

Abstract
RNA processing and transport are mediated by cotranscriptionally assembled ribonucleoprotein (RNP) complexes. RNPs have been postulated to help specify coordinated gene expression, but the requirements for specific RNP complexes in mammalian development and tissue homeostasis have not been extensively evaluated. THO is an evolutionarily conserved RNP complex that links transcription with nuclear export. THO is not essential for Saccharomyces cerevisiae viability, but it is essential for early mouse embryonic development. Embryonic lethality has limited the characterization of THO requirements in adult tissues. To overcome this limitation, a mouse model has been generated that allows widespread inducible deletion of Thoc1, which encodes an essential protein subunit of THO. Widespread Thoc1 deletion disrupts homeostasis within the small intestine but does not have detectable effects in other epithelial tissues such as the related mucosa of the large intestine. Thoc1 loss compromises the proliferation and lineage-generating capacity of small intestinal stem cells, disrupting the supply of differentiated cells in this rapidly renewing tissue. These findings demonstrate that the effects of THO deficiency in the adult mouse are tissue and cell type dependent.
INTRODUCTION
Numerous RNA processing and transport events are required before nascent transcripts mature into functional mRNA available for translation in the cytoplasm. These RNA processing and transport events are regulated by ribonucleoprotein (RNP) complexes that begin forming cotranscriptionally (1). RNPs are composed of a diverse array of proteins and noncoding RNAs with combinatorial complexity sufficient to accommodate unique RNP-mediated processing mechanisms for different subsets of transcripts (2). Since transcripts that encode functionally related proteins can be regulated by a common RNP processing pathway, RNP-mediated mechanisms have been hypothesized to provide an additional layer of regulation that is necessary for specifying coordinated gene expression (3). The observation that mutations in some RNPs can cause specific developmental defects in humans supports this hypothesis (4), but the contribution of most RNP complexes to normal development and tissue homeostasis is unknown.
THO is an evolutionarily conserved, cotranscriptionally assembled RNP complex that is important for coupling transcription with nuclear RNA export (5–9). THO associates with the transcribing RNA polymerase II, binds nascent RNA, and recruits additional proteins to form larger complexes such as TREX that facilitate interaction with and activation of the nuclear export apparatus (10, 11). Loss of THO compromises multiple events that are dependent on RNP-mediated mechanisms, including transcriptional elongation (12–14), transcript 3′ end formation (15), and nuclear export (16). In Saccharomyces cerevisiae, THO associates with most actively transcribed genes, but THO deficiency affects the expression of only a subset of those genes (17). Thus, THO is not absolutely required for gene expression and is not required for yeast viability (18). The fraction of transcripts affected by THO deficiency appears to be restricted even further in higher eukaryotes (7, 14, 19, 20), perhaps because multiple nuclear export pathways exist (21), because different mechanisms are utilized to recruit THO to nascent transcripts (22), or because the subunit composition of metazoan THO differs from that seen with yeast. It is also noteworthy that different subsets of transcripts are dependent on THO depending on the biological context, suggesting the potential for cell type-specific transcript regulation.
Mammalian THO contains six proteins encoded by the Thoc1, Thoc2, Thoc5, Thoc6, Thoc7, and Tex1 genes (23). Thoc1 and Thoc5 have each been demonstrated to be required for mouse embryonic development (20, 24), but the requirements for THO in adult tissue development and homeostasis are not well characterized. Inducible Cre recombinase-mediated Thoc5 gene deletion has been reported to affect hematopoiesis, but effects on other tissues are unknown because of the limited distribution of tissues suffering Thoc5 deficiency (20). Mice homozygous for a hypomorphic Thoc1 allele, which expresses reduced Thoc1 protein in many tissues, are viable but sterile (19, 25). This supports the hypothesis that different tissues have different requirements for Thoc1 during development. To compare the effects of Thoc1 deficiency on the homeostasis of adult tissues, a mouse model has been engineered to allow inducible, widespread deletion of floxed Thoc1 alleles using the RosaCreERT2 knock-in allele (26). While a number of tissues do not exhibit detectable phenotypes upon Thoc1 deletion, significant disruption of stem/progenitor cell homeostasis is observed in the small intestinal crypt. This defect severely compromises the integrity of the tissue, eventually causing death.
MATERIALS AND METHODS
Mice, genotyping, and taxoxifen treatment.
The creation and genotyping of the floxed Thoc1 allele has been previously described (25). Mice containing the Rosa26CreERT2 (26), Lgr5GFP-IRES-CreERT2 (27), and RosamT/mG (28) alleles have been obtained from The Jackson Laboratories. Mouse mammary tumor virus (MMTV)-Cre mice were obtained from the NCI Mouse Models of Human Cancer Consortium (29). Mice used were on a mixed genetic background, typically C57BL/6::129SvJ. DNA from an ear or tail clipping of 7- to 9-day-old pups was used for genotyping. Tamoxifen (Sigma-Aldrich, St. Louis, MO) at 20 mg/ml was prepared and administered to 9- to 12-week-old mice via intraperitoneal injection at a dose of 2 mg/day. For survival studies, tamoxifen was given daily for 3 to 6 consecutive days. Weight was monitored during and after treatment. Survival curves were generated using the Kaplan-Meier method. For time course studies, 2 mg/day tamoxifen was given daily for 1 to 6 days. Mice were euthanized 24 h after each day of treatment. All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at the Roswell Park Cancer Institute (RPCI).
Tissue collection, histology, and immunostaining.
All examined tissues were dissected immediately following euthanization, and sections of each tissue were snap-frozen in liquid nitrogen for protein/RNA extraction or fixed in 4% paraformaldehyde (J. T. Baker, Center Valley, PA) overnight at 4°C. Following fixation, tissues were washed in phosphate-buffered saline (PBS) (three times for 20 min each time) and in 65% ethyl alcohol (EtOH) (once for 30 min) and stored in 70% EtOH. “Swiss rolls” were prepared for examining the histology of the small and large intestines (30). Tissue was processed by incubation in 70% EtOH, 95% EtOH, 100% EtOH, xylene, paraffin no. 1 for 30 min, paraffin no. 2 for 30 min, and paraffin no. 3 for 14 h. Five-micrometer-thick sections were cut from paraffin-embedded blocks and were mounted on charged microscope slides. Prior to staining, slides were deparaffinized and rehydrated with xylene and graded alcohol and equilibrated with Tris-phosphate buffer. Slides were stained with hematoxylin and eosin (H&E) using standard procedures. Image J was used to measure the length and width of 30 to 50 intestinal crypts and villi from 4 to 5 test and control mice at each time point examined. Mammary gland whole mounts were prepared and stained as described previously (31). For immunohistochemical and diaminobenzidine staining, a citrate-PBST (0.1% Tween 20–1× PBS) system was used at pH 6.0 according to the manufacturer's recommendations (Vectastain ABC rabbit kit; Vector Laboratories, Burlingame, CA). The following primary antibodies were used: rabbit polyclonal cleaved caspase-3 (Cell Signaling; catalog no. 9661), rabbit polyclonal Ki67 (1:1,000), rabbit polyclonal lysozyme (Dako; catalog no. A0099) (1:1,000), and rabbit polyclonal green fluorescent protein (GFP) (Cell Signaling; catalog no. 2555S) (1:500). Ki67 and activated caspase-3 immunostaining was quantified in the colon, in the small intestinal crypt base, and in the small intestinal transit-amplifying region of four to five test and control mice at each time point. The cells below the +4 position were designated the small intestinal crypt base. Everything above the +4 position with respect to the crypt-villus junction was designated the transit-amplifying region. A total of 30 to 50 crypts were counted per mouse. Positive-cell counts in tissue sections were compared by the Holm-Sidak t test to correct for multiple comparisons.
