. 2011;117:419–428. doi: 10.1182/blood-2009-06-230359. [DOI] [PubMed] [Google Scholar]
- Toledano MB, Delaunay A, Monceau L, Tacnet F. Microbial H2O2 sensors as archetypical redox signaling modules. Trends Biochem Sci. 2004;29:351–357. doi: 10.1016/j.tibs.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Tothova Z, Gilliland DG, Fox O. Transcription factors and stem cell homeostasis: insights from the hematopoietic system. Cell Stem Cell. 2007;1:140–152. doi: 10.1016/j.stem.2007.07.017. [DOI] [PubMed] [Google Scholar]
- Urao N, Ushio-Fukai M. Redox regulation of stem/progenitor cells and bone marrow niche. Free Radic Biol Med. 2013;54:26–39. doi: 10.1016/j.freeradbiomed.2012.10.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urao N, McKinney RD, Fukai T, Ushio-Fukai M. NADPH oxidase 2 regulates bone marrow microenvironment following hindlimb ischemia: role in reparative mobilization of progenitor cells. Stem Cells. 2012;30:923–934. doi: 10.1002/stem.1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ushio-Fukai M, Urao N. Novel role of NADPH oxidase in angiogenesis and stem/progenitor cell function. Antioxid Redox Signal. 2009;11:2517–2533. doi: 10.1089/ars.2009.2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandervelde S, van Luyn MJ, Tio RA, Harmsen MC. Signaling factors in stem cell-mediated repair of infarcted myocardium. J Mol Cell Cardiol. 2005;39:363–376. doi: 10.1016/j.yjmcc.2005.05.012. [DOI] [PubMed] [Google Scholar]
- Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem. 1995;270:1230–1237. doi: 10.1074/jbc.270.3.1230. [DOI] [PubMed] [Google Scholar]
- Wang N, Xie K, Huo S, Zhao J, Zhang S, Miao J. Suppressing phosphatidylcholine-specific phospholipase C and elevating ROS level, NADPH oxidase activity and Rb level induced neuronal differentiation in mesenchymal stem cells. J Cell Biochem. 2007;100:1548–1557. doi: 10.1002/jcb.21139. [DOI] [PubMed] [Google Scholar]
- von Wangenheim KH, Peterson HP. Control of cell proliferation by progress in differentiation: clues to mechanisms of aging, cancer causation and therapy. J Theor Biol. 1998;193:663–678. doi: 10.1006/jtbi.1998.0731. [DOI] [PubMed] [Google Scholar]
- Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol. 2010;11:872–884. doi: 10.1038/nrm3013. [DOI] [PubMed] [Google Scholar]
- Wilson DF, Rumsey WL, Green TJ, Vanderkooi JM. The oxygen dependence of mitochondrial oxidative phosphorylation measured by a new optical method for measuring oxygen concentration. J Biol Chem. 1988;263:2712–2718. [PubMed] [Google Scholar]
- Winterbourn CC, Hampton MB. Thiol chemistry and specificity in redox signaling. Free Radic Biol Med. 2008;45:549–561. doi: 10.1016/j.freeradbiomed.2008.05.004. [DOI] [PubMed] [Google Scholar]
- Wittenberg JB. Myoglobin-facilitated oxygen diffusion: role of myoglobin in oxygen entry into muscle. Physiol Rev. 1970;50:559–636. doi: 10.1152/physrev.1970.50.4.559. [DOI] [PubMed] [Google Scholar]
- Woolley JF, Naughton R, Stanicka J, Gough DR, Bhatt L, Dickinson BC, et al. H2O2 production downstream of FLT3 is mediated by p22phox in the endoplasmic reticulum and is required for STAT5 signalling. PLoS ONE. 2012;7:e34050. doi: 10.1371/journal.pone.0034050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Xiao Q, Luo Z, Pepe AE, Margariti A, Zeng L, Xu Q. Embryonic stem cell differentiation into smooth muscle cells is mediated by Nox4-produced H2O2. Am J Physiol Cell Physiol. 2009;296:C711–C723. doi: 10.1152/ajpcell.00442.2008. [DOI] [PubMed] [Google Scholar]
- Xie Y, Yin T, Wiegraebe W, He XC, Miller D, Stark D, et al. Detection of functional haematopoietic stem cell niche using real-time imaging. Nature. 2009;457:97–101. doi: 10.1038/nature07639. [DOI] [PubMed] [Google Scholar]
- Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, Wu H, et al. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature. 2006;44:475–482. doi: 10.1038/nature04703. [DOI] [PubMed] [Google Scholar]
- Yu WM, Liu X, Shen J, Jovanovic O, Pohl EE, Gerson SL, et al. Metabolic regulation by the mitochondrial phosphatase PTPMT1 is required for hematopoietic stem cell differentiation. Cell Stem Cell. 2013;12:62–74. doi: 10.1016/j.stem.2012.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Xu J, Liu N, Wang Y, Li X, Pallast S, et al. Mitochondrial distribution of neuroglobin and its response to oxygen-glucose deprivation in primary-cultured mouse cortical neurons. Neuroscience. 2012;218:235–242. doi: 10.1016/j.neuroscience.2012.05.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497–5510. doi: 10.1038/onc.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283:10892–10903. doi: 10.1074/jbc.M800102200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zhang J, Grindley JC, Yin T, Jayasinghe S, He XC, Ross JT, et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature. 2006;44:518–522. doi: 10.1038/nature04747. [DOI] [PubMed] [Google Scholar]
- Zinkevich NS, Gutterman DD. ROS-induced ROS release in vascular biology: redox-redox signaling. Am J Physiol Heart Circ Physiol. 2011;301:H647–H653. doi: 10.1152/ajpheart.01271.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial ROS-induced ROS release: an update and review. Biochim Biophys Acta. 2006;1757:509–517. doi: 10.1016/j.bbabio.2006.04.029. [DOI] [PubMed] [Google Scholar]