

Abstract
Aberrant expression and function of retinoic acid receptor γ (RARγ) are often involved in the progression of several cancers. However, the role of RARγ in cholangiocarcinoma (CCA), chemoresistant bile duct carcinoma with a poor prognosis, remains unclear. In the present study, we found that RARγ was frequently overexpressed in human CCA specimens. Its overexpression was associated with poor differentiation, lymph node metastasis, high serum carbohydrate antigen 19-9 level, and poor prognosis of CCA. Downregulation of RARγ reduced CCA cell proliferation, migration, invasion, and colony formation ability in vitro and tumorigenic potential in nude mice. RARγ knockdown resulted in upregulation of cell cycle inhibitor P21, as well as downregulation of cyclin D1, proliferating cell nuclear antigen, and matrix metallopeptidase 9, in parallel with suppression of the Akt/NF-κB pathway. Furthermore, overexpression of RARγ contributed to the multidrug chemoresistance of CCA cells, at least in part due to upregulation of P glycoprotein via activation of the Wnt/β-catenin pathway. Molecular mechanism studies revealed that RARγ interacted with β-catenin and led to β-catenin nuclear translocation. Taken together, our results suggested that RARγ plays an important role in the proliferation, metastasis, and chemoresistance of CCA through simultaneous activation of the Akt/NF-κB and Wnt/β-catenin pathways, serving as a potential molecular target for CCA treatment.
INTRODUCTION
Cholangiocarcinoma (CCA) is the second most common primary hepatic malignancy, next to hepatocellular carcinoma (HCC) originating from bile duct epithelia (1). The incidence of this deadly neoplasm has increased rapidly in the past 3 decades. CCA is characterized by poor prognosis and a 5-year survival rate of less than 5% because of its remarkably high malignancy, early metastasis, and multidrug resistance (2). Thus, it is urgent to address the mechanisms underlying CCA proliferation, metastasis, and chemoresistance for the development of novel therapeutic strategies.
Like other nuclear receptors, retinoic acid receptors (RARs) are transcription factors that are essential in embryonic development, maintenance of differentiated cellular phenotypes, metabolism, and cell death (3, 4). There are three RAR subtypes: α, β, and γ. Among them, RARγ plays unique and uncharacterized roles in many different cell types and specific cell microenvironments. For example, RARγ is critical for maintaining a balance between hematopoietic stem cell self-renewal and differentiation (5). It also mediates the retinoic acid (RA)-induced growth arrest and differentiation of S91 murine melanoma cells (6). Overexpression of RARγ increases death of SH-SY5Y neuroblastoma cells in response to RA (7), suppresses the progression of non-hematopoietic-cell-intrinsic myeloproliferative syndromes (8), and inhibits the invasiveness of melanoma by RARγ-inducible gene carbohydrate sulfotransferase 10 (9). RARγ also shows an antiproliferative property in mouse keratinocytes (10).
One of the mechanisms that underlie the above-mentioned diverse biological activities of RARγ in the regulation of differentiation, proliferation, apoptosis, and survival is genomic regulation. However, a considerable number of studies have now shown that RARs can nongenomically regulate some rapid biological responses, such as activation of GTPase Rac, protein kinase C, extracellular signal-regulated protein kinase 2, and cyclic AMP (cAMP) response element-binding protein, to play a different role than genomic regulation (11–13). RARγ can activate cellular SRC (c-SRC) in the cytoplasm through hormone-dependent direct binding to c-SRC, a process required for neuritogenesis (14). In particular, a recent report revealed that RARγ plays an oncogenic role via activation of the Akt/NF-κB pathway in the progression of HCC (15). However, the expression profile of RARγ protein and its roles and mechanisms in CCA progression remain unknown.
In the current study, we found increased RARγ expression in a subset of human CCAs and a significant correlation of the RARγ level with the progression of CCA. RARγ knockdown in CCA cells decreased the cell proliferation rate, cell metastatic abilities, and oncogenic potential both in vitro and in vivo. Moreover, overexpression of RARγ contributed to multidrug resistance of CCA cells via upregulation of P glycoprotein (P-gp). The activation of the Akt/NF-κB and Wnt/β-catenin pathways in CCA is likely responsible for the carcinogenic activities of RARγ.
MATERIALS AND METHODS
Reagents.
Goat anti-rabbit and anti-mouse secondary antibodies conjugated to horseradish peroxidase, donkey anti-mouse antibody–Alexa Fluor 488 or rabbit antibody–Alexa Fluor 594, normal mouse IgG, Lipofectamine RNAi MAX, Lipofectamine 2000, and Stealth small interfering RNA (siRNA) were from Invitrogen (Carlsbad, CA, USA). The BioCoat Matrigel invasion chamber was from BD Biosciences, Inc. (Rockville, MD, USA). LY294002, BMS345541, 5-fluorouracil (5-FU), 12-O-tetradecanoylphorbol-13-acetate (TPA), methyl thiazolyl tetrazolium (MTT), 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI), tumor necrosis factor alpha (TNF-α), Wnt3a, and RA were from Sigma-Aldrich (Indianapolis, IN, USA). Monoclonal antibodies against cyclin A, cyclin B1, cyclin D1, cyclin E1, P21, P27, P-gp, PCNA, matrix metalloproteinase 9 (MMP-9), β-actin, and siRNA targeting β-catenin (siβ-catenin) were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Polyclonal antibodies against RARγ, AKT, phosphorylated AKT (p-AKT), IκB, and phosphorylated IκB (p-IκB); monoclonal antibodies against β-catenin; and P65 antibodies were from Abcam Ltd. (Cambridge, United Kingdom). The EliVision Plus kit was from Maixin Bio (Fuzhou, China).
Patients and tumor specimens.
Informed consent was obtained from each patient. The study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki and was approved by the Institute Research Ethics Committee of the First Affiliated Hospital of Xiamen University. Fresh surgical samples from CCA tissues (T) (n = 42) and the adjacent noncancerous tissues (A) (n = 42; <3 to 5 cm distant from the edges of tumor nodules), cholangitis tissues (C) (n = 27); and normal bile duct tissues (N) (n = 13) were collected between 2007 and 2011. All patients enrolled into the study did not receive preoperative treatment, such as radiation or chemotherapy. Metastatic tumors from other tissues were excluded from the study. The clinical data are shown in Table 1. Follow-up was performed through outpatient clinic interview and telephone communication for an average period of 1.6 years (range, 1 to 3 years).
Table 1.
Expression of RARγ protein in four different bile duct specimens
| Tissue type | n | No. with RARγ expressiona |
High-expression rate (%) | |||
|---|---|---|---|---|---|---|
| ±/− | + | ++ | +++ | |||
| T | 42 | 3 | 8 | 12 | 19 | 73.8 |
| A | 42 | 16 | 20 | 4 | 2 | 14.3 |
| C | 27 | 15 | 8 | 2 | 2 | 14.8 |
| N | 13 | 9 | 4 | 0 | 0 | 0 |
χ2, 56.52; P, 0.000.