Isolation of epithelial cell fractions.
Crypt and villus fractions of small intestinal epithelial tissue were isolated as described previously (32). Briefly, freshly harvested small intestinal tissue was rinsed with ice-cold phosphate-buffered saline, cut open along longitudinally, and cut into 2- to 3-cm lengths. Small intestinal segments were then washed in ice-cold Hanks' balanced salt solution-dithiothreitol (DTT) for 10 min at 4°C and in ice-cold chelating buffer (27 mM trisodium citrate, 5 mM Na2HPO4, 96 mM NaCl, 8 mM KH2PO4, 1.5 mM KCl, 0.5 mM DTT, 55 mM d-sorbitol, 44 mM sucrose) for 20 min. Crypt and villus epithelial cells were obtained by sequential washing (inverting tubes by hand 20 times per fraction) with the ice-cold chelating buffer in 50-ml tubes. Washes were combined to generate 3 fractions of villus epithelial cells (V1 to V3) and 2 fractions of crypt cells (C1 and C2). For routine validation of fraction purity, the presence of the crypt cell marker CcnD1 was analyzed by Western blotting. Mammary epithelial cells were isolated for protein analysis as described previously (33).
QRT-PCR.
RNA samples were extracted from mouse tissues using TRIzol reagent (Invitrogen, Grand Island, NY), treated with DNase (Fermentas, Hanover, MD), and converted into cDNA using an iScript cDNA synthesis kit (Bio-Rad, Hercules, CA). Quantitative RT-PCR (QRT-PCR) was performed using 2× iTaq Universal SYBR green Supermix (Bio-Rad, Hercules, CA) and the 7300 real-time PCR system (Applied Biosystems, Foster City, CA). A 20-μl amplification mixture consisted of 1,000 to 5,000 ng template cDNA, 10 μl of 2× iTaq Universal SYBR green Supermix, and 100 nM forward and reverse primers (Integrated DNA Technologies, Coralville, IA). Assays were run in triplicate for each target gene with no-template and no-reverse-transcriptase controls. For analysis of Thoc1, data from exon 6 (Ex6)- and Ex7-specific primers were normalized to the internal control exon 16- and 17-specific primers. All other target genes were normalized to the 18S rRNA. The ΔCT method (where “CT” represents the threshold cycle) was used to compare fold changes in gene expression. Statistical comparisons were performed using the Holm-Sidak t test to correct for multiple comparisons. The primers used were as follows: for Thoc1 Ex16 and Ex17, forward, 5′-AGA CTT CTT AGG CAA AGG GCC CAA-3′, reverse, 5′-CAG GAC AGA GAT TCC AAA GCC T-3′; and for Thoc1 Ex6 and Ex7, forward, 5′-CAT TCT ATT CTG CTG GCA AAA ATT AT-3′, reverse, 5′-AAA GAG TTG AAT TCT TCC ACA GAA AAC-3′.
Protein analysis.
Protein extracts from snap-frozen mouse tissue were obtained by extraction in Lysis 250 buffer and protein concentrations determined using the Bradford assay as previously described (34). Equal amounts of protein were resolved by SDS-PAGE and blotted onto nitrocellulose membranes, and membranes were blocked with 5% nonfat dry milk–1× PBST (0.1% Tween 20–1× PBS) for 1 h at room temperature before overnight incubation with primary antibodies. Primary antibodies used included Thoc1 (GeneTex Inc., Irvine, CA), CcnD1 (RM-9104-S; Neomarkers, Fremont, CA), and β-actin (CalBiochem, Billerica, MA). Following incubation with primary antibodies, membranes were washed with 1× PBST and incubated with with horseradish peroxidase-conjugated anti-mouse secondary antibodies (Amersham Pharmacia [GE], Pittsburgh, PA) and washed, and signal was developed with Pierce ECL Western blot substrate (Thermo Scientific, Rockford, IL).
Lineage-tracing analysis.
Tamoxifen was administered to five 9- to 12-week-old Rosa26CreERT2/mTmG and five Thoc1F/F::Rosa26CreERT2/mTmG mice via intraperitoneal injection at a dose of 2 mg/day for 2 consecutive days. This dose is sublethal in mice homozygous for the floxed Thoc1 allele. Small intestinal (jejunum section) and colon tissue was harvested either 24 h or 10 weeks after tamoxifen administration. Tissue was processed either for paraffin or optimal-cutting-temperature (OCT) embedding, and GFP was detected using either immunohistochemistry (IHC) or direct visualization of fluorescence. At 24 h, sporadic GFP-positive cells are detectable throughout the epithelial tissue of both the small and large intestines. At 10 weeks, stripes corresponding to contiguous GFP-positive cells are detectable that reflect the presence of GFP-labeled self-renewing stem cells and their progeny. Eight to 10 microscope images of each tissue were captured from each of 5 mice for each genotype, and the fractions of crypts containing stripes of GFP-positive cells were determined by counting.
RESULTS
Thoc1 is required for the viability of adult mice.
To characterize the postnatal requirements for Thoc1, we previously created a floxed allele of Thoc1 (Thoc1F); loxP sites flank exons 6 and 7, and Cre-mediated deletion creates a null allele (25). Thoc1F alleles were combined with a transgene expressing the Cre recombinase-estrogen receptor fusion protein (Rosa26CreERT2) to allow widespread, tamoxifen-inducible Thoc1 gene deletion (26). Untreated and thus undeleted Thoc1F/F::Rosa26CreERT2 mice were viable and did not exhibit phenotypes distinguishable from those of wild-type mice. Nine- to 12-week-old Thoc1F/F::Rosa26CreERT2 test mice or Thoc1+/+::Rosa26CreERT2 control mice were administered tamoxifen daily for 6 days to drive high levels of Cre-mediated Thoc1 gene deletion. The extent of Thoc1 gene deletion in different tissues was determined by assaying the fraction of undeleted Thoc1 transcripts remaining in bulk tissue. Thoc1 deletion was widespread in tamoxifen-treated Thoc1F/F::Rosa26CreERT2 mice, with Thoc1 transcript levels ranging from about 10% to 50% of those of control mice, depending on the tissue (Fig. 1A). Thoc1-depleted mice showed a significant decline in weight beginning 6 days after the start of tamoxifen treatment (Fig. 1B), and they died 5 to 7 days later (Fig. 1C). These data demonstrated that Thoc1 is essential for the viability of adult mice. This phenotype is dependent on the dose of tamoxifen administered, as mice treated with a lower concentration or for fewer days were viable and did not exhibit significant changes in weight. This suggests that Thoc1 must be successfully deleted in a threshold number of cells for the lethal phenotype to be realized.
Fig 1.
