

Abstract
Background and Purpose
Atherosclerosis is associated with reduced vascular hydrogen sulfide (H2S) biosynthesis. GYY4137 is a novel slow-releasing H2S compound that may effectively mimic the time course of H2S release in vivo. However, it is not known whether GYY4137 affects atherosclerosis.
Experimental Approach
RAW 264.7 cells and human blood monocyte-derived macrophages were incubated with oxidized low density lipoprotein (ox-LDL) with/without GYY4137. ApoE−/− mice were fed a high-fat diet for 4 weeks and administered GYY4137 for 30 days. Lipid and atherosclerotic lesions were measured by oil red O staining. Endothelium-dependent relaxation was assessed in response to acetylcholine. Superoxide production was detected by dihydroethidium staining. Expression of mRNA and protein were evaluated by quantitative real-time PCR and Western blot.
Key Results
GYY4137 inhibited ox-LDL-induced foam cell formation and cholesterol esterification in cultured cells. GYY4137 decreased the expression of lectin-like ox-LDL receptor-1, iNOS, phosphorylated IκBα, NF-κB, ICAM-1, VCAM-1 and chemokines, including CXCL2, CXCR4, CXCL10 and CCL17, but increased the scavenger protein CD36, in ox-LDL-treated RAW 264.7 cells. In vivo, GYY4137 decreased aortic atherosclerotic plaque formation and partially restored aortic endothelium-dependent relaxation in apoE−/− mice. GYY4137 decreased ICAM-1, TNF-α and IL-6 mRNA expression as well as superoxide (O2−) generation in aorta. In addition, GYY4137 increased aortic eNOS phosphorylation and expression of PI3K, enhanced Akt Ser473 phosphorylation and down-regulated the expression of LOX-1.
Conclusion and Implications
GYY4137 inhibits lipid accumulation induced by ox-LDL in RAW 264.7 cells. In vivo, GYY4137 decreased vascular inflammation and oxidative stress, improved endothelial function and reduced atherosclerotic plaque formation in apoE−/− mice.
Keywords: arteriosclerosis, hydrogen sulfide, inflammation, oxidative stress, endothelial dysfunction
Introduction
Atherosclerosis, a leading cause of mortality and morbidity in Western countries, is a complex process involving multiple cell types, mediators and a combination of pathogenic factors, which includes chronic inflammation, endothelial dysfunction and oxidative stress (Libby, 2002; Davignon and Ganz, 2004; Stocker and Keaney, 2004). While the role of NO in atherosclerosis has been intensively studied in recent years (Gkaliagkousi and Ferro, 2011), the part played by another naturally occurring gaseous mediator, hydrogen sulfide (H2S), in this disease has received less attention. Like NO, H2S biosynthesis from cysteine via the enzyme, cystathionine γ lyase (CSE), occurs in vascular endothelial, smooth muscle cells as well as in macrophages (Yang et al., 2004; 2008; Bełtowski et al., 2010; Zhu et al., 2010), all of which play crucial roles in atherosclerosis. Moreover, H2S donor drugs exhibit vasodilator, pro-angiogenic and anti-inflammatory activity (Zhao et al., 2001; Zhao and Wang, 2002; Papapetropoulos et al., 2009; Ekundi-Valentim et al., 2010). As such, like NO, H2S is becoming widely regarded as a vasculoprotective agent. Moreover, the possibility that atherosclerosis is associated with changes in vascular H2S production has been suggested (Wang et al., 2009) and NaHS has recently been shown to inhibit the oxidized LDL (ox-LDL)-evoked rise in macrophage cholesterol levels (Zhao et al., 2011).
However, the precise role of H2S in atherosclerosis in vivo and macrophage foam cell formation in vitro remains unclear as does the possibility that atherosclerotic disease can be influenced by H2S donors. One reason for the lack of clarity is the reliance on NaHS as an H2S donor in studies of this type. NaHS has been widely used to evaluate the biology of H2S and has provided useful information about the pharmacological effects of this gas. However, when dissolved in water, NaHS releases copious amounts of H2S over a short time frame (seconds) and as such does not effectively mimic the time course of H2S release in vivo. With this in mind, we have been evaluated the biological effects of GYY4137, which, as a novel H2S donor, releases low concentrations of H2S slowly (hours) in aqueous solution at physiological pH and temperature and may effectively mimic the time course of H2S release in vivo (Li et al., 2008). Furthermore, GYY4137 reduces the formation of pro-inflammatory cytokines in LPS-challenged RAW 264.7 macrophages (Whiteman et al., 2009) and is anti-inflammatory in LPS-treated mice (Li et al., 2009). In addition, GYY4137 does not cause apoptosis or cell death in normal cells such as fibroblasts (Lee et al., 2011). However, it is not known whether GYY4137 affects foam cell formation in vitro or atherosclerosis in vivo.
In the present study we obtained evidence that the slow-releasing H2S donor, GYY4137, not only reduces biochemical and histological markers of atherosclerosis in an isolated cell system but, importantly, retards the development of an ongoing atherosclerotic response in the mouse. The mechanisms involved probably include inhibition of macrophage foam cell formation, reduced O2− and inflammation, and enhanced endothelial cell function. The molecular basis of these effects includes inhibition of NF-κB transcription leading to a range of beneficial downstream effects including activation of eNOS. Thus, the present data raise the possibility that, treatment with slow-releasing H2S donors, such as GYY4137, can provide a novel therapeutic approach to reduce the development, and hence limit the deleterious cardiovascular consequences of this most pervasive disease.
The nomenclature used for receptors conforms to BJP's Guide to Receptors and Channels (Alexander et al., 2011).
Methods
Macrophage culture: mouse and human cells
Murine macrophages (RAW 264.7 cells), purchased from the American Type Culture Collection (Rockville, MD, USA), were cultured in DMEM with 10% v/v FBS, penicillin (100 U·mL−1) and streptomycin (100 mg·mL−1) in a 95% O2:5% CO2 humidified atmosphere at 37°C. Confluent cells (85–90%) were incubated (six well plates) with ox-LDL (100 μg mL−1, Yiyuan biotechnology, Guangzhou, China) with or without different concentrations of GYY4137 (50–400 μmol·L−1) or ZYJ1122 (200 μmol·L−1), which is a structural analogue of GYY4137 lacking sulfur (Lee et al., 2011) and unable to release H2S or influence the lipid accumulation in macrophages (Supporting Information Figure S1).
Human peripheral blood monocytes were isolated from each subject by density gradient centrifugation as described previously (Zhao et al., 2006). Cells were thereafter differentiated into macrophages using macrophage colony-stimulating factor (M-CSF, 1 ng·mL−1) for 5 days in X-Vivo 15 medium (BioWhittaker Ltd, Walkerville, MD, USA) and supplemented with 10% v/v human serum. After 5 days, cells were incubated (48 h) with ox-LDL (50 μg mL−1) in the presence or absence of either GYY4137 (50–400 μmol·L−1) or ZYJ1122 (200 μmol·L−1). The investigation conforms to the principles outlined in the Declaration of Helsinki. The study protocol was approved by the ethics committees of Nanjing Medical University. Written informed consent was obtained from all patients and volunteers (See supplementary material online for detailed methods).
Animals
Eight-week-old male C57BL/6J mice (wild-type mice, WT) and homozygous apoE−/− mice were purchased from the Animal Center of Beijing University, Beijing, China. To accelerate the development of spontaneous atherosclerotic lesions in apoE−/− mice, animals were fed a high-fat diet (15% fat plus 1.25% cholesterol) for 4 weeks. Thereafter, mice were randomly divided into three groups: (i) WT, (ii) apoE−/− + NS (normal saline, 0.1 mL·day−1, i.p.) for 30 days, and (iii) apoE−/− + GYY4137 (133 μmol·kg−1·day−1, i.p.) for 30 days. The dose of GYY4137 used in this work was based upon previous reports of the anti-inflammatory effect of this compound in other animal models (Li et al., 2009; 2013). At the end of the treatment period, animals were killed by an overdose of pentobarbital (100 mg·kg−1, i.v.).