Cell culture and transfection.
The human CCA cell lines QBC939, SK-ChA-1, and MZ-ChA-1 were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin at 37°C in an atmosphere of 5% CO2. Stealth siRNA targeting RARγ (siRARγ; CAGCTATGAGCTGAGCCCTCAGTTA) and nonspecific Stealth siRNA (siCtrl) were transfected with Lipofectamine RNAi Max according to the manufacturer's instructions. To obtain stably transfected cells, QBC939 cells were transfected with pll3.7 control vector (shCtrl) or pll3.7-RARγ vector (shRARγ) with Lipofectamine 2000.
Cell proliferation assay.
Cell proliferation was analyzed by MTT assay, as previously described (16). Briefly, a total of 3 × 103 cells were seeded in 96-well plates, and MTT was added to each well every 24 h. The absorbance was measured with a microplate reader (model 680; Bio-Rad, Hercules, CA, USA) at 490 nm.
Cell cycle analysis.
Cells were synchronized to the G0/G1 phases by serum starvation and then cultured for 48 h. The cells were washed twice with ice-cold phosphate-buffered saline (PBS), harvested, fixed with ice-cold PBS in 70% ethanol, and stored at 4°C overnight. After centrifugation, the cells were resuspended in 100 μg/ml RNase A at 37°C for 30 min and subsequently stained with 50 μg/ml propidium iodide (PI) at 4°C for 30 min in the dark. The cells were analyzed with a FACScan flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA) at 488 nm.
Colony formation.
Five hundred cells of the QBC939, SK-ChA-1, or MZ-ChA-1 cell line were cultured in six-well plates for 14 days and then fixed and stained with 0.005% crystal violet. Colonies more than 100 μm in diameter were counted.
Tumor xenografts.
Nude mice (BALB/c; specific-pathogen-free [SPF] grade; 4 to 5 weeks old) were injected subcutaneously with 200 μl (2 × 106) cells. The tumor size was measured with a vernier caliper every 3 days. Tumor volumes were determined according to the following formula: A × B2/2, where A is the largest diameter and B is the perpendicular diameter. Mice were sacrificed at day 21 after cell injection, and tumors were taken for future use. The experiments on drug susceptibility were divided into two groups (shCtrl and shRARγ). The dosage of 5-FU was 80 mg/kg of body weight/day (using PBS for the control), administered on day 8 posttransplantation. Mice were also sacrificed at day 21 after cell injection. The inhibition rate was calculated according to the following formula: [1 − tumor weight (5-FU)/tumor weight (PBS)] × 100%. All manipulations involving living mice were approved by the Animal Care and Use Committee of the First Affiliated Hospital of Xiamen University.
Cell migration and invasion assay.
Cell migration was examined by wound-healing and transwell assays, and in vitro invasion was evaluated by using a Matrigel invasion chamber. For wound-healing assays, cells were cultured in 24-well plates with 100% confluence and then wounded in a line across the slides with a micropipette tip. The medium was removed, and the monolayer was washed with PBS. Graphics were obtained by 0.05% crystal violet staining after 0, 24, and 48 h of incubation. For transwell assays, 2.5 × 104 cells were seeded per upper chamber in serum-free RPMI 1640, whereas the lower chambers were loaded with RPMI 1640 containing 5% FBS. After 24 h, the nonmigrating cells in the upper chambers were removed with a cotton swab, and cells migrating through the membrane to the underside of the membrane were stained with 0.05% crystal violet and counted. Cell in vitro invasion assays were performed similarly, but with Matrigel.
Gelatin zymography.
Cells were seeded in a 6-cm plate and cultured in serum-free medium with or without 100 nM TPA for 24 h. Conditioned medium was collected and subjected to electrophoresis on a 10% SDS-PAGE gel containing 1 mg/ml gelatin. The gels were washed in buffer I (50 mM Tris-HCl, pH 7.5, and 2.5% Triton X-100) for 30 min to remove the SDS and incubated in buffer II (0.2 M NaCl, 5 mM CaCl2, 50 mM Tris-HCl, pH 7.5, 0.02% NaN3) for 48 h. The gels were stained with 0.25% Coomassie blue R-250 for 30 min and then washed. The gel was visualized under a chemiluminescence imaging system (Bio-Rad, Hercules, CA, USA).
qPCR.
Quantitative real-time reverse transcription-PCR (qPCR) was carried out using a Light Cycler 480 (Roche Molecular Biochemicals, Mannheim, Germany) as previously described (16). Human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a control. The primer sequences are listed in Table 2.
Table 2.
Primer sequences for qPCR study
| Gene | Primer sequence (5′–3′) | |
|---|---|---|
| GAPDH | Sense | TCACCCACACTGTGCCCATCTACGA |
| Antisense | CAGCGGAACCGCTCATTGCCAATGG | |
| RARγ | Sense | AAAACTGTATCATCAACAAGG |
| Antisense | CTTCACCTCTTTCTTCTTCTTG | |
| MMP-9 | Sense | TGACAGCGACAAGAAGTGG |
| Antisense | CTCAGTGAAGCGGTACATAGG | |
| P-gp | Sense | GACATCCCAGTGCTTCAGG |
| Antisense | GCCACTGAACATTCAGTCG | |
| Cyclin A | Sense | AAGCACTCCCTGACTGTGG |
| Antisense | CAGGTCTGACTTGAGTGTG | |
| Cyclin B1 | Sense | CCGAGTCACCAGGAACTCGAA |
| Antisense | CTGTTCTTGGCCTCAGTCC | |
| Cyclin D1 | Sense | CCTCAACCATCCTGGCTGCG |
| Antisense | AGGACAGACTCCGCTGTGC | |
| Cyclin E1 | Sense | CACTTTCTTGAGCAACACC |
| Antisense | ATTTCCTCAAGTTTGGCTGCA | |
| P21 | Sense | CTAGTTCTACCTCAGGCAGCT |
| Antisense | GTCGCTGGACGATTTGAGG | |
| P27 | Sense | GTTAGCGGAGCAATGCGC |
| Antisense | CAGGCTTCTTGGGCGTCTG | |
Western blotting.
Western blot analyses were performed as previously described (16). Aliquots containing 20 μg of protein underwent SDS-10% PAGE and then were transferred onto a polyvinylidene difluoride (PVDF) membrane. The membrane was incubated with primary and secondary antibodies, and the signal was finally visualized with an enhanced chemiluminescence (ECL) system.
IHC.