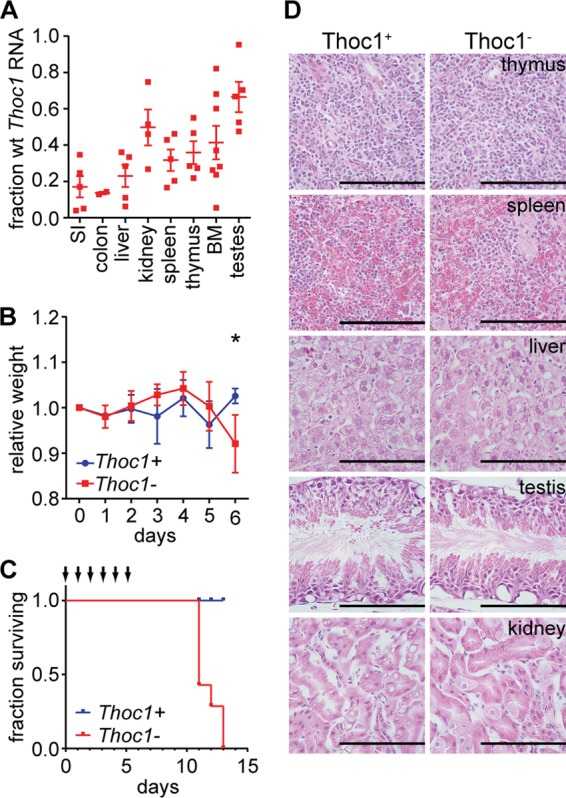
Widespread Thoc1 gene deletion compromises mouse viability but does not affect most tissues. (A) The fraction of wild-type (wt) Thoc1 transcripts remaining in the indicated tissues after tamoxifen treatment of Thoc1F/F::Rosa26CreERT2 mice (Thoc1−) in comparison to the fraction seen with tamoxifen-treated Thoc1+/+::Rosa26CreERT2 mice (Thoc1+) was assayed by real-time RT-PCR. Each data point represents analysis of tissue from an individual mouse. The error bars represent the standard errors of the means. SI, small intestine; BM, bone marrow. (B) The weights of mice with the indicated genotype were measured over the course of tamoxifen treatment relative to their weights prior to treatment. The data points represent the means and the standard errors from measurements of 4 mice. The asterisk indicates a significant difference between genotypes (t test, P < 0.01). (C) The survival of mice of the indicated genotypes is shown. Arrows represent the timing of tamoxifen treatments. The cohort monitored included 7 mice of each genotype. (D) The indicated tissues were dissected from mice of the indicated genotypes 1 day following the last tamoxifen treatment, and histology of H&E-stained tissue sections was examined. Representative images are shown. Scale bars represent 200 μm.
We euthanized a cohort of Thoc1F/F::Rosa26CreERT2 mice 6 days after the first tamoxifen treatment at the lethal dose and subjected them to necropsies to identify affected tissues. The histology of most tissues examined, including the thymus, spleen, liver, kidney, skin, and testis, was not distinguishable from that of control mice by examination of H&E-stained tissue sections (Fig. 1D). These observations suggest that tissues in a diverse collection were relatively insensitive to the effects of Thoc1 loss over the time course examined. In contrast, a marked phenotype was observed in the small intestine. Thoc1-deficient small intestines appeared thinner than the wild type, were distended due to fluid retention, and were largely devoid of solid fecal material. Thoc1-deficient small intestine histology was characterized by disruption of normal tissue architecture and considerable loss of epithelial cellularity in both crypts and villi (Fig. 2A). Surprisingly, no phenotype was detected upon Thoc1 deletion in the related mucosa of the large intestine (Fig. 2B). Nuclear Thoc1 protein was normally expressed in crypt epithelium of the small intestine as detected by immunostaining; Thoc1 immunostaining appeared weaker in villus epithelium (Fig. 2C). This Thoc1 expression pattern was confirmed by Western blot analysis of crypts and villi isolated by differential tissue dissociation. Higher Thoc1 protein levels were present in crypt compared with villus fractions, with the purest villus tip fractions lacking detectable Thoc1 protein (Fig. 2D). Nuclear Thoc1 protein immunostaining was also observed in the crypts of the large intestine (Fig. 2E). The immunostaining intensity was greater near the crypt base than in the differentiated epithelium. Thoc1 protein levels were similarly depleted by tamoxifen treatment in both the small and large intestines (Fig. 2G and H). Thus, the differential effects of Thoc1 gene deletion on the small and large intestines were not readily explained by differences in Thoc1 gene expression or by differences in the efficiency of Thoc1 gene deletion.
Fig 2.

Thoc1 gene deletion affects the histology of the small intestine but not that of the large intestine. (A) Representative H&E-stained small intestine tissue sections from tamoxifen-treated Thoc1F/F::Rosa26CreERT2 (Thoc1−) or Thoc1+/+::Rosa26CreERT2 (Thoc1+) mice are shown. Scale bars represent 200 μm. (B) Representative H&E-stained large intestine tissue sections from mice of the indicated genotypes treated as described for panel A. (C) Small intestine tissue sections from wild-type mice were immunostained for Thoc1 protein. A representative image is shown. The boxes represent the portion of the image magnified in the panels on the right to highlight staining of the crypt and villi. (D) The relative expression of Thoc1 protein in small intestinal villi (fractions V1 to V3, descending from villus tip) or crypts (fractions C1 and C2, descending to crypt bottom) isolated by differential tissue dissociation was assayed by Western blotting. Successful isolation of crypts and villi was verified by the presence of the crypt-specific marker CcnD1. Actin is the protein loading control. (E) Large intestine tissue sections from wild-type mice were immunostained for Thoc1 protein. A representative image is shown. The boxes represent the portion of the image magnified in the panels on the right to highlight staining of the crypt and differentiated epithelium. (F) The fraction of Thoc1 transcripts remaining at the indicated times from the start of tamoxifen treatment in the indicated tissues from Thoc1F/F::Rosa26CreERT2 mice relative to Thoc1+/+::Rosa26CreERT2 mice was quantitated by real-time RT-PCR. Each data point represents analysis of tissue from an individual mouse. The error bars represent the standard errors of the means. (G) Thoc1 protein levels in the small intestine of tamoxifen-treated Thoc1F/F::Rosa26CreERT2 (−) or Thoc1+/+::Rosa26CreERT2 (+) mice were assayed at the indicated times from the start of tamoxifen treatment by Western blotting. Actin is the protein loading control. (H) Thoc1 protein levels in the large intestine of tamoxifen-treated Thoc1F/F::Rosa26CreERT2 (−) or Thoc1+/+::Rosa26CreERT2 (+) mice were assayed as described for panel G.
We examined tissue histology at different time points to characterize how the small intestine phenotype develops. The first abnormalities in tissue architecture were observed as early as day 3 of tamoxifen treatment, coincident with the nadir in Thoc1 protein levels (Fig. 3A). Villus length was significantly shorter in Thoc1-deficient small intestine at day 3 and declined further with time (Fig. 3B). Crypt cells with morphology reminiscent of apoptotic cells were also apparent by day 3 (Fig. 3A). Immunostaining for activated caspase-3, a marker for apoptosis, confirmed the appearance of apoptotic cells in the crypts of Thoc1-deficient small intestine (Fig. 3C). The number of apoptotic cells per crypt increased with time (Fig. 3D). No activated caspase-3-immunopositive cells were observed in Thoc1-deficient villi or in Thoc1-deficient large intestine. We also examined proliferation by immunostaining with the proliferation marker Ki67. In control small intestine, nuclear Ki67 immunostaining was observed in crypt cells but not in differentiated cells of the villus. A noticeable decline in Ki67 immunostaining was observed by day 3 of tamoxifen treatment in Thoc1-deficient crypts (Fig. 3E). The decline in Ki67 immunostaining among stem cells at the crypt base preceded the decline in Ki67 among progenitor cells within the transit-amplifying region by about a day (Fig. 3F and G). Thoc1 loss did not affect Ki67 immunostaining in the large intestine (Fig. 3H). These observations suggested that the Thoc1 deficiency directly affects the growth and viability of stem and progenitor cells within the small intestinal crypt. Since the rapid turnover of the differentiated villus epithelium is dependent on a continuous supply of new cells, the degeneration of the small intestine and, ultimately, the death of Thoc1-deficient mice were probably caused by this stem/progenitor cell defect.
Fig 3.