A total of 32 male homozygous apoE−/− mice and 16 male C57BL/6J mice (wild-type mice) were used in the experiments. The animals were housed individually in the specific pathogen free barrier facility at constant temperature (20-22°C) and humidity (45%-55%) with a 12 hour light-dark cycle. ApoE−/− mice were fed with a high-fat diet (15% fat plus 1.25% cholesterol), and C57BL/6J mice with rodent diet for 4 weeks.
The animal experiments were approved by the Committee on Animal Care of Nanjing Medical University (NJMU-ERLAUA-20100112) and were conducted according to NIH Guidelines for the Care and Use of Laboratory Animals. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Measurement of H2S, lipid staining and cholesterol in macrophages
Briefly, medium (75 μL) was mixed 1% (w/v) zinc acetate (250 μL) and distilled water (425 μL). N,N,dimethyl-p-phenylenediamine sulfate (20 mmol·L−1) in 7.2 M HCl (133 μL) and FeCl3 (30 mmol·L−1) in 1.2 mM HCl (133 μL) were added and incubated (10 min) at room temperature. Protein was precipitated by adding 10% v/v trichloroacetic acid (250 μL) to the reaction mixture. The absorbance of the resulting solution was measured by spectrophotometry at 670 nm. NaHS (2.5–200 μmol·L−1) was used as a standard (Zhang et al., 2003; Zhu et al., 2007; Sowmya et al., 2010).
Lipid staining was assessed histologically using 0.5% v/v oil red O for 1 h. Cell lipid was also extracted in order to measure cholesterol esterification (CE). The difference between total cholesterol and free cholesterol is defined as esterified cholesterol (Gamble et al., 1978). (See supplementary material online for detailed methods).
Measurement of atherosclerotic lesions
Hearts were embedded in tissue optimal cutting temperature compound, serially sectioned from the heart to the root of aortic valve (5 μm, Leica CM1900 cryostat, Solms, Germany), mounted on slides and then stained with Oil-Red-O solution for analysis of atherosclerotic lesions as described previously (Wang et al., 2009). Total plasma cholesterol (TC), triglycerides (TG), high-density lipoprotein cholesterol (HDL-C) and low-density lipoprotein-cholesterol (LDL-C) concentration were determined using commercially available kits (Zhong Sheng Bei Kong, Peking, China) as per the manufacturer's instructions.
Assessment of endothelium-dependent vasodilatation
Vasorelaxation of isolated aortic ring segments were determined in organ baths containing oxygenated Kreb's solution. After an equilibration period of 60 min, aortic rings were precontracted with noradrenaline (NA) at a concentration (0.1 μmol·L−1). Endothelium-dependent relaxation was then assessed in response to cumulative addition of acetylcholine (ACh, 0.001–10 μmol·L−1). Relaxation at each concentration was measured and expressed as the percentage of force generated in response to NA.
Measurement of H2S in plasma and tissue in apoE−/− mice
Plasma H2S was measured as described previously using NaHS (2.5–200 μmol·L−1) as a standard (Xu et al., 2009). H2S synthesizing activity was determined using tissue homogenates. Briefly, the assay mixture (500 μl) contained 460 μL of tissue homogenate, 10 mol·L−1 L-cysteine and 2 mmol·L−1 pyridoxal 5′-phosphate. Incubations were carried out tightly sealed Eppendorf vials. After incubation (37°C, 30 min), zinc acetate (1% w/v, 250 μL) was added to trap the H2S generated, followed by trichloroacetic acid (10% w/v, 25 μL) to stop the reaction. N,N-dimethyl-p-phenylenediamine sulfate (20 mmol·L−1, 133 μL) and FeCl3 (30 mmol·L−1, 133 μL) were added and the mixture centrifuged (12 000 x g). The absorbance of aliquots (200 μL) was determined 10 min thereafter at 670 nm.
Measurement of superoxide formation in mouse aorta
Superoxide production in tissue sections of mouse aorta was detected by fluorescence microtopography using the fluorescent probe dihydroethidium (DHE) as described previously (Ali et al., 2008). Sections (5 μm) were subsequently incubated (30 min, 37°C) in Kreb's HEPES buffer containing DHE (2 μmol·L−1) in a light-protected chamber. The slides were examined with a Nikon TE2000 Inverted Microscope (Nikon Ltd, Tokyo, Japan), using excitation and emission wavelengths of 480 and 610 nm respectively.
Quantitative real-time PCR
For tissues, total RNA was extracted using Trizol reagent (Invitrogen, Carlsbad, CA, USA) and cDNA was synthesized using Rever Tra Ace qPCR RT kit (Toyobo, Osaka, Japan) using the TaqMan system based on real-time detection of accumulated fluorescence. Gene expression was normalized to 18S mRNA and reported as ratios compared to the level of expression in control mice (given an arbitrary value of 1). For macrophages, total RNA was extracted with Trizol and purified using the PureLink RNA mini-kit (Invitrogen). RNA (1 μg) was added as a template to reverse-transcriptase reactions carried out using SuperScript® III First-Strand Synthesis Kit (Invitrogen). Quantitative real-time PCRs (qRT-PCRs) were carried out with the resulting cDNAs in triplicate using iQ SYBR Green Supermix (Bio-Rad, Hercules, CA, USA) and Biorad CFX384 RealTime System. Experimental Ct values were normalized to r12s and relative mRNA expression was calculated in comparison to a reference sample. Each sample was run and analysed in triplicate. (Structures of all primers used are listed in Supporting Information Table S1).
Western blot analysis
Protein samples were separated on 10% v/v SDS-PAGE, transferred onto PVDF membrane (Millipore, Billerica, MA, USA) and then immunoblotted with antibodies (Detailed in Supporting Information Table S2). Proteins were visualized by enhanced chemiluminescence substrate (Thermo Fisher Scientific Inc., Rockford, IL, USA).
Measurement of NF-κB binding activity
NF-κB binding activity was measured using an electrophoretic mobility shift assays kit (Pierce Chemical, Rockford, IL, USA) according to the manufacturer's instructions (See supplementary material online for detailed methods).
Statistical analysis
All data are expressed as mean ± SD and were analysed by Student's paired t test or one-way anova followed by Newman–Keuls multiple comparison test as appropriate (GraphPad Prism version 5 software). A value of P < 0.05 was considered statistically significant.
Results
Effect of GYY4137 on lipid accumulation and H2S system in ox-LDL-treated mice macrophages in vitro
Incubation of cultured RAW 264.7 cells with ox-LDL caused foam cell formation, as evidenced, histologically, by enhanced oil red O staining and an almost threefold rise in cell CE (Figure 1A,C). Treatment of ox-LDL-stimulated RAW 264.7 cells with GYY4137 (50–400 μmol·L−1), but not ZYJ1122 (200 μmol·L−1), inhibited foam cell formation determined both histologically (Figure 1A,B) and by monitoring cell CE (Figure 1C,D). Exposure of RAW 264.7 cells to ox-LDL decreased the H2S concentration detected in the medium (Figure 1E). In contrast, CSE expression in treated cells was increased (Figure 1F). Concomitant exposure of RAW 264.7 cells to both ox-LDL and GYY4137 (50–400 μmol·L−1) reversed the ox-LDL-induced change in macrophage H2S concentration (Figure 1E) and CSE protein expression (Figure 1F); these effects were not observed with ZYJ1122 (200 μmol·L−1) (Figure 1G).
Figure 1.