Immunohistochemistry (IH) was performed as previously described (16). Human CCA tissue sections were immunostained with antibody against RARγ (1:200). The IHC scoring was carried out blindly and independently by two pathologists.
Immunofluorescence (IF).
Cells were fixed with 10% paraformaldehyde for 30 min, permeabilized using 0.5% Triton X-100 for 15 min, and blocked using normal donkey serum for 30 min. The primary antibodies (RARγ, 1:100; β-catenin, 1:100) were added and incubated at 4°C overnight. The slides were washed and incubated with Alexa Fluor 488- and Alexa Fluor 594-conjugated secondary antibodies (1:500) at room temperature for 1 h and then costained with DAPI (0.1 μg/ml) to visualize the nuclei. Negative-control experiments were carried out without primary antibody. The staining was examined by using a Leica TCS SP5 II laser confocal microscope (Leica, Barcelona, Spain).
Luciferase reporter assay.
QBC939 cells (1.0 × 105 cells/well) were seeded in 24-well plates for 24 h before transfection. Then, cells in each well were cotransfected with 500 ng TOP Flash or FOP Flash plasmid and 200 ng β-galactosidase (β-Gal) (for transfection efficiency normalization) expression vectors using Lipofectamine 2000. The indicated cells treated with Wnt3a (50 ng/ml) or RA (20 μM) for 6 h were analyzed for luciferase activity by using a dual-luciferase reporter assay system (Promega, Madison, WI, USA), according to the manufacturer's instructions, with a Modulus single-tube-type multifunction detector (YuanPingHao Bio, Beijing, China).
Coimmunoprecipitation (co-IP) assay.
The cells were lysed in 500 μl of lysis buffer (10 mM Tris-HCl, pH 8.0, 100 mM NaC1, 10 mM EDTA, and 1% NP-40) containing protease inhibitors. The lysates were incubated with 20 μl of anti-RARγ monoclonal antibody at 4°C for 2 h. Immunocomplexes were then precipitated with 20 μl of protein A-Sepharose and incubated for 3 h at 4°C with gentle rotation. After extensive washing with lysis buffer, the beads were boiled in 40 μl lysis buffer and 10 μl 5× loading buffer and analyzed by Western blotting. Normal mouse IgG was used as a negative control.
Statistical analysis.
Data from IHC were analyzed by Pearson's χ2 test, and the overall survival rate was evaluated by the Kaplan-Meier method. The significance of differences between groups was evaluated by Student's t test or one-way analysis of variance (ANOVA) when applicable. Data are represented as means and standard errors of the mean (SEM) from at least three independent experiments. All data were processed with the SPSS 16.0 statistical software package (SPSS Inc., Chicago, IL, USA).
RESULTS
Clinical significance and overexpression of RARγ in CCA.
To assess the expression of RARγ in CCA, qPCR and IHC were performed in a set of 42 tumor tissues (T) and their adjacent noncancerous tissues (A), as well as 27 cholangitis (C) and 13 normal tissues (N). As shown in Fig. 1A, RARγ mRNA expression was significantly elevated in 42 T specimens compared with A, C, and N specimens (P < 0.05). The IHC results showed that RARγ protein, predominantly present in the cell cytoplasm, was strongly stained in CCA tissues but weakly or not stained in other tissues (Fig. 1B, top). The staining intensity of RARγ protein in four types of bile duct tissues was categorized as low (± to +) or high (++ to +++) (Fig. 1B, bottom), and the high-expression rate of RARγ protein in CCA tissues (73.8%) was much higher than that in other tissues (Table 1) (P < 0.01). These data demonstrated that RARγ is markedly overexpressed in CCA patients.
Fig 1.
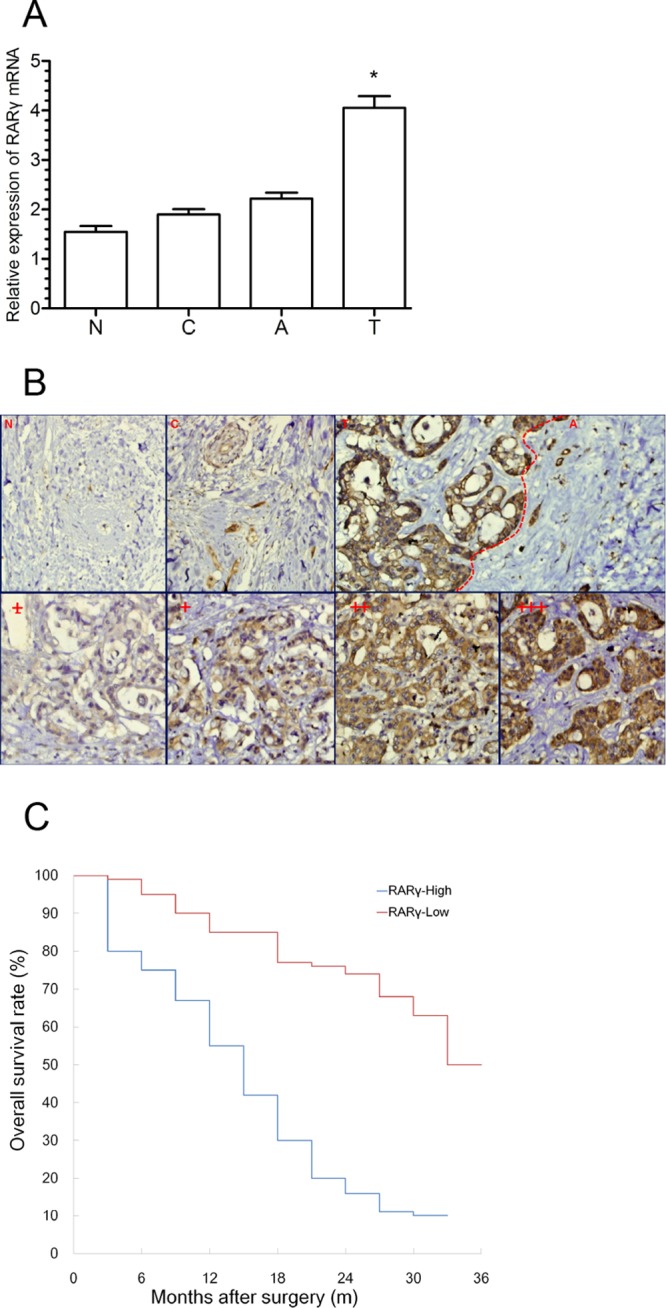
Clinical significance and overexpression of RARγ in CCA. (A) Expression of RARγ mRNA in different bile duct tissues as assessed by qPCR. T, n = 42; A, n = 42; C, n = 27; N, n = 13. The data were normalized to GAPDH. The error bars indicate SEM. *, P < 0.05. (B) Expression of RARγ protein in different bile duct tissues as detected by IHC. (Top) Expression of RARγ protein in N, C, T, and A. (Bottom) Four different grades of staining intensity according to their different positive rates of RARγ expression. The scoring criteria were as follows: +++, >60%; ++, 30 to 60%; +, 10 to 30%; and ±, <10% positive cells. Magnification, ×200. (C) Association between RARγ expression and overall survival rate analyzed by the Kaplan-Meier method.