Thoc1 loss compromises cell proliferation and viability in the small intestinal crypt. (A) The histology of the small intestine in Thoc1F/F::Rosa26CreERT2 (Thoc1−) or Thoc1+/+::Rosa26CreERT2 (Thoc1+) mice was characterized at the indicated times from the start of tamoxifen treatment. Representative images are shown. The lower panels magnify the indicated crypts from the upper panels. Arrows highlight cells with apoptotic morphology. Scale bars represent 200 μm. (B) The length of the small intestinal villus was measured at the indicated times from the start of tamoxifen treatment in mice of the indicated genotypes. The data points represent the means and standard errors of the results deteermined for approximately 300 villi from 3 mice for each genotype. Asterisks indicate statistically significant differences between genotypes (t test, P < 0.01). (C) The tissue sections described for panel A were immunostained for the activated form of caspase-3. Representative images are shown. The arrow highlights immunopositive cells. Scale bars represent 200 μm. (D) The number of activated caspase-3 immunopositive cells per small intestinal crypt from tamoxifen-treated Thoc1F/F::Rosa26CreERT2 mice was determined by counting. Each data point represents the number determined from a different mouse. Error bars are the standard errors of the means. No immunopositive cells were detected in control mice. (E) The tissue sections described for panel A were immunostained for the proliferation marker Ki67. Representative images are shown. The arrow highlights an immunopositive cell from the crypt base. (F) The number of Ki67-immunopositive cells per small intestinal crypt base (at or below the +4 position) was determined by counting cells at the indicated time from the start of tamoxifen treatment from Thoc1F/F::Rosa26CreERT2 or Thoc1+/+::Rosa26CreERT2 mice. Each data point represents the number determined from a different mouse. Error bars represent the standard errors of the means. Asterisks indicate statistically significant differences between genotypes (t test, P < 0.01). (G) The Ki67-immunopositive cells in the transit-amplifying region of the small intestinal crypt (above the +4 position) were counted as described for panel B. (H) The Ki67-immunopositive cells were counted in large intestinal crypts as described for panel F.
Thoc1 is required for normal stem cell homeostasis in the small intestine.
We have performed lineage tracing to test whether there are differential requirements for Thoc1 in small and large intestinal stem cells. Cells undergoing Cre-mediated recombination are marked irreversibly by green fluorescent protein (GFP), while unrecombined cells are marked by red fluorescent protein (RFP) using the Rosa26mT/mG Cre reporter allele (28). Thoc1F/F::Rosa26CreERT2/mTmG and Thoc1+/+::Rosa26CreERT2/mTmG mice were treated with a sublethal dose of tamoxifen sufficient to GFP mark sporadic cells throughout the intestinal epithelium (Fig. 4A and B). The numbers of marked cells per crypt were similar in tissues of the two genotypes at 24 h after tamoxifen treatment, indicating that the efficiencies of GFP marking were equivalent (Fig. 4C). Given that the entire population of mouse intestinal epithelium cells turns over within days, the only GFP-marked cells remaining 10 weeks later were self-renewing stem cells and their progeny. The lineage-generating potential of GFP-marked small intestinal stem cells, defined as the percentage of crypt/villus units with stripes of GFP-marked cells, was significantly reduced upon Thoc1 deletion (Fig. 4A and D). In contrast, the lineage-generating potential of large intestinal stem cells was not significantly affected by Thoc1 deletion (Fig. 4B and D). As Cre-mediated GFP marking and Thoc1 deletion are independent events, not every GFP-marked cell would lack Thoc1. We have examined Thoc1 expression in GFP-marked cells to assess this. GFP-positive small intestinal cells remaining 6 weeks after tamoxifen treatment of Thoc1F/F::Rosa26CreERT2/mTmG mice expressed wild-type Thoc1 RNA at levels comparable to those seen with unrecombined RFP cells (Fig. 4E). This indicates that most of the GFP-marked cells had escaped Cre-mediated Thoc1 deletion. In contrast, GFP-marked large intestinal cells express very low levels of Thoc1 RNA relative to unrecombined RFP-marked cells. A significant fraction of large intestinal stem cells, therefore, retained self-renewing and lineage-generating potential in the absence of Thoc1. In sum, these data suggest that small intestinal stem cells, but not large intestinal stem cells, require Thoc1 to maintain self-renewal and lineage-generating potential.
Fig 4.
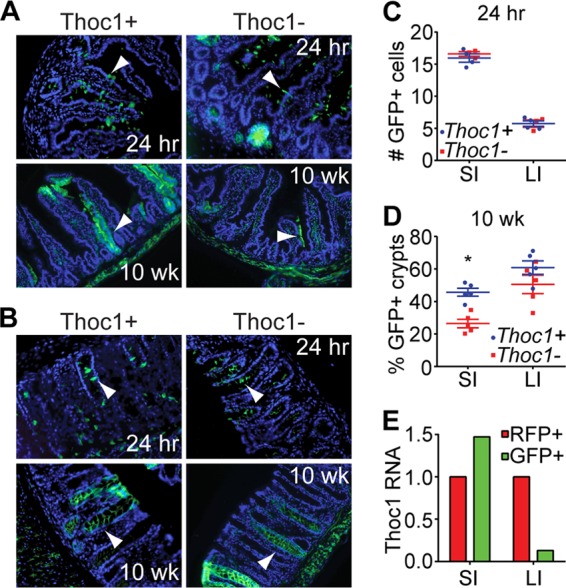
The number of stem cells with long-term lineage-generating capacity is reduced in Thoc1-deficient small intestine. (A) Thoc1F/F::Rosa26CreERT2/mTmG (Thoc1−) or Thoc1+/+::Rosa26CreERT2/mTmG (Thoc1+) mice were treated with tamoxifen and small intestine tissue sections examined for the presence of GFP-marked cells at the indicated times posttreatment by fluorescence microscopy. Representative images are shown. Arrowheads indicate sporadic GFP-positive cells at 24 h or ribbons of GFP-positive cells at 10 weeks (wk). (B) Large intestine tissue sections from tamoxifen-treated mice were examined as described for panel A. (C) The numbers of GFP-positive cells per crypt-villus unit of the small intestine (SI) or per crypt of the large intestine (LI) were determined by counting cells at 24 h after tamoxifen treatment in Thoc1F/F::Rosa26CreERT2/mTmG (Thoc1−) or Thoc1+/+::Rosa26CreERT2/mTmG (Thoc1+) mice. Each data point represents the mean number determined from 25 crypts from a single mouse. The bars represent the means and standard errors for all mice of the given genotype. (D) The percentages of crypts containing ribbons of GFP-positive cells at 10 weeks after tamoxifen treatment were determined by counting as described for panel C. The asterisk marks a statistically significant difference between genotypes (t test < 0.01). (E) Small intestine (SI) or large intestine (LI) cells remaining 6 weeks after tamoxifen treatment were sorted for GFP (Cre recombined) or RFP (not Cre recombined) and wild-type Thoc1 RNAs measured by real-time RT-PCR. The data represent means of the results of triplicate experiments determined for a Thoc1F/F::Rosa26CreERT2/mTmG mouse versus a Thoc1+/+::Rosa26CreERT2/mTmG mouse. Data are normalized to the RFP sample.
Terminally differentiated Paneth cells support and mark the small intestinal stem cell niche, as they are interspersed among stem cells at the crypt base (35). We stained Paneth cells over the time course of tamoxifen administration to examine the structure of the stem cell niche. In control crypts, Paneth cells resided exclusively at the crypt base, as expected (Fig. 5A). As early as day 3 of tamoxifen administration, cells immunostaining for the Paneth cell marker lysozyme were present above the +4 cell position from the crypt base that normally marks the boundary of the stem cell niche. By day 6 of tamoxifen administration, immunopositive cells were found in the transit-amplifying region well above the +4 position, attesting to the structural breakdown of the stem cell niche.
Fig 5.