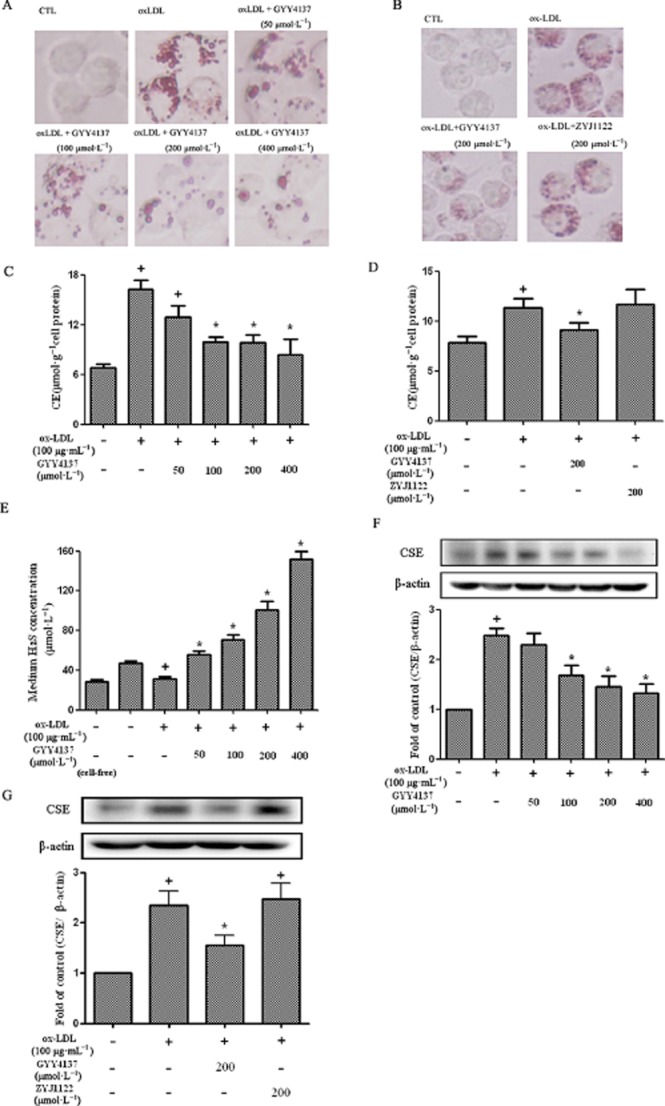
Effect of GYY4137 on lipid accumulation and H2S in RAW264.7 cells. RAW 264.7 cells were exposed to ox-LDL (100 μg·mL−1) in the presence or absence of GYY4137 (50–400 μmol·L−1) or ZYJ1122 (200 μmol·L−1) for 24 h. Representative photographs showing RAW 264.7 cells stained with Oil Red O (A and B). Measurement of CE in RAW 264.7 cells (C and D). H2S concentration in culture medium (E). Western blot analysis and quantification of CSE protein expression (F and G). +P < 0.05, compared with no treatment, *P < 0.05, compared with treatment with ox-LDL (n = 4).
Effect of GYY4137 on lipid accumulation in ox-LDL-treated human macrophages in vitro
Essentially identical experiments were also conducted using human peripheral blood monocyte-derived macrophages isolated from either healthy, human volunteers or patients with coronary heart disease. Exposure to ox-LDL (50 μg·mL−1, 48 h) again resulted in increased cell oil red O staining combined with a marked rise in cell CE in both populations. As before, pretreatment with GYY4137, but not ZYJ1122 (both 200 μmol·L−1), reduced the ox-LDL-induced rise in lipid staining and CE in blood monocyte-derived macrophages from both healthy individuals and patients with coronary artery disease (Figure 2A-D).
Figure 2.

Effect of GYY4137 on lipid accumulation in human macrophages in vitro; the macrophages were exposed to ox-LDL (50 μg·mL−1) for 48 h. Monocyte-derived macrophages from healthy donors (A) and patients with coronary heart disease (B) stained with Oil Red O. Measurement of CE in monocyte-derived macrophages from healthy donors (C) and patients with coronary heart disease (D). +P < 0.05, compared with no treatment, *P < 0.05, compared with treatment with ox-LDL (n = 4–9).
Effect of GYY4137 on LOX-1, iNOS expression and chemokine release in ox-LDL-treated RAW 264.7 cells
LOX-1, a scavenger receptor for ox-LDL that facilitates its cellular uptake, is expressed in macrophages, smooth muscle cells and vascular endothelial cells. Incubation of RAW 264.7 cells with ox-LDL resulted in marked up-regulation of LOX-1expression; this effect was inhibited by co-treatment with GYY4137 (50–400 μmol·L−1) (Figure 3A).
Figure 3.
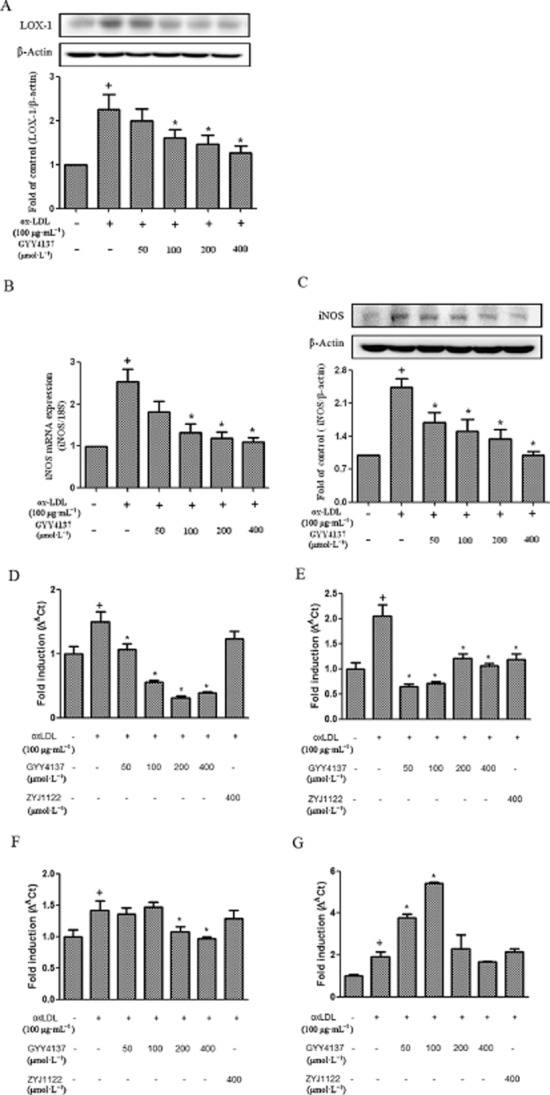
Effect of GYY4137 on LOX-1, iNOS expression and chemokine release in RAW264.7 cells. RAW 264.7 cells were exposed to ox-LDL (100 μg·mL−1) in the presence or absence of GYY4137 (50–400 μmol·L−1) or ZYJ1122 (400 μmol·L−1) for 24 h. Western blot analysis and quantification of LOX-1 protein expression (A). Quantification of iNOS mRNA by real-time PCR (B). Western blot analysis and quantification of iNOS protein expression (C). Quantification of CXCL2 (D), CXCR-4 (E), CXCL10 (F) and CD36 (G) as carried out by real-time PCR. +P < 0.05, compared with no treatment, *P < 0.05, compared with treatment with ox-LDL (n = 4).
The effect of GYY4137 on the generation of a range of pro-inflammatory mediators/enzymes in ox-LDL-challenged macrophages was also assessed in an attempt to gain insight into the cellular mechanisms involved. Thus, incubation of RAW 264.7 cells with ox-LDL resulted in a marked up-regulation of iNOS mRNA and protein expression and these effects were inhibited by co-treatment with different doses of GYY4137 (50–400 μmol·L−1) (Figure 3B-C).