The clinical data in Table 3 show that RARγ overexpression was significantly associated with poor differentiation, lymph node metastasis, and a high serum carbohydrate antigen 19-9 (CA19-9) level (P < 0.05), whereas no association was found with age, sex, tumor location, and a high serum carcinoembryonic antigen (CEA) level (P > 0.05). Furthermore, the postoperative overall survival rate in patients with high RARγ expression (median survival time, 9.2 months; cumulative 3-year survival rate, 0%) was lower than that in patients with low RARγ expression (median survival time, 27.6 months; cumulative 3-year survival rate, 36%; P < 0.05) (Fig. 1C). These results indicated the clinical significance of RARγ in the diagnosis and prognosis of CCA patients.
Table 3.
Associations between RARγ and clinical features of CCA
| Featurea | n | No. with RARγ expression |
P | |
|---|---|---|---|---|
| Low | High | |||
| Age (yr) | ||||
| <50 | 8 | 2 | 6 | 0.932 |
| ≥50 | 34 | 9 | 25 | |
| Sex | ||||
| Male | 32 | 9 | 23 | 0.610 |
| Female | 10 | 2 | 8 | |
| Tumor differentiation | ||||
| W + WM | 7 | 5 | 2 | 0.003b |
| M | 11 | 4 | 7 | |
| MP + P | 24 | 2 | 22 | |
| Tumor localization | ||||
| Intrahepatic | 13 | 4 | 9 | 0.541 |
| Extrahepatic | 29 | 7 | 22 | |
| Lymph node metastasis | ||||
| Positve | 27 | 3 | 24 | 0.003b |
| Negative | 15 | 8 | 7 | |
| CEA (U/ml) | ||||
| <37 | 18 | 5 | 13 | 0.839 |
| ≥37 | 24 | 6 | 18 | |
| CA 19-9 (U/ml) | ||||
| <37 | 9 | 5 | 4 | 0.024b |
| ≥37 | 33 | 6 | 27 | |
W, well differentiated; WM, well to moderately differentiated; M, moderately differentiated; MP, moderately to poorly differentiated; P, poorly differentiated.
A P value of less than 0.05 was considered statistically significant.
Downregulation of RARγ inhibits CCA cell proliferation and cell cycle progression.
To study the role of RARγ in the progression of CCA, RNA interference was employed. As shown in Fig. 2A, the endogenous RARγ but not RARα or RARβ mRNA levels in three CCA cell lines were reduced after transfection with siRARγ compared with their respective controls. Furthermore, RARγ knockdown resulted in suppression of cell proliferation at 3 days to 5 days. QBC939 cells stably transfected with shRARγ also showed decreased RARγ expression and significantly reduced proliferation rates compared to those with shCtrl (Fig. 2B). These data indicated that RARγ expression contributes to CCA cell proliferation.
Fig 2.

RARγ knockdown inhibited tumor cell growth and cell cycle progression. (A) Expression of RARα, RARβ, and RARγ mRNAs detected by qPCR in three human CCA cell lines after transfection with siRARγ or siCtrl on day 5. (B) Growth of three CCA cell lines after transfection with siRARγ or siCtrl was measured by MTT assays. The growth of QBC939 cells stably transfected with shRARγ or shCtrl was measured by direct cell counting. Downregulation of RARγ protein expression on day 5 was confirmed by Western blotting (insets). (C) Oncogenic potential of RARγ assessed by colony formation assays. The number of cells was normalized with that of QBC939 siCtrl group. (D) RARγ knockdown decreased QBC939 cell xenograft tumor growth in nude mice. (Top) Tumor sections. (Bottom left) Tumor growth curve. (Bottom right) Quantitative analysis of tumor weight. (E) Western blot of RARγ and PCNA protein expression in shRARγ and shCtrl QBC939 cell xenograft tumor tissues (left) and quantitative results (right). (F) Cell cycle analysis of shRARγ and shCtrl QBC939 cells by flow cytometry. (G) Expression of cell cycle-associated proteins in shRARγ and shCtrl QBC939 cells detected by Western blotting (left) and quantitative results (right). n = 3. *, P < 0.05; **, P < 0.01. The error bars indicate SEM.
To investigate the oncogenic potential of RARγ, colony formation assays were performed in the cell culture plates. The abilities of three CCA cell lines to form foci were significantly impaired after transfection with siRARγ compared to that with siCtrl (Fig. 2C). We further assessed the effects of RARγ knockdown on the growth of CCA xenograft tumors in nude mice. RARγ knockdown led to slower growth, lighter tumor weight (Fig. 2D), and decreased expression of PCNA protein in QBC939 tumors (Fig. 2E). Therefore, RARγ seems to be important for CCA cell tumorigenesis.
Cell cycle analysis was performed to examine whether there is cell cycle arrest at a specific phase in RARγ knockdown cells. Flow cytometry results showed that RARγ knockdown increased the number of cells in G1 phase and decreased th number of cells in S and G2/M phases (Fig. 2F). Western blotting results showed that P21 was increased and cyclin D1 was decreased in shRARγ cells compared with shCtrl cells, whereas the levels of other proteins were comparable (Fig. 2G). These results were in line with the qPCR results (data not shown). The data suggested that RARγ might promote CCA cell cycle procession by regulating P21 and cyclin D1.
RARγ knockdown inhibits metastatic abilities and enhances drug sensitivity of CCA cells.
In the clinical study, overexpression of RARγ was associated with lymph node metastasis, suggesting that RARγ may play a role in CCA metastasis. To address this issue, the effects of RARγ knockdown on the migratory and invasive abilities of CCA cells were evaluated by using wound-healing and transwell assays. RARγ knockdown inhibited the migration of QBC939 cells compared with that of the groups treated with TPA (promoting tumor cell invasion via MMP-9 induction [17]) or shCtrl cells (Fig. 3A). The data from transwell assays also showed that the migratory and invasive abilities of QBC939 cells were inhibited by RARγ knockdown (Fig. 3B). Additionally, the mRNA expression and enzymatic activity of MMP-9 were significantly decreased by RARγ knockdown in the TPA-treated group (Fig. 3C and D). Hence, MMP-9 might be involved in the suppression of CCA cell migration and invasion induced by RARγ knockdown.
Fig 3.