Thoc1 loss disrupts homeostasis of the stem cell niche in the small intestine. (A) Small intestine tissue sections from Thoc1F/F::Rosa26CreERT2 (Thoc1−) or Thoc1+/+::Rosa26CreERT2 (Thoc1+) mice were characterized at the indicated times from the start of tamoxifen treatment by immunostaining for the Paneth cell marker lysozyme. Representative images are shown. Scale bars represent 200 μm. (B) The expression of the indicated stem cell markers in small intestine crypt epithelium from Thoc1F/F::Rosa26CreERT2 (red) or Thoc1+/+::Rosa26CreERT2 (blue) mice was quantitated at the indicated times from the start of tamoxifen treatment by real-time RT-PCR. Each data point is from an individual mouse normalized to a single control mouse at each time point. The bars represent the means and standard errors. Asterisks indicate statistically significant differences between genotypes (t test, P ≤ 0.01). (C) Large intestine tissue was examined for the expression of stem cell marker genes as described for panel B. (D) The expression of the indicated Wnt signaling regulatory genes in small intestine crypt epithelium tissue was assayed at 2 days from the start of tamoxifen treatment as described for panel B. (E) The expression of Wnt regulatory genes was assayed in small intestine crypt epithelium at 5 days from the start of tamoxifen treatment as described for panel B.
The expression of stem cell marker genes was also examined in isolated small intestinal crypts. By day 3 from the start of tamoxifen administration, the expression of Lgr5, Lrig1, and Tert in Thoc1-deficient crypts increased to levels significantly higher than those seen with control crypts (Fig. 5B). The increased expression was unlikely to have been due to the presence of more stem cells given the decline in crypt cell proliferation observed by Ki67 immunostaining (Fig. 3F and G). The relative expression of these genes then declined to very low levels by day 6 of tamoxifen treatment in Thoc1-deficient crypts, probably reflecting the loss of cells observed at this time. However, the expression of Bmi1 was not significantly affected by Thoc1 loss at any time point examined, suggesting that Bmi1 marks a cell subpopulation distinct from that marked by the other genes examined. Consistent with the lack of a detectable phenotype in Thoc1-deficient large intestine cells, Thoc1 loss did not affect the expression of these stem cell marker genes (Fig. 5C).
Wnt signaling is a major regulator of homeostatic self-renewal within the intestinal crypt (36). Lgr5 and Tert are regulated directly by Wnt signaling (27, 37), while Lrig1 is a transcriptional target of Myc, itself directly regulated by Wnt. Alteration in the expression of these genes in Thoc1-deficient small intestinal crypts may reflect disruption of normal Wnt signaling. The expression of four genes that regulate Wnt signaling has been examined to explore this possibility. Axin2 expression is significantly reduced at 2 days from the start of tamoxifen administration in Thoc1-deficient crypts, while the expression levels of Wif1, Nkd1, and Apcdd1 were not significantly different (Fig. 5D). The expression of all of these genes was reduced at day 5 from the start of tamoxifen treatment, relative to control tissue, consistent with the loss of cellularity at this time point (Fig. 5E). Axin2 encodes an inhibitor of Wnt signaling that contributes to feedback regulation. Reduced Axin2 expression on day 2 could conceivably have contributed to increased expression of the Wnt-regulated genes Lgr5 and Tert observed on day 3 in Thoc1-deficient small intestinal crypts.
The Trp53 stress response pathway is integrated into feedback regulation of Wnt signaling. The Trp53 pathway is activated by Wnt signaling via β-catenin-mediated downregulation of the Trp53 inhibitor Mdm2 (38). In turn, Trp53 utilizes several mechanisms to attenuate Wnt signaling activity (39). The evidence of deregulated Wnt signaling noted above, as well as decreased cell proliferation and increased apoptosis, suggests that a Trp53 stress response may be activated in Thoc1-deficient small intestinal crypts. Consistent with this hypothesis, the RNA levels for Trp53 target genes Cdkn1a, Mdm2, and Bax were increased in Thoc1-deficient small intestinal crypts (Fig. 6A). Western blot analysis indicated that Trp53 and Cdkn1a protein levels were initially higher in Thoc1-deficient small intestinal crypts (Fig. 6B). At later times, a lower-molecular-weight isoform of Trp53 protein accumulated in favor of full-length protein. The size of this protein is consistent with the Δ40p53 isoform that is known to accumulate in response to some forms of cellular stress (40). Δ40p53 suppresses the expression of Trp53 target genes such as Cdkn1a (41, 42), and the Cdkn1a protein level was suppressed in Thoc1-deficient small intestinal crypt epithelium upon accumulation of this lower-molecular-weight Trp53 isoform. Cdkn1a, Mdm2, and Bax RNA levels were also higher in Thoc1-deficient large intestine cells, although the increases occurred at later times after the start of tamoxifen treatment (Fig. 6C).
Fig 6.
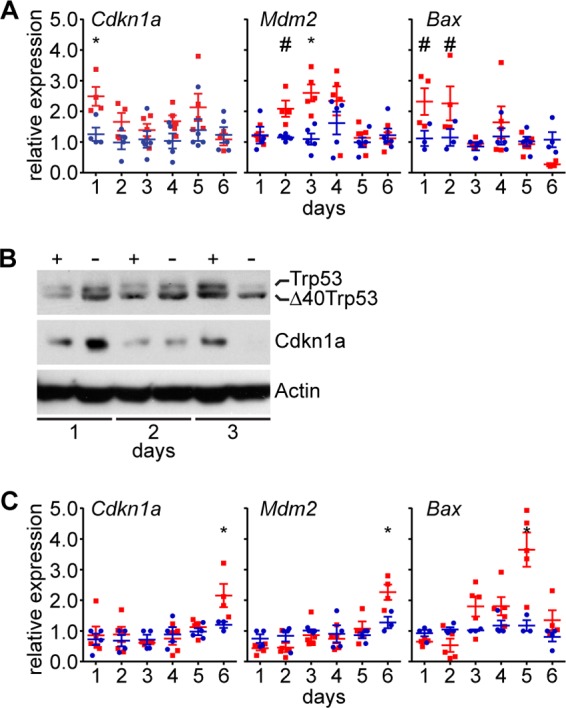
Thoc1 deficiency activates a Trp53 response in intestinal tissue. (A) Thoc1F/F::Rosa26CreERT2 (red) or Thoc1+/+::Rosa26CreERT2 (blue) small intestinal crypt epithelium tissue was assayed at the indicated times from the start of tamoxifen for RNA expression from the indicated Trp53 target genes by real-time RT-PCR. Each data point is from an individual mouse normalized to a single control mouse at each time point. The bars represent the means and standard errors. Asterisks (t test, P ≤ 0.01) and number signs (t test, P ≤ 0.05) indicate statistically significant differences between genotypes. (B) Thoc1F/F::Rosa26CreERT2 (−) or Thoc1+/+::Rosa26CreERT2 (+) small intestinal crypt epithelium tissue was assayed for the indicated proteins at the indicated times from the start of tamoxifen treatment by Western blot analysis. Actin is the protein loading control. (C) Large intestine tissue was assayed for RNA levels expressed from the indicated genes as described for panel A.
Given the observed effects of Thoc1 deficiency on the small intestinal stem cell niche, Thoc1 was deleted specifically in Lgr5-expressing stem cells using the Lgr5-enhanced GFP (EGFP)-internal ribosome entry site (IRES)-CreERT2 knock-in allele (27). Mice containing this allele and homozygous for the floxed Thoc1 allele did not exhibit a detectable small intestine phenotype even at the highest doses of tamoxifen used. The lack of phenotype was likely due to the previously noted variegated expression of CreERT2 from this allele which results in reduced efficiency of gene deletion. Consistent with the lack of phenotype in Thoc1F/F::Rosa26CreERT2 mice treated with lower doses of tamoxifen, the small intestine could tolerate loss of Thoc1 in a subthreshold fraction of Lgr5-positive stem cells.