Exposure of RAW 264.7 cells to ox-LDL also resulted in a significant (P < 0.05) release into the medium of several chemokines including CXCL2 (macrophage inflammatory protein-2, MIP-2), CXCR4 (fusin), CXCL10 (interferonγ-induced protein 10, IP-10) as well as the scavenger protein, CD36. Co-treatment with GYY4137 (50–400 μmol·L−1) inhibited chemokine release in all cases. ZYJ1122 (400 μmol·L−1) also reduced the concentration of CXCR4 but had no effect on the other chemokines (Figure 3D-G). Interestingly, GYY4137 increased CD36 at lower concentrations (i.e. 50 and 100 μmol·L−1), but this effect was not seen at higher concentrations (i.e. 200 and 400 μmol·L−1). In contrast, ox-LDL alone failed to affect the concentration, in the culture medium, of a range of other chemokines including CCL2 (monocyte chemotactic protein-1, MCP-1), CCL5 (RANTES), CXCL9 (macrophage inflammatory protein-1) and CXCL11 (interferon-inducible T-cell α chemoattractant, I-TAC) (data not shown).
Effect of GYY4137 on NF-κB activation in RAW 264.7 cells
RAW 264.7 cells challenged with ox-LDL cells showed increased expression of NF-κB p65 (Figure 4A), IκBα phosphorylation and degradation (Figure 4B-D). GYY4137 (50–400 μmol·L−1) suppressed the increased expression of NF-κB p65 protein following exposure of RAW 264.7 cells to ox-LDL (Figure 4A) and inhibited IκBα phosphorylation and degradation (Figure 4B-D).
Figure 4.

Effect of GYY4137 on NF-κB activation in RAW 264.7 cells. RAW 264.7 cells were exposed to ox-LDL (100 μg·mL−1) in the presence or absence of GYY4137 (50–400 μmol·L−1) or ZYJ1122 (200 μmol·L−1) for 24 h. Western blots and quantification of NF-κB p65 protein expression (A). Representative examples of Western blots and quantification of IκBα phosphorylation (B), IκBα degradation (C) and ratio of IκBα phosphorylation to IκBα (D). NF-κB binding activity was evaluated by EMSA: (E-a) 1. Control, 2. ox-LDL alone, 3. ox-LDL+GYY4137 (50 μmol·L−1), 4. ox-LDL+GYY4137 (100 μmol·L−1), 5. ox-LDL+GYY4137 (200 μmol·L−1), 6. ox-LDL+GYY4137 (400 μmol·L−1). (E-b) Bands quantified by densitometric analysis. (F-a) 1. Control, 2. ox-LDL alone, 3. ox-LDL+GYY4137 (200 μmol·L−1), 4. ox-LDL+ZYJ1122 (200 μmol·L−1). Bands quantified by densitometric analysis (F-b). Quantification of ICAM-1 and VCAM-1 mRNA by real-time PCR (G-H). Western blot analysis and quantification of ICAM-1 and VCAM-1 protein expression (I-J). +P < 0.05, compared with no treatment, *P < 0.05, compared with treatment with ox-LDL (n = 3–5).
In separate experiments, NF-κB binding activity in nuclear protein extracts was assessed using a gel mobility shift assay. Ox-LDL increased NF-κB binding activity in RAW 264.7 cells and this effect was reduced by GYY4137 (Figure 4E), but ZYJ1122 (200 μmol·L−1) failed to reduce this increase in NF-κB binding activity (Figure 4F). Ox-LDL also increased ICAM-1 and VCAM-1 expression in RAW 264.7 cells and this effect was reduced by different concentrations of GYY4137 (Figure 4G-J).
Effect of GYY4137 on atherosclerotic lesion size in intact apoE−/− mice
Further experiments were undertaken to determine the effect of GYY4137 treatment on the development of atherosclerosis in high-fat fed apoE−/− mice. Plasma TC, TG and LDL-C concentrations were elevated while plasma HDL-C concentration was reduced in high-fat fed apoE−/− mice when compared with control animals (Supporting Information Table S3). GYY4137 (133 μmol·kg−1·day−1, 30 days) did not affect the plasma lipid profile or body weight of these animals.
ApoE−/− mice fed a high-fat diet for 4 weeks showed significant aortic oil red O staining (Figure 5A). Interestingly, subsequent daily administration of vehicle for a further 30 days to a separate group of apoE−/− animals resulted in an additional, marked aortic lipid deposition (approx. twofold increase compared to animals on high fat diet for 4 weeks, Figure 5B). In contrast, daily administration of GYY4137 over the same period abolished this further increase in atherosclerotic lesion size.
Figure 5.
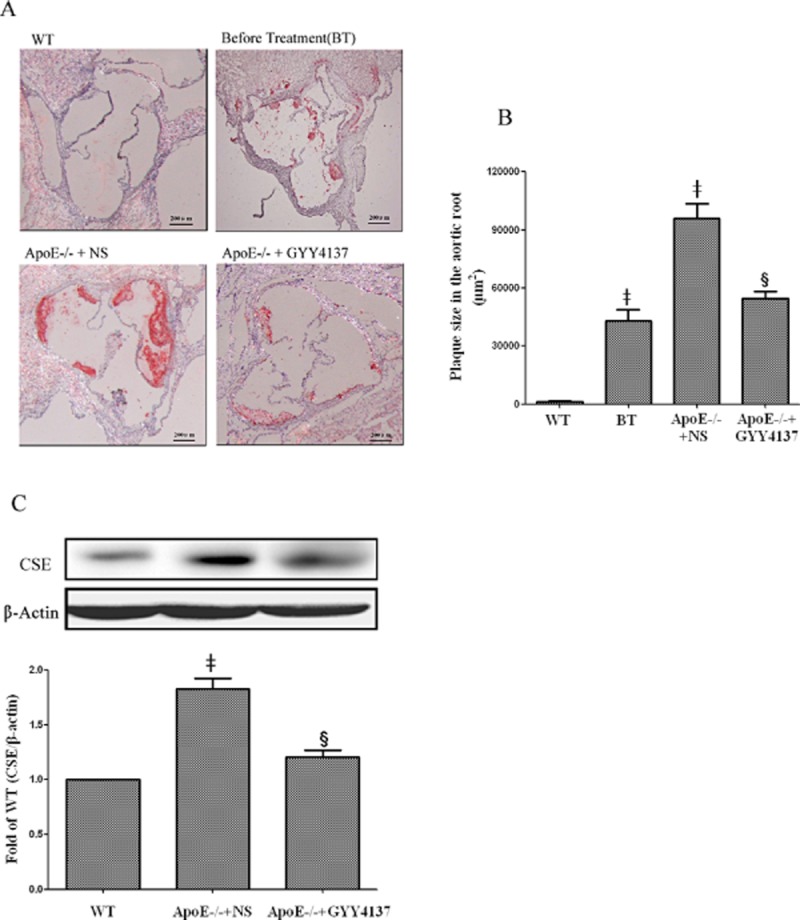
Effect of GYY4137 on atherosclerotic lesion formation and CSE expression in aorta of apoE−/− mice. Effect of GYY4137 (133 μmol·kg−1·day−1) on atherosclerotic lesion formation determined by Oil-Red O-staining of the aortic sinus (A, 40 × magnification). Quantitation of lesion area in Oil Red O-stained aortic sections by Image-Pro Plus software (B). Western blot analysis and quantification of CSE protein expression (C). ‡P < 0.05, compared with WT mice, §P < 0.05, compared with apoE−/− + NS (n = 5).
Effect of GYY4137 on H2S system in apoE−/− mice
In separate experiments, it was observed that the plasma H2S concentration of apoE−/− mice (24.80 ± 9.46 μmol·L−1, n = 5) was significantly (P < 0.05) less than that detected in WT controls (38.42 ± 5.84 μmol·L−1, n = 6); administration of GYY4137 to apoE−/− mice for 30 days increased plasma H2S concentration to values similar to those detected in controls (38.71 ± 5.34 μmol·L−1, n = 9, P > 0.05). In contrast, the expression of CSE in aorta from apoE−/− mice was increased compared to WT controls, but this rise was also reduced by GYY4137 treatment (Figure 5C).