(A) Cell migration was detected by wound-healing assay. QBC939-shCtrl and -shRARγ cells were wounded with a 10-μl plastic pipette tip and cultured in serum-free medium for 48 h. One group of QBC939-shCtrl cells was treated with TPA as a positive control. (B) Effects of RARγ knockdown on migration and invasion of CCA cells. A representative cell migration and invasion assay from transwell analysis (top) and the calculated number from triplicates (bottom) are shown. shCtrl cells treated with TPA for 2 h served as a positive control. Magnification, ×100. (C) mRNA expression of MMP-9 measured by qPCR. (D) MMP-9 enzymatic activity assessed by gelatin zymography. (E) The sensitivity of three CCA cells to 5-FU was significantly enhanced after RARγ knockdown. (F) Effects of RARγ knockdown on drug sensitivity of QBC939 cell xenograft tumors in nude mice: tumor sections (left) and growth inhibition rates of 5-FU in shRARγ and shCtrl tumors (right). (G) Expression of P-gp protein in QBC939 cells and xenograft tumor tissues detected by IHC. Magnification, ×400. TPA, 100 nM. *, P < 0.05. The error bars indicate SEM.
The influence of RARγ knockdown on the drug sensitivity of CCA cells was studied as well. The results showed that RARγ knockdown enhanced the sensitivity of all three CCA cell lines to 5-FU (Fig. 3E). The growth rates of shRARγ and shCtrl QBC939 xenograft tumors in nude mice with 5-FU treatment were compared. There was no obvious difference in tumor weights between the PBS and 5-FU treatments in the shCtrl group (Fig. 3F). However, the growth inhibition rate of 5-FU in shRARγ tumors (56.7%) was higher than that in the shCtrl group (5.6%). IHC staining showed reduced expression of P-gp in both shRARγ cells and tumors compared to their respective controls (Fig. 3G). These data suggested that downregulation of P-gp may be involved in the enhanced sensitivity of CCA cells to 5-FU induced by RARγ knockdown.
RARγ knockdown inhibits the activation of the Akt/NF-κB pathway.
To determine the mechanisms by which RARγ elicits oncogenic activity, we investigated the effects of RARγ on the regulation of the Akt and NF-κB signaling pathways in CCA, which have been reported in HCC (15). Figure 4A shows that the expression of P21 was upregulated but PCNA was downregulated in the shRARγ cells, as well as in cells treated with LY294002 or BMS345541, specific inhibitors of Akt and NF-κB, respectively. The expression of the P-gp protein was markedly downregulated by shRARγ or Akt inhibitor compared with that of the control. In addition, RARγ knockdown, as well as that of the inhibitors, significantly reduced TPA-induced MMP-9 mRNA expression (Fig. 4B). These data indicated that PCNA, P21, and MMP-9 expression regulated by RARγ may be linked with activation of the Akt and NF-κB pathways and that the downregulation of P-gp in shRARγ cells may be in part through other pathways.
Fig 4.
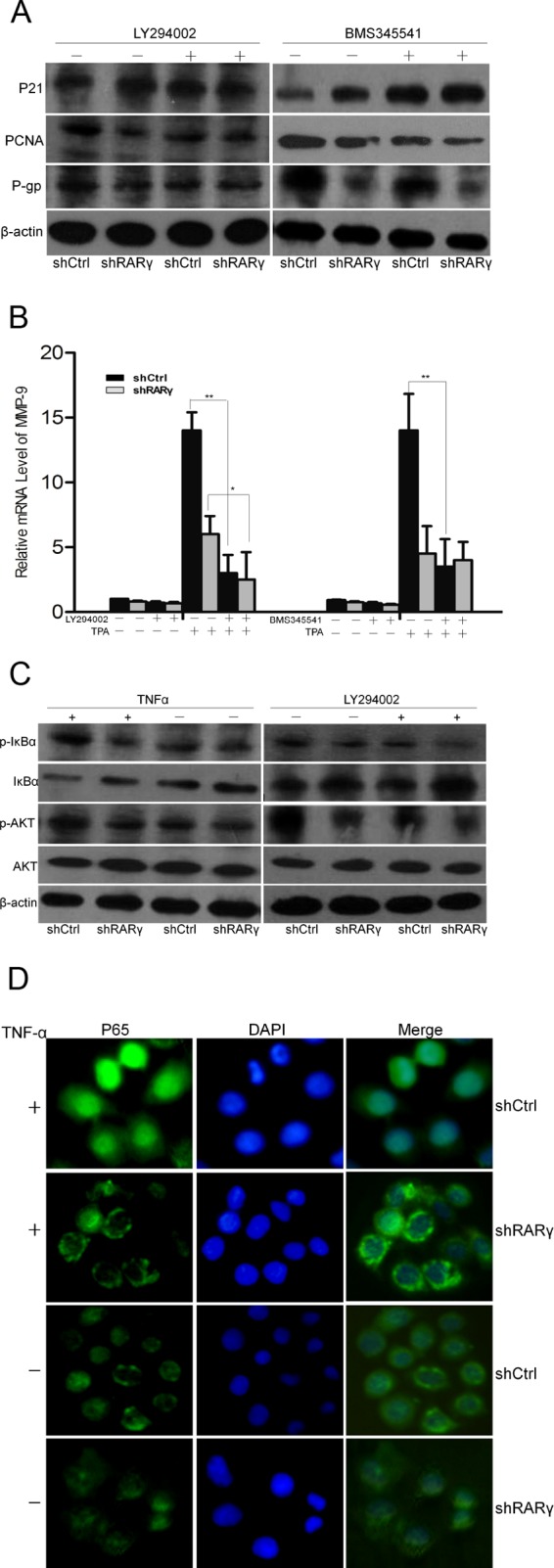
Activation of the Akt/NF-κB pathway in shCtrl and shRARγ cells in the presence or absence of inhibitors. (A) Western blot analysis of P21, PCNA, and P-gp protein expression. (B) Expression of MMP-9 mRNA detected by qPCR in cells with or without TPA treatment. The error bars indicate SEM. (C) Phosphorylation of key signaling proteins for the Akt/NF-κB pathway measured by Western blotting. (D) Cellular localization of p65 after TNF-α treatment detected by IF. LY294002, 1 μM; BMS345541, 1 μM; TPA, 100 nM; TNF-α, 30 μg/liter. Magnification, ×200. *, P < 0.05; **, P < 0.01.
RARγ-mediated activation of the Akt/NF-κB pathway was further illustrated by results showing that TNF-α-induced phosphorylation of Akt and IκB was inhibited by RARγ knockdown and that IκB phosphorylation was inhibited by LY294002 (Fig. 4C). Therefore, RARγ-mediated activation of Akt is required for the activation of NF-κB. Furthermore, IF results revealed that TNF-α-induced p65 nuclear translocation was suppressed by RARγ knockdown (Fig. 4D). Collectively, these results demonstrated that RARγ is involved in the regulation of the Akt/NF-κB pathway.