Mammary gland expansion during pregnancy and lactation is not detectably affected by pThoc1 depletion.
Small intestinal stem cells are some of the most rapidly proliferating tissue stem cells known, and Thoc1 loss compromises their proliferation and viability. It is possible, therefore, that Thoc1 is required generally for rapidly proliferating cells. The lack of phenotype in the large intestine, whose rate of turnover and proliferation is also quite high, argues against this hypothesis. To test this hypothesis in another rapidly proliferating tissue with distinct developmental origin, Thoc1 has been deleted in the rapidly proliferating mammary epithelium during pregnancy and lactation. The MMTV-Cre transgene is bred into mice homozygous for the floxed Thoc1 allele to drive widespread Cre expression throughout the mammary epithelium, including stem and progenitor cells (43). Mammary glands were dissected from female mice at different stages of pregnancy and whole mounts or tissue sections examined. Mammary gland epithelium from Thoc1F/F::MMTV-Cre mice appearsed similar to that from Thoc1+/+::MMTV-Cre mice (Fig. 7A and B), despite efficient Thoc1 protein depletion (Fig. 7C). Female Thoc1F/F::MMTV-Cre were able to successfully nurse normal-size litters through multiple pregnancies, indicating that mammary gland function was not significantly impaired. While subtle phenotypes in the mammary gland may go undetected, these results indicate that highly proliferative mammary gland epithelium is relatively insensitive to Thoc1 deletion.
Fig 7.
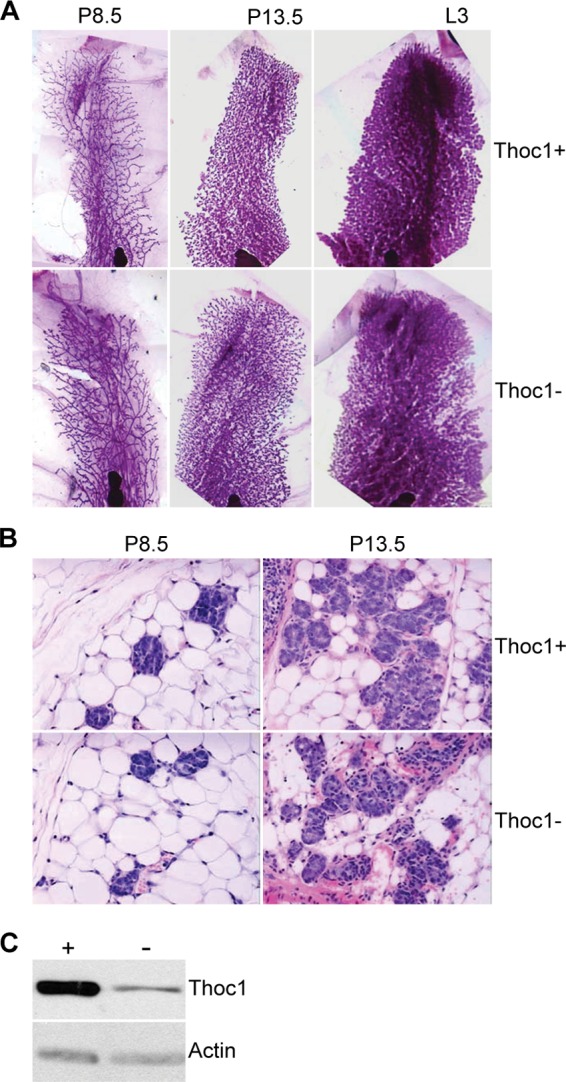
Thoc1 deficiency does not have detectable effects on the lactating mammary gland. (A) Mammary glands from Thoc1F/F::MMTV-Cre (Thoc1−) or Thoc1+/+::MMTV-Cre (Thoc1+) mice at 8.5 (P8.5) and 13.5 (P13.5) days of pregnancy or at 3 days of lactation (L3) were isolated, processed for whole mount, and stained with carmine. Representative images are shown. (B) Mammary glands from the experiment described for panel A were sectioned and H&E stained. Representative images are shown. (C) Thoc1 protein levels in mammary epithelial cells isolated from MMTV-Cre:Thoc1F/F (−) or MMTV-Cre:Thoc1+/+ (+) mice were analyzed by Western blotting. Actin is the protein loading control.
DISCUSSION
Thoc1 encodes an essential protein component of the evolutionarily conserved THO RNP complex that physically couples nascent transcripts with the nuclear export apparatus. Thoc1 null mice exhibit an early block to embryonic development (24), so the contribution of Thoc1 and THO to normal tissue homeostasis in the adult mouse is not well characterized. Here we have engineered a mouse strain facilitating widespread, inducible, Cre-mediated Thoc1 gene deletion in order to characterize these contributions. Thoc1 deletion causes acute defects in the small intestine that ultimately compromise tissue function and mouse viability. This phenotype is dependent on highly efficient Thoc1 deletion, indicating that the tissue can tolerate Thoc1 loss in some fraction of cells. Phenotypes are not observed in other epithelial tissues where Thoc1 is efficiently deleted, including the large intestine. It is possible that phenotypes in these tissues develop over times that exceed the length of the current experiments. However, the acute effects of Thoc1 loss on cell proliferation and apoptosis are clearly different in the small and large intestine. Further, the lineage-tracing experiments indicate that Thoc1 loss has little effect on large intestine stem cells over a period of 10 weeks, enough time for the population of cells in this tissue to completely turn over multiple times. These observations indicate that Thoc1 is required for homeostasis in some tissues of adult mice, and the effects of Thoc1 deficiency are context dependent.
The biological basis for this context dependency is unknown; we propose three, non-mutually exclusive explanations that may account for it. One, Thoc1 may be preferentially required in highly proliferative cells. Highly proliferative cells require increased transcriptional output to support biosynthesis, and Thoc1 deficiency may limit transcriptional output generally, as suggested by studies in yeast (17). However, this model does not account for the relative insensitivity of other rapidly regenerating tissues such as the large intestine or the mammary gland to Thoc1 loss. Two, different tissues may respond differently to stress induced by Thoc1 deficiency. For example, the mouse large intestine is more resistant to radiation-induced stress than the small intestine due, in part, to differences in the expression of apoptotic regulators (44, 45). That both small and large intestines deficient for Thoc1 trigger a Trp53 response but only the small intestine exhibits effects on cell proliferation and viability is consistent with this hypothesis. Three, Thoc1 may regulate different subsets of transcripts in different tissues. The observation the Thoc1 loss affects Wnt signaling target and regulatory genes in the small intestine but not in the large intestine is consistent with this possibility. Additional experiments that identify Thoc1-regulated transcripts and characterize the stress responses induced by Thoc1 deficiency will be required to rigorously distinguish between these hypotheses.