Effect of GYY4137 on aortic superoxide formation in apoE−/− mice
Aortic endothelial superoxide production was measured by dihydroethidium oxidative fluorescence microtopography. Compared with WT mice, endothelial fluorescence was increased in apoE−/− mice. In contrast, endothelial superoxide production in apoE−/− mice was restored to close to normal levels following chronic GYY4137 administration (Figure 6A).
Figure 6.

Effect of GYY4137 on superoxide production, LOX-1 expression, cytokines and ICAM-1 in aortas of apoE−/− mice. Dihydroethidium (DHE) staining of aortic sections for superoxide production (A-a) and quantification of superoxide production(A-b). Western blot analyses and quantification of LOX-1 protein expression (B). Quantification of aortic TNF-α mRNA (C), IL-6 mRNA (D) and ICAM-1mRNA (E) was carried out by real-time PCR. ‡P < 0.05, compared with WT mice, §P < 0.05, compared with apoE−/− + NS (n = 5).
Effect of GYY4137 on aortic LOX-1 expression, cytokines and ICAM-1 in apoE−/− mice
As noted previously, GYY4137 inhibited LOX-1 expression and the production of a range of pro-inflammatory mediators in RAW 264.7 cells exposed to ox-LDL possibly by an effect on the NF-κB pathway. It was thus of interest to determine whether a similar effect might occur in apoE−/− mice administered GYY4137 over a 30 day period. Firstly, aortic LOX-1 protein expression was up-regulated in apoE−/− mice; an effect that was reduced by GYY4137 administration (Figure 6B). Secondly, atherosclerosis in these animals was characterized by a marked increase in aortic TNF-α, IL-6 and ICAM-1 mRNA and the up-regulation of these cytokines was successfully suppressed by GYY4137 treatment (Figure 6C-E).
Effect of GYY4137 on aortic endothelium-dependent relaxation in apoE−/− mice
Endothelium-dependent relaxation, assessed in response to cumulative addition of acetylcholine, was reduced in apoE−/− mice and treatment of these animals with GYY4137 for 30 days significantly (P < 0.05) reversed this deficit in acetylcholine-induced relaxation almost to that observed in controls (Figure 7A). Further work was carried out to investigate the molecular mechanisms by which GYY4137 restores endothelium-dependent vasodilatation in apoE−/− mice. Western blot analysis showed that phosphorylation of eNOS at Ser1177 was diminished in aorta from apoE−/− mice (c.f. WT controls). Intriguingly, treatment of apoE−/− mice with GYY4137 normalized this decreased aortic eNOS phosphorylation (Figure 7B). Since eNOS activity is governed to some extent by upstream regulators such as PI3K and Akt, we also demonstrated that GYY4137 treatment increased the expression of the p85α subunit of PI3K and enhanced the phosphorylation of Akt Ser473 (Figure 7C,D).
Figure 7.
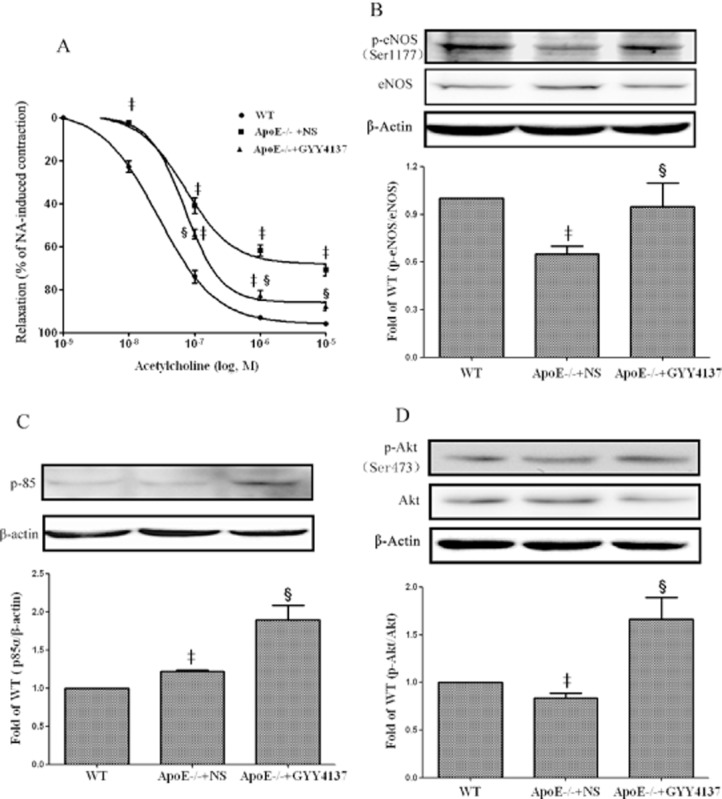
Effect of GYY4137 on aortic endothelium-dependent relaxation in intact apoE−/− mice. Endothelium-dependent vasodilatation of aortic segments induced by acetylcholine (A). Western blots and quantification of phospho-eNOS protein (B), p85α subunit of PI3K (C) and phospho-Akt (D) protein. ‡P < 0.05, compared with WT mice, §P < 0.05, compared with apoE−/− + NS. n = 5.
Discussion and conclusions
Atherosclerosis predisposes patients to numerous cardiovascular diseases including hypertension, coronary heart disease and stroke and, as such, considerable attention has been focused on identifying novel mediators of one or more of the component parts of the underlying pathology with a view to developing new therapeutic approaches. In this context, H2S donors may be an option worth considering since this gas exhibits a wide range of beneficial effects in the vasculature including dilation of blood vessels (Zhao et al., 2001; Zhao and Wang, 2002), inhibition of smooth muscle cell proliferation both in vitro and following balloon injury-induced neointimal hyperplasia in vivo (Meng et al., 2007) and reduction of atherogenic modification of purified LDL by hypochlorite in vitro (Laggner et al., 2007). In addition, H2S has profound anti-inflammatory activity (Zanardo et al., 2006) as evidenced by changes in leukocyte adhesion to microvessels and the associated altered expression of adhesion molecules coupled with potent antioxidant and free radical quenching activity (Jha et al., 2008; Pan et al., 2011). Such a spectrum of biological effects would be expected to be ‘vasculoprotective’ that is able to preserve normal vascular function, and thus be of value in atherosclerosis.
Recently, a defect in the cysteine/CSE/H2S pathway, has been suggested to play a part in some of the manifestations of atherosclerosis. In the present study, we noted that ox-LDL promotes CSE expression but reduced H2S generation in macrophages. Moreover, in whole animals, we observed increased aortic CSE mRNA in atherosclerotic mice whereas plasma and aortic H2S synthesis were reduced. These findings appear counterintuitive. Interestingly, an identical inverse relationship between CSE expression and H2S concentration has been reported previously in ox-LDL challenged macrophages (Wang et al., 2009). These authors attributed this apparent discrepancy to the existence of a positive compensatory feedback mechanism, presumably operative both in vitro and in vivo, whereby low concentrations of H2S serve to promote CSE expression. Certainly, in our experiments, administration of GYY4137 (which elevates H2S concentration) reversed the rise in CSE expression. Further experiments are required to examine the nature of this possible feedback loop in more detail. A number of other questions also remain to be addressed. For example, the identity of the cell type(s) involved in the deficient H2S biosynthesis in these animals is not known. Vascular endothelial and smooth muscle cells as well as macrophages contain CSE and can synthesize H2S (Yang et al., 2004; Bełtowski et al., 2010; Zhu et al., 2010). Macrophages, activated by inflammatory stimuli such as LPS, consistently show evidence of CSE up-regulation. However, it is not clear whether macrophages challenged with ox-LDL to form foam cells also exhibit altered H2S biosynthesis (Wang et al., 2009). Taken together, these observations suggest that atherosclerosis is associated with vascular H2S deficiency. Whether a lack of H2S predisposes to atherosclerosis or is a consequence thereof is also not clear. One approach to this conundrum is to assess whether H2S ‘replacement therapy’ with a slow-releasing donor is beneficial in this condition.