RARγ upregulates P-gp through activation of the Wnt/β-catenin pathway.
Cyclin D1, a target of the Wnt/β-catenin pathway (18–21), was downregulated in shRARγ cells (Fig. 2G). Interestingly, RA treatment upregulated RARγ in 4 h and downregulated β-catenin phosphorylation in 16 h, as well as upregulating total β-catenin in 8 h (Fig. 5A). Luciferase reporter assays showed that activation of the Wnt/β-catenin pathway was remarkably enhanced by RA or Wnt3a treatment and was abolished by RARγ or β-catenin knockdown (Fig. 5B). Western blotting revealed that expression of P-gp protein was upregulated by RA or Wnt3a treatment and retarded by siβ-catenin or siRARγ (Fig. 5C). These data suggested that the activation of the Wnt/β-catenin pathway is necessary for RARγ-induced upregulation of P-gp.
Fig 5.

RARγ knockdown inhibited activation of the Wnt/β-catenin pathway. (A) Western blot analysis of RARγ, β-catenin, and phosphorylated β-catenin (p-β-catenin) expression in QBC939 cells with RA treatment. (B) Activation of Wnt signaling assessed by β-catenin/T-cell factor (TCF)-responsive luciferase reporter activity, normalized with β-Gal. (C) Western blot analysis of β-catenin and P-gp expression in QBC939 cells with different treatments. (D) Subcellular colocalization of endogenous RARγ and β-catenin detected by IF. Magnification, ×200. (E) Interaction of endogenous RARγ and β-catenin in QBC939 whole-cell extract, cytoplasm, and nuclear extracts assessed by co-IP. No interaction between RARγ and β-catenin was seen in the whole-cell extract without RA treatment (left), whereas the interaction was seen in both the cytoplasm and nucleus of the RA-treated cells (right).WB, Western blot. (F) Schematic model for the mechanism by which RARγ promotes the growth and chemoresistance of CCA through simultaneous activation of the Akt/NF-κB and Wnt/β-catenin pathways. The small arrows indicate the net effects of actions of signal transduction. RA, 20 μM; Wnt3a, 50 μg/liter. n = 3. *, P < 0.05; **, P < 0.01.
In order to explain the mechanisms by which RARγ activates the Wnt/β-catenin pathway, the precise cellular localization of β-catenin was observed under a laser scanning confocal microscope with IF. As shown in Fig. 5D, β-catenin and RARγ were predominantly expressed in the cytoplasm in shCtrl cells. Preincubation with RA significantly increased nuclear translocation of β-catenin, which could be abolished by RARγ knockdown (shRARγ). Additionally, the interaction of endogenous RARγ and β-catenin in QBC939 cells was evaluated by co-IP assays, and the results suggested that when treated with RA, RARγ regulated the localization pattern of β-catenin through physical interaction with β-catenin in both the cytoplasm and the nucleus (Fig. 5E). Collectively, these data demonstrated that RARγ served as an essential coactivator for Wnt/β-catenin pathway activation by physically interacting with β-catenin and thus enhances its transcriptional activity for P-gp.
DISCUSSION
RARγ shows various biological functions through both genomic and nongenomic regulation, which are associated with not only different cell types but also an aberrant expression profile for itself (22). Altered expression and subcellular localization of retinoid receptors are closely linked with cancer progression (23–25). In the present study, we revealed that RARγ was significantly elevated and abnormally expressed in the cytoplasm in human CCA. Our results are in concert with a previous study regarding the expression profile of RARγ in HCC, which suggests that the overexpression of RARγ may be a common event in patients with primary hepatobiliary malignancy (15). Expression of RARγ might be a valuable factor in estimating malignancy, tumor aggressiveness, and chemotherapy efficacy, which are highly associated with the prognosis of CCA (26). Therefore, RARγ could be a potential predictor of poor prognosis of CCA patients after curative resection, and its overexpression should be taken into account in following the optimal treatment scheme.
Previous reports have demonstrated that RARγ is an important regulator of embryonic development, cell differentiation, metabolism, and apoptosis. RARγ acts as a tumor suppressor or oncogene in different cancers, depending on the cell-specific context (6–10, 15). In the present study, we provided strong evidence that RARγ is a pivotal oncogene in CCA. First, RARγ plays an active role in promoting cell proliferation and tumorigenicity, enhancing migration and invasion, and inducing multidrug resistance of CCA cells. Second, RARγ facilitated the G1/S transition; regulated P21 and cyclin D1, two critical cell cycle regulators (27); and upregulated the expression of the proliferation marker PCNA (28), which may contribute to tumor growth. Third, CCA is characterized by early metastasis (1), in which MMP-9 plays a critical role (29). RARγ knockdown downregulated MMP-9, supporting a clinical observation that overexpression of RARγ is frequent in patients with lymph node metastasis. Last but not least, due to multidrug resistance in medical treatment, CCA has a poor overall survival rate despite advances in chemotherapy (1), and the mechanisms behind this have been only partially elucidated (30). One of the best-studied mechanisms is the increased P-gp expression (31). We also found that overexpression of RARγ enhanced the multidrug resistance of CCA cells via upregulation of P-gp. This may partially explain the reason for the short overall survival time of patients with high RARγ expression. Taken together, RARγ could potently facilitate tumorigenesis and metastasis in many respects throughout the progression of CCA.
Our findings regarding the molecular mechanism of the oncogenic activity of RARγ in the progression of CCA was in agreement with a previous study showing that the oncogenic effects of RARγ could be due to Akt/NF-κB pathway activation in HCC (15). Cytoplasmic localization of nuclear receptors not only regulates gene transcription by modulating their availability in the nucleus but also plays an active role in the cross talk with other pathways (32). For example, RAR subtypes are able to activate the phosphatidylinositol 3-kinase (PI3K)/Akt pathway (13). The Akt/NF-κB pathway, a potent regulator of many biological functions, including cell proliferation, survival, metabolism, and metastasis, is one of the major survival pathways, while its altered activation is common in many cancers, including CCA (33, 34). Our study revealed that RARγ knockdown, as well as an Akt or NF-κB pathway inhibitor, could regulate the expression of PCNA, P21, cyclin D1, and MMP-9. These results elucidated the molecular mechanism by which RARγ plays oncogenic roles in the cell proliferation and metastasis of CCA.