Several pieces of evidence support the conclusion that Thoc1 is required for stem cell function in the small intestine. Expression of Thoc1 is higher in the intestinal crypts where the stem cell niche resides than in the villi composed of differentiated epithelium. Thoc1 loss coincides with decreased cell proliferation and viability in the crypt and crypt base, whereas villus epithelium is initially unaffected. The number of stem cells capable of generating lineages of differentiated daughter cells is significantly reduced by Thoc1 loss. This defect eventually affects the replenishment of the differentiated villus epithelium, severely compromising villus length and function, as indicated by absorption defects. Changes in the levels of transcripts that mark stem cell populations indicate that stem cell homeostasis is disrupted by Thoc1 loss. Lgr5, Tert, and Lrig1 transcript levels initially increase after Thoc1 deletion but then decline to barely detectable levels. Bmi1 transcript levels, on the other hand, are not significantly affected by Thoc1 loss, suggesting that they mark subpopulations of cells distinct from those marked by Lgr5, Tert, and Lrig1. Bmi1 has been proposed to mark label-retaining cells in the intestinal crypt that function as Paneth cell precursors and also have stem cell properties under some conditions (46–48). Lgr5, Tert, and Lrig1 are directly or indirectly regulated by Wnt signaling, suggesting that Wnt signaling may be disrupted by Thoc1 deficiency. Indeed, the expression of the negative Wnt signaling regulator Axin2 declines after Thoc1 loss, potentially contributing to the transient increase in expression of Wnt target genes such as Lgr5 and Tert. Additional experiments will be required to determine whether different intestinal stem and progenitor cell populations have different requirements for Thoc1 and whether Thoc1 regulates different subsets of transcripts in these cells.
Cotranscriptional RNP biogenesis is important for RNA processing and transport. Indeed, the fate of mRNA in the cytoplasm appears to be predetermined by RNP complex formation in the nucleus, indicating that the different stages of a transcript's life cycle are physically coupled to one another (21). It is increasingly appreciated that RNP-mediated mechanisms contribute an additional layer of regulation for the specification of the coordinated gene expression program (3). Consistent with this hypothesis, the data presented here indicate that Thoc1 deficiency causes context-dependent effects on adult tissue homeostasis in the mouse. In particular, the THO complex makes critical contributions to stem cell homeostasis in the small intestine but not in other tissues such as the large intestine. Context-dependent effects have also been observed upon deletion of other RNP-encoding genes such as Elav1 (49). Interestingly, the tissues affected by Elav1 deficiency differed from those reported here despite use of the same Rosa26CreERT2 gene deletion system. For example, the small intestine, the large intestine, and the spleen were all affected by Elav1 deletion. Similar observations have been made in humans, where mutations in RNP-encoding genes can cause context-dependent developmental defects (4). These findings validate the importance of co- and posttranscriptional RNP-mediated mechanisms in specifying coordinated gene expression during normal growth and development.
ACKNOWLEDGMENTS
This work was supported by Public Health Service grant R01 CA125665 from the National Cancer Institute (D.W.G.) and by University at Buffalo Mark Diamond Fund grant SU-11-14 (L.P.). Core facilities utilized in this work were supported by NCI grant P30 CA016056.
Footnotes
Published ahead of print 1 July 2013
REFERENCES
- 1.Tutucci E, Stutz F. 2011. Keeping mRNPs in check during assembly and nuclear export. Nat. Rev. Mol. Cell Biol. 12:377–384 [DOI] [PubMed] [Google Scholar]
- 2.Glisovic T, Bachorik JL, Yong J, Dreyfuss G. 2008. RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett. 582:1977–1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Keene JD. 2007. RNA regulons: coordination of post-transcriptional events. Nat. Rev. Genet. 8:533–543 [DOI] [PubMed] [Google Scholar]
- 4.Cooper TA, Wan L, Dreyfuss G. 2009. RNA and disease. Cell 136:777–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luna R, Rondon AG, Aguilera A. 2012. New clues to understand the role of THO and other functionally related factors in mRNP biogenesis. Biochim. Biophys. Acta 1819:514–520 [DOI] [PubMed] [Google Scholar]
- 6.Chávez S, Beilharz T, Rondon AG, Erdjument-Bromage H, Tempst P, Svejstrup JQ, Lithgow T, Aguilera A. 2000. A protein complex containing Tho2, Hpr1, Mft1 and a novel protein, Thp2, connects transcription elongation with mitotic recombination in Saccharomyces cerevisiae. EMBO J. 19:5824–5834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rehwinkel J, Herold A, Gari K, Kocher T, Rode M, Ciccarelli FL, Wilm M, Izaurralde E. 2004. Genome-wide analysis of mRNAs regulated by the THO complex in Drosophila melanogaster. Nat. Struct. Mol. Biol. 11:558–566 [DOI] [PubMed] [Google Scholar]
- 8.Strässer K, Masuda S, Mason P, Pfannstiel J, Oppizzi M, Rodriguez-Navarro S, Rondon AG, Aguilera A, Struhl K, Reed R, Hurt E. 2002. TREX is a conserved complex coupling transcription with messenger RNA export. Nature 417:304–308 [DOI] [PubMed] [Google Scholar]
- 9.Furumizu C, Tsukaya H, Komeda Y. 2010. Characterization of EMU, the Arabidopsis homolog of the yeast THO complex member HPR1. RNA 16:1809–1817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Viphakone N, Hautbergue GM, Walsh M, Chang CT, Holland A, Folco EG, Reed R, Wilson SA. 2012. TREX exposes the RNA-binding domain of Nxf1 to enable mRNA export. Nat. Commun. 3:1006. 10.1038/ncomms2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gwizdek C, Iglesias N, Rodriguez MS, Ossareh-Nazari B, Hobeika M, Divita G, Stutz F, Dargemont C. 2006. Ubiquitin-associated domain of Mex67 synchronizes recruitment of the mRNA export machinery with transcription. Proc. Natl. Acad. Sci. U. S. A. 103:16376–16381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huertas P, Aguilera A. 2003. Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Mol. Cell 12:711–721 [DOI] [PubMed] [Google Scholar]
- 13.Li Y, Wang X, Zhang X, Goodrich DW. 2005. Human hHpr1/p84/Thoc1 regulates transcriptional elongation and physically links RNA polymerase II and RNA processing factors. Mol. Cell. Biol. 25:4023–4033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Domínguez-Sánchez MS, Barroso S, Gomez-Gonzalez B, Luna R, Aguilera A. 2011. Genome instability and transcription elongation impairment in human cells depleted of THO/TREX. PLoS Genet. 7:e1002386. 10.1371/journal.pgen.1002386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saguez C, Schmid M, Olesen JR, Ghazy MA, Qu X, Poulsen MB, Nasser T, Moore C, Jensen TH. 2008. Nuclear mRNA surveillance in THO/sub2 mutants is triggered by inefficient polyadenylation. Mol. Cell 31:91–103 [DOI] [PubMed] [Google Scholar]