We show here, for the first time, that ox-LDL-challenged macrophages release less H2S into the medium but, paradoxically, express more CSE protein. It is possible that the rise in CSE expression reflects a compensatory change in these cells in an attempt to restore the deficient H2S generation. In the apoE−/− atherosclerotic mouse, we also show that plasma H2S concentration is reduced compared to WT mice, although care should be taken in interpreting plasma H2S concentrations particularly using the spectrophotometric assay, which detects not only H2S but also other thiols and, as such, probably overestimates the absolute value of H2S. Nevertheless, this assay has been widely used to detect changes in plasma H2S in different disease states and, as such, it seems reasonable to conclude that reduced levels of H2S (defined here as H2S, HS- and S-) occur in atherosclerotic mouse plasma. Thus, both in vitro and in vivo data support the notion that atherosclerosis is associated with a decline in bioavailable H2S and raise the possibility that an H2S donor may exhibit anti-atherosclerotic activity.
As noted earlier, considerable emphasis has been placed in the literature on the use of NaHS as a ‘tool’ to model the biological effects of endogenous H2S. NaHS is not an ‘ideal’ H2S donor in this respect since its biological effects are not only transient but the concentrations of H2S generated are high (probably far beyond physiological concentrations) and likely to cause a fall in blood pressure in normotensive animals (Yan et al., 2004) and perhaps also to trigger cell apoptosis in normal cells (Yang et al., 2004). With this in mind, we used GYY4137, which releases H2S slowly over a period of several hours and has previously been shown to inhibit LPS-evoked release of pro-inflammatory mediators from macrophages and to reduce LPS-evoked endotoxic shock in the mouse (Li et al., 2009; Whiteman et al., 2009).
In the present work, GYY4137 caused an inhibition of ox-LDL-evoked macrophage foam cell formation, as measured by oil red O staining and inhibition of cholesterol ester accumulation. These effects were not mimicked by the inactive structural analogue, ZYJ1122, suggesting that they were secondary to H2S release induced by GYY4137. Furthermore, GYY4137 reduced the ox-LDL-mediated up-regulation of LOX-1, a scavenger receptor for ox-LDL expressed in macrophages, smooth muscle cells and vascular endothelial cells which facilitates its cellular uptake (Vohra et al., 2006). This effect may be especially significant for the anti-atherosclerotic activity of GYY4137 since LOX-1 knockout mice have recently been shown to exhibit markedly reduced atherosclerotic lesions when grown in a pro-atherogenic background under a high-cholesterol diet (Mehta et al., 2007). Interestingly, lower concentrations of GYY4137 increased CD36 mRNA, an alternative ox-LDL scavenger receptor, while higher concentrations were without effect. The precise role of CD36 and its interrelationship with LOX-1 in the effect of GYY4137 on atherosclerosis thus requires further study. In subsequent experiments, GYY4137 treatment also reduced the ox-LDL-evoked increase in a range of pro-inflammatory chemoattractant chemokines such as CXCL2 and its receptor (CXCR4) (Zernecke et al., 2008). Taken together, these data suggest that GYY4137 exhibits significant vascular anti-inflammatory activity in apoE−/−- mice.
The molecular mechanisms underlying the anti-atherosclerotic effect of GYY4137 in isolated macrophages most likely centre upon inhibition of NF-κB transcription, since GYY4137 reduced the increased expression of NF-κB in ox-LDL-exposed macrophages and also inhibited IκBα phosphorylation and degradation and prevented nuclear translocation of NF-κB p65. We have previously reported that the anti-inflammatory effect of GYY4137 in LPS-challenged macrophages is dependent on the inhibition of NF-κB transcription (Whiteman et al., 2009). Such an effect on macrophages would account for the ability of GYY4137 to down-regulate iNOS and chemokine expression in RAW 264.7 cells. Interestingly, the expression of LOX-1 is also linked to NF-κB activation in that inhibition of LOX-1 is associated with reduced activation of NF-κB (Chen et al., 2007). Previous researchers have reported that H2S-linked sulfhydration of NF-κB mediates anti-apoptotic actions. Thus, H2S sulfhydrates the p65 subunit of NF-κB at cysteine-38 and this is likely to be a critical physiological determinant of the anti-apoptotic activity of this gas in macrophages (Sen et al., 2012). Moreoever, NaHS significantly inhibited NF-κB activation, as indicated by the attenuated breakdown of the NFκB inhibitor (IκBα) and nuclear translocation of NF-κB (Wang et al., 2009). Since activation of NF-κB is involved in up-regulating the expression of iNOS, ICAM-1 and VCAM-1 and we showed here that GYY4137 suppresses the ox-LDL-induced increase in iNOS, ICAM-1 and VCAM-1 mRNA and protein; it seems likely that GYY4137 acts by inhibiting NF-kB transcription. However, the details of the molecular mechanism(s) by which H2S interacts with NF-κB are not yet clearly understood.
The effect of GYY4137 treatment on the development of atherosclerosis in vivo was also assessed in the high-fat fed, apoE−/− mouse model. This is the first report of the use of a slow releasing H2S donor in this way. Treatment of animals with GYY4137 abolished the development of further atherosclerotic plaque during the 30 day treatment period and also partially restored aortic endothelium-dependent relaxation responses. Both enhanced vascular lipid deposition and endothelial dysfunction are considered to be early markers of atherosclerosis, as highlighted by the observation that fatty streak progression is associated with increasingly impaired vascular relaxation (Murphy et al., 2005). Endothelial dysfunction is associated with reduced eNOS enzyme activity leading to a reduced generation of NO, which is a key regulator of vascular homeostasis. eNOS is activated by phosphorylation of a serine amino acid residues (Ser1177) achieved via a number of protein kinases including PI3K, which is an upstream kinase from Akt (Dimmeler et al., 1999). Thus, activation of either of these protein kinases would, in turn, activate eNOS. We found that the phosphorylation of eNOS was decreased in apoE−/− mice on a high fat diet. It is well-documented that there are different pathways in various cells or tissues involved in eNOS activation and in a previous study it was shown that in the process of endothelial dysfunction, phosphorylation of eNOS by ADP and other purine nucleotides is dependent on PKCδ, but not classical Akt and AMP-dependent protein kinase (Vita, 2011). Thus, we presume that it reasonable to conclude that Akt phosphorylation might not necessarily be reduced during endothelial dysfunction in apoE−/− mice, the mechanism of which is still not completely understood. Indeed, previous studies have demonstrated that H2S protects the heart against the damaging effects of ischaemia-reperfusion by activation of the Akt and eNOS pathways (Yong et al., 2008). Moreover, GYY4137 treatment of these apoE−/− mice did increase the expression of the p85α subunit of PI3K and Akt phosphorylation along with restoring the deficient phosphorylation of eNOS at Ser1177. Thus, the up-regulation of eNOS phosphorylation by GYY4137 is likely to contribute to the improvement in vascular endothelial function detected in apoE−/− mice treated with this H2S donor.