The survival of the patients with high RARγ expression was worse than that of patients with low RARγ expression, suggesting that RARγ may regulate the efficacy of chemotherapy. Importantly, our results demonstrated that the Wnt/β-catenin pathway was involved in the RARγ-induced upregulation of P-gp in CCA cells. As previous studies reported, many oncogenic factors lead to sustained activation of the Wnt/β-catenin pathway in the development of cancers, including CCA (33, 35, 36). Consequently, instead of being degraded in the ubiquitin proteasome, β-catenin translocates to the nucleus, where it interacts with LEF/TCF transcription factors to regulate target gene expression (37). It has recently been reported that RARγ interacts with β-catenin and induces dissociation of β-catenin from lymphoid enhancer factor in chondrocyte nuclei (38). However, our study showed that the interaction of RARγ and β-catenin in the cytoplasm of CCA cells might result in upregulation of β-catenin and/or facilitate β-catenin nuclear translocation and subsequently lead to the activation of the Wnt/β-catenin pathway. The paradoxical roles of RARγ in the regulation of β-catenin might depend on its particular cellular location. Additionally, the activation of Wnt/β-catenin signaling contributes to multidrug resistance of tumor or endothelial cells through upregulation of P-gp (18, 19, 21). Indeed, we also found that RARγ upregulated P-gp expression through activation of the Wnt/β-catenin pathway.
In conclusion, the present work not only identified RARγ as a pivotal oncogene in the progression of CCA but also clarified one of the mechanisms by which RARγ could simultaneously activate the Akt/NF-κB and Wnt/β-catenin pathways (Fig. 5F). Our findings are potentially beneficial for future development of novel therapeutic strategies in CCA.
ACKNOWLEDGMENTS
This work was supported by grants from the National Nature Science Foundation of China (81101502) and the Youth Foundation of Fujian Health Department, China (2011-2-58).
We thank Xiaokun Zhang (Xiamen University, Xiamen, China) for providing the pll3.7-RARγ plasmid and Yabing Chen (University of Alabama at Birmingham, Birmingham, AL, USA) for providing SK-ChA-1 and MZ-ChA-1 cells.
We have no conflicting interests to disclose.
Footnotes
Published ahead of print 24 June 2013
REFERENCES
- 1.Aljiffry M, Walsh MJ, Molinari M. 2009. Advances in diagnosis, treatment and palliation of cholangiocarcinoma: 1990–2009. World J. Gastroenterol. 15:4240–4262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Khan SA, Toledano MB, Taylor-Robinson SD. 2008. Epidemiology, risk factors, and pathogenesis of cholangiocarcinoma. HPB 10:77–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gronemeyer H, Gustafsson JA, Laudet V. 2004. Principles for modulation of the nuclear receptor superfamily. Nat. Rev. Drug Discov. 3:950–964 [DOI] [PubMed] [Google Scholar]
- 4.Marinelli A, Bossi D, Pelicci PG, Minucci S. 2007. A redundant oncogenic potential of the retinoic receptor (RAR) alpha, beta and gamma isoforms in acute promyelocytic leukemia. Leukemia 21:647–650 [DOI] [PubMed] [Google Scholar]
- 5.Purton LE, Dworkin S, Olsen GH, Walkley CR, Fabb SA, Collins SJ, Chambon P. 2006. RARgamma is critical for maintaining a balance between hematopoietic stem cell self-renewal and differentiation. J. Exp. Med. 203:1283–1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao X, Demary K, Wong L, Vaziri C, McKenzie AB, Eberlein TJ, Spanjaard RA. 2001. Retinoic acid receptor-independent mechanism of apoptosis of melanoma cells by the retinoid CD437 (AHPN). Cell Death Differ. 8:878–886 [DOI] [PubMed] [Google Scholar]
- 7.Goranov BB, Campbell Hewson QD, Pearson AD, Redfern CP. 2006. Overexpression of RARgamma increases death of SH-SY5Y neuroblastoma cells in response to retinoic acid but not fenretinide. Cell Death Differ. 13:676–679 [DOI] [PubMed] [Google Scholar]
- 8.Walkley CR, Olsen GH, Dworkin S, Fabb SA, Swann J, McArthur GA, Westmoreland SV, Chambon P, Scadden DT, Purton LE. 2007. A microenvironment-induced myeloproliferative syndrome caused by retinoic acid receptor gamma deficiency. Cell 129:1097–1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao X, Graves C, Ames SJ, Fisher DE, Spanjaard RA. 2009. Mechanism of regulation and suppression of melanoma invasiveness by novel retinoic acid receptor-gamma target gene carbohydrate sulfotransferase 10. Cancer Res. 69:5218–5225 [DOI] [PubMed] [Google Scholar]
- 10.Chen CF, Goyette P, Lohnes D. 2004. RARgamma acts as a tumor suppressor in mouse keratinocytes. Oncogene 23:5350–5359 [DOI] [PubMed] [Google Scholar]
- 11.Aggarwal S, Kim SW, Cheon K, Tabassam FH, Yoon JH, Koo JS. 2006. Nonclassical action of retinoic acid on the activation of the cAMP response element-binding protein in normal human bronchial epithelial cells. Mol. Biol. Cell 17:566–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hughes PJ, Zhao Y, Chandraratna RA, Brown G. 2006. Retinoid-mediated stimulation of steroid sulfatase activity in myeloid leukemic cell lines requires RARalpha and RXR and involves the phosphoinositide 3-kinase and ERK-MAP kinase pathways. J. Cell. Biochem. 97:327–350 [DOI] [PubMed] [Google Scholar]
- 13.Masia S, Alvarez S, de Lera AR, Barettino D. 2007. Rapid, nongenomic actions of retinoic acid on phosphatidylinositol-3-kinase signaling pathway mediated by the retinoic acid receptor. Mol. Endocrinol. 21:2391–2402 [DOI] [PubMed] [Google Scholar]
- 14.Dey N, De PK, Wang M, Zhang H, Dobrota EA, Robertson KA, Durden DL. 2007. CSK controls retinoic acid receptor (RAR) signaling: a RAR-c-SRC signaling axis is required for neuritogenic differentiation. Mol. Cell. Biol. 27:4179–4197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yan TD, Wu H, Zhang HP, Lu N, Ye P, Yu FH, Zhou H, Li WG, Cao X, Lin YY, He JY, Gao WW, Zhao Y, Xie L, Chen JB, Zhang XK, Zeng JZ. 2010. Oncogenic potential of retinoic acid receptor-gamma in hepatocellular carcinoma. Cancer Res. 70:2285–2295 [DOI] [PubMed] [Google Scholar]