- 16.Schneiter R, Guerra CE, Lampl M, Gogg G, Kohlwein SD, Klein HL. 1999. The Saccharomyces cerevisiae hyperrecombination mutant hpr1Delta is synthetically lethal with two conditional alleles of the acetyl coenzyme A carboxylase gene and causes a defect in nuclear export of polyadenylated RNA. Mol. Cell. Biol. 19:3415–3422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gómez-González B, Garcia-Rubio M, Bermejo R, Gaillard H, Shirahige K, Marin A, Foiani M, Aguilera A. 2011. Genome-wide function of THO/TREX in active genes prevents R-loop-dependent replication obstacles. EMBO J. 30:3106–3119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aguilera A, Klein HL. 1990. HPR1, a novel yeast gene that prevents intrachromosomal excision recombination, shows carboxy-terminal homology to the Saccharomyces cerevisiae TOP1 gene. Mol. Cell. Biol. 10:1439–1451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang X, Chinnam M, Wang J, Wang Y, Zhang X, Marcon E, Moens P, Goodrich DW. 2009. Thoc1 deficiency compromises gene expression necessary for normal testis development in the mouse. Mol. Cell. Biol. 29:2794–2803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mancini A, Niemann-Seyde SC, Pankow R, El Bounkari O, Klebba-Farber S, Koch A, Jaworska E, Spooncer E, Gruber AD, Whetton AD, Tamura T. 2010. THOC5/FMIP, an mRNA export TREX complex protein, is essential for hematopoietic primitive cell survival in vivo. BMC Biol. 8:1. 10.1186/1741-7007-8-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Müller-McNicoll M, Neugebauer KM. 2013. How cells get the message: dynamic assembly and function of mRNA-protein complexes. Nat. Rev. Genet. 14:275–287 [DOI] [PubMed] [Google Scholar]
- 22.Masuda S, Das R, Cheng H, Hurt E, Dorman N, Reed R. 2005. Recruitment of the human TREX complex to mRNA during splicing. Genes Dev. 19:1512–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chi B, Wang Q, Wu G, Tan M, Wang L, Shi M, Chang X, Cheng H. 2013. Aly and THO are required for assembly of the human TREX complex and association of TREX components with the spliced mRNA. Nucleic Acids Res. 41:1294–1306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang X, Chang Y, Li Y, Zhang X, Goodrich DW. 2006. Thoc1/Hpr1/p84 is essential for early embryonic development in the mouse. Mol. Cell. Biol. 26:4362–4367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang X, Li Y, Zhang X, Goodrich DW. 2007. An allelic series for studying the mouse Thoc1 gene. Genesis 45:32–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, Newman J, Reczek EE, Weissleder R, Jacks T. 2007. Restoration of p53 function leads to tumour regression in vivo. Nature 445:661–665 [DOI] [PubMed] [Google Scholar]
- 27.Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, Clevers H. 2007. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449:1003–1007 [DOI] [PubMed] [Google Scholar]
- 28.Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. 2007. A global double-fluorescent Cre reporter mouse. Genesis 45:593–605 [DOI] [PubMed] [Google Scholar]
- 29.Wagner KU, Wall RJ, St-Onge L, Gruss P, Wynshaw-Boris A, Garrett L, Li M, Furth PA, Hennighausen L. 1997. Cre-mediated gene deletion in the mammary gland. Nucleic Acids Res. 25:4323–4330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moolenbeek C, Ruitenberg EJ. 1981. The “Swiss roll”: a simple technique for histological studies of the rodent intestine. Lab. Anim. 15:57–59 [DOI] [PubMed] [Google Scholar]
- 31.Wagner KU, Young WS, III, Liu X, Ginns EI, Li M, Furth PA, Hennighausen L. 1997. Oxytocin and milk removal are required for post-partum mammary-gland development. Genes Funct. 1:233–244 [DOI] [PubMed] [Google Scholar]
- 32.Flint N, Cove FL, Evans GS. 1991. A low-temperature method for the isolation of small-intestinal epithelium along the crypt-villus axis. Biochem. J. 280(Pt 2):331–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ip MM, Masso-Welch PA, Shoemaker SF, Shea-Eaton WK, Ip C. 1999. Conjugated linoleic acid inhibits proliferation and induces apoptosis of normal rat mammary epithelial cells in primary culture. Exp. Cell Res. 250:22–34 [DOI] [PubMed] [Google Scholar]
- 34.Li Y, Lin AW, Zhang X, Wang Y, Wang X, Goodrich DW. 2007. Cancer cells and normal cells differ in their requirements for Thoc1. Cancer Res. 67:6657–6664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sato T, van Es JH, Snippert HJ, Stange DE, Vries RG, van den Born M, Barker N, Shroyer NF, van de Wetering M, Clevers H. 2011. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 469:415–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schepers A, Clevers H. 2012. Wnt signaling, stem cells, and cancer of the gastrointestinal tract. Cold Spring Harb. Perspect. Biol. 4:a007989. 10.1101/cshperspect.a007989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoffmeyer K, Raggioli A, Rudloff S, Anton R, Hierholzer A, Del Valle I, Hein K, Vogt R, Kemler R. 2012. Wnt/beta-catenin signaling regulates telomerase in stem cells and cancer cells. Science 336:1549–1554 [DOI] [PubMed] [Google Scholar]
- 38.Damalas A, Kahan S, Shtutman M, Ben-Ze'ev A, Oren M. 2001. Deregulated beta-catenin induces a p53- and ARF-dependent growth arrest and cooperates with Ras in transformation. EMBO J. 20:4912–4922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim NH, Kim HS, Kim NG, Lee I, Choi HS, Li XY, Kang SE, Cha SY, Ryu JK, Na JM, Park C, Kim K, Lee S, Gumbiner BM, Yook JI, Weiss SJ. 2011. p53 and microRNA-34 are suppressors of canonical Wnt signaling. Sci. Signal. 4:ra71. 10.1126/scisignal.2001744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bourougaa K, Naski N, Boularan C, Mlynarczyk C, Candeias MM, Marullo S, Fahraeus R. 2010. Endoplasmic reticulum stress induces G2 cell-cycle arrest via mRNA translation of the p53 isoform p53/47. Mol. Cell 38:78–88 [DOI] [PubMed] [Google Scholar]
- 41.Yin Y, Stephen CW, Luciani MG, Fahraeus R. 2002. p53 stability and activity is regulated by Mdm2-mediated induction of alternative p53 translation products. Nat. Cell Biol. 4:462–467 [DOI] [PubMed] [Google Scholar]
- 42.Courtois S, Verhaegh G, North S, Luciani MG, Lassus P, Hibner U, Oren M, Hainaut P. 2002. DeltaN-p53, a natural isoform of p53 lacking the first transactivation domain, counteracts growth suppression by wild-type p53. Oncogene 21:6722–6728 [DOI] [PubMed] [Google Scholar]
- 43.Yamaji D, Na R, Feuermann Y, Pechhold S, Chen W, Robinson GW, Hennighausen L. 2009. Development of mammary luminal progenitor cells is controlled by the transcription factor STAT5A. Genes Dev. 23:2382–2387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pritchard DM, Potten CS, Korsmeyer SJ, Roberts S, Hickman JA. 1999. Damage-induced apoptosis in intestinal epithelia from bcl-2-null and bax-null mice: investigations of the mechanistic determinants of epithelial apoptosis in vivo. Oncogene 18:7287–7293 [DOI] [PubMed] [Google Scholar]
- 45.Merritt AJ, Potten CS, Watson AJ, Loh DY, Nakayama K, Hickman JA. 1995. Differential expression of bcl-2 in intestinal epithelia. Correlation with attenuation of apoptosis in colonic crypts and the incidence of colonic neoplasia. J. Cell Sci. 108(Pt 6):2261–2271 [DOI] [PubMed] [Google Scholar]
- 46.Buczacki SJ, Zecchini HI, Nicholson AM, Russell R, Vermeulen L, Kemp R, Winton DJ. 2013. Intestinal label-retaining cells are secretory precursors expressing Lgr5. Nature 495:65–69 [DOI] [PubMed] [Google Scholar]
- 47.Tian H, Biehs B, Warming S, Leong KG, Rangell L, Klein OD, de Sauvage FJ. 2011. A reserve stem cell population in small intestine renders Lgr5-positive cells dispensable. Nature 478:255–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yan KS, Chia LA, Li X, Ootani A, Su J, Lee JY, Su N, Luo Y, Heilshorn SC, Amieva MR, Sangiorgi E, Capecchi MR, Kuo CJ. 2012. The intestinal stem cell markers Bmi1 and Lgr5 identify two functionally distinct populations. Proc. Natl. Acad. Sci. U. S. A. 109:466–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ghosh M, Aguila HL, Michaud J, Ai Y, Wu MT, Hemmes A, Ristimaki A, Guo C, Furneaux H, Hla T. 2009. Essential role of the RNA-binding protein HuR in progenitor cell survival in mice. J. Clin. Invest. 119:3530–3543 [DOI] [PMC free article] [PubMed] [Google Scholar]