In addition to a decreased production of NO, endothelial dysfunction is also associated with an increase in vascular oxidant stress due to the production of endogenous ROS. Increasing evidence indicates that ROS can both initiate and accelerate the development of atherosclerosis (Li and Shah, 2004). The finding that GYY4137 treatment reduced endothelial superoxide production in the aortic wall suggests an additional possible mechanism underlying the protective effect of this compound in atherosclerosis. Interestingly, LOX-1 activation can cause oxidative stress, and oxidative stress, in turn, can stimulate LOX-1 expression, suggesting a positive feedback loop between oxidative stress and LOX-1 expression (Cominacini et al., 2000). In these experiments, aortic LOX-1 expression was up-regulated in apoE−/− mice and this effect was reduced by GYY4137, highlighting an additional site by which GYY4137, acting as an anti-oxidant, may reduce atherosclerotic disease.
In conclusion, our study provides evidence that the novel slow-releasing H2S donor, GYY4137, not only reduces biochemical and histological markers of atherosclerosis in an isolated cell system but, importantly, retards the development of an ongoing atherosclerotic response in the mouse. The mechanisms involved probably include inhibition of macrophage foam cell formation, reduced O2− production and inflammation, and enhanced endothelial cell function. The molecular basis of these effects includes inhibition of NF-κB transcription leading to a range of beneficial downstream effects including activation of eNOS. Thus, the present data raise the possibility that treatment with slow-releasing H2S donors, such as GYY4137, can provide a novel therapeutic approach to reduce the development of atherosclerosis and hence limit the deleterious cardiovascular consequences of this most pervasive disease.
Acknowledgments
This work was supported by National Basic Research Program of China (973) (refs. 2011CB503903), National Natural Science Foundation of China (refs. 81170083, 81200196, 81200197) and PAPD, to QC by the National Basic Research Program of China (973) (ref. 2012CB517503) and by a grant to PKM from the Biomedical Research Council (BMRC) of Singapore (10/1/21/19/646).
Glossary
- CCL
chemokine (C-C motif) ligands
- CE
cholesterol
- CSE
cystathionine γ lyase
- CXCL
chemokine (C-X-C motif) ligand
- CXCR
chemokine (C-X-C motif) receptor
- GYY4137
morpholin-4-ium-4-methoxyphenyl(morpholino) phosphinodithioate
- ICAM-1
intercellular adhesion molecule-1
- iNOS
inducible NOS
- ox-LDL
oxidized low density lipoprotein
- VCAM-1
vascular cell adhesion molecule-1
Conflict of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 Effect of ZYJ1122 on lipid accumulation and H2S release in ox-LDL-treated mice macrophages in vitro. RAW 264.7 cells were exposed to ox-LDL (100 μg·mL−1) in the presence of ZYJ1122 (50 μmol·L−1–400 μmol·L−1) for 24 h or with ZYJ1122 (400 μmol·L−1) for 6 h to 48 h. Representative photographs showing RAW 264.7 cells stained with Oil Red O (A and B). Measurement of cholesterol (CE) in RAW 264.7 cells (C and D). Measurement of H2S concentration in culture medium (E and F). +P < 0.05, c.f. no treatment, *P < 0.05, c.f. treatment with ox-LDL (n = 4).
Table S1 Sequences of primers.
Table S2 Antibodies for Western blotting.
Table S3 Changes in body weight and plasma lipids profiles of experimental mice.
References
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 5th Edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali ZA, Bursill CA, Douglas G, McNeill E, Papaspyridonos M, Tatham AL, et al. CCR2-mediated antiinflammatory effects of endothelial tetrahydrobiopterin inhibit vascular injury-induced accelerated atherosclerosis. Circulation. 2008;118:S71–S77. doi: 10.1161/CIRCULATIONAHA.107.753558. [DOI] [PubMed] [Google Scholar]
- Bełtowski J, Jamroz-Wiśniewska A, Tokarzewska D. Hydrogen sulfide and its modulation in arterial hypertension and atherosclerosis. Cardiovasc Hematol Agents Med Chem. 2010;8:173–186. doi: 10.2174/187152510792481207. [DOI] [PubMed] [Google Scholar]
- Chen XP, Zhang TT, Du GH. Lectin-like oxidized low-density lipoprotein receptor-1, a new promising target for the therapy of atherosclerosis? Cardiovasc Drug Rev. 2007;25:146–161. doi: 10.1111/j.1527-3466.2007.00009.x. [DOI] [PubMed] [Google Scholar]
- Cominacini L, Pasini AF, Garbin U, Davoli A, Tosetti ML, Campagnola M, et al. Oxidized low density lipoprotein (ox-LDL) binding to ox-LDL receptor-1 in endothelial cells induces the activation of NF-kappa B through an increased production of intracellular reactive oxygen species. J Biol Chem. 2000;275:12633–12638. doi: 10.1074/jbc.275.17.12633. [DOI] [PubMed] [Google Scholar]
- Davignon J, Ganz P. Role of endothelial dysfunction in atherosclerosis. Circulation. 2004;109:III27–III32. doi: 10.1161/01.CIR.0000131515.03336.f8. [DOI] [PubMed] [Google Scholar]
- Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- Ekundi-Valentim E, Santos KT, Camargo EA, Denadai-Souza A, Teixeira SA, Zanoni CI, et al. Differing effects of exogenous and endogenous hydrogen sulphide in carrageenan-induced knee joint synovitis in the rat. Br J Pharmacol. 2010;159:1463–1474. doi: 10.1111/j.1476-5381.2010.00640.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamble W, Vaughan M, Kruth HS, Avigan J. Procedure for determination of free and total cholesterol in micro- or nanogram amounts suitable for studies with cultured cells. J Lipid Res. 1978;199:1068–1070. [PubMed] [Google Scholar]
- Gkaliagkousi E, Ferro A. Nitric oxide signaling in the regulation of cardiovascular and platelet function. Front Biosci. 2011;16:1873–1897. doi: 10.2741/3828. [DOI] [PubMed] [Google Scholar]
- Jha S, Calvert JW, Duranski MR, Ramachandran A, Lefer DJ. Hydrogen sulfide attenuates hepatic ischemia-reperfusion injury: role of antioxidant and antiapoptotic signaling. Am J Physiol Heart Circ Physiol. 2008;295:H801–H806. doi: 10.1152/ajpheart.00377.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laggner H, Muellner MK, Schreier S, Sturm B, Hermann M, Exner M, et al. Hydrogen sulphide: a novel physiological inhibitor of LDL atherogenic modification by HOCl. Free Radic Res. 2007;41:741–747. doi: 10.1080/10715760701263265. [DOI] [PubMed] [Google Scholar]
- Lee ZW, Zhou J, Chen CS, Zhao Y, Tan CH, Li L, et al. The slow-releasing hydrogen sulfide donor, GYY4137, exhibits novel anti-cancer effects in vitro and in vivo. PLoS ONE. 2011;6:e21077. doi: 10.1371/journal.pone.0021077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JM, Shah AM. Endothelial cell superoxide generation: regulation and relevance for cardiovascular pathophysiology. Am J Physiol Regul Integr Comp Physiol. 2004;287:R1014–R1030. doi: 10.1152/ajpregu.00124.2004. [DOI] [PubMed] [Google Scholar]
- Li L, Whiteman M, Guan YY, Neo KL, Cheng Y, Lee SW, et al. Characterization of a novel, water-soluble hydrogen sulfide-releasing molecule (GYY4137): new insights into the biology of hydrogen sulfide. Circulation. 2008;117:2351–2360. doi: 10.1161/CIRCULATIONAHA.107.753467. [DOI] [PubMed] [Google Scholar]