- 16.Shen DY, Zhan YH, Wang QM, Rui G, Zhang ZM. 2013. Oncogenic potential of cyclin kinase subunit-2 in cholangiocarcinoma. Liver Int. 33:137–148 [DOI] [PubMed] [Google Scholar]
- 17.Zhou X, Yang J, Wang Y, Li W, Li-Ling J, Deng Y, Zhang M. 2012. Cucurbitacin B inhibits 12-O-tetradecanoylphorbol 13-acetate-induced invasion and migration of human hepatoma cells through inactivating mitogen-activated protein kinase and PI3K/Akt signal transduction pathways. Hepatol. Res. 42:401–411 [DOI] [PubMed] [Google Scholar]
- 18.Chikazawa N, Tanaka H, Tasaka T, Nakamura M, Tanaka M, Onishi H, Katano M. 2010. Inhibition of Wnt signaling pathway decreases chemotherapy-resistant side-population colon cancer cells. Anticancer Res. 30:2041–2048 [PubMed] [Google Scholar]
- 19.Flahaut M, Meier R, Coulon A, Nardou KA, Niggli FK, Martinet D, Beckmann JS, Joseph JM, Muhlethaler-Mottet A, Gross N. 2009. The Wnt receptor FZD1 mediates chemoresistance in neuroblastoma through activation of the Wnt/beta-catenin pathway. Oncogene 28:2245–2256 [DOI] [PubMed] [Google Scholar]
- 20.Shtutman M, Zhurinsky J, Simcha I, Albanese C, D'Amico M, Pestell R, Ben-Ze'ev A. 1999. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc. Natl. Acad. Sci. U. S. A. 96:5522–5527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang H, Zhang X, Wu X, Li W, Su P, Cheng H, Xiang L, Gao P, Zhou G. 2012. Interference of Frizzled 1 (FZD1) reverses multidrug resistance in breast cancer cells through the Wnt/beta-catenin pathway. Cancer Lett. 323:106–113 [DOI] [PubMed] [Google Scholar]
- 22.Fukunaka K, Saito T, Wataba K, Ashihara K, Ito E, Kudo R. 2001. Changes in expression and subcellular localization of nuclear retinoic acid receptors in human endometrial epithelium during the menstrual cycle. Mol. Hum. Reprod. 7:437–446 [DOI] [PubMed] [Google Scholar]
- 23.Albrechtsson E, Ohlsson B, Axelson J. 2002. The expression of retinoic acid receptors and the effects in vitro by retinoids in human pancreatic cancer cell lines. Pancreas 25:49–56 [DOI] [PubMed] [Google Scholar]
- 24.Chakravarti N, Lotan R, Diwan AH, Warneke CL, Johnson MM, Prieto VG. 2007. Decreased expression of retinoid receptors in melanoma: entailment in tumorigenesis and prognosis. Clin. Cancer Res. 13:4817–4824 [DOI] [PubMed] [Google Scholar]
- 25.Kumar A, Kaur J, Chattopadhyay TK, Mathur M, Ralhan R. 2004. Differential expression of retinoic acid receptors in normal and malignant esophageal tissues. J. Exp. Ther. Oncol. 4:1–8 [PubMed] [Google Scholar]
- 26.Briggs CD, Neal CP, Mann CD, Steward WP, Manson MM, Berry DP. 2009. Prognostic molecular markers in cholangiocarcinoma: a systematic review. Eur. J. Cancer 45:33–47 [DOI] [PubMed] [Google Scholar]
- 27.Hahnvajanawong C, Ketnimit S, Pattanapanyasat K, Anantachoke N, Sripa B, Pinmai K, Seubwai W, Reutrakul V. 2012. Involvement of p53 and nuclear factor-kappaB signaling pathway for the induction of G1-phase cell cycle Arrest of cholangiocarcinoma cell lines by isomorellin. Biol. Pharm. Bull. 35:1914–1925 [DOI] [PubMed] [Google Scholar]
- 28.Batheja N, Suriawinata A, Saxena R, Ionescu G, Schwartz M, Thung SN. 2000. Expression of p53 and PCNA in cholangiocarcinoma and primary sclerosing cholangitis. Mod. Pathol. 13:1265–1268 [DOI] [PubMed] [Google Scholar]
- 29.Itatsu K, Sasaki M, Yamaguchi J, Ohira S, Ishikawa A, Ikeda H, Sato Y, Harada K, Zen Y, Sato H, Ohta T, Nagino M, Nimura Y, Nakanuma Y. 2009. Cyclooxygenase-2 is involved in the up-regulation of matrix metalloproteinase-9 in cholangiocarcinoma induced by tumor necrosis factor-alpha. Am. J. Pathol. 174:829–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sirica AE. 2005. Cholangiocarcinoma: molecular targeting strategies for chemoprevention and therapy. Hepatology 41:5–15 [DOI] [PubMed] [Google Scholar]
- 31.Liu ZH, He YP, Zhou Y, Zhang P, Qin H. 2011. Establishment and identification of the human multi-drug-resistant cholangiocarcinoma cell line QBC939/ADM. Mol. Biol. Rep. 38:3075–3082 [DOI] [PubMed] [Google Scholar]
- 32.Sadana P. 2012. Noncanonical mechanisms to regulate nuclear receptor signaling. Future Med. Chem. 4:1307–1333 [DOI] [PubMed] [Google Scholar]
- 33.Prakobwong S, Gupta SC, Kim JH, Sung B, Pinlaor P, Hiraku Y, Wongkham S, Sripa B, Pinlaor S, Aggarwal BB. 2011. Curcumin suppresses proliferation and induces apoptosis in human biliary cancer cells through modulation of multiple cell signaling pathways. Carcinogenesis 32:1372–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schmitz KJ, Lang H, Wohlschlaeger J, Sotiropoulos GC, Reis H, Schmid KW, Baba HA. 2007. AKT and ERK1/2 signaling in intrahepatic cholangiocarcinoma. World J. Gastroenterol. 13:6470–6477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sugimachi K, Taguchi K, Aishima S, Tanaka S, Shimada M, Kajiyama K, Tsuneyoshi M. 2001. Altered expression of beta-catenin without genetic mutation in intrahepatic cholangiocarcinoma. Mod. Pathol. 14:900–905 [DOI] [PubMed] [Google Scholar]
- 36.Tokumoto N, Ikeda S, Ishizaki Y, Kurihara T, Ozaki S, Iseki M, Shimizu Y, Itamoto T, Arihiro K, Okajima M, Asahara T. 2005. Immunohistochemical and mutational analyses of Wnt signaling components and target genes in intrahepatic cholangiocarcinomas. Int. J. Oncol. 27:973–980 [PubMed] [Google Scholar]
- 37.Clevers H, Nusse R. 2012. Wnt/beta-catenin signaling and disease. Cell 149:1192–1205 [DOI] [PubMed] [Google Scholar]
- 38.Yasuhara R, Yuasa T, Williams JA, Byers SW, Shah S, Pacifici M, Iwamoto M, Enomoto-Iwamoto M. 2010. Wnt/beta-catenin and retinoic acid receptor signaling pathways interact to regulate chondrocyte function and matrix turnover. J. Biol. Chem. 285:317–327 [DOI] [PMC free article] [PubMed] [Google Scholar]