- Li L, Salto-Tellez M, Tan CH, Whiteman M, Moore PK. GYY4137, a novel hydrogen sulfide-releasing molecule, protects against endotoxic shock in the rat. Free Radic Biol Med. 2009;47:103–113. doi: 10.1016/j.freeradbiomed.2009.04.014. [DOI] [PubMed] [Google Scholar]
- Li L, Fox B, Keeble J, Salto-Tellez M, Winyard PG, Wood ME, et al. The complex effects of the slow-releasing hydrogen sulfide donor GYY4137 in a model of acute joint inflammation and in human cartilage cells. J Cell Mol Med. 2013;17:365–376. doi: 10.1111/jcmm.12016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta JL, Sanada N, Hu CP, Chen J, Dandapat A, Sugawara F, et al. Deletion of LOX-1 reduces atherogenesis in LDLR knockout mice fed High cholesterol diet. Circ Res. 2007;100:1634–1642. doi: 10.1161/CIRCRESAHA.107.149724. [DOI] [PubMed] [Google Scholar]
- Meng QH, Yang G, Yang W, Jiang B, Wu L, Wang R. Protective effect of hydrogen sulphide on balloon injury-induced neointima hyperplasia in rat carotid arteries. Am J Pathol. 2007;170:1407–1414. doi: 10.2353/ajpath.2007.060939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JE, Tedbury PR, Homer-Vanniasinkam S, Walker JH, Ponnambalam S. Biochemistry and cell biology of mammalian scavenger receptors. Atherosclerosis. 2005;182:1–15. doi: 10.1016/j.atherosclerosis.2005.03.036. [DOI] [PubMed] [Google Scholar]
- Pan LL, Liu XH, Gong QH, Wu D, Zhu YZ. Hydrogen sulfide attenuated tumor necrosis factor-α-induced inflammatory signaling and dysfunction in vascular endothelial cells. PLoS ONE. 2011;6:e19766. doi: 10.1371/journal.pone.0019766. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Papapetropoulos A, Pyriochou A, Altaany Z, Yang G, Marazioti A, Zhou Z, et al. Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proc Natl Acad Sci U S A. 2009;106:21972–21977. doi: 10.1073/pnas.0908047106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen N, Paul BD, Gadalla MM, Mustafa AK, Sen T, Xu R, et al. Hydrogen sulfide-linked sulfhydration of NFκB mediates its antiapoptotic actions. Mol Cell. 2012;45:13–24. doi: 10.1016/j.molcel.2011.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowmya S, Swathi Y, Yeo AL, Shoon ML, Moore PK, Bhatia M. Hydrogen sulfide: regulatory role on blood pressure in hyperhomocysteinemia. Vasc Pharmacol. 2010;53:138–143. doi: 10.1016/j.vph.2010.05.004. [DOI] [PubMed] [Google Scholar]
- Stocker R, Keaney JF., Jr Role of oxidative modifications in atherosclerosis. Physiol Rev. 2004;84:1381–1478. doi: 10.1152/physrev.00047.2003. [DOI] [PubMed] [Google Scholar]
- Vita JA. Endothelial function. Circulation. 2011;124:e906–e912. doi: 10.1161/CIRCULATIONAHA.111.078824. [DOI] [PubMed] [Google Scholar]
- Vohra RS, Murphy JE, Walker JH, Ponnambalam S, Homer-Vanniasinkam S. Atherosclerosis and the lectin-like oxidized low-density lipoprotein scavenger receptor. Trends Cardiovasc Med. 2006;16:60–64. doi: 10.1016/j.tcm.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Wang Y, Zhao X, Jin H, Wei H, Li W, Bu D, et al. Role of hydrogen sulfide in the development of atherosclerotic lesions in apolipoprotein E knockout mice. Arterioscler Thromb Vasc Biol. 2009;29:173–179. doi: 10.1161/ATVBAHA.108.179333. [DOI] [PubMed] [Google Scholar]
- Whiteman M, Li L, Rose P, Tan CH, Parkinson DB, Moore PK. The effect of hydrogen sulfide donors on lipopolysaccharide-induced formation of inflammatory mediators in macrophages. Antioxid Redox Signal. 2009;12:1147–1154. doi: 10.1089/ars.2009.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Prathapasinqhe G, Wu N, Hwanq SY, Siow YL, O K. Ischemia-reperfusion reduces cystathionine-beta-synthase-mediated hydrogen sulfide generation in the kidney. Am J Physiol Renal Physiol. 2009;297:F27–F35. doi: 10.1152/ajprenal.00096.2009. [DOI] [PubMed] [Google Scholar]
- Yan H, Du J, Tang C. The possible role of hydrogen sulfide on the pathogenesis of spontaneous hypertension in rats. Biochem Biophys Res Commun. 2004;313:22–27. doi: 10.1016/j.bbrc.2003.11.081. [DOI] [PubMed] [Google Scholar]
- Yang G, Sun X, Wang R. Hydrogen sulfide-induced apoptosis of human aorta smooth muscle cells via the activation of mitogen-activated protein kinases and caspase-3. FASEB J. 2004;18:1782–1784. doi: 10.1096/fj.04-2279fje. [DOI] [PubMed] [Google Scholar]
- Yang G, Wu L, Jiang W, Qi J, Cao K, Meng Q, et al. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science. 2008;322:587–590. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong QC, Lee SW, Foo CS, Neo KL, Chen X, Bian JS. Endogenous hydrogen sulphide mediates the cardioprotection induced by ischemic postconditioning. Am J Physiol Heart Circ Physiol. 2008;295:H1330–H1340. doi: 10.1152/ajpheart.00244.2008. [DOI] [PubMed] [Google Scholar]
- Zanardo RC, Brancaleone V, Distrutti E, Fiorucci S, Cirino G, Wallace JL. Hydrogen sulfide is an endogenous modulator of leukocyte-mediated inflammation. FASEB J. 2006;20:2118–2120. doi: 10.1096/fj.06-6270fje. [DOI] [PubMed] [Google Scholar]
- Zernecke A, Shagdarsun E, Weber C. Chemokines in atherosclerosis: an update. Arterioscler Thromb Vasc Biol. 2008;28:1897–1908. doi: 10.1161/ATVBAHA.107.161174. [DOI] [PubMed] [Google Scholar]
- Zhang C, Du J, Bu D, Yan H, Tang X, Tang C. The regulatory effect of hydrogen sulfide on hypoxic pulmonary hypertension in rats. Biochem Biophys Res Commun. 2003;302:810–816. doi: 10.1016/s0006-291x(03)00256-0. [DOI] [PubMed] [Google Scholar]
- Zhao C, Thibault S, Messier N, Ouellette M, Papadopoulou B, Tremblay MJ. In primary human monocyte-derived macrophages exposed to Human immunodeficiency virus type 1, does the increased intracellular growth of Leishmania infantum rely on its enhanced uptake? J Gen Virol. 2006;87:1295–1302. doi: 10.1099/vir.0.81647-0. [DOI] [PubMed] [Google Scholar]
- Zhao W, Wang R. H(2)S-induced vasorelaxation and underlying cellular and molecular mechanisms. Am J Physiol Heart Circ Physiol. 2002;283:H474–H480. doi: 10.1152/ajpheart.00013.2002. [DOI] [PubMed] [Google Scholar]
- Zhao W, Zhang J, Lu Y, Wang R. The vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP) channel opener. EMBO J. 2001;20:6008–6016. doi: 10.1093/emboj/20.21.6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao ZZ, Wang Z, Li GH, Wang R, Tang JM, Cao X, et al. Hydrogen sulfide inhibits macrophage-derived foam cell formation. Exp Biol Med. 2011;236:169–176. doi: 10.1258/ebm.2010.010308. [DOI] [PubMed] [Google Scholar]
- Zhu XY, Liu SJ, Liu YJ, Wang S, Ni X. Glucocorticoids suppress cystathionine gamma-lyase expression and H2S production in lipopolysaccharide-treated macrophages. Cell Mol Life Sci. 2010;67:1119–1132. doi: 10.1007/s00018-009-0250-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu YZ, Wang ZJ, Ho P, Loke YY, Zhu YC, Huang SH, et al. Hydrogen sulfide and its possible roles in myocardial ischemia in experimental rats. J Appl Physiol. 2007;102:261–268. doi: 10.1152/japplphysiol.00096.2006